

Abstract
Objectives
Hepatitis C virus (HCV) infection causes chronic liver disease and is a major public health problem worldwide. The aim of this study was to evaluate the potential of Monascus pigment derivatives, which were derived from a microbial secondary metabolite synthesized from polyketides by Monascus spp., as HCV antiviral agents.
Methods
We performed an in vitro RNA-dependent RNA polymerase (RdRp) assay to screen for HCV RdRp inhibitors. The anti-HCV activity of RdRp inhibitors in HCV-replicating cells was evaluated by quantification of the RNA viral genome. Molecular docking analysis was performed to predict the binding sites of the selected RdRp inhibitors.
Results
We have identified a Monascus pigment and its derivatives as inhibitors of the HCV NS5B RdRp. A group of Monascus orange pigment (MOP) amino acid derivatives, in which the reactive oxygen moiety was changed to amino acids, significantly inhibited HCV replication. Further, combination of the MOP derivatives (Phe, Val or Leu conjugates) with interferon (IFN)-α inhibited HCV replication more than IFN-α treatment alone. Lastly, molecular docking studies indicate the inhibitors may bind to a thumb subdomain allosteric site of NS5B. The antiviral activity of the MOP derivatives was related to a modulation of the mevalonate pathway, since the mevalonate-induced increase in HCV replication was suppressed by the MOP compounds.
Conclusions
Our results identify amino acid derivatives of MOP as potential anti-HCV agents and suggest that their combination with IFN-α might offer an alternative strategy for the control of HCV replication.
Keywords: HCV, RNA-dependent RNA polymerase, HMG-CoA reductase
Introduction
Hepatitis C virus (HCV) infects ∼170 million people worldwide, and is often associated with chronic hepatitis, leading to liver cirrhosis and hepatocellular carcinoma.1 Currently, pegylated interferon (IFN) and the nucleoside analogue ribavirin are used as the standard therapy to treat chronic HCV infection. However, IFN-α alone or in combination with ribavirin often leads to a range of side effects, and the sustained virological response rate after combination therapy of pegylated IFN-α and ribavirin is <50%, particularly for those infected with HCV genotype 1 or 4.2 Therefore, there is an urgent need for the development of alternative anti-HCV agents, especially for patients who do not respond to IFN-α therapy.
HCV is an enveloped RNA virus with a positive-sense single-strand RNA genome of ∼9.6 kb. The viral genome consists of one long open reading frame (ORF) flanked by untranslated regions (UTRs) at both the 5′ and 3′ ends of the genome. The ORF encodes a single polyprotein of 3010 amino acids that is proteolytically processed by cellular and viral proteases into ≥10 polypeptides corresponding to the viral structural and non-structural (NS) proteins. The 65 kDa HCV NS5B protein has RNA-dependent RNA polymerase (RdRp) activity and is a key player in HCV RNA replication.3 RdRp activity is not present in mammalian cells, offering the opportunity to identify selective inhibitors of the HCV RdRp. The crystal structure of HCV NS5B resembles a right hand shape with finger, thumb and palm domains similar to other polymerases.4 Over the last decade, researchers have begun developing nucleoside and non-nucleoside analogue inhibitors, which target the active site in the palm subdomain and the allosteric sites in the thumb subdomain of NS5B.5
Microbial secondary metabolites have a variety of biological properties that make them useful as antibiotics, anticancer drugs and antiviral drugs, and in other applications.6Monascus-fermented products, first mentioned in a monograph of Chinese medicine in 1590, are produced by Monascus species, and historically have been used to treat indigestion, muscle bruises and dysentery.7 Monacolin K, also known as lovastatin, is the major secondary metabolite produced by Monascus spp. and a potent inhibitor of 3′-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase.8,9Monascus pigments are also secondary metabolites synthesized from polyketides by Monascus spp.10 There are six major Monascus pigments, including the yellow pigments monascin and ankaflavin, the orange pigments monascorubin (C23H26O5) and rubropunctatin (C21H22O5), and the red pigments monascropunctamine and rubropunctamine.11Monascus pigments have been used as food additives and traditional medicines in East Asian countries, including China, Korea, Japan and Taiwan.12 Further, the pigments have many useful biological activities, such as antimicrobial,13 tumour suppressive and immunosuppressive activities,14 as well as hypolipidaemic activities;8 however, their mechanisms of action have not been well defined.
