

Abstract
Cell-free protein synthesis is a useful method for preparing proteins for functional or structural analyses. However, batch-to-batch variability with regard to protein synthesis activity remains a problem for large-scale production of cell extract in the laboratory. To address this issue, we have developed a novel procedure for large-scale preparation of bacterial cell extract with high protein synthesis activity. The developed procedure comprises cell cultivation using a fermentor, harvesting and washing of cells by tangential flow filtration, cell disruption with high-pressure homogenizer and continuous diafiltration. By optimizing and combining these methods, ∼100 ml of the cell extract was prepared from 150 g of Escherichia coli cells. The protein synthesis activities, defined as the yield of protein per unit of absorbance at 260 nm of the cell extract, were shown to be reproducible, and the average activity of several batches was twice that obtained using a previously reported method. In addition, combinatorial use of the high-pressure homogenizer and diafiltration increased the scalability, indicating that the cell concentration at disruption varies from 0.04 to 1 g/ml. Furthermore, addition of Gam protein and examinations of the N-terminal sequence rendered the extract prepared here useful for rapid screening with linear DNA templates.
Keywords: cell-free protein synthesis, diafiltration, Escherichia coli, S30 extract, tangential flow filtration
Studies for cell-free protein synthesis have started from examinations of processes underlying protein metabolism and turned to a fundamental question, how genetic information emerges in living cells (1, 2). Intensive studies over two decades have elucidated an overview of translation process through which ribosomes create proteins and of which features of the translation process are conserved among all organisms (3–6). Sophisticated protocols of bacterial cell-free protein synthesis system have been established as coupled transcription-translation systems with exogenous DNA (7, 8). Due to advanced developments, such as a continuous-flow system that provides reaction solutions with essential substrates and energy sources (9) and the PURE (protein synthesis using recombinant elements) system that is reconstituted by purified ribosomes and well-known translation-related factors (10, 11), the cellular machinery of protein synthesis can now be prepared and reconstituted from various organisms in the laboratory or obtained from commercial suppliers (3, 12).
Cell-free protein synthesis systems have become an important tool in laboratory for preparing proteins that are not easily synthesized using in vivo systems, e.g. membrane protein synthesis with nanolipoprotein particles, and for introductions of non-standard amino acids to investigate roles of post-translational modifications (13–18). The PURE system is employed as ribosome-display systems for in vitro evolution of enzyme and for in vitro selection of drugs (17, 19–23). Cell-free protein synthesis systems have also been used in structural genomics studies. For example, through the structural genomics project ‘Protein 3000’ that was conducted from 2003 to 2007, many proteins were synthesized by using an Escherichia coli (E. coli)-based cell-free system, resulting in >1000 protein structures, as determined by nuclear magnetic resonance analysis, being deposited in the Protein Data Bank by RIKEN (Japan) (24–28). Cell-free protein synthesis systems have also been used as a powerful means of preparing samples for X-ray crystallographic studies (29–34). Furthermore, E. coli-based cell-free systems have been successfully used for industrial production of single-chain Fv antibody fragment and cytokines (35, 36). These industrial applications were achieved by using what is known as the ‘Cytomim’ (or ‘mimicking of the cytoplasmic environment’) approach (37, 38). This approach started from the use of genetically modified E. coli strains to modulate amino acid consumption in the cell-free protein synthesis system (39). Then, it was extended to solve the problems associated with energetic metabolism: for example, regeneration of ATP by the intrinsic activity of oxidative phosphorylation in addition to phosphorylation of nucleotide diphosphates can supply enough amount of ATP and GTP required for the cell-free protein synthesis (40, 41). These approaches using genetic information and quantification of metabolites could provide clues for simulation studies to investigate the growth rate and replication accuracy of cells, which could lead to elucidation of homeostatic mechanisms (42–46).
Thus, cell-free protein synthesis systems will be further improved by the development of a novel strain and the adaptation of in vitro conditions to produce proteins in the amounts required for biochemical analyses. Proteins that are difficult to produce using in vivo systems require multiple examinations of cell-free reaction conditions and thus consume a large amount of the cell extract: ∼1,500 ml per year in our group. Therefore, in the present study, we focused on the establishment of a reproducible process for producing bacterial cell extracts with high productivity on a large scale in the laboratory. First, we attempted to use tangential flow filtration (TFF) to harvest and wash cells on a laboratory scale. TFF is used for viral vaccine production and rapid concentration of bacterial culture or marine waters (41, 47, 48). However, only the concentrated cell viability has been examined and no enzymatic activity of the cell extract has been tested; hence, this served as a starting point for designing a new procedure. After an examination of the cell disruption methods, we also applied the TFF technique during dialysis of the cell extract. Thereafter, TFF was utilized to prepare bacterial extracts for cell-free protein synthesis, which contributed to the reproducibility and the scalability of the process.
Materials and Methods
Strains, plasmids and chemicals
E. coli strain BL21 (Stratagene, CA, USA) was transformed with pMINOR2 plasmid, which, like the pMINOR plasmid, encodes a kanamycin resistance gene and tRNA genes cognate with minor codons (49). A T7 RNA polymerase expression vector (50) was kindly provided by Dr. J. Studier (51). The corresponding plasmid pAR1219 (ATCC 39563TM) can be obtained from ATCC (https://www.atcc.org/). Bacto tryptone, yeast extract, NaCl, rhamnose, isopropyl-β-D-thiogalactopyranoside (IPTG), DTT and all the chemicals for cell extract preparation and the cell-free protein synthesis reactions were purchased from Nacalai Tesque Inc. (Japan).
Construction and preparation of vectors
We constructed expression vector for green fluorescent protein (GFP) mutant, pCR2.1-GFPS2, using by the two-step PCR method (52). The second PCR product contains the transcription promoter of T7 RNA polymerase, ‘N11-tag’ (MKDHLIHNHHKHEHAHAEH, affinity tag for nickel resin) (31), protease cleavage sites (TEV and PreScission proteases), GFPS2 gene, stop codons and T7 transcription terminator. The coding region was deposited to DDBJ as ‘GFPS2’ (accession No. LC185343). Furthermore, DNA fragments were amplified with the universal U2 primer at the both termini and ligated into pCR2.1-TOPO vector (Life Technologies, USA) (52). To construct GST-GFPS2 vector, we replaced the N11-tag and protease cleavage sites with the gene of glutathione S-transferase (GST) from Schistosoma japonicum (UniProt ID P08515) in pCR2.1-GFPS2. The N-terminal amino acids of the GST-GFPS2 vector were substituted by using a PCR-mutagenesis method with PrimeStar Max DNA polymerase (TAKARA, Japan) and the restriction enzyme Dpn I (Takara, Japan).
A Gam fragment corresponding to the residues 40–138 (PDB entry 2UUZ: NC_001416.1) was cloned into the pET-11a vector without His-tagging to produce pET-Gam vector.
The plasmids used for the cell-free protein synthesis reactions as templates were purified by using NucleoBond Xtra Midi Plus kit (Macherey-Nagel, Germany) and quantified by measuring absorbance at a wavelength of 260 nm (1 A260 = 50 µg/ml).
Cultivation and harvesting of E. coli cells
The culture medium for E. coli strain BL21/pMINOR2 contained 10 mg/l kanamycin. To establish a large-scale cell culture, at least two successive inoculations are required. Firstly, 5 ml of LB medium was inoculated with a single colony picked from an agar plate and incubated at 30 °C for 7–8 h in an incubator shaker (BR-40LF; TAITEC Co., Japan). After incubation, 100 µl of the 5-ml pre-culture was transferred to 500 ml of 2 × YT medium in a 2-l flask and incubated overnight at 30 °C in an incubator shaker (Innova 4430, New Brunswick Scientific Co., Inc., USA). Next day, the pre-culture was inoculated to 20 l of 2 × YT medium whose optical density at 600 nm (OD600) became ∼0.1. Cultivation was performed in a fermentor (TS-MW-30, TAKASUGI MFG. Co., Ltd., Japan) under the control of temperature (25 °C), aeration (20 l/min), pressure (0.20 MPa, near atmospheric pressure) and agitation (220 rpm). The time courses of pH and dissolved oxygen concentration were measured by using the devices on the fermentor. The optical density, OD600 was assessed manually by using a Novaspec II spectrophotometer (GE Healthcare, Japan).
Cells were harvested after the mid-log phase, when the OD600 exceeded 2.5, by centrifugation at 4,300 rpm (6,100 × g) for 10 min by using a H-12000 rotor with RC12BP (Sorvall) at 4 °C. The harvested cells were then suspended in S30 buffer containing 10 mM Tris–acetate (pH 8.2), 14 mM magnesium acetate, 60 mM potassium acetate and 1 mM DTT with Polytron PT3100 Homogenizer (Kinematica, Switzerland) for 10–20 s at 3,000 rpm. Then the suspension was centrifuged at 6,100 × g for 10 min. The suspension and centrifugation process was repeated three times totally. A final centrifugation was performed at 7,000 rpm (7,500 × g) for 23 min at 4 °C by using an R12A3 rotor with CR21G, (Hitachi Koki Co., Ltd., Japan).
