

Abstract
Objectives
To determine the antiviral activity of phosphorodiamidate morpholino oligomers (PMO) and peptide-conjugated PMO (PPMO) in AG129 mice infected with dengue 2 virus (DENV-2).
Methods
Antisense PMO and PPMO were designed against the 5′ terminal region (5′SL) or the 3′-cyclization sequence region (3′CS) of DENV genomic RNA and administered to AG129 mice before and/or after infection with DENV-2. In addition, cell culture evaluations designed to determine optimum PPMO length, and pharmacokinetic and toxicity analysis of PPMO were also carried out.
Results
Mock-treated AG129 mice lived for 9–17 days following intraperitoneal (ip) infection with 104–106pfu of DENV-2 (strain New Guinea C). Intraperitoneal administration of 5′SL or 3′CS PPMO before and after DENV infection produced an increase in the average survival time of up to 8 days. Animals receiving only post-infection PPMO treatment did not benefit significantly. Cell culture studies showed that PPMO of 22–24 bases long produced substantially higher DENV titre reductions than did PPMO that were either shorter or longer. Pharmacokinetic and toxicology analysis with non-infected animals showed that nine consecutive once‐daily ip treatments of 10 mg/kg PPMO resulted in high concentrations of PPMO in the liver and caused little impact on overall health.
Conclusions
The data indicate that PPMO had considerable antiviral efficacy against DENV-2 in the AG129 mouse model and that PPMO treatment early in the course of an infection was critical to extending the survival times of DENV-2-infected mice in the AG129 model system.
Keywords: flavivirus, antiviral, PMO, PPMO, antisense, dengue virus, morpholino oligomers
Introduction
An estimated 50–100 million people are infected with dengue virus (DENV) yearly, mostly in southern Asia and northern Latin America. Infection with DENV causes a spectrum of outcomes that can be grouped into four categories of increasing clinical severity: infection with no symptoms, undifferentiated fever, classic dengue fever (DF) and dengue haemorrhagic fever (DHF).1 Those afflicted with the latter two categories exhibit overt symptoms and usually seek medical attention. The WHO estimates that DF and DHF together result in ~500 000 hospitalizations and 15 000 deaths per year (http://www.who.int/mediacentre/factsheets/fs117/en/). However, the total number of people with debilitating illness caused by DENV is likely in the tens of millions annually. Currently, no vaccine or effective therapeutic to prevent or treat DF/DHF is commercially available. Several promising DENV vaccine approaches have been reported recently2–4 and some are currently being evaluated in human clinical trials (http://www.clinicaltrials.gov). Several small molecules and monoclonal antibodies have produced promising results in drug development studies against DENV.2 In addition, nucleic-acid-based strategies employing C-5 propyne-substituted phosphorothioate oligonucleotides5 and small interfering RNA6 have both been shown to suppress DENV replication in mammalian cell culture. However, at the time of writing, no clinical trials registered with the United States Food and Drug Administration for a therapeutic agent to treat DENV infections are in progress (http://www.clinicaltrials.gov).
Dengue viruses occur in a complex of four distinct serotypes (DENV-1, -2, -3 and -4), each of which is composed of many genotypic strains. Various strains of each serotype can cause severe disease. The four serotypes of DENV form a single species in the genus Flavivirus of the family Flaviviridae.7 The DENV genome is an ∼11 kb single-stranded RNA molecule of positive polarity, containing a single open reading frame (ORF) flanked by 5′ and 3′ untranslated regions (UTRs). The ORF is translated into a single polyprotein that is co- and post-translationally cleaved to produce 10 mature viral proteins. The UTRs contain conserved sequences and structures that have important roles in the regulation of many aspects of the viral life cycle, including translation, RNA synthesis and virion assembly.8,9
Dengue viruses are generally not pathogenic for mice. However, a number of murine models for the study of DENV exist, although none are thought to accurately mirror DENV pathogenesis in humans.10 AG129 mice lack α- and β-interferon and gamma interferon receptor genes, and therefore have a crippled innate immune system. Their adaptive immune system is considered to be functional, however, and they have been used successfully as a model system for DENV vaccine development.11,12 Certain strains of mouse‐adapted DENV-2 can cause viraemia and can be detected at high titres in a number of tissues after intraperitoneal (ip) administration in AG129 mice.11,13,14 AG129 mice infected with neuroadapted DENV-2 strain New Guinea C (NGC) or DENV-1 Mochizuki strain typically suffer 100% mortality by day 18 post-infection.11,12,15 Recently, Schul et al.14 showed that ip administration of unadapted DENV-2 can produce transient viraemia in AG129 mice and that antiviral drug candidates could be evaluated for their effect on viraemia and cytokine levels through blood testing.
Phosphorodiamidate morpholino oligomers (PMO) are non-ionic oligonucleotide analogues that contain the same four bases as DNA but possess a backbone composed of morpholine rings and phosphorodiamidate inter-subunit linkages instead of the deoxyribose and phosphodiester components of DNA.16 Antisense PMO have been reported to be effective against both Ebola virus17 and feline calicivirus18in vivo. PMO delivery into cells can be much improved through covalent conjugation to arginine-rich cell-penetrating peptides (CPP).19,20 CPP-conjugated PMO (PPMO) have been shown to generate potent inhibition of DENV infections21,22 in cell cultures, and of West Nile virus (WNV),23 poliovirus (PV),24 murine coronavirus25 and influenza A virus26 infections, in both cell culture and mouse models. Experiments with various CPPs conjugated to PMO indicate that a 14 residue peptide, named P7, containing four repeats of arginine:6-aminohexanoic acid:arginine, appears to have characteristics that allow its consideration for potential therapeutic applications.27–30
Previous reports showed that PPMO designed against either the 5′ terminal nucleotides (nt) (5′SL) or against the 3′ cyclization-sequence (3′CS) region of the DENV genome were highly effective at inhibiting DENV amplification in cell cultures.21,22 Other reports have documented that PPMO designed against corresponding regions in WNV and other flaviviruses were effective both in cell cultures and in vivo.20,23 The intention of the study reported here was to extend those previous observations and explore the efficacy of PPMO against DENV in vivo. We found that PPMO targeting the DENV 5′-terminal and 3′-cyclization sequence region were effective in vivo, extending the average survival of DENV-2-infected AG129 mice by up to 8 days. Further, we compared PPMO of various lengths in cell cultures designed against either the 5′SL- or 3′CS-regions for anti-DENV efficacy, and found that PPMO of 22–24 bases were more effective than shorter or longer PPMO targeting the same regions. Pharmacokinetic analysis indicated that PPMO distributes readily into liver and can persist there for at least 5 days. Several toxicological analyses indicated that repeated ip dosing of PPMO, at 10 mg/kg, did not have deleterious effects on the health of non-infected mice.