Here, we report that Monascus pigment derivatives have anti-HCV activity. These compounds were identified from a screen of microbial secondary metabolites for HCV NS5B RdRp inhibitors. We demonstrated that a group of Monascus orange pigment (MOP) derivatives effectively inhibited NS5B RdRp activity and interfered with the mevalonate synthesis pathway, thereby suppressing HCV replication in cells harbouring an HCV genotype 1b subgenomic replicon and in cells infected with genotype 2a HCV.
Materials and methods
Cell culture
The Huh7 human hepatoma cell line was grown in Dulbecco's modified Eagle's medium (DMEM; BioWhittaker, Walkersville, MA, USA) with supplements, as described previously.15 The Huh7-derived cell line R-1, which supports stable, autonomous replication of a genotype 1b HCV subgenomic replicon, was maintained in DMEM with 1 mg/mL G418, as described previously.15
Monascus pigments and reagents
MOP amino acid derivatives (AADs) were produced using Monascus sp. KCCM 10093 and purified from thin layer chromatography, as described previously.11,16 The purity of the MOP AADs was evaluated by HPLC, as described previously.11 The purified MOP AAD compounds were stored as a 10 mM stock solution in DMSO at −20°C and diluted in serum-free medium for use such that the final DMSO concentration did not exceed 0.05%. IFN-α was purchased from Sigma–Aldrich (I-4276, St Louis, MO, USA).
HCV infection and pigment treatment
Infectious HCV RNA of genotype 2a HCV JFH117 was prepared by in vitro transcription using the MEGAscript T7 kit (Ambion, Austin, TX, USA) and electroporated into Huh7 cells, as described previously.18 Huh7 cells were infected with JFH1 virus at a multiplicity of infection of 0.3, as described previously.18 For evaluation of antiviral activity, the indicated doses of IFN-α and/or MOP AADs were added to DMEM containing 5% fetal bovine serum. After 3 days, cells were harvested and the relative HCV genomic RNA levels were assessed by quantitative reverse transcription real-time PCR (qRT–PCR). The half-maximal inhibitory concentration (IC50) value was determined by fitting the data to a three-parametric sigmoidal function using SigmaPlot software (version 10.0; Systat Software Inc., Richmond, CA, USA).
Cell viability assay
The cytotoxicity of MOP derivatives was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent, as described previously.19 Briefly, Huh7 cells grown on a 96-well plate to 70% confluence were incubated for 72 h with MOP derivatives at various concentrations in complete DMEM. Formazan formation was measured by reading the optical absorbance at 570 nm on a microplate reader (FLUOstar Optima; BMG Labtech GmbH, Offenburg, Germany).
RdRp assay
Recombinant HCV NS5B protein with an N-terminal hexahistidine tag was expressed in Escherichia coli and purified, as described previously.15In vitro RNA polymerase activity assays were performed in a streptavidin-coated FlashPlate (PerkinElmer Life and Analytical Science, Waltham, MA, USA), as described previously.18 In brief, the reaction was performed in a 25 μL total volume mixture containing 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 5 mM MgCl2, 1 mM DTT, 20 U of RNase inhibitor (Promega, Madison, WI, USA), 6 μM UTP, 1 μg of poly(A) RNA, 10 pmol of biotinlyated oligo(U)12, 5 μCi [α-32P]UTP (3000 Ci/mmol; Amersham Pharmacia Biotech, Piscataway, NJ, USA) and 3.75 pmol of purified NS5B. The reaction mixture was incubated at 32°C for 2 h. To stop the reaction, 25 μL of 100 mM EDTA was added. After 30 min incubation at room temperature, the reaction mixture was removed and the plates were washed with phosphate-buffered saline. The captured, labelled RNA products were measured in a PerkinElmer TopCount scintillation counter.