Alternatively, cells were harvested by using a tangential flow filtration (TFF) system comprising a SartoJet Pump and Micofiltration Set (Sartorius, Germany) with a filtration membrane, Sartocon Hydrosart® Cassette (3021860706W-SG; pore size, 0.2 µm, filtration area, 0.6 m2). The 20-l culture was transferred to a stainless-steel tank chilled on ice and then concentrated to 1 l by using the TFF system. The cell suspension was then diluted 2-fold with chilled S30 buffer and then concentrated again; the concentration–dilution cycle was repeated four times. A final centrifugation was performed at 7,000 rpm (7,500 × g) for 23 min at 4 °C by using an R12A3 rotor with CR21G, (Hitachi Koki Co., Ltd., Japan).
In preliminary experiments, a crossflow filtration system, AKTAcrossflow (GE Healthcare) equipped with Sartocon Slice 200 Hydrosart® Cassette (3081860702W–SG; pore size, 0.2 μm, filtration area, 200 cm2) and ProFlux M12 (Merck Millipore, USA) equipped with Sartocon Slice Hydrosart® Cassette (3051860701W–SG; pore size, 0.2 μm, filtration area, 0.1 m2) were used for harvesting from 3-l and 10-l cultures, respectively.
Cells harvested by either method were transferred to a plastic bag (Clean Resealable Bag in Standard Size, AC-3, ASO Co., Ltd.) and pressed by hand in the form of a thin plate and stored at -80 °C. A typical yield from a 20-l culture was 130–150 g cells (Table I).
Table I.
Methods and conditions for cultivation and yields of cells
| Instrument, capacity | Medium, volume | Harvested cells (g) | Yield*a(g/l) | Strain, temperature, cultivation time |
|---|---|---|---|---|
| 2 l flask | 0.5 l, 2xYT | 3.5 | 7.0 | BL21/pMINOR2, 30 °C, 4.5 h |
| 30 l fermentor | 20 l, 2xYT | 154 | 7.7 | BL21/pMINOR2, 25 °C, 6.3 h |
| 30 l fermentor | 20 l, 2xYT | 164 | 8.2 | BL21/pMINOR2, 25 °C, 6.6 h |
| 30 l fermentor | 20 l, 2xYT | 147 | 7.4 | BL21/pMINOR2, 25 °C, 6.4 h |
| 30 l fermentor | 20 l, 2xYT | 142 | 7.1 | BL21/pMINOR2, 25 °C, 6.7 h |
| 30 l fermentor | 20 l, 2xYT | 149 | 7.5 | BL21/pMINOR2, 25 °C, 6.7 h |
| 30 l fermentor | 20 l, 2xYT | 152 | 7.6 | BL21/pMINOR2, 25 °C, 6.8 h |
aCell weight per culture volume.
After use, the filtration membrane, Sartocon Hydrosart® Cassette was flushed on the TFF system with 5 l of 0.9% (w/v) NaCl, and then rinsed with 15 l of distilled water. For further cleanup, 2 l of 0.5 N NaOH heated at 50 °C was circulated for 60 min and the cassette was rinsed with 15 l of distilled water. This alkaline cleanup was repeated twice. After NaOH was rinsed with the distilled water thoroughly, the membrane was removed from the system and stored in 200 ml of 0.1 N NaOH at 4 °C. Before use, the membrane was set on the TFF system and the alkaline cleanup was performed once. Then the system was reinitialized with 5 l of 0.9% (w/v) NaCl. The volumes of solutions and water for cleanup and re-initialization were determined based on the filtration area of the membrane.
Preparation of E. coli S30 extract
The overall method for preparing S30 extract was in accordance with previously published reports (25, 27), except we used Stansted laboratory-scale high-pressure cell disruptor (Stansted Fluid Power Ltd., UK) instead of Multi-beads Shocker (Yasui Kikai). One-hundred grams of frozen cells were suspended in 100 ml of S30 buffer in a 700 ml stainless-steel open container using BeadBeater (Bio Spec Products Inc., USA) without glass beads. The pressure cell was chilled to 2 °C. The suspension was processed at a needle valve pressure of 30 psi and a process pressure of 260 MPa (actual pressure exerted on the sample solution was ∼200 MPa).
After cell disruption, the homogenate was centrifuged at 30,000 × g for 30 min two times at 4 °C by using a JLA 16.250 rotor with Avanti HP30I (Beckman, USA), and the supernatant was transferred to a new tube and incubated with or without pre-incubation buffer (27) at 37 °C for 80 min. The composition of pre-incubation buffer is listed in Supplementary Table SI. After incubation, the extract was centrifuged at 16,000 × g for 30 min and the supernatant was processed by diafiltration using a QuixStand system (QSM-02S; GE Healthcare). The molecular weight cut-off (MWCO) value of the ultrafiltration membrane was 10 kDa (Kvick Lab cassette UFELA0010010ST 56-4113-25; GE Healthcare). Pump output was adjusted to keep the inlet pressure below 0.2 MPa. Discontinuous diafiltration was performed by 4 or 5 times sequential dilution with an equal volume of S30 buffer and concentrated back to the original volume by ultrafiltration. Alternatively, in the case of continuous diafiltration S30 buffer was supplied to the concentrate at the same rate of permeating until the absorbance at 260 nm (A260) of the permeating solution was <1.0.
After diafiltration, the extract was centrifuged at 30,000 × g for 30 min, and the supernatant was dispensed as small aliquots, frozen in liquid nitrogen and stored at −80 °C until use.
Measurement of the protein synthesis activity of the extract
Composition of the cell-free protein synthesis reactions were basically the same as previously reported (31). Dialysis method (53) was performed by using a 24-well plate filled with 1 ml feeding solution per well. The list of chemicals and the concentrations used here is shown in Supplementary Table SII. The concentration of pCR2.1-GFPS2 plasmid (described above) in 30 µl of reaction mixture was 2 ng/µl and the reaction ran at 25 °C for 4 h with shaking at 240 rpm (IKA KS130 basic orbital shaker). After the reaction, 5 µl of reaction mixture was diluted to 200 µl with 20 mM Tris–HCl buffer (pH 8.0) containing 150 mM NaCl. Serial 2-fold dilutions were conducted from the 40-fold diluted solution (200 µl), and the fluorescence intensity of each dilution at a wavelength of 535 nm when excited at a wavelength of 485 nm was measured by ARVO SX multi-label counter (PerkinElmer, USA) with 96-well black flat-bottom microplates (Cat. No. 3650; Corning Inc., USA).
When using a linear DNA template, the reaction solution (30 µl) contained 3.6 OD260 unit of S30 extract and 120 ng of DNA.
Expression and purification of GFPS2 as a quantitative standard
E. coli strain KRX (Promega KK, USA) was transformed with the pCR2.1-GFPS2 plasmid and stored at −80 °C as glycerol stocks. The glycerol stock was inoculated into 100 ml of LB medium containing 100 µg/ml ampicillin and incubated overnight at 37 °C with shaking at 160 rpm. Then, 15 ml of the overnight culture was transferred to 1.5 l of LB medium and cultivated at 37 °C until the OD600 of the culture reached ∼0.6. The expression of GFPS2 was induced by the addition of 0.1% rhamnose to the culture medium. After incubation for further 3 h at 37 °C, cells were harvested by means of centrifugation and stored at −80 °C until use.
To purify expressed GFPS2, frozen cells (wet weight, 5.3 g) were suspended in a lysis buffer containing 20 mM Tris–HCl (pH 7.5), 500 mM NaCl and 20 mM imidazole and disrupted by using an ultrasonic homogenizer VP-30S (duty cycle 50%, 9 repeats at output 6 for 40 s; TAITEC Co., Japan). After centrifugation at 15,000 rpm (32,000 × g) for 20 min at 4 °C by using an R15A rotor with CR22N (Hitachi Koki Co., Ltd.), the supernatant was passed through a syringe filter (pore size, 0.45 µm; Millipore) and loaded onto a HisTrap HP column (bed volume, 5 ml; GE Healthcare) equilibrated with the lysis buffer. The column was rinsed with 20 column volumes of lysis buffer and eluted by a linear gradient of imidazole from 20 to 500 mM in 20 column volumes. The fractions containing GFPS2 were pooled and dialyzed against 20 mM Tris–HCl buffer (pH 7.5) containing 100 mM NaCl and 5 mM 2-mercaptoethanol (2-ME). After dialysis, the protein solution was diluted 2-fold in 20 mM Tris–HCl buffer (pH 8.0) containing 5 mM 2-ME and loaded onto a HiTrap Q column (bed volume, 5 ml; GE Healthcare) equilibrated with 20 mM Tris–HCl buffer (pH 8.0) containing 95 mM NaCl and 5 mM 2-ME. The column was washed with 4 column volumes of the same buffer, and eluted with a linear gradient of 95–200 mM NaCl in 15 column volumes. The fraction containing GFPS2 was concentrated by using an ultrafiltration unit (AmiconUltra; MWCO, 10 kDa; Merck Millipore, Germany) and further purified by means of size exclusion chromatography with HiLoad 16/60 Superdex75 column (GE Healthcare) equilibrated with a buffer of 20 mM Tris–HCl (pH 8.0) containing 150 mM NaCl. The purified protein was concentrated to 1 mg/ml, frozen in liquid nitrogen and stored at −80 °C until use. The typical yield of GFPS2 was 24 mg from 5.3 g of cells (1.5-l culture).