Materials and methods
Cells and viruses
For cell culture experiments, DENV-2 strain D2/IC-30P-A, derived as previously described from an infectious cDNA clone of wild-type DENV-2 16681 virus,31 was used. For in vivo studies, stocks of mouse-neuroadapted DENV-2 strain New Guinea C (NGC) (CDC, Fort Collins, CO) were used. Viral infectious titres were determined by plaque assay in Vero cells as previously described.31 The LD50 was not determined, but 100 pfu of the DENV-2 NGC was found to be 100% lethal for 7- to 9-week-old AG129 mice. For cell culture experiments, Vero cells were grown in Iscove′s modified Dulbecco′s medium (IMDM) (HyClone, Logan, UT, USA) supplemented with 9% heat-inactivated FBS (HyClone), sodium bicarbonate (0.75 g/L), penicillin G (100 U/mL) and streptomycin sulphate (100 mg/L) (indicated as IMDM-9%FBS medium) in 12-well plates at 37°C and 5% CO2.
Synthesis of PMO compounds
All PMO were synthesized at AVI BioPharma Inc. (Corvallis, OR, USA) by methods described previously.16 The CPP (RXR)4XB (designated ‘P7’) (where R = arginine, X = 6-aminohexanoic acid and B = beta-alanine) was covalently conjugated to the 5′ end of each PMO. P7-PMO (often abbreviated PPMO in this report) compounds were synthesized, purified and analysed at AVI BioPharma, Inc. by methods described previously.27 Sixteen PPMO were prepared (Figure 1). Three of these PPMO were also prepared as PMO, and 3 were prepared with fluorescein (Fl) conjugated to their 3′ end, for use in cell uptake evaluation. Eight of the antisense PPMO compounds were designed to duplex, through complementary base-pairing, with the 5′-terminal region (nt 1-30), while seven of the compounds were designed to duplex with the 3′ cyclization sequence (CS)-region32 (nt 10 610–10 640) of DENV-2 (GenBank Accession no. U87411) genomic RNA. Both of these DENV regions have previously been shown to be productive target sites for PMO technology.21,22 A random sequence of 50% G/C content (DScr) was prepared as both PMO and PPMO, for use as a negative control for the non-sequence-specific effects of the respective chemistries. Prior to use, lyophilized PMO and PPMO were resuspended to a concentration of 2 mM in PBS, and stored in the dark at 4°C. For in vivo studies, all PMO and PPMO were constituted in endotoxin-free PBS.
Figure 1.

Sequences of PMO compounds and their targets in DENV-2 RNA.
PPMO treatment of DENV-infected cell cultures
All cell culture incubations took place at 37°C and 5% CO2. Vero cells were seeded into 12-well plates at 5.0–5.5 log10 cells per well and grown to confluence. The cells were washed twice with PBS, and were then incubated with 1 mL of IMDM lacking FBS (IMDM-0%FBS) alone or with this medium containing the appropriate concentration of PPMO compound for 12 h. The medium was then removed, and the cells were infected with DENV-2 D2/IC-30P-A at an m.o.i. of 1.0 pfu/cell in 0.1 mL of IMDM-0%FBS. After a 2 h infection period, the inoculum was removed, the cells washed twice with PBS and fresh IMDM-0% FBS with or without PPMO (at the same concentration as before infection) added. At 4 and 8 days after infection, a 20 µL aliquot of medium was removed from each well and stored as previously described22 until plaque titration. Plaque titrations were performed under agarose overlay in Vero cell monolayers grown in 6-well plates, as described previously.33
Flow cytometry
To investigate the relative uptake of PPMO of various lengths, Vero cells were cultured in IMDM-9%FBS in 12-well plates. At 95% confluence, cells were washed and treated with IMDM-0% FBS containing 1 µM of 5′SL 18-mer, 24-mer, or 30-mer fluorescein-labelled PPMO for 4 h at 37 ºC and 5% CO2. After treatment, cells were washed with PBS and treated with 0.25% trypsin for 10 min at 37ºC. The trypsin was inactivated by the addition of medium containing 10% FBS. Cells were then pelleted at 1000 g, washed twice with cold PBS and resuspended in PBS containing 1% FBS. These suspensions were then analysed with a Beckman Coulter FC-500 Cytometer, and data were processed using FCS Express 2 (DeNovo Software, Thornhill, Ontario, Canada).
Experiments using AG129 Mice
AG129 mice were bred and housed at CDC Fort Collins, CO, USA and were 7–9 weeks old at the beginning of all experiments. All mice were allowed food and water ad libitum throughout the studies. All studies were approved by the Institutional Animal Care and Use Committee of the CDC at Fort Collins.
Toxicity evaluation of PPMO in non-infected AG129 mice
Prior to the initiation of either of the two in vivo viral inhibition experiments in this study, the effect of prospective dosing regimens was assessed for toxicity in non-infected AG129 mice. Before the first antiviral study, groups of four mice received two ip doses, separated by 24 h, of 200 µL of PBS, or 200 µL of PBS containing either 380 µg (∼19.1 mg/kg) of PMO DScr or 240 µg (∼12.1 mg/kg) of PPMO DScr. Preliminary to the second antiviral study, groups of six mice received ip doses of 200 µL of PBS or 200 µL of PBS containing 200 µg (∼10 mg/kg) of PPMO 5′SL 22-mer once a day for 9 days. Mice were observed daily for abnormal appearance or behaviour (including lethargy, hunching, ruffled fur and diarrhoea) and weighed once per week (and also on days 4 and 10 in the second experiment) for the 5 week duration of each experiment.
Evaluation of PMO and PPMO antiviral efficacy in AG129 mice
For the first in vivo antiviral study, groups of four mice received ip administration of PBS, PMO or PPMO at 6 h before and 24 h after ip inoculation with 106 pfu of DENV-2 NGC. All mice were observed daily for signs of severe disease and survivors were weighed at 7 and 14 days post-infection (dpi). For the second experiment, groups of six mice (except the PBS control group which had only five mice) received a total of nine ip treatments each of PBS or PPMO at 24 h and 6 h before virus challenge and once a day on days +1 to +7 post-challenge with 104 pfu of DENV-2 NGC. One mouse group received the seven post-challenge PPMO 5′SL treatments, but not the two PPMO treatments at 24 and 6 h before virus challenge. All injections used 200 µL PBS as vehicle. Both AG129 antiviral experiments included non-infected, non-treated mice as controls.
