

Abstract
Although primary genomic analysis has revealed that severe acute respiratory syndrome coronavirus (SARS CoV) is a new type of coronavirus, the different protein trees published in previous reports have provided no conclusive evidence indicating the phylogenetic position of SARS CoV. To clarify the phylogenetic relationship between SARS CoV and other coronaviruses, we compiled a large data set composed of 7 concatenated protein sequences and performed comprehensive analyses, using the maximum-likelihood, Bayesian-inference, and maximum-parsimony methods. All resulting phylogenetic trees displayed an identical topology and supported the hypothesis that the relationship between SARS CoV and group 2 CoVs is monophyletic. Relationships among all major groups were well resolved and were supported by all statistical analyses.
In the short amount of time since a novel coronavirus was identified as being the cause of the ongoing outbreak of severe acute respiratory syndrome (SARS) around the world [1], several SARS coronavirus (CoV) isolates have been cloned, and several complete genomic sequences have been determined [2–4]. Preliminary sequence analyses have indicated that SARS CoV is a new type of coronavirus that does not belong to any group of coronaviruses yet characterized [2, 3]. However, the phylogenetic position and origin of SARS CoV remain elusive. Most reported phylogenetic analyses have been based on either individual proteins [2, 3, 5], short nucleotide sequences [1], or whole-genome similarity [6]. Although these analyses have not yielded conflicting results, the phylogenetic relationship between SARS CoV and its relatives remained inconclusive. Whereas most reported phylogenetic trees have placed SARS CoV between group 2 CoVs and group 3 CoVs, a few trees have indicated that there is a relationship between SARS CoV and group 3 CoVs [1–3, 5].
It is notable that both SARS CoV and infectious bronchitis virus (IBV), a group 3 CoV, form long branches in all reported phylogenetic trees, because it implies that theremight have been a problem associated with a long-branch attraction (LBA) artifact during the tree reconstructions [7]. An LBA artifact might be caused by limited taxa, a small number of amino acid or nucleotide positions, or highly variable regions of nucleotide sequences, any of which could generate misleading phylogenetic information during the tree-reconstruction process. To further define the phylogenetic relationship between SARS CoV and other coronaviruses, we took advantage of recently published SARS CoV genome sequences and constructed a single, large data set composed of 3364 well-aligned amino acid positions. Because the 7 proteins used to construct the data set appeared to have different long branches [1–3, 5], the effect of an LBA artifact in tree reconstruction should be minimized. The aim of the present study was to apply reliable analytical methods to the construction of a robust hypothesis about the phylogeny of SARS CoV in relation to other coronaviruses.
Materials and methods. We retrieved the following protein sequences (accession numbers) from GenBank: SARS CoV (NC_004718), human CoV 229E (AF304460), porcine epidemic diarrhea virus (AF353511), transmissible gastroenteritis virus (AJ271965), bovine CoV (AF220295), murine hepatitis virus (AF201929), and IBV (M95169). The amino acid sequences of the 3CLpro, POL, HEL, S, E, M, and N proteins were individually aligned by the Clustral X program (version 1.83). Gaps and unambiguous alignments were excluded from each alignment. After a parsimony-based partition-homogeneity test revealed no significant incongruence between trees derived from different proteins, 7 protein alignments were concatenated to form a large data set of 3364 aa positions for subsequent phylogenetic analysis. The parsimony-based partition-homogeneity test was performed by the PAUP* program [8]. The homogeneity of the structural proteins (S, E, M, and N) and of the enzymatic (3CLpro, POL, and HEL) proteins, as well as the homogeneity of the 2 protein categories (structural vs. enzymatic), were tested using a heuristic search algorithm with 100 replicates; it showed no statistically significant incongruence (among enzymatic proteins, P = 1; among structural proteins, P = .26; between enzymatic proteins and structural proteins, P = 1).
For phylogenetic analysis, first the Tree-Puzzle program (version 5.0) was used to generate approximate quartet-likelihood trees, with 1000 puzzling steps [9]. Parameters were estimated on the basis of the topology of a neighbor-joining tree, and the amino acid frequencies were estimated on the basis of the concatenated protein data set, by use of a Jones-Taylor-Thornton (JTT) model of amino acid substitution, a model that included the consideration of rate heterogeneity (i.e., the fraction of invariance and 4-rate gamma distributions [JTT + Finv + Г]). Parameters that had been established on the basis of the puzzling analysis were then applied to a true maximum-likelihood (ML) analysis by the ProML program included in the PHYLIP package [10], with the sequence input order randomized and with global rearrangements enabled during the tree search. In addition, phylogenetic trees were also reconstructed using the aforementioned JTT model of amino acid substitution, by a Bayesian-inference (BI) method and the MrBayes program (version 3.0) [11]. A total of 100,000 generations of searches were performed, with 4 chains running simultaneously. Stable ML values were quickly reached before 1000 generations of searches, indicating that the Markov chain Monte Carlo analysis had been allowed to run for sufficient generations. Posterior probabilities at tree nodes were obtained by calculating the consensus tree from the best 901 BI trees, by the 50% majority ruling method. The bootstrapping test was performed, with 1000 replicates, using the maximum-parsimony (MP) method, by the PAUP* program [8]; the input order of each search was randomized, in 100 replicates. A full heuristic algorithm was used to search for the best trees, and tree-bisection reconnection was applied for branch swapping. The statistical significance for the difference between each resulting best tree and all alternative trees were tested by both the Kishino-Hasegawa (KH) method [12] and the Shimodaira-Hasegawa (SH) method [13].