Expression and purification of Gam protein
E. coli strain Rosetta (DE3) was transformed with pET-Gam and stored at −80 °C as glycerol stocks. The glycerol stock was inoculated into 100 ml of LB medium containing 100 µg/ml ampicillin and incubated overnight at 37 °C with shaking at 160 rpm. Fifteen ml of the overnight culture was transferred to 1.5 l of LB medium in a 5-l baffled flask, and cultivated at 37 °C until the OD600 of the culture reached ∼0.7. After addition of 0.5 mM IPTG, the culture was further incubated overnight at 20 °C. The cells were harvested by centrifugation at 7,500 × g for 20 min at 4 °C and stored at −80 °C until use.
To purify expressed Gam protein, frozen cells (wet weight, 10.6 g) were suspended in 35 ml of a lysis buffer comprising 50 mM Tris–HCl (pH 8.0), 200 mM NaCl, 1 mM EDTA, 1 mM DTT and 10% sucrose and then disrupted by using an ultrasonic homogenizer VP-30S (duty cycle, 50%, 5 repeats at output 7 for 2 min). The cell lysate was centrifuged at 32,000 × g for 30 min at 4 °C. The supernatant was added to 65% saturated ammonium sulfate, and the solution was stirred for 30 min at 4 °C. Then, the solution was centrifuged at 32,000 × g for 30 min at 4 °C and the precipitate was dissolved in 35 ml of the lysis buffer. After 4-h dialysis against the lysis buffer, the protein solution was loaded onto HiTrap Q column (bed volume, 5 ml) equilibrated with 50 mM Tris–HCl buffer (pH 8.0) containing 2 mM DTT. After washing with 20 column volumes of 50 mM Tris–HCl buffer (pH 8.0) containing 100 mM NaCl and 2 mM DTT, Gam protein was eluted with a linear gradient of 100–500 mM NaCl in 15 column volumes. The fractions were pooled, diluted 3-fold with 50 mM Tris–HCl buffer (pH 8.0) containing 2 mM DTT, and loaded onto a Mono Q 10/100 GL column (GE Healthcare). After washing with 4 column volumes of the same buffer containing 200 mM NaCl, Gam protein was eluted with a linear gradient of 200–450 mM NaCl in 15 column volumes. The eluted fractions were concentrated by using an ultrafiltration unit (AmiconUltra; MWCO, 10 kDa). Further purification for homogeneity was performed by using a HiLoad 16/60 Superdex 75 column equilibrated with 50 mM Tris–HCl (pH 7.5) buffer containing 200 mM NaCl, and 2 mM DTT. The dimeric Gam fractions estimated from the retention time were collected and concentrated to 12 mg/ml, frozen in liquid nitrogen, and stored at −80 °C until use. The typical yield of Gam was 9.8 mg from 10.6 g of cells (1.5-l culture).
Expression and purification of T7 RNA polymerase
Preparation of T7 RNA polymerase (T7RNP) was conducted as previously reported with minor modifications (54). E. coli strain BL21-Gold(DE3) was transformed with pAR1219. Colonies were picked from an LB agar plate and inoculated in 5 ml of LB medium at 37 °C. After a few hours incubation, the 5-ml pre-culture was transferred to 6 l of LB medium and cultivated at 37 °C until the OD600 of the culture reached ∼0.7. After addition of 0.3 mM IPTG, the culture was incubated for a further 3 h. The cells were harvested by centrifugation at 7,500 × g for 20 min at 4 °C and stored at −80 °C until use.
To purify expressed T7RNP, frozen cells (wet weight, 30.2 g) were suspended in 90 ml of lysis buffer containing 20 mM Tris–HCl (pH 8.0), 20 mM NaCl, 2 mM EDTA and 1 mM DTT. The cells were first lysed on ice by using lysozyme solution at a concentration of 0.3 mg/ml for 20 min, after which 10 ml of sodium deoxycholate (0.8% v/v) was added to the suspension. The suspension was set on ice for 20 min and then disrupted by using an ultrasonic homogenizer VP-30S (duty cycle 50%, 6 repeats at output control 7 for 30 s). The homogenate was centrifuged at 30,000 × g for 20 min at 4 °C and the supernatant (∼125 ml) was added to 20 ml of 2 M ammonium sulfate. After the solution was brought to 200 ml with lysis buffer, 20 ml of polyethylenimine (10% v/v) was added, and the mixture was stirred at 4 °C for 20 min. After centrifugation at 30,000 × g for 20 min, the supernatant was mixed with 0.82 volume of cold saturated ammonium sulfate and stirred for 15 min at 4 °C. The suspension was centrifuged at 30,000 × g for 20 min and the collected precipitate containing T7RNP was dissolved in 60 ml of buffer C [20 mM sodium phosphate (pH 7.7), 1 mM EDTA (pH 8.0), 5% glycerol and 1 mM DTT] containing 100 mM NaCl. Benzamidine (1 mM) and bacitracin (0.01 mg/ml) were then added to the solution, and the protein solution was dialyzed against 3 l of buffer C containing 100 mM NaCl for overnight at 4 °C. After centrifugation at 30,000 × g for 20 min, the supernatant was diluted with an equal volume of buffer C and loaded onto a HiLoad 26/10 SP Sepharose HP column (GE Healthcare) equilibrated with buffer C containing 100 mM NaCl. Elution was performed by using a linear gradient of 70–170 mM NaCl in 5 column volumes. The fractions containing T7RNP were pooled and concentrated to 40 ml by using an ultrafiltration unit (Amicon Ultra-15; MWCO, 30 kDa), and then dialyzed against 1 l of buffer C containing 100 mM NaCl and 50% glycerol at 4 °C for overnight. After dialysis, the concentration of the purified T7RNP was adjusted to 10 mg/ml and its transcription activity was checked by GFPS2 production in the cell-free system. The purified T7RNP was frozen in liquid nitrogen and stored at −80 °C until use. The typical yield of T7RNP was 470 mg of protein from 30 g of cells (6-l culture).
PCR assay for quantification of linear DNA templates
To quantify the amount of linear DNA templates remaining in the cell-free reaction mixture, a quantitative PCR assay was performed by using SYBR Green Realtime PCR Master Mix-Plus (Code No. QPK-211; TOYOBO, Japan). The 91-residue fragment region of pCR2.1-GFPS2, which is upstream of the initiation codon containing the T7RNP promoter, was amplified by using QuantStudio6 Flex Real-Time PCR system (Applied Biosystems, USA).
Cell-free reactions were stopped by 100-fold dilution with distilled water followed by boiling at 95 °C for 5 min. Samples were stored at −20 °C until analysis. Each reaction mixture (20 µl) contained 0.2 µl of sample solution, 10 µl of SYBR Green Realtime PCR Master Mix-Plus, 2 µl of Plus solution, 1.2 µl of 10 µM forward primer (5′-ATTGTGCTTCGCATGATTACG-3′), 1.2 µl of 10 µM reverse primer (5′-TTCTAGAGGGAAACCGTTGTGG-3′) and water. The linear DNA templates were denatured at 95 °C for 20 s and then amplified for data collection with 40 cycles at 95 °C for 1 s then 60 °C for 20 s.
Results and Discussions
Optimization of cultivation condition of 20 l culture
Small-scale cultivation (0.5 l) using a 2-l flask and shaking is a well-established method for preparing E. coli cell extract (25, 27). Therefore, we first determined the optimal cultivation conditions using a fermentor to obtain comparable yield with a 2-l flask cultivation. In the early log phase of cultivation at 30 °C, the growth curve of 2-l flask cultivation by using a shaker at 160 rpm was similar to that obtained by using a fermentor (aeration, 20 l/min; agitation, 220 rpm) (Fig. 1A and B). However, after the middle log phase, the concentration of dissolved oxygen (DO) in the fermentor rapidly decreased, probably because of the rapid increase in the number of E. coli cells, which would reduce growth in the late-middle log phase and make it difficult to harvest cells at the same condition every time. To address this issue, we lowered the cultivation temperature to 25 °C to prolong the growth condition until the late log phase. Although cultivation at lower temperatures slowed the growth rate, the culture condition in the late-log phase became reproducible (Fig. 1C). In practice, harvesting according to the OD600 exceeding 2.5 yielded almost the same weight of cells in repeated cultivations (Table I). The 2.5 OD600 is usable as an indicator for the mid-late log phase. The pH of the culture medium was comparable between the two temperatures examined.
Fig. 1.

Growth curves of Escherichia coli strain BL21/pMINOR2 under different conditions. (A) Growth curve of a culture of E. coli grown in 0.5 l of 2× YT medium in a 2-l flask at 30 °C with shaking. The doubling time in the mid-log phase was 52 min. (B) Growth curves of two cultures of E. coli grown in 20 l of 2× YT medium in a 30-l fermentor at 30 °C. Optical density of the culture at 600 nm (OD600) and concentration of dissolved oxygen (DO) are shown as circles and triangles, respectively. The doubling times in the mid-log phase of the two cultures were 57 min (open circles) and 48 min (filled circles). (C) Growth curves of three cultures of E. coli grown in 20 l of 2× YT in a 30-l fermentor at 25 °C. The doubling times in the mid-log phase of the three cultures (open, closed and gray circles) were all ∼70 min.