Pharmacokinetic and toxicity study of PPMO using non-infected BALB/c mice
Experimentally naive BALB/c mice (Simonsen Laboratories Inc, Gilroy, CA, USA) were used for this study. All procedures were carried out in the Laboratory Animal Resource Center at Oregon State University (OSU), Corvallis, OR, USA, in accordance with and monitored by the Institutional Animal Care and Use Committee of OSU.
Each group in all of the pharmacokinetic and toxicology studies below contained three animals. In one arm of the study, the brain, liver and spleen were harvested at 0.2, 0.5, 1, 2, 4, 8, 12 and 24 h time points following a 10 mg/kg single-dose ip administration of 5′SL 24-mer PPMO-Fl. In a separate experiment, 10 mg/kg ip injections of 5′SL 24-mer PPMO-Fl were administered once a day for 9 consecutive days. PPMO-Fl concentrations were evaluated in the above tissues at 24 h following the last dose and at 3 and 5 days following the end of the 9 day regimen. Animals were observed for any abnormal behaviour or appearance (as defined above for AG129 mice) during and after the treatment period.
Sample preparation and analytical method
Whole blood samples were collected in clean 2 mL tubes containing EDTA and immediately centrifuged at 14 000 rpm for 10 min. The plasma portion was then collected in fresh amber tubes. The organs were homogenized immediately after collection in tissue-PELB™ buffer (G-Biosciences, St Louis, MO, USA) and centrifuged. The supernatant was collected into fresh amber tubes. Both plasma and tissue homogenates were immediately analysed fluorometrically at excitation and emission wavelengths of 485 and 528 nm, respectively, using an FLx800 Microplate Fluorescence Reader (Bio-Tek Instruments Inc, Winooski, VT, USA).
Pharmacokinetic analysis
Pharmacokinetic (PK) profile was characterized by compartmental analysis from the blood samples collected at 0.2, 0.5, 1, 2, 4, 8, 12 and 24 h time points following the single-dose ip injection. The plasma concentration versus time profile was fit into a two-compartmental open model using PK Solutions Software (Summit Research Services, Montrose, CO, USA). The fitted PK parameters include the apparent elimination half-life (T1/2β), plasma clearance (Clpl), volume of distribution (Vd) and the area under the curve versus time curve (AUC). The AUC was estimated using the log-linear trapezoidal method. The plasma data were fitted to equation 1, C (t) = Ae−αt+Be−βt, where C(t) is the plasma concentration at time t; A and B are intercept terms; α is the distribution rate constant; and β is the elimination rate constant.
Statistical analysis
Error bars in this report represent means ± standard deviations. Statistical analysis consisted of Student′s t‐test and linear regression analysis and was calculated with Graph-Pad 4 Prism software (Graph-Pad Software, San Diego, CA, USA). P values <0.05 were considered statistically significant.
Results
Initial in vivo study of PPMO against DENV-2 in AG-129 mice
It was previously shown that two treatments of cell cultures with 20-mer 5′SL or 18-mer 3′CS PPMO, with one treatment starting 12 h before and another starting at 2 h after an inoculation period with DENV-2 D2/IC-30P-A, could produce titre reductions of several orders of magnitude, compared with controls, for several days after inoculation.22 We sought to investigate the antiviral effect of a similar dosing regimen in an in vivo setting. First, we evaluated the effect of the prospective dosing regimen on the health of non-infected animals. Intraperitoneal administration of PMO to mice has been reported to produce no detectable toxicity, even at high doses.23 It is generally accepted that toxicity caused by PPMO is due to the peptide component, and therefore we considered evaluation of a single PPMO compound as representative of the off-target effects likely to result from any PPMO under the employed regimen. Groups of mice were treated twice (24 h apart) with PBS, 380 µg of DScr PMO or 240 µg of DScr PPMO, and found to suffer no weight loss or apparent ill-effects for the ensuing 35 days of monitoring (data not shown). After establishing this subtoxic dosing regimen with non-infected animals, the same regimen with PMO or PPMO was employed in an in vivo viral inhibition challenge. Figure 2(a) shows the survival of mice treated once before and once after infection with 106pfu DENV-2 NGC. Of the six PMO or PPMO evaluated, two generated a statistically significant extension of survival. Compared with the PBS-treated group, members of the 5′SL 20-mer and 3′CS 18-mer PPMO-treated groups survived an average of 8 and 5 days longer (P = 0.009 and 0.006), respectively (Figure 2a and b). Three of the four mice treated with the 5′SL 20-mer PPMO were still alive on day 16, whereas three of four mice in the PBS- or DScr PPMO-treated groups had died by day 9. Furthermore, the 5′SL PPMO group showed little, and 3′CS-PPMO group only moderate, weight-loss through day 14 (Figure 2c), reflective of the enhanced survival observed.
Figure 2.
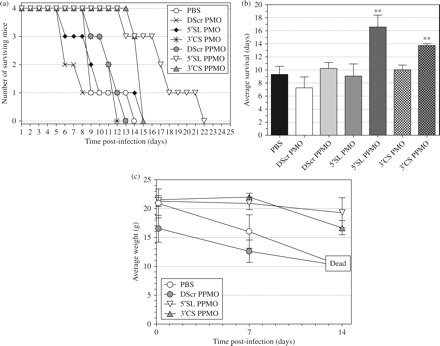
Antisense PPMO treatment extended survival of AG129 mice infected with DENV-2 NGC. Groups of four AG129 mice received ip administration of 380 µg PMO, 250 µg PPMO or mock-treatment (PBS) at 6 h before and 24 h after ip inoculation with 106 pfu of DENV-2 strain NGC. (a) Kaplan–Meier survival chart showing day to day survival in mock-, PMO- or PPMO-treated groups. (b) Comparison of average survival times for the groups in (a). Asterisks indicate a statistically significant difference in comparison to PBS-treated group (P = 0.009 for 5′SL PPMO and 0.006 for 3′CS PPMO). (c) Average body weight values at days 0, 7, and 14 of mice groups treated with PBS or PPMO.
Effect of PPMO length on antiviral activity in cell cultures
In an effort to improve the potency of PPMO targeting the DENV 5′SL and 3′CS regions, panels of PPMO of varying lengths, directed at these two regions, were synthesized for use in cell culture trials against DENV-2 D2/IC-30PA. Eight PPMO of 16–30 bases long and 7 PPMO of 14–30 bases were designed against the 5′SL- and 3′CS-region, respectively (Figure 1). Cells were incubated in 1 µM PPMO for 12 h prior to and again after infection with DENV-2. Viral titres were assessed at 4 and 8 dpi. Figure 3(a) shows that for the 5′SL PPMO, the 22 and 24 base sequences were most effective, reducing viral titre 3 and 4 log10, respectively, compared with the mock- and DScr-treated controls at 4 dpi. For the 3′CS-PPMO, the 22-mer was the most effective, resulting in more than a 2 log10 reduction in DENV-2 titre at 4 dpi. Of note, the viral titre reductions produced by the effective compounds were considerably greater on day 4 than they were on day 8, indicating that the virus was rebounding from the effect of the drug, in a manner consistent with that reported in a similar previous experiment evaluating the effect of PPMO against DENV-2 over time in cell cultures.22
Figure 3.