Results. The data set was composed of 7 concatenated protein sequences (i.e., 3CLpro, POL, HEL, S, E, M, and N) obtained from 7 coronavirus isolates, and a partition-homogeneity test [14] revealed no significant incongruence between phylogenetic trees derived from different proteins. The best trees were inferred from the data set by the ML, BI, and MP methods [8–11], all of which yielded the same tree topology and supported the hypothesis that the relationship between SARS CoV and group 2 CoVs is monophyletic (figure 1). The statistical supporting values at all nodes were 100%, by ML quartet-puzzling, BI posterior-probability, and MP bootstrapping analyses. Pairwise comparison of protein distances, corrected by the ML method, also showed that the intergroup distance between SARS CoV and group 2 CoVs was the shortest, compared with those between SARS CoV and other coronaviruses (table 1). The SARS CoV + group 2 CoVs clade was subsequently joined by IBV, a group 3 CoV. The separation of SARS CoV and IBV was more evident in the trees resulting from the present study than in previously reported protein trees, in which SARS CoV and IBV were either minimally separated by very short branches or artificially joined at deep branches [1–3]. The hypothesis that the relationship between SARS CoV and group 2 CoVs is monophyletic was fully supported by the KH test, in which the ML values of all the other 944 possible trees were shown to be significantly worse than that of the present best tree. When a more conservative SH test was employed, only 19 suboptimal trees did not show significant differences in their ML values, compared with the tree shown in figure 1. Among these 19 trees, 13 supported the hypothesis that the relationship between SARS CoV and group 2 CoVs is monophyletic, and only 6 either placed SARS CoV at the base of group 2 CoVs/group 3 CoVs or identified it as a sister to group 3 CoVs. Therefore, the SH test did not reject the best tree but, rather, implied that there are strong links between SARS CoV, group 2 CoVs, and group 3 CoVs. In addition, the hypothesis that the relationship between SARS CoV and group 2 CoVs is monophyletic is supported by a recently reported phylogenetic analysis using the replicase gene [15].
Figure 1.

Unrooted best maximum-likelihood (ML) tree (-lnL = 42,346.53), inferred from 3364 amino acid positions of 7 concatenated protein sequences obtained from 7 taxa, including severe acute respiratory syndrome coronavirus (SARS CoV) and 6 other coronaviruses. The supporting values—by ML quartet-puzzling (MLQP), Bayesian-inference posterior-probability (BIPP), and maximum-parsimony bootstrapping analyses (MPPA)—are indicated. BCoV, bovine coronavirus; HCoV-229E, human coronavirus 229E; IBV, infectious bronchitis virus; MHV, murine hepatitis virus; PEDV, porcine epidemic diarrhea virus; TGEV, transmissible gastroenteritis virus.
Table 1.
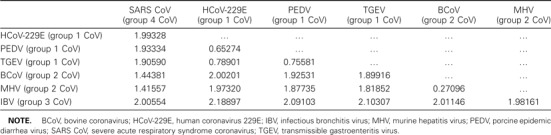
Pairwise distance among coronaviruses (CoVs), corrected by maximum-likelihood model.
Discussion. Despite the evidence supporting a monophyletic relationship between SARS CoV and group 2 CoVs, the data currently available still support the preliminary conclusion that SARS CoV might be a new type of coronavirus (i.e., group 4). The problem of the origin of SARS CoV cannot be resolved here—it may require the identification and sequencing of additional, closely related coronaviruses from humans and/or animals. Nonetheless, the establishment of a solid phylogenetic relationship between SARS CoV and other coronaviruses may provide us with valuable information to use in the development of vaccines and therapeutics and may, in the near future, help shed light on the true origin of SARS CoV.
Acknowledgments
We thank all investigators who were involved in the identification, cloning, and sequencing of the entire SARS CoV genome.
References
- 1.Drosten C, Gunther S, Preiser W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1947–58. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Marra MA, Jones SJ, Astell CR, et al. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 3.Rota PA, Oberste MS, Monroe SS, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–9. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 4.Leung FC. Hong Kong SARS sequence. Science. 2003;301:309–10. doi: 10.1126/science.301.5631.309c. [DOI] [PubMed] [Google Scholar]
- 5.Ksiazek TG, Erdman D, Goldsmith CS, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–66. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 6.Ruan YJ, Wei CL, Ee AL, et al. Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. Lancet. 2003;361:1779–85. doi: 10.1016/S0140-6736(03)13414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Philippe H, Laurent J. How good are deep phylogenetic trees? Curr Opin Genet Dev. 1998;8:616–23. doi: 10.1016/s0959-437x(98)80028-2. [DOI] [PubMed] [Google Scholar]
- 8.Swofford DL. PAUP*: phylogenetic analysis using parsimony (*and other methods) Sunderland, Massachusetts: Sinauer Associates; 2000. Versions 4.0b4a and 4.0b6. [Google Scholar]
- 9.Strimmer K, von Haeseler A. Quartet puzzling: a quartet maximum-likelihood method for reconstructing tree topologies. Mol Biol Evol. 1996;13:964–9. [Google Scholar]
- 10.Felsenstein J. PHYLIP: phylogeny inference package. Seattle: Department of Genetics, University of Washington; 2002. Version 3.6a3. [Google Scholar]
- 11.Huelsenbeck JP, Ronquist F. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 12.Kishino H, Hasegawa M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea. J Mol Evol. 1989;29:170–9. doi: 10.1007/BF02100115. [DOI] [PubMed] [Google Scholar]
- 13.Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. 1999;16:1114–6. [Google Scholar]
- 14.Farris JS, Kallersjo M, Kluge AG, Bult C. Testing significance of incongruence. Cladistics. 1995;10:315–9. [Google Scholar]
- 15.Snijder EJ, Bredenbeek PJ, Dobbe JC, et al. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]