Utilization of tangential flow filtration for harvesting and washing cells
Next, we optimized the process of harvesting and washing cells, which is traditionally accomplished by repeated centrifugation (25, 27). Because tangential flow filtration (TFF) is a well-established technique for the rapid concentration of bacterial culture or marine waters (48), we attempted to use it to harvest and wash cells for preparing cell extract with high efficiency of protein synthesis on a laboratory scale. The TFF system comprises a pump, pressure valves and a filtration membrane (Fig. 2A). The required operation time is estimated by considering the flow rate of the permeate through a given area of membrane per unit time (flux), commonly expressed by using the unit l/m2/h (LMH). Flux depends on transmembrane pressure (TMP), which is the difference between the pressures on the upstream (applied pressure) and the downstream (permeate) surfaces of the membrane. It is calculated using the equation TMP = [Pfeed (P1) + Pretentate (P2)]/2 − Ppermeate (P3). In the TFF system, the tangential flow of the circulating cell solution on the surface of the filtration membrane should be large enough, so that the permeate passes continuously through the membrane without clogging it. Circulating flow rate is controlled by the pump output, affected by the module architecture and pressure resistance of intended targets, and indicated as the difference between pressure Pfeed (P1) and Pretentate (P2). It is represented as delta-P. The system is configured by modulating TMP and delta-P, the values of which should be determined experimentally within the range where flux increases linearly with respect to TMP. Once the effect of TMP on the flux of the membrane is characterized, the system can be easily scaled up by increasing the filtration area.
Fig. 2.
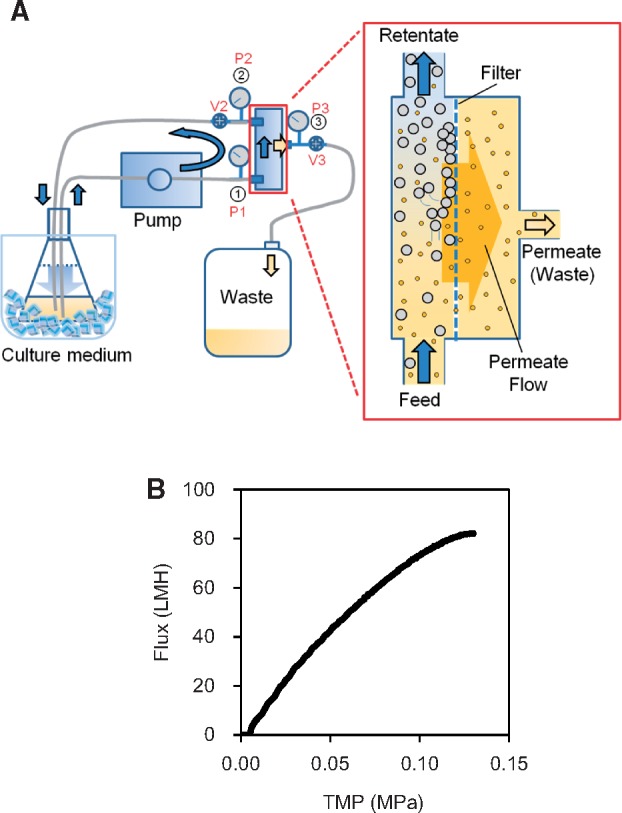
Schematic representation of the tangential flow filtration (TFF) system used in the present study. (A) The system was manually controlled by using the pump output and adjustable valves 2 and 3 (V2 and V3) with modulating transmembrane pressure (TMP). TMP is the difference between the pressure upstream (applied pressure) and downstream (permeate) of the membrane, and is calculated using the equation TMP = [Pfeed (P1) + Pretentate (P2)]/2 − Ppermeate (P3). The three pressure indicators are labeled as P1, P2 and P3. (B) Characterization of the membrane. The permeate flow (flux) of E. coli cell culture (OD600, ∼3.0) was measured by setting delta-P (P1 − P2) at 0.1 MPa and altering TMP in the range of 0–0.13 MPa.
We examined a dose-response relation between flux and TMP of the filtration membrane, Sartocon Slice 200 Hydrosart® Cassette (pore size, 0.2 µm, filtration area, 200 cm2) on a small scale using AKTAcrossflow as shown in Table II. Based on the results, we measured the flux by setting delta-P at 0.1 MPa and altering TMP in the range of 0–0.13 MPa (Fig. 2B). As shown in Fig. 2B, the slope of flux against TMP decreased after TMP exceeded 0.09 MPa. Then we concluded that the Hydrosart® Cassette (pore size, 0.2 mm) should be used by setting TMP at <0.09 MPa. During these examinations, we also measured the pH and conductivity of the permeate to estimate the required amounts of cell washing buffer. To ensure that the TFF system is comparable to the traditional method in terms of the yield of cells and the required time, a 20-l culture whose OD600 was ∼3.0 was divided into two 10-l cultures, which were processed by the traditional repeated centrifugation and TFF methods, respectively. The repeated centrifugation method required about 4 h. On the other hand, TFF system required about 110 min by using ProFlux M12 (Merck Millipore) equipped with Sartocon Slice Hydrosart® Cassette (filtration area, 0.1 m2). The conductivities of the supernatants obtained after the final centrifugation were 6.58 and 7.25 mS/cm by the repeated centrifugation and TFF methods, respectively. The yield of cells by each method was ∼53 g.
Table II.
Characterization of the Hydrosart® Cassette (pore size, 0.2 μm)
| Pump system | AKTA crossflow | ProFlux M12 | SartoJet pump and micofiltration set |
|---|---|---|---|
| Filtration area (m2) | 0.02 | 0.1 | 0.6 |
| delta-P*a(MPa) | 0.05–0.08 | 0.1–0.16 | 0.14 |
| 0.08–0.12 | 0.16 | 0.14 | |
| TMP*a(MPa) | 0.03–0.05 | 0.06 | 0.03 |
| 0.05–0.08 | 0.06 | 0.03 | |
| Flux*a (l/m2/h) | 150 | 100 | 45*b |
| 85 | 100 | 21*b | |
| Operation time (min) | – | 110 | 63*b |
| Culture (l) | – | 10 | 20 |
aValues for the concentration (upper) and washing (lower) steps are represented.
bThe average of five operations.
In practice, we used Sartocon Hydrosart® Cassette (filtration area, 0.6 m2), set the pump output at 70−75% (P1 = 0.14 MPa), opened valve 2 fully, and adjusted valve 3 to maintain the permeate pressure (P3) to a value higher than or equal to 0.05 MPa. The resultant TMP was 0.03 MPa and the total process time was ∼60 min (Table II). Although this TMP setting was lower than that of the preliminary experiment, the flux satisfied the estimated harvesting time. A lower permeate pressure (P3) would briefly increase the flux but would also result in a higher TMP and quicker clogging of the membrane.
With appropriate rinsing and re-initialization (see Materials and Methods), we found that the membrane could be reused at least five times (data not shown). By using TFF, we were able to harvest about 150 g of cells from 20 l of 2× YT medium within 2 h (Tables I and II), and we were able to process three batches of the medium per week for a total of about 450 g of cells.
A scalable method for the preparation of S30 extract
There are two major difficulties in large-scale preparation of cell extract for cell-free protein synthesis. One is scalability of the cell disruption step and the other is efficient dialysis of a large volume of extract. A previously reported method using a Multi-beads Shocker cell disrupter is reproducible, but the throughput capacity is low, requiring 16 tubes dedicated for Multi-beads Shocker to be centrifuged for every 100 ml of S30 extract prepared (25, 27).
An alternative method to disrupt cells on a large scale is to use a bead-based homogenizer, BeadBeater (Bio Spec Products Inc.). A standard protocol for using the BeadBeater to disrupt cells uses an equal volume of a lysis buffer to cells and 3.2-fold of glass beads per batch. However, the minimum required cell weights for complete disruption are 50 g for a large batch and 10–20 g for a small batch. Therefore, this machine cannot be applied for a test-scale preparation from <5 g of cells. Although glass-bead-based homogenization is useful for processing a given amount of cells, this method is unsuitable for intended scalable preparation.
To address the issue of scalability, we examined to use a high-pressure homogenizer to disrupt cells. The Stansted laboratory-scale high-pressure cell disruptor (Stansted Fluid Power Ltd.) is equipped with a 10-ml processing cell. Once the optimal condition for a single stroke has been determined, that processes ∼7 ml of cell solution, the process can be scaled up to the required volume simply by repeating the strokes.
We sought the minimum pressure for adequate cell disruption. When the pellet after the first centrifugation forms layers with different colors, cells would not be fully disrupted because of insufficient pressure. We gradually adjusted the process pressure set by a dial on the front of the machine. For reproducible processing, we determined to set the needle valve pressure at 30 psi and the process pressure at ∼260 MPa. The actual pressure exerted on the sample solution is shown digitally on the front and it should be kept at ∼200 MPa by manual control during cell disruption.
To determine whether high-pressure cell disruption can be scaled, we prepared S30 extracts from 3.5 g of cells. To minimize the loss of extract, cells were suspended in 10-fold the volume of S30 buffer and rinsed out of the disrupter with the same buffer. The extract was 24-fold diluted as compared with the volume in a previously reported method (25, 27). Even under these heavily diluted conditions, after high-speed centrifugation and concentration by dialysis and ultrafiltration, the protein synthesis activity was sufficient (‘GFPS2 yield per cell weigh’ in Table IV).
Table IV.