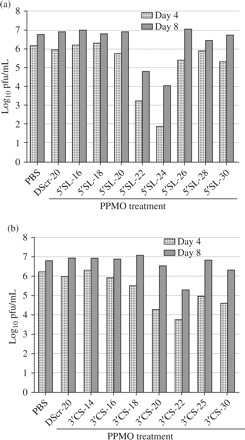
PPMO of 22–24 bases in length have the highest activity against DENV-2 D2/IC-30P-A in cell cultures. Vero cells were incubated in 1 µM PPMO for 12 h prior to, and again after, the adsorption period with DENV-2 at m.o.i of 1.0 pfu/cell. The log10 viral titres at 4 and 8 days p.i. are shown. All compounds shown are PPMO, and DScr is included as a control (see Figure 1 for exact PPMO sequences). The two graphs show PPMO of varying lengths that target (a) the 5′-terminal nucleotides or (b) the 3′CS region, of the DENV-2 genome. In each graph, the average of two experiments is shown.
Uptake of PPMO by cells in culture evaluated by flow-cytometry
The results in Figure 2 showed that PPMO of 22–24 bases in length had considerably higher antiviral activity than shorter or longer PPMO targeting the same regions. To determine whether the PMO-transporting capacity of the P7-peptide component of PPMO was affected by PMO length, we evaluated the uptake of PPMO-Fl of different lengths into non-infected Vero cells. Cells were incubated with 1 µM 5′SL PPMO-Fl of 18, 24 or 30 bases in length (Figure 1) for 4 h under experimental conditions similar to those used in the antiviral experiments of Figure 3, and the presence of fluorescein was then evaluated by flow cytometry. We observed that all three PPMO entered over 95% of the cells and that each PPMO-Fl produced nearly identical signal intensity. No differences between the cell entry characteristics of the three PPMO-Fl were observed (data not shown).
Second in vivo study of PPMO against DENV-2 in AG129 mice
For a second in vivo experiment, the 5′SL 22-mer and 3′CS 22-mer PPMO were used, and the number of doses increased to nine. As for the first antiviral in vivo experiment, a preliminary toxicity evaluation of a prospective dosing regimen was carried out with non-infected AG129 mice. Groups of six AG129 mice were treated ip once a day for 9 days consecutively with PBS or 5′SL 22-mer PPMO, and their weight and appearance monitored for 5 weeks. No weight loss occurred in either group over the duration of the monitoring period (Figure 4a). For the second viral inhibition challenge, all groups of mice, except one, received two PBS- or PPMO-treatments before and seven treatments after infection with 104pfu of DENV-2 NGC. One group received no pre-infection treatments, but only the seven post-infection treatments with 5′SL 22-mer PPMO. Mice receiving pre- and post-infection treatments of 5′SL 22-mer lived, on average, 7 days longer, and those on 5′SL and 3′CS combination treatment lived ~8 days longer than PBS-treated controls (P = 0.03 and 0.006, respectively). The 3′CS 22-mer treatment group and the group receiving the 5′SL 22-mer with only post-infection treatments outlived the PBS controls by ~6 and 4 days, respectively; the results that were not statistically significant (Figure 4b). The average survival of the PBS-treated mice was several days longer in this experiment (Figure 4b) than in the first in vivo experiment (Figure 2b), probably because of the lower challenge dose of the NGC virus used. In a previous unrelated experiment, AG129 mice survived an average of 9.0 ± 1.0 and 13.8 ± 2.5 days following ip challenge with DENV-2 NGC at doses of 6 and 4 log10 pfu, respectively (Richard Kinney, unpublished results). All the mice in the PBS- or DScr PPMO-treated groups died by day 17, whereas half the mice in each of the 5′SL, 3′CS, and 5′SL and 3′CS combination groups lived until day 22–24. Body weight measurements reflected the relatively stable health of the surviving animals at day 21 (Figure 4d).
Figure 4.
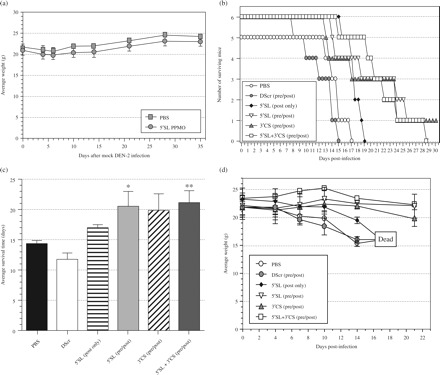
Multiple PPMO treatments are subtoxic and extend survival time of DENV-2 NGC-infected AG129 mice. (a) Non-infected AG129 mice (n = 6) were treated ip once per day for 9 days with PBS or 200 µg (∼10 mg/kg) of 5′SL 22-mer PPMO. Whole body weight was monitored at indicated time points for 5 weeks starting from the first day of treatment. (b–d) AG129 mice (n = 6, except for the PBS group where n = 5) were treated with PBS or 200 µg of the indicated 22-mer PPMO before (at -24 h and -6 h) and after (once per day at +1 to +7 dpi) (+pre/+post), or only after (post only), ip inoculation with 104pfu DENV-2. One group received an equal combination of two PPMO (5′SL+3′CS; 100 µg of each). (b) Kaplan–Meier survival plot. (c) Comparison of average survival times for the groups shown in (b). A single mouse in the 3′CS group survived the experiment and was ascribed a survival time of 30 days for this calculation. Asterisks indicate a statistically significant difference in comparison with PBS-treated group (P = 0.03 for 5′SL PPMO and P = 0.006 for 5′SL and 3′CS combination treatment). (d) Average body weight values, measured every 3–4 days, of indicated groups. At day 22, three of the groups had half (3) of their members alive, and the average weight of those survivors is shown.