Comparison of S30 extracts prepared by different methods
| Cell and buffer | Machine condition | S30 extract (vol, conc.) | GFPS2 yield*e [mg/ml] (conc.)*f | GFPS2 yield*g (10−3mg/A260) | GFPS2 yield (mg/g cell) | |
|---|---|---|---|---|---|---|
| 1 | 3.5 g | HPCD*a | 3.1 ml | 1.9 | 15.8 | 5.6 |
| 35 ml | 200 MPa | 400 OD260/ml | (120 OD260/ml) | |||
| 2 | 136 g | HPCD*a | 114 ml | 1.5 | 11.4 | 4.2 |
| 140 ml | 200 MPa | 437 OD260/ml | (131 OD260/ml) | |||
| 3 | 137 g | HPCD*a | 121 ml | 1.0 | 8.0 | 2.9 |
| 140 ml | 200 MPa | 415 OD260/ml | (124 OD260/ml) | |||
| 4 | 145 g | HPCD*a | 112 ml | 1.3 | 10.8 | 3.3 |
| 140 ml | 200 MPa | 400 OD260/ml | (120 OD260/ml) | |||
| 5 | 146 g | HPCD*a | 112 ml | 1.3 | 10.8 | 3.3 |
| 140 ml | 200 MPa | 400 OD260/ml | (120 OD260/ml) | |||
| 6 | 144 g | HPCD*a | 93 ml | 1.3 | 9.8 | 2.8 |
| 140 ml | 200 MPa | 442 OD260/ml | (133 OD260/ml) | |||
| 7 | 144 g | HPCD*a | 93 ml | 1.1 | 8.3 | 2.4 |
| 140 ml | 200 MPa | 442 OD260/ml | (133 OD260/ml) | |||
| 8 | 7 g | MBS*b | 8 ml | 0.4 | 5.3 | 1.5 |
| 8.9 ml | 252 OD260/ml | (76 OD260/ml) | ||||
| 9 | 7 g | MBS*b | 8 ml | 0.4 | 5.4 | 1.5 |
| 8.9 ml | 248 OD260/ml | (74 OD260/ml) | ||||
| 10 | 14 g | MBS*b | 15.5 ml | 0.7 | 8.5 | 2.6 |
| 17.8 ml | 275 OD260/ml | (83 OD260/ml) | ||||
| 11 | 6 g | BB*c | 5 ml*d | 0.2 | 4.0 | 0.6 |
| 6 ml | 3 min | 168 OD260/ml | (50 OD260/ml) | |||
| 12 | 6 g | BB*c | 4 ml*d | 0.4 | 6.6 | 0.9 |
| 6 ml | 4.5 min | 201 OD260/ml | (60 OD260/ml) | |||
| 13 | 6 g | BB*c | 5 ml*d | 0.5 | 7.1 | 1.4 |
| 6 ml | 7.5 min | 236 OD260/ml | (71 OD260/ml) | |||
| 14 | 50 g | BB | 42 ml | 1.1 | 9.3 | 3.1 |
| 50 ml | 12min | 396 OD260/ml | (119 OD260/ml) |
aHPCD means High-pressure cell disrupter.
bMBS means Multi-beads Shocker using at 2,700 rpm for 1.5 min.
cBB means BeadBeater by repeats of the run for 30 s and the interval for 30 s.
dAliquots corresponding to 6 g cells in 20 ml of suspension were sampled from 200-ml suspension including 60 g cells and 190 g glass beads and progressed for further preparation steps of S30 extract (k, l and m).
eGFPS2 yield per reaction volume.
f‘conc.’ means the concentration of S30 extract in the reaction solution.
gGFPS2 yield per amount of S30 extract estimated by absorbance at 260 nm (A260).
Since the S30 extract prepared under these heavily diluted conditions exhibited no loss of production efficiency, we attempted to use the TFF system to dialyze a large volume of S30 extract. In other words, the dialysis step for solvent exchange and removal of low-molecular chemicals was replaced with diafiltration (55, 56). An ultrafiltration membrane (filtration area, 0.11 m2; MWCO, 10 kDa) was applied to a TFF system, and the pump and valves were configured in a manner similar to that during cell harvesting, in fact, valves 2 and 3 were fully opened. More than 2 l of buffer was required for continuous diafiltration of 300 ml of S30 extract (Fig. 3). By using this method, we were able to prepare 90–100 ml of S30 extract from 120 to 150 g cells. Then we are able to scale the process up to prepare 300-ml batches of S30 extract. This method has the advantage that the absorbance of the permeate at a wavelength of 260 nm (A260) can be measured to assess the removal of nucleotides and nucleosides, which ensures batch-to-batch reproducibility with regard to protein synthesis activity.
Fig. 3.

Diafiltration of S30 extract by means of tangential flow filtration. Representative results from a typical experiment using 300 ml of S30 extract preparation are shown. The MWCO value of the ultrafiltration membrane was 10 kDa. Optical density of the permeate was monitored by measuring absorbance at 260 nm. The dotted line shows the cumulative volume of permeate.
Determination of protein synthesis activity of S30 extract
To compare protein production efficiencies of each S30 extract prepared under various conditions easily, we employ one of the green fluorescent protein mutants, GFPS2, which has some amino acid replacements to fold quickly after synthesis on ribosomes, as a standard product (53, 57). Since a linear correlation between GFPS2 concentration and fluorescence intensity was confirmed in the range of 3–60 µg/ml of purified recombinant GFPS2, the yield of GFPS2 in our cell-free system could be quantified by using a 40-fold diluted reaction solution (Fig. 4A). The measurement protocol of GFPS2 production is described in Materials and Methods.
Fig. 4.

Protein productivity of S30 extract. (A) Fluorescence intensities of a dilution series of purified recombinant GFPS2. Fluorescence intensities at 535 nm when excited at 485 nm were calculated as 9 points average of 0.1 s measurement. (B) Protein synthesis activities of S30 extracts prepared by using a BeadBeater (white) and the high-pressure cell disrupter (gray), as assessed by determining yields of green fluorescence protein (GFPS2). (C) Relationship between GFPS2 synthesis activity and the amount of S30 extract in the reaction solution (30 µl), as assessed by using optical density units (OD260). The reactions ran for 4 h. (D) Chloramphenicol acetyltransferase (CAT) assay of S30 extracts prepared by using a BeadBeater (white) and the high-pressure cell disrupter (gray). The reaction solution contained 2% PEG8000 additionally as previously reported (53). Each point on the graph represents the average of three independent experiments.
Firstly, the activities of S30 extracts prepared by using BeadBeater and high-pressure cell disruption methods were compared as shown in Fig. 4B. The S30 extract prepared by both methods exhibited sufficient activities. However, concerning the reaction time for the S30 extract prepared by the present new method, when the cell-free reactions were left to run for longer than 8-h, the yields of GFPS2 obtained were highly variable (Fig. 4B). The S30 extract with enough high activity is supposed to consume requisite compounds within 8 h. Since the similar tendency was observed in the assay of chloramphenicol acetyltransferase (CAT) synthesis (Fig. 4D), we decided to measure the production efficiency of S30 extract by the GFPS2 yield of 4-h reaction to ensure batch-to-batch consistency.
Next, we examined the relationship between GFPS2 synthesis activity and the amount of S30 extract in the reaction solution, which is determined by measuring absorbance at 260 nm (A260) of S30 extract and expressed by using OD260 unit. The yield of GFPS2 increases with increasing S30 extract content in the reaction solution (Fig. 4C). The S30 extract content of ∼6 OD260 unit in 30 µl of reaction solution had almost maximal efficiency. In this case, S30 extract prepared in the concentration of 667 OD260/ml was added at 30% volume of the reaction solution and the final concentration of S30 extract was 200 OD260/ml. Although the S30 extract could be concentrated up to 740 OD260/ml in a small scale, the extract is usually prepared in the concentration of 500 OD260/ml in large scales and the content in the normal reaction condition is 4.5 OD260 unit per 30 µl.
Thirdly, we assessed protein synthesis activities of the S30 extracts by using a CAT assay in which CAT activity is used as an index of the amount of CAT synthesized to compare with that of the extract prepared by Multi-beads Shocker (25, 53). The yields of CAT in both preparations were confirmed to be comparable with that of the S30 extract previously reported (Table III).
Table III.