Pharmacokinetics evaluation of PPMO-Fl
The linear and log plots of the plasma concentration versus time curves for a single 10 mg/kg injection of the 5′SL 24-mer PPMO-Fl in BALB/c mice are shown in Figure 5(a and b). A summary of the key PK estimates is presented in Table 1. Each data point represents mean ± SD of triplicate values. The peak plasma concentration (Cmax) was observed to be 0.48 mg/L at a Tmax of 0.67 h following administration. The PPMO-Fl had a distribution half-life of 2.79 ± 0.47 h, and elimination half-life was observed to be 7.31 ± 0.78 h. The AUC was estimated to be 1.11 ± 0.05 µg·h/mL with a corresponding volume of distribution (Vd) of 1.42 ± 0.16 L/kg body weight and a plasma clearance (Clp1) of 0.03 ± 0.01 L/h.
Figure 5.
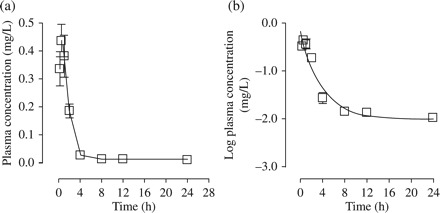
Plasma concentration versus time profiles of 5′SL PPMO-Fl following single-dose ip injection of Balb/c mice with 10 mg/kg. (a and b) Linear and log plots of plasma concentration versus time, respectively. Data expressed as μg PPMO/mL plasma. Each data point represents the mean ± SD of triplicate values.
Table 1.
Plasma pharmacokinetic parameter estimates for 5′SL 24mer PPMO-Fl in BALB/c mice
| Dose (mg/kg) | Route | AUC (μg·h/mL) | C max (µg/mL) | T max (h) | Clp1 (L/h) | V d (L/kg) | T 1/2α (h) | T 1/2β (h) |
|---|---|---|---|---|---|---|---|---|
| 10.0 | ip | 1.11 ± 0.05 | 0.48 ± 0.08 | 0.67 ± 0.29 | 0.03 ± 0.0 | 1.42 ± 0.16 | 2.79 ± 0.47 | 7.31 ± 0.78 |
Tissue distribution of PPMO-Fl
Figure 6(a) represents tissue PPMO-Fl levels in the brain, liver and spleen of BALB/c mice at different time points following a single-dose administration. Figure 6(b) shows tissue distribution following single daily injections for 9 consecutive days and at 3 and 5 day washout time points following the 9 day treatment. Of the three organs evaluated, the liver was observed to be the site of highest accumulation, with the brain accumulating the least amount of PPMO-Fl over the entire period (Figure 6a). The corresponding tissue concentrations (μM) are shown in Table 2. The linear regression analysis showed that liver accumulated the PPMO-Fl fastest, with a positive slope of 0.226 ± 0.09 µg/g/h compared with brain (slope = 0.0005 ± 0.002 µg/g/h) and spleen (slope = 0.021 ± 0.02 µg/g/h). The presence of PPMO-Fl in tissues at detectable levels as early as 0.2 h post‐dose is indicative of a rapid tissue distribution from the vascular space into peripheral tissues. Also, tissue to plasma concentration ratio at Tmax was 0.08, 2.43 and 0.72 for brain, liver and spleen, respectively. PPMO-Fl levels did not change significantly during the 5 day washout period after the last dose (Figure 6b).
Figure 6.
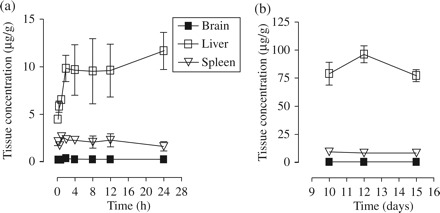
Profiles of 5′SL PPMO-Fl concentration in Balb/c mice tissues versus time. (a) Tissue recovery over a 24 h period following a single 10 mg/kg ip injection. (b) Tissue recovery on day 9 following nine consecutive daily injections of 10 mg/kg and at 3 and 5 day washout time points after termination of treatment. Data expressed as μg PPMO/g tissue. Each data point represents the mean ± SD of triplicate values.
Table 2.
Tissue recovery following ip administration of 5′SL PPMO-Fl in BALB/c mice
| Time post-treatment (days) | Recovery (µM) |
||
|---|---|---|---|
| brain | liver | spleen | |
| 1 | 0.01 ± 0.0 | 7.67 ± 1.72 | 0.88 ± 0.16 |
| 3 | 0.02 ± 0.004 | 9.34 ± 1.27 | 0.78 ± 0.19 |
| 5 | 0.01 ± 0.01 | 7.51 ± 0.86 | 0.80 ± 0.13 |
Safety assessment of PPMO-Fl
Balb/c mice received 9 ip doses (once per day for 9 days) of 10 mg/kg 5′SL 24-mer PPMO-Fl, and were weighed and observed daily for signs of clinical illness during the course of the 15 day study. Serum chemistry, haematology and body weight were also monitored as part of the safety evaluation. No changes in animal behaviour or appearance during and after treatment were observed. As shown in Figure 7(a–d), the only indices that showed significant change during and after treatment were an increase in serum cholesterol (P < 0.01), and a decrease in haematocrit (P < 0.05), haemoglobin (P < 0.01), packed cell volume (P < 0.05) and red blood cell (RBC) count (P < 0.05) during treatment. All these indices reverted to levels comparable to the untreated control during the 5 day washout period, as shown in Figure 7. Body weights of the animals increased by ~1 g during the duration of the study, with little variation between individuals (data not shown).
Figure 7.
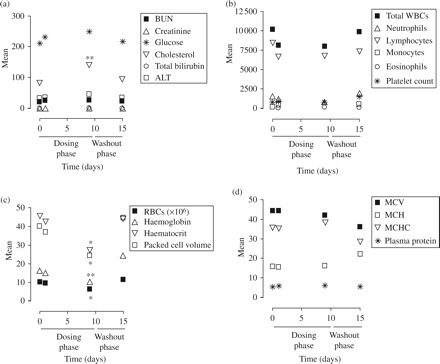
Toxicity analysis of PPMO in Balb/c mice. Safety assessment data following 10 mg/kg single daily injections of 5′SL PPMO-Fl for 9 consecutive days. (a) Serum chemistry and (b–d) haematology data. *P < 0.05, haematocrit, packed cell volume and red blood cell count at 9 day time point of treatment phase were significantly less than control, **P < 0.01 serum cholesterol at 9 day time point of treatment phase was significantly greater than control, haemoglobin at 9 day time point of treatment phase was significantly less than control (n = 3). For serum chemistry and haematology, the units are as follows: BUN (mg/dL), creatinine (mg/dL), glucose (mg/dL), cholesterol (mg/dL), total bilirubin (mg/dL), ALT (U/L), total WBCs (# of cells/μL), neutrophils (# of cells/μL), lymphocytes (# of cells/μL), monocytes (# of cells/μL), eosinophils (# of cells/μL), platelet count (× 1000/μL), RBCs (× 106/μL), haemoglobin (g/dL), haematocrit (%), packed cell volume (%), MCV (fL), MCH (pg), MCHC (g/dL).
Discussion
DENV is a major public health problem in tropical and subtropical regions, for which no highly effective therapy exists. Advances in the development of therapeutic approaches to treating DENV infections are a public health priority not just in tropical and subtropical regions, but in temperate regions as well. This study showed that PPMO targeting the 5′ terminal- or 3′ cyclization sequence region of the DENV-2 genome can significantly extend the survival time of AG129 mice infected with a lethal dose of DENV-2 NGC. Pharmacological and toxicological evaluation in mice showed that PPMO distributes to the liver and persists there for at least several days, and produces little toxicity at a dosing regimen demonstrated to be effectively antiviral. The overall profile of PPMO appears to be favourable for consideration for possible human drug development.
PPMO are effective against DENV-2 in vivo
In the first of two in vivo experiments carried out in this study, PMO was compared with PPMO for antiviral efficacy in DENV-infected AG129 mice. A 20-mer 5′SL or 18-mer 3′CS PMO produced no significant treatment benefit, whereas 20-mer 5′SL or 18-mer 3′CS PPMO treated mice showed a statistically significant increase in survival time (Figure 1). 5′SL and 3′CS PPMO of various lengths were then compared in cell culture for anti-DENV efficacy. PPMO of 22–24 bases were found to be the most effective, and able to reduce viral titres by several orders of magnitude when present at 1 µM in the cell culture medium. This antiviral effect was also greater than that reported previously for 5′SL 20-mer and 3′CS 18-mer PPMO that had been made with a different CPP (‘P4’). In that earlier report, at 1 µM concentration, the P4-PMO exhibited no antiviral activity against DENV-2 D2/IC-30P-A virus in Vero cell cultures.22 5′SL and 3′CS 22-mer PPMO, which had produced high efficacy in the length versus activity evaluation, were then used in a second mouse study. With a higher number of doses and a longer time period over which dosing occurred than were employed in the first in vivo experiment, survival benefit of the antisense PPMO-treated groups again extended to an average of ~1 week. For the second in vivo experiment, we had hypothesized that employing a greater number of doses with PPMO of more effective length during the first week post-infection, concomitant with a lower (but still severe) DENV-2 NGC challenge dose, would limit viral replication sufficiently to permit development of a fully protective anti-DENV-2 immune response. The PBS-treated group lived an average of 14 days in the second experiment compared with 9 days in the first experiment. In both experiments the effective PPMO produced up to a 7–8 day extension of survival time, and protection from DENV-induced weight loss for a period of about a week compared with controls (Figures 1 and 3).
It is difficult to compare the in vivo results from this study with those obtained with other potential anti-DENV drugs, as there have been no reports in the literature (at the time of writing) of studies in which survival time was employed as an end-point in the evaluation of potential drugs using AG129 mice. The results here strongly suggest that 5′SL and 3′CS PPMO treatments suppressed viral titres in vivo for a period of several days, but that residual virus was able to rebound a number of days after treatment cessation. A similar pattern was also observed in the cell culture studies reported here and previously.22 An alternate, and likewise untested, hypothesis is that virus resistant to the effects of the PPMO arose over time in response to the treatments. To date, however, there have been no reports in the literature of viral mutations arising in response to PPMO treatment in animals. Other investigators have sequenced virus from mice that had been treated with PPMO highly active in vivo against WNV23 and PV,24 and found no mutations in the PPMO-targeted regions. Our current in vivo experiments did not include enough animals to evaluate the amount or sequence of virus in blood or tissues.
The biology of DENV infection in the AG129 mouse is considered to be quite different from that of natural DENV infection in a human. Nevertheless, the benefit afforded the mice by PPMO in this study is a promising result. The level of viraemia affects the duration and severity of dengue disease in humans,34–36 and a drug capable of limiting viral replication could reasonably be expected to be beneficial to some patients. The second in vivo experiment here showed that post-infection treatment alone was not as effective as pre- and post-infection treatment with 5′SL 22-mer PPMO (Figure 4b–d). Although this result indicates that early intervention with PPMO is necessary for maximum protective effect, it is not possible from these experiments to conclude whether this type of technology will ultimately be clinically useful. The pathogenesis of DF/DHF in humans is characterized by a wide variety of clinical progressions, including subsets of patients who remain viraemic and harbour virus in their tissues for periods of time beyond the onset of symptoms.3 Considering the large number of total cases of DF/DHF, it is likely that an agent that effectively reduces virus replication in a non-toxic manner would find utility.
PPMO length affects activity
Reports in the literature indicate that highly effective PPMO in other studies have varied in length between 9 and 25 bases.37,38 To determine the relative antiviral efficacy of PPMO of various lengths, we analysed two panels of seven or eight PPMO. The most effective PPMO from both panels were 22–24 bases in length. Although the binding affinity of an oligonucleotide to single strand nucleic acid, as reflected by melting point temperature, increases as a function of oligonucleotide length, a number of other design factors can affect the functional performance of an antisense oligomer.39,40 It was previously shown that PPMO targeted to the DENV 5′ terminus can interfere with translation,21 and that the corresponding PPMO can interfere with WNV translation20 and RNA 5′ capping reactions.41 3′CS-directed PPMO have been shown to interfere with RNA synthesis in DENV21 and WNV.20 Thus, PPMO targeting the 5′SL and 3′CS regions likely work by interfering in different aspects of the viral life cycle. Despite the different mechanisms of action, the most effective PPMO from both panels were of similar length.