Comparison of features of preparation methods
| Reference | This study | Kigawa et al. (25) | Kwon and Jewett (45) | Fujiwara and Doi (67) | |
|---|---|---|---|---|---|
| Strain | BL21/pMINOR2 | BL21-Codon Plus-RIL | BL21 StarTM (DE3) | BL21-Codon Plus (DE3)-RIL | |
| Culture vol. for a batch | 500 ml–60 l | 2–8 l | 10 ml–10 l | 1–9 l | |
| Cell conc. at disruption | Variable from 1 to 0.04 g/ml | Fixed at 7 g per 8.9 ml | Optimized at 1 g/ml | Optimized at 0.67 g/ml | |
| Cell disruption | High pressure cell homogenizer | Multi-beads shocker | Q125 Sonicator | LoFT*a | |
| Centrifugation | 30k × g, 30 min | 30k × g, 30 min | 12k × g, 30 min | 25k × g, 60 min | |
| Pre-incubation | 37 °C, 80 min | 37 °C, 80 min with chemicals*b | 37 °C, 60 min | – | |
| Removal of low-molecular chemicals | Diafiltration by TFF system*c | Dialysis | – | Diafiltration*d | |
| Adjustment of conc. of extract | Based on A260/ml | – | – | Based on volume | |
| Protein yield*e | GFPS2 | CAT | CAT | sfGFP | sfGFP |
| batch mode (mg/ml) | – | – | 0.8 (37 °C, 1 h) | 0.5 (37 °C, 4 h) 1.0 (20 h) | 0.5 (29 °C, 14 h) |
| dialysis mode (mg/ml) | 1.1 (25 °C, 4h) | 6.6 (30 °C, 16 h) | 5.4 (30 °C, 16 h)*f | – | – |
aLysozyme treatment, osmotic shock and freeze-thawing method.
bAll chemicals are listed in Supplementary Table SI.
cCentrifugal ultrafiltration was used for <100-ml extract.
d1 ml of extract was diluted by 13 ml of buffer and concentrated by centrifugal ultrafiltration to be 1 ml.
eYields are shown in mg of protein per reaction solution (mg/ml) and the reaction temperature and time are shown in parentheses.
fThe yield of dialysis mode was reported by Seki et al. (53).
Currently, it is unclear whether the addition of pre-incubation buffer is necessary in the incubation step of cell homogenate during preparation of S30 extract (58). Because pre-incubation buffer is expensive, the cost of the process can be reduced if pre-incubation buffer is unnecessary. Therefore, we compared the activities of S30 extracts prepared with and without pre-incubation buffer. Both S30 extracts produced yields of GFPS2 of 1.1–1.2 mg/ml at 4 h, suggesting that pre-incubation buffer does not need to be used in our developed process (data not shown).
Application of our developed process for rapid screening with PCR fragments
One of the major advantages of cell-free protein synthesis is the ability to use PCR products as linear DNA templates. This makes it particularly suitable for screening expressed regions and selecting tags for the detection, purification and folding of synthesized proteins. Unfortunately, the S30 extract prepared by our developed process did not exhibit a high protein synthesis efficiency when PCR products were used. It has been reported that cultivation at low temperatures and the use of glass-bead homogenizer for the preparation of S30 extract reduced the degradation of linear DNA templates (53). Alternatively, the introduction of a stem-loop structure at the 3′-end of the DNA template and the use of an E. coli strain deficient in the endo-ribonuclease E have also been shown to reduce the degradation of linear DNA templates (59).
In the present study, we used recombinant Gam protein, an inhibitor of deoxyribonuclease V, to reduce the degradation of linear DNA and improve protein synthesis activity (60). We purified Gam protein expressed by E. coli and added it to the cell-free reactions. Productivity was markedly improved by the addition of Gam, even when it was added a low concentration (1 µg/ml) (Fig. 5A). To examine the effect of Gam as a nuclease inhibitor, the amount of linear DNA template remaining in the reaction solution was quantified by using a quantitative PCR method as described in Materials and Methods. The addition of Gam reduced the degradation of linear DNA template in a dose-dependent manner (Fig. 5B). In the absence of Gam protein, the almost all of the template DNA was digested within 1 h, whereas 40–50% of it remained when Gam was added. The addition of 10 µg/ml Gam protein is enough for screening experiments terminated at 4 h.
Fig. 5.

Effects of the addition of Gam protein on protein synthesis activity with a linear DNA template. (A) Micro-dialysis reaction (30 µl) containing 120 ng of linear DNA template was performed with 1 ml of feed solution at 25 °C with shaking at 240 rpm (IKA KS130 basic orbital shaker). Purified Gam protein was added at a concentration of 0 (diamonds), 1 (squares), 10 (triangles), or 20 ng/µl (circles). (B) The amount of linear DNA template remaining in the reaction solution was determined by means of quantitative PCR. The linear template of GFPS2 was generated by means of PCR by using the universal U2 primer (52). The first measurement was recorded at 5 min. Each result represents the average of at least three independent experiments.
Fusion with Glutathione S-transferase (GST) is one of the techniques useful for a rapid detection of synthesized proteins and an affinity purification with glutathione ligand (61). However, we were unable to efficiently synthesize GST-GFPS2 fusion protein by using our cell-free system (MSP_tcc_cct in Fig. 6). The addition of ‘N11-tag’, whose sequence is similar to that of the poly-histidine affinity tag (HAT) (31, 62), to the N-terminus exhibited 20-fold increase in the yield of GST-GFPS2 fusion protein (N11GST-GFPS2 in Fig. 6), suggesting that this problem could be overcome by altering the N-terminal sequence of GST. Therefore, the N-terminal three amino acids, MSP, were replaced with MKP, MSD and MKD to examine the productivities. The replacement of the second amino acid, serine, with lysine improved the efficiency either with AAA or AAG codon, and the third amino acid proline substitution for aspartate showed moderate and additional effects on the productivity (Fig. 6). The sequence MKD encoded by ATGAAAGAT improved productivity the most significantly. These results are consistent with previous reports that translation efficiency in vivo and in vitro is affected by the second codon in an open reading frame (63–65). Although an optimum reaction condition for synthesis of a target protein fused to GST would be a little different from those for GST-GFPS2, the replacement of the N-terminal amino acids with MKD is an enough improvement for detection of a target protein in the first screening with linear DNA templates.
Fig. 6.

Improvement in protein synthesis activity by replacement of amino acids in the N-terminus of glutathione S-transferase (GST). The N-terminal amino acid sequence of wild-type GST is MSP and the corresponding nucleotide sequence is atg-tcc-cct. The N-terminal sequences of amino acids and the second and third codons of mutants are shown in uppercase and lowercase, respectively. N11-GST-GFPS2 means the addition of the N11-tag to the N terminus of the GST-green fluorescent protein (GFPS2) fusion protein. Protein yields were estimated by using the fluorescence intensity of GFPS2 fused to the C terminus of GST. Purified GFPS2 was used as the internal standard. The micro-dialysis reaction (30 µl) was performed at 25 °C for 4 h. The reaction solutions contained 120 ng of linear DNA template and 0.6 µg of Gam protein. Each result represents the average of three independent experiments.
Overall discussion
Our new process for the preparation of S30 extract can be used for previously developed cell-free protein synthesis systems as summarized in Table III (27, 28). That is, rapid screening with linear DNA templates and Gam protein can be used to examine an expression region and tags. After the screening, the target sequence could be cloned into the pCR2.1-TOPO vector and the resulting plasmid used either in cell-free reactions or in expression systems using E. coli strain KRX. This S30 extract can be used for antibody fragment production such as scFv and Fab under oxidative condition (66) and the yield is ∼1 mg/ml in a preliminary preparation (data not shown).
We also examined the ribosome content of the S30 extract by measuring A260. If nucleotides and oligonucleotides are not removed completely from the S30 extract during the diafiltration step, the protein synthesis activity, defined as protein yield (mg) per A260, would be significantly reduced. Using TFF in this step makes presents the A260 of the permeate as a direct indicator of the nucleotide concentration that can be used to ensure adequate removal of low molecular weight chemicals and batch-to-batch consistency with regard to the protein synthesis efficiency. S30 extract prepared with our new procedure exhibited ∼2-fold higher activity per cell weight compared with those of Multi-beads Shocker, probably because the high-pressure homogenizer disrupted cells more effectively than did Multi-beads Shocker (Table IV). The average activity of six preparations was 9.9 µg/A260, which corresponded to 3.1 mg/g of cells.
Recently, high-throughput and scalable preparation methods of S30 extract have been reported as shown in Table III (45, 67). Although they appear to be useful, for example, for screening of E. coli strain suitable for cell-free protein synthesis, it is uncertain whether they could be applied for repeated large-scale productions. As being the other approaches, inorganic pyrophosphate, a metabolite of ATP regeneration by creatine phosphate and phosphoenolpyruvate, that inhibits protein synthesis, could be utilized as a phosphate donor to regenerate ATP by addition of maltose as a carbon source (68, 69). It improved the cell-free protein synthesis efficiency in a batch mode reaction significantly. The other group has reported that a decrease of the concentration of magnesium ion leads to 16S rRNA degradation in a cell-free protein synthesis system using E. coli A19 strain (70), which may be related to the accumulation of pyrophosphate (71). Therefore, not only improvement of procedures for cell extract but also further analyses of the metabolic processes during in vitro protein synthesis reaction are needed. Furthermore, we intend to use the S30 extract prepared in this procedure as a basic condition and to expand applicable products by addition of purified enzymes useful for production efficiency and detergents for membrane proteins.
Supplementary Data
Supplementary Data are available at JB Online.
Supplementary Material
Acknowledgements
We would like to thank Asahi Glass Co. Ltd. (Tokyo, Japan) for their assistance with preliminary experiments using the tangential flow filtration systems.
Conflict of Interest
None declared.