Pharmacokinetic and safety profiles of DENV-specific PPMO
Because of the limited number of AG129 mice available to us, we used BALB/c mice for PK/toxicity analysis. A P7-PMO-Fl compound has previously been shown to be stable in human serum for at least 2 h.30 Here, the PK profile was based on plasma concentration data monitored over 24 h following a single ip injection. Peak plasma concentration was observed at a Tmax of 0.67 h following injection, which is indicative of rapid transfer from the site of injection to the vascular space. The observed distribution half-life of ~3 h, a high volume of distribution (Vd) and AUC, and detectable levels of PPMO-Fl observed in brain, liver and spleen at 0.2 h (10 min) post-dose all indicate that the PPMO-Fl distributed fairly rapidly into tissues. Tissue to plasma concentration ratios in liver (2.43) and spleen (0.72) at the Tmax provided a further indication that the PPMO-Fl rapidly penetrated into these tissues. Of the three tissues tested, liver was observed to be the primary site of accumulation, with brain accumulating minimal amounts of the compound. We assume that the higher perfusion of liver (11.8 mL/min in rats) compared to spleen (0.6 mL/min) and brain (1.3 mL/min)42 contributed to the observed pattern. We attribute the low cerebral uptake to the presence of the blood brain barrier. As shown in Figure 6(b), the level of PPMO-Fl in brain, liver and spleen did not change significantly for 3–5 days following the cessation of the 9 day treatment period. This observation indicates that these tissues retained the compound, and provides useful information for prospective dosing frequency in future studies. Although the liver is not considered a major target organ for DENV replication, the involvement of the liver in the pathogenesis of DENV infection has been documented.43,44 DENV antigens and RNA have been detected in various cells of the liver in humans.43,44
Safety assessment of the PPMO was based on changes in behaviour, appearance, body weight, serum chemistry panel and haematology in BALB/c mice after nine daily treatments. The only significant changes in serum chemistry or haematology during or after treatment were elevated serum cholesterol and a decrease in haematocrit, haemoglobin, packed cell volume and red blood cell (RBC) count. However, these changes did not appear to impact the overall health of the animals, and all of these indices reverted to levels comparable with the untreated control during the 5 day washout period, as shown in Figure 7. The fact that total bilirubin was not altered during the treatment period suggests that the observed decrease in red blood cell count, haematocrit and packed cell volume was a result of interference with red blood cell production rather than haemolysis of these cells. These data demonstrate that the DENV 5′SL PPMO possesses a favourable safety profile and kinetic behaviour. The data from this study will likely prove useful during the design of future in vivo studies with PPMO.
The results of the two in vivo antiviral experiments here and the PK analysis showing a residence time of PPMO of several days suggest that an extended treatment period, perhaps with less frequent dosing, could produce longer extensions of survival time than those observed in this study.
Funding
This study was conducted under a Contract Research and Development Agreement between AVI BioPharma and the Centers for Disease Control.
Transparency declarations
D. A. S., A. A., S. C., R. E. B. and P. L. I. work or worked for the company that manufactures the antisense oligomers used in this report. The remaining authors have none to declare.
Acknowledgements
The authors wish to express their gratitude to the Chemistry Group at AVI Biopharma for the expert production of all PMO compounds used in this study, and to Carla London for expert technical assistance.
References
- 1.Deen JL, Harris E, Wills B, et al. The WHO dengue classification and case definitions: time for a reassessment. Lancet. 2006;368:170–3. doi: 10.1016/S0140-6736(06)69006-5. [DOI] [PubMed] [Google Scholar]
- 2.Damonte EB, Pujol CA, Coto CE. Prospects for the therapy and prevention of dengue virus infections. Adv Virus Res. 2004;63:239–85. doi: 10.1016/S0065-3527(04)63004-1. [DOI] [PubMed] [Google Scholar]
- 3.Whitehead SS, Blaney JE, Durbin AP, et al. Prospects for a dengue virus vaccine. Nat Rev Microbiol. 2007;5:518–28. doi: 10.1038/nrmicro1690. [DOI] [PubMed] [Google Scholar]
- 4.Edelman R. Dengue vaccines approach the finish line. Clin Infect Dis. 2007;45(Suppl 1):S56–60. doi: 10.1086/518148. [DOI] [PubMed] [Google Scholar]
- 5.Raviprakash K, Liu K, Matteucci M, et al. Inhibition of dengue virus by novel, modified antisense oligonucleotides. J Virol. 1995;69:69–74. doi: 10.1128/jvi.69.1.69-74.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang W, Singam R, Hellermann G, et al. Attenuation of dengue virus infection by adeno-associated virus-mediated siRNA delivery. Genet Vaccines Ther. 2004;2:8. doi: 10.1186/1479-0556-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gubler DJ, Kuno G, Markoff L. Flaviviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia, PA: Lippincott, Williams, and Wilkins; 2007. Chapter 34. [Google Scholar]
- 8.Lindenbach BD, Theil HJ, Rice CM. Flaviviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. Philidelphia, PA: Lippincott, Williams, and Wilkins; 2007. Chapter 33. [Google Scholar]
- 9.Clyde K, Kyle JL, Harris E. Recent advances in deciphering viral and host determinants of dengue virus replication and pathogenesis. J Virol. 2006;80:11418–31. doi: 10.1128/JVI.01257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charlier N, De Clercq E, Neyts J. Mouse and hamster models for the study of therapy against flavivirus infections. Novartis Found Symp. 2006;277:218–29. discussion 29–32, 51–3. [PubMed] [Google Scholar]
- 11.Johnson AJ, Roehrig JT. New mouse model for dengue virus vaccine testing. J Virol. 1999;73:783–6. doi: 10.1128/jvi.73.1.783-786.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calvert AE, Huang CY, Kinney RM, et al. Non-structural proteins of dengue 2 virus offer limited protection to interferon-deficient mice after dengue 2 virus challenge. J Gen Virol. 2006;87:339–46. doi: 10.1099/vir.0.81256-0. [DOI] [PubMed] [Google Scholar]
- 13.Shresta S, Sharar KL, Prigozhin DM, et al. Murine model for dengue virus-induced lethal disease with increased vascular permeability. J Virol. 2006;80:10208–17. doi: 10.1128/JVI.00062-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schul W, Liu W, Xu HY, et al. A dengue fever viremia model in mice shows reduction in viral replication and suppression of the inflammatory response after treatment with antiviral drugs. J Infect Dis. 2007;195:665–74. doi: 10.1086/511310. [DOI] [PubMed] [Google Scholar]