Glossary
Abbreviations
- 2-ME
2-mercaptoehanol
- CAT
chloramphenicol acetyltransferase
- DO
dissolved oxygen
- Fab
fragment antigen-binding
- GFP
green fluorescent protein
- GST
Glutathione S-transferase
- IPTG
isopropyl β-D-thiogalactopyranoside
- MWCO
molecular weight cut-off
- OD
optical density
- T7RNP
T7 RNA polymerase
- TEV
tobacco etch virus
- TFF
tangential flow filtration
- TMP
transmembrane pressure
References
- 1. Zamecnik P. C., Frantz I. D. J., Loftfield R. B., Stephenson M. L. (1948) Incorporation in vitro of radioactive carbon from carboxyl-labeled DL-alanine and glycine into proteins of normal and malignant rat livers. J. Biol. Chem. 175, 299–314 [PubMed] [Google Scholar]
- 2. Spirin A.S. (eds.) (2002) Cell-Free Translation Systems, pp. 3–20, Springer, New York [Google Scholar]
- 3. Chong S. (2014) Overview of cell-free protein synthesis: Historic landmarks, commercial systems, and expanding applications. Curr. Protoc. Mol. Biol. 2014, 16.30.1–16.30.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lucas-Lenard J. (1971) Protein biosynthesis. Annu. Rev. Biochem. 40, 409–48 [DOI] [PubMed] [Google Scholar]
- 5. Haselkorn R., Rothman-Denes L. B. (1973) Protein synthesis. Annu. Rev. Biochem. 42, 397–438 [DOI] [PubMed] [Google Scholar]
- 6. Nirenberg M. W., Matthaei J. H. (1961) The dependence of cell-free protein synthesis in E. coli upon naturally occurring or synthetic polyribonucleotides. Proc. Natl. Acad. Sci. U. S. A. 47, 1588–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zubay G. (1973) In vitro synthesis of protein in microbial systems. Annu. Rev. Genet. 7, 267–87 [DOI] [PubMed] [Google Scholar]
- 8. Gold L. M., Schweiger M. (1969) Synthesis of phage-specific alpha- and beta-glucosyl transferases directed by T-even DNA in vitro. Proc. Natl. Acad. Sci. U. S. A. 62, 892–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spirin A. S., Baranov V. I., Ryabova L. A., Ovodov S. Y., Alakhov Y. B. (1988) A continuous cell-free translation system capable of producing polypeptides in high yield. Science 242, 1162–4 [DOI] [PubMed] [Google Scholar]
- 10. Shimizu Y., Inoue A., Tomari Y., Suzuki T., Yokogawa T., Nishikawa K., Ueda T. (2001) Cell-free translation reconstituted with purified components. Nat. Biotechnol. 19, 751–5 [DOI] [PubMed] [Google Scholar]
- 11. Shimizu Y., Kanamori T., Ueda T. (2005) Protein synthesis by pure translation systems. Methods 36, 299–304 [DOI] [PubMed] [Google Scholar]
- 12. Endo Y., Takai K., Ueda T. (eds.) (2010) Cell-Free Protein Production, Humana Press, New York [Google Scholar]
- 13. Quast R. B., Mrusek D., Hoffmeister C., Sonnabend A., Kubick S. (2015) Cotranslational incorporation of non-standard amino acids using cell-free protein synthesis. FEBS Lett. 589, 1703–12 [DOI] [PubMed] [Google Scholar]
- 14. Katzen F., Fletcher J. E., Yang J. P., Kang D., Peterson T. C., Cappuccio J. A., Blanchette C. D., Sulchek T., Chromy B. A., Hoeprich P. D., Coleman M. A., Kudlicki W. (2008) Insertion of membrane proteins into discoidal membranes using a cell-free protein expression approach. J. Proteome Res. 7, 3535–42 [DOI] [PubMed] [Google Scholar]
- 15. Katzen F., Peterson T. C., Kudlicki W. (2009) Membrane protein expression: no cells required. Trends Biotechnol. 27, 455–60 [DOI] [PubMed] [Google Scholar]
- 16. Oza J. P., Aerni H. R., Pirman N. L., Barber K. W., ter Haar C. M., Rogulina S., Amrofell M. B., Isaacs F. J., Rinehart J., Jewett M. C. (2015) Robust production of recombinant phosphoproteins using cell-free protein synthesis. Nat. Commun. 6, 8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuruma Y., Ueda T. (2015) The PURE system for the cell-free synthesis of membrane proteins. Nat. Protoc. 10, 1328–44 [DOI] [PubMed] [Google Scholar]
- 18. Wakamori M., Fujii Y., Suka N., Shirouzu M., Sakamoto K., Umehara T., Yokoyama S. (2015) Intra- and inter-nucleosomal interactions of the histone H4 tail revealed with a human nucleosome core particle with genetically-incorporated H4 tetra-acetylation. Sci. Rep. 5, 17204.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fujii S., Matsuura T., Sunami T., Kazuta Y., Yomo T. (2013) In vitro evolution of α-hemolysin using a liposome display. Proc. Natl. Acad. Sci. U. S. A. 110, 16796–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fujii S., Matsuura T., Sunami T., Nishikawa T., Kazuta Y., Yomo T. (2014) Liposome display for in vitro selection and evolution of membrane proteins. Nat. Protoc. 9, 1578–91 [DOI] [PubMed] [Google Scholar]
- 21. Passioura T., Katoh T., Goto Y., Suga H. (2014) Selection-based discovery of druglike macrocyclic peptides. Ann. Rev. Biochem. 83, 727–52 [DOI] [PubMed] [Google Scholar]
- 22. Maini R., Umemoto S., Suga H. (2016) Ribosome-mediated synthesis of natural product-like peptides via cell-free translation. Curr. Opin. Chem. Biol. 34, 44–52 [DOI] [PubMed] [Google Scholar]
- 23. Lyukmanova E. N., Shenkarev Z. O., Khabibullina N. F., Kopeina G. S., Shulepko M. A., Paramonov A. S., Mineev K. S., Tikhonov R. V., Shingarova L. N., Petrovskaya L. E., Dolgikh D. A., Arseniev A. S., Kirpichnikov M. P. (2012) Lipid–protein nanodiscs for cell-free production of integral membrane proteins in a soluble and folded state: Comparison with detergent micelles, bicelles and liposomes. Biochim. Biophys. Acta 1818, 349–58 [DOI] [PubMed] [Google Scholar]
- 24. Yokoyama S. (2003) Protein expression systems for structural genomics and proteomics. Curr. Opin. Chem. Biol. 7, 39–43 [DOI] [PubMed] [Google Scholar]
- 25. Kigawa T., Yabuki T., Matsuda N., Matsuda T., Nakajima R., Tanaka A., Yokoyama S. (2004) Preparation of Escherichia coli cell extract for highly productive cell-free protein expression. J. Struct. Funct. Genomics 5, 63–8 [DOI] [PubMed] [Google Scholar]
- 26. Matsuda T., Koshiba S., Tochio N., Seki E., Iwasaki N., Yabuki T., Inoue M., Yokoyama S., Kigawa T. (2007) Improving cell-free protein synthesis for stable-isotope labeling. J. Biomol. NMR 37, 225–9 [DOI] [PubMed] [Google Scholar]
- 27. Kigawa T. (2010) Cell-free protein preparation through prokaryotic transcription-translation methods. Methods Mol. Biol. 607, 1–10 [DOI] [PubMed] [Google Scholar]
- 28. Terada T., Yokoyama S. (2015) Escherichia coli Cell-Free Protein Synthesis and Isotope Labeling of Mammalian Proteins in Methods in Enzymology (Kelman Z., ed.) Vol. 565, pp. 311–345, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 29. Muramatsu T., Kim Y. T., Nishii W., Terada T., Shirouzu M., Yokoyama S. (2013) Autoprocessing mechanism of severe acute respiratory syndrome coronavirus 3C-like protease (SARS-CoV 3CLpro) from its polyproteins. FEBS J. 280, 2002–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tanabe H., Motoyama K., Ikeda M., Wakiyama M., Terada T., Ohsawa N., Hosaka T., Hato M., Fujii Y., Nakamura Y., Ogasawara S., Hino T., Murata T., Iwata S., Okada-Iwabu M., Iwabu M., Hirata K., Kawano Y., Yamamoto M., Kimura-Someya T., Shirouzu M., Yamauchi T., Kadowaki T., Yokoyama S. (2015) Expression, purification, crystallization, and preliminary X-ray crystallographic studies of the human adiponectin receptors, AdipoR1 and AdipoR2. J. Struct. Funct. Genomics 16, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shinoda T., Shinya N., Ito K., Ishizuka-Katsura Y., Ohsawa N., Terada T., Hirata K., Kawano Y., Yamamoto M., Tomita T., Ishibashi Y., Hirabayashi Y., Kimura-Someya T., Shirouzu M., Yokoyama S. (2016) Cell-free methods to produce structurally intact mammalian membrane proteins. Sci. Rep. 6, 30442.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shimizu H., Miyazaki H., Ohsawa N., Shoji S., Ishizuka-Katsura Y., Tosaki A., Oyama F., Terada T., Sakamoto K., Shirouzu M., Sekine S., Nukina N., Yokoyama S. (2016) Structure-based site-directed photo-crosslinking analyses of multimeric cell-adhesive interactions of voltage-gated sodium channel β subunits. Sci. Rep. 6, 26618.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Terada T., Murata T., Shirouzu M., Yokoyama S. (2014) Cell-free expression of protein complexes for structural biology. Methods Mol. Biol. 1091, 151–9 [DOI] [PubMed] [Google Scholar]
- 34. Suzuki K., Mizutani K., Maruyama S., Shimono K., Imai F. L., Muneyuki E., Kakinuma Y., Ishizuka-Katsura Y., Shirouzu M., Yokoyama S., Yamato I., Murata T. (2016) Crystal structures of the ATP-binding and ADP-release dwells of the V1 rotary motor. Nat. Commun. 7, 13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kanter G., Yang J., Voloshin A., Levy S., Swartz J. R., Levy R. (2007) Cell-free production of scFv fusion proteins: an efficient approach for personalized lymphoma vaccines. Blood 109, 3393–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zawada J. F., Yin G., Steiner A. R., Yang J., Naresh A., Roy S. M., Gold D. S., Heinsohn H. G., Murray C. J. (2011) Microscale to manufacturing scale-up of cell-free cytokine production-a new approach for shortening protein production development timelines. Biotechnol. Bioeng. 108, 1570–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jewett M. C., Swartz J. R. (2004) Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnol. Bioeng. 86, 19–26 [DOI] [PubMed] [Google Scholar]
- 38. Swartz J. R., Jewett M. C., Woodrow K. A. (2004) Cell-free protein synthesis with prokaryotic combined transcription-translation. Methods Mol. Biol. 267, 169–82 [DOI] [PubMed] [Google Scholar]
- 39. Calhoun K. A., Swartz J. R. (2006) Total amino acid stabilization during cell-free protein synthesis reactions. J. Biotechnol. 123, 193–203 [DOI] [PubMed] [Google Scholar]
- 40. Jewett M. C., Miller M. L., Chen Y., Swartz J. R. (2009) Continued protein synthesis at low [ATP] and [GTP] enables cell adaptation during energy limitation. J. Bacteriol. 191, 1083–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cai Q., Hanson J. a., Steiner A. R., Tran C., Masikat M. R., Chen R., Zawada J. F., Sato A. K., Hallam T. J., Yin G. (2015) A simplified and robust protocol for immunoglobulin expression in Escherichia coli cell-free protein synthesis systems. Biotechnol. Prog. 31, 823–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jewett M. C., Calhoun K. a., Voloshin A., Wuu J. J., Swartz J. R. (2008) An integrated cell-free metabolic platform for protein production and synthetic biology. Mol. Syst. Biol. 4, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tadmor A. D., Tlusty T. (2008) A coarse-grained biophysical model of E. coli and its application to perturbation of the rRNA operon copy number. PLoS Comput. Biol. 4, e1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weiße A. Y., Oyarzún D. A., Danos V., Swain P. S. (2015) Mechanistic links between cellular trade-offs, gene expression, and growth. Proc. Natl. Acad. Sci. U. S. A. 112, E1038–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kwon Y. C., Jewett M. C. (2015) High-throughput preparation methods of crude extract for robust cell-free protein synthesis. Sci. Rep. 5, 8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Caschera F., Lee J. W., Ho K. K. Y., Liu A. P., Jewett M. C. (2016) Cell-free compartmentalized protein synthesis inside double emulsion templated liposomes with in vitro synthesized and assembled ribosomes. Chem. Commun. 52, 5467–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tapia F., Vázquez-Ramírez D., Genzel Y., Reichl U. (2016) Bioreactors for high cell density and continuous multi-stage cultivations: options for process intensification in cell culture-based viral vaccine production. Appl. Microbiol. Biotechnol. 100, 2121–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tanny G. B., Mirelman I. D., Pistolet T. (1980) Filtration technique. Appl. Environ. Microbiol. 40, 269–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chumpolkulwong N., Sakamoto K., Hayashi A., Iraha F., Shinya N., Matsuda N., Kiga D., Urushibata A., Shirouzu M., Oki K., Kigawa T., Yokoyama S. (2006) Translation of “rare” codons in a cell-free protein synthesis system from Escherichia coli. J. Struct. Funct. Genomics 7, 31–6 [DOI] [PubMed] [Google Scholar]
- 50. Davanloo P., Rosenberg a. H., Dunn J. J., Studier F. W. (1984) Cloning and expression of the gene for bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 81, 2035–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ueda T., Tohda H., Chikazumi N., Eckstein F., Watanabe K. (1991) Phosphorothioate-containing RNAs show mRNA activity in the prokaryotic translation systems in vitro. Nucleic Acids Res. 19, 547–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yabuki T., Motoda Y., Hanada K., Nunokawa E., Saito M., Seki E., Inoue M., Kigawa T., Yokoyama S. (2007) A robust two-step PCR method of template DNA production for high-throughput cell-free protein synthesis. J. Struct. Funct. Genomics 8, 173–91 [DOI] [PubMed] [Google Scholar]
- 53. Seki E., Matsuda N., Yokoyama S., Kigawa T. (2008) Cell-free protein synthesis system from Escherichia coli cells cultured at decreased temperatures improves productivity by decreasing DNA template degradation. Anal. Biochem. 377, 156–61 [DOI] [PubMed] [Google Scholar]
- 54. Zawadzki V., Gross H. J. (1991) Rapid and simple purification of T7 RNA polymerase. Nucleic Acids Res. 19, 1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Meier K., Carstensen F., Scheeren C., Regestein L., Wessling M., Büchs J. (2014) In situ product recovery of single-chain antibodies in a membrane bioreactor. Biotechnol. Bioeng. 111, 1566–76 [DOI] [PubMed] [Google Scholar]
- 56. Neal G., Francis R., Shamlou P. A., Keshavarz-Moore E. (2004) Separation of immunoglobulin G precipitate from contaminating proteins using microfiltration. Biotechnol. Appl. Biochem. 39, 241–8 [DOI] [PubMed] [Google Scholar]
- 57. Ito Y., Suzuki M., Husimi Y. (1999) A novel mutant of green fluorescent protein with enhanced sensitivity for microanalysis at 488 nm excitation. Biochem. Biophys. Res. Commun. 264, 556–60 [DOI] [PubMed] [Google Scholar]
- 58. Liu D. V., Zawada J. F., Swartz J. R. (2005) Streamlining Escherichia coli S30 extract preparation for economical cell-free protein synthesis. Biotechnol. Prog. 21, 460–5 [DOI] [PubMed] [Google Scholar]
- 59. Ahn J. H., Chu H. S., Kim T. W., Oh I. S., Choi C. Y., Hahn G. H., Park C. G., Kim D. M. (2005) Cell-free synthesis of recombinant proteins from PCR-amplified genes at a comparable productivity to that of plasmid-based reactions. Biochem. Biophys. Res. Commun. 338, 1346–52 [DOI] [PubMed] [Google Scholar]
- 60. Sitaraman K., Esposito D., Klarmann G., Le Grice S. F., Hartley J. L., Chatterjee D. K. (2004) A novel cell-free protein synthesis system. J. Biotechnol. 110, 257–63 [DOI] [PubMed] [Google Scholar]
- 61. GST Gene Fusion System Handbook, Amersham Biosciences, (2002). http://www.gelifesciences.com/file_source/GELS/Service%20and%20Support/Documents%20and%20Downloads/Handbooks/pdfs/GST_gene_fusion_system_handbook.pdf
- 62. Chaga G., Bochkariov D. E., Jokhadze G. G., Hopp J., Nelson P. (1999) Natural poly-histidine affinity tag for purification of recombinant proteins on cobalt(II)-carboxymethylaspartate crosslinked agarose. J. Chromatogr. A 864, 247–56 [DOI] [PubMed] [Google Scholar]
- 63. Sato T., Terabe M., Watanabe H., Gojobori T., Hori-Takemoto C., Miura K. (2001) Codon and base biases after the initiation codon of the open reading frames in the Escherichia coli genome and their influence on the translation efficiency. J. Biochem. 129, 851–60 [DOI] [PubMed] [Google Scholar]
- 64. Zamora-Romo E., Cruz-Vera L. R., Vivanco-Domíed;nguez S., Magos-Castro M. A., Guarneros G. (2007) Efficient expression of gene variants that harbour AGA codons next to the initiation codon. Nucleic Acids Res. 35, 5966–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bandmann N., Nygren P.-A. (2007) Combinatorial expression vector engineering for tuning of recombinant protein production in Escherichia coli. Nucleic Acids Res. 35, e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Matsuda T., Watanabe S., Kigawa T. (2013) Cell-free synthesis system suitable for disulfide-containing proteins. Biochem. Biophys. Res. Commun. 431, 296–301 [DOI] [PubMed] [Google Scholar]
- 67. Fujiwara K., Doi N. (2016) Biochemical preparation of cell extract for cell-free protein synthesis without physical disruption. PLoS One 11, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Caschera F., Noireaux V. (2014) Synthesis of 2.3 mg/ml of protein with an all Escherichia coli cell-free transcription-translation system. Biochimie 99, 162–8 [DOI] [PubMed] [Google Scholar]
- 69. Garamella J., Marshall R., Rustad M., Noireaux V. (2016) The All E. coli TX-TL Toolbox 2.0: A Platform for Cell-Free Synthetic Biology. ACS Synth. Biol. 5, 344–55 [DOI] [PubMed] [Google Scholar]
- 70. Failmezger J., Nitschel R., Sánchez-Kopper A., Kraml M., Siemann-Herzberg M. (2016) Site-specific cleavage of ribosomal RNA in Escherichia coli-based cell-free protein synthesis systems. PLoS One 11, e0168764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim T. W., Kim D. M., Choi C. Y. (2006) Rapid production of milligram quantities of proteins in a batch cell-free protein synthesis system. J. Biotechnol. 124, 373–80 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.