- 15.Huang CY, Butrapet S, Tsuchiya KR, et al. Dengue 2 PDK-53 virus as a chimeric carrier for tetravalent dengue vaccine development. J Virol. 2003;77:11436–47. doi: 10.1128/JVI.77.21.11436-11447.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Summerton J, Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997;7:187–95. doi: 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]
- 17.Warfield KL, Swenson DL, Olinger GG, et al. Gene-specific countermeasures against Ebola virus based on antisense phosphorodiamidate morpholino oligomers. PLoS Pathog. 2006;2:e1. doi: 10.1371/journal.ppat.0020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith AW, Iversen PL, O'Hanley PD, et al. Virus-specific antiviral treatment for controlling severe and fatal outbreaks of feline calicivirus infection. Am J Vet Res. 2008;69:23–32. doi: 10.2460/ajvr.69.1.23. [DOI] [PubMed] [Google Scholar]
- 19.Moulton HM, Nelson MH, Hatlevig SA, et al. Cellular uptake of antisense morpholino oligomers conjugated to arginine-rich peptides. Bioconjug Chem. 2004;15:290–9. doi: 10.1021/bc034221g. [DOI] [PubMed] [Google Scholar]
- 20.Deas TS, Binduga-Gajewska I, Tilgner M, et al. Inhibition of flavivirus infections by antisense oligomers specifically suppressing viral translation and RNA replication. J Virol. 2005;79:4599–609. doi: 10.1128/JVI.79.8.4599-4609.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holden KL, Stein DA, Pierson TC, et al. Inhibition of dengue virus translation and RNA synthesis by a morpholino oligomer targeted to the top of the terminal 3′ stem-loop structure. Virology. 2006;344:439–52. doi: 10.1016/j.virol.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 22.Kinney RM, Huang CY, Rose BC, et al. Inhibition of dengue virus serotypes 1 to 4 in vero cell cultures with morpholino oligomers. J Virol. 2005;79:5116–28. doi: 10.1128/JVI.79.8.5116-5128.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deas TS, Bennett CJ, Jones SA, et al. In vitro resistance selection and in vivo efficacy of morpholino oligomers against West Nile virus. Antimicrob Agents Chemother. 2007;51:2470–82. doi: 10.1128/AAC.00069-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stone JK, Rijnbrand R, Stein DA, et al. A morpholino oligomer targeting highly conserved internal ribosome entry site sequence is able to inhibit multiple species of picornavirus. Antimicrob Agents Chemother. 2008;52:1970–81. doi: 10.1128/AAC.00011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burrer R, Neuman BW, Ting JP, et al. Antiviral effects of antisense morpholino oligomers in murine coronavirus infection models. J Virol. 2007;81:5637–48. doi: 10.1128/JVI.02360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gabriel G, Nordmann A, Stein DA, et al. Morpholino oligomers targeting the PB1 and NP genes enhance the survival of mice infected with highly pathogenic influenza A H7N7 virus. J Gen Virol. 2008;89:939–48. doi: 10.1099/vir.0.83449-0. [DOI] [PubMed] [Google Scholar]
- 27.Abes S, Moulton HM, Clair P, et al. Vectorization of morpholino oligomers by the (R-Ahx-R)(4) peptide allows efficient splicing correction in the absence of endosomolytic agents. J Control Release. 2006;116:304–13. doi: 10.1016/j.jconrel.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 28.Nelson MH, Stein DA, Kroeker AD, et al. Arginine-rich peptide conjugation to morpholino oligomers: effects on antisense activity and specificity. Bioconjug Chem. 2005;16:959–66. doi: 10.1021/bc0501045. [DOI] [PubMed] [Google Scholar]
- 29.Amantana A, Moulton HM, Cate ML, et al. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug Chem. 2007;18:1325–31. doi: 10.1021/bc070060v. [DOI] [PubMed] [Google Scholar]
- 30.Youngblood DS, Hatlevig SA, Hassinger JN, et al. Stability of cell-penetrating peptide-morpholino oligomer conjugates in human serum and in cells. Bioconjug Chem. 2007;18:50–60. doi: 10.1021/bc060138s. [DOI] [PubMed] [Google Scholar]
- 31.Kinney RM, Butrapet S, Chang GJ, et al. Construction of infectious cDNA clones for dengue 2 virus: strain 16681 and its attenuated vaccine derivative, strain PDK-53. Virology. 1997;230:300–8. doi: 10.1006/viro.1997.8500. [DOI] [PubMed] [Google Scholar]
- 32.Khromykh AA, Meka H, Guyatt KJ, et al. Essential role of cyclization sequences in flavivirus RNA replication. J Virol. 2001;75:6719–28. doi: 10.1128/JVI.75.14.6719-6728.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Butrapet S, Huang CY, Pierro DJ, et al. Attenuation markers of a candidate dengue type 2 vaccine virus, strain 16681 (PDK-53), are defined by mutations in the 5′ noncoding region and nonstructural proteins 1 and 3. J Virol. 2000;74:3011–9. doi: 10.1128/jvi.74.7.3011-3019.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vaughn DW, Green S, Kalayanarooj S, et al. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J Infect Dis. 2000;181:2–9. doi: 10.1086/315215. [DOI] [PubMed] [Google Scholar]
- 35.Wang WK, Chao DY, Kao CL, et al. High levels of plasma dengue viral load during defervescence in patients with dengue hemorrhagic fever: implications for pathogenesis. Virology. 2003;305:330–8. doi: 10.1006/viro.2002.1704. [DOI] [PubMed] [Google Scholar]
- 36.Wang WK, Chen HL, Yang CF, et al. Slower rates of clearance of viral load and virus-containing immune complexes in patients with dengue hemorrhagic fever. Clin Infect Dis. 2006;43:1023–30. doi: 10.1086/507635. [DOI] [PubMed] [Google Scholar]
- 37.Deere J, Iversen P, Geller BL. Antisense phosphorodiamidate morpholino oligomer length and target position effects on gene-specific inhibition in Escherichia coli. Antimicrob Agents Chemother. 2005;49:249–55. doi: 10.1128/AAC.49.1.249-255.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fletcher S, Honeyman K, Fall AM, et al. Dystrophin expression in the mdx mouse after localised and systemic administration of a morpholino antisense oligonucleotide. J Gene Med. 2006;8:207–16. doi: 10.1002/jgm.838. [DOI] [PubMed] [Google Scholar]
- 39.Pan WH, Clawson GA. Identifying accessible sites in RNA: the first step in designing antisense reagents. Curr Med Chem. 2006;13:3083–103. doi: 10.2174/092986706778521788. [DOI] [PubMed] [Google Scholar]
- 40.Schiavone N, Donnini M, Nicolin A, et al. Antisense oligonucleotide drug design. Curr Pharm Des. 2004;10:769–84. doi: 10.2174/1381612043452956. [DOI] [PubMed] [Google Scholar]
- 41.Dong H, Ray D, Ren S, et al. Distinct RNA elements confer specificity to flavivirus RNA cap methylation events. J Virol. 2007;81:4412–21. doi: 10.1128/JVI.02455-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hosseini-Yeganeh M, McLachlan AJ. Physiologically based pharmacokinetic model for terbinafine in rats and humans. Antimicrob Agents Chemother. 2002;46:2219–28. doi: 10.1128/AAC.46.7.2219-2228.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paes MV, Pinhao AT, Barreto DF, et al. Liver injury and viremia in mice infected with dengue-2 virus. Virology. 2005;338:236–46. doi: 10.1016/j.virol.2005.04.042. [DOI] [PubMed] [Google Scholar]
- 44.Jessie K, Fong MY, Devi S, et al. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J Infect Dis. 2004;189:1411–8. doi: 10.1086/383043. [DOI] [PubMed] [Google Scholar]