HMG-CoA reductase assay and cholesterol analysis
Assays were performed using the HMG-CoA reductase assay kit (Sigma–Aldrich), according to the manufacturer's protocol. The enzyme activity based on NADPH oxidation was determined by measuring the absorbance at 340 nm for 10 min (37°C) using a spectrophotometer (FLUOstar Optima; BMG Labtech GmbH). The reaction was performed in the presence of each derivative of MOP (10 μM) or with pravastatin (1 μM; Sigma–Aldrich). Total cholesterol was extracted from the cells as described previously.20 The cholesterol content was determined with the AmplexRed cholesterol assay kit (Invitrogen, Carlsbad, CA, USA). Mevalonate was prepared by the hydrolysis of mevalonolactone (Sigma–Aldrich) with KOH, as previously described.21
Viral RNA genome quantification
Total RNA was extracted from R-1 cells or HCV-infected cells by using the Trizol LS reagent (Invitrogen). The HCV RNA level in each sample was quantified by qRT–PCR using a primer pair and the TaqMan probe targeting a region within the HCV 5′-UTR, as described previously.18 Cellular glyceraldehyde-3-phosphate dehydrogenase mRNA from the same extracts was used as an internal control.
Western blot analysis
R-1 cells or HCV-infected Huh7 cells were resuspended in lysis buffer containing 25 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100 and an EDTA-free protease inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany). Cell lysates containing equal amounts of proteins were resolved by SDS–PAGE, transferred to nitrocellulose membranes and immunoblotted with anti-NS5A (ViroGen, Watertown, MA, USA) or anti-NS5B18 antibodies, as described previously.22 To demonstrate equal loading of cell lysates, α-tubulin was measured by western blot analysis using an anti-α-tubulin antibody (Oncogene Research Products, Cambridge, MA, USA). Blots were developed using the Enhanced Chemiluminescence Western Blot System (GE Healthcare Life Sciences, Piscataway, NJ, USA), as described previously.22 Quantitative analyses of western blots were performed using the Scion image software (Scion Co., Frederick, MD, USA).
Molecular docking analysis
Crystal structures of HCV NS5B protein (Protein Data Bank codes 1NHU, 1NHV, 2GIR, 2BRK, 2QE2 and 3FQK) were used for molecular docking simulation analyses. The NS5B structures were kept rigid, whereas the torsional bonds in MOP AADs were set free to flexible docking. Affinity grids on the binding pocket were constructed using AutoGrid423 with a grid spacing of 0.375 Å. Each grid map consisted of 40 × 40 × 40 grid points. AutoDock423 was used to find the binding positions for MOP AADs on the HCV NS5B by molecular docking simulation. Molecular docking was performed by a global genetic algorithm combined with local minimization, Lamarckian genetic algorithm to explore the compound conformational space. After each docking job with 100 trials, the final docked conformations were clustered using a tolerance of 1 Å root-mean-square deviation. All other parameters were set to default values. The docking conformation properly oriented towards the HCV NS5B inhibitor binding pocket was selected and the free energy of binding was estimated. The molecular graphics for the inhibitor binding pocket and refined docking model for the selected MOP AADs were generated using the PyMol software package.24
Results
Novel HCV RdRp inhibitory activity of Monascus pigments
The HCV NS5B RdRp is an essential enzyme for HCV replication and is therefore a promising antiviral drug target for blocking viral genome replication.3,5 We screened an in-house microbial secondary metabolite library and identified the MOP secondary metabolite (Figure 1a) as an inhibitor of HCV RdRp activity. To screen the library, we performed an in vitro RdRp assay using recombinant NS5B. The in vitro RdRp assay was carried out in a FlashPlate coated with streptavidin using poly(A) RNA/biotinylated oligo(U)12 as a substrate in the presence of 10 μM of each library compound. As shown in Figure 1(b), the natural MOP inhibited NS5B activity by ∼28% at 10 μM concentration. In an attempt to improve the inhibitory potency, we produced various AADs of MOP in which the aromatic ring oxygen of MOP was changed to different amino acids (Figure 1a). These derivatives were tested for RdRp inhibitory activity. Among the resulting MOP AADs, we found that derivatives containing Asn, Lys, Phe, Gly, Ala, Val, Leu or Ile side chains had greater inhibitory potency (up to ∼45%) than the parent MOP. However, the activity was still ∼50-fold lower than that of benzothiadiazinylquinone (42% inhibition at 200 nM), a potent HCV inhibitor designed to target the palm site of the NS5B polymerase.25 Considering the novel potential of these microbial secondary metabolites as anti-HCV agents, we proceeded to characterize their structure–function relationships.
Figure 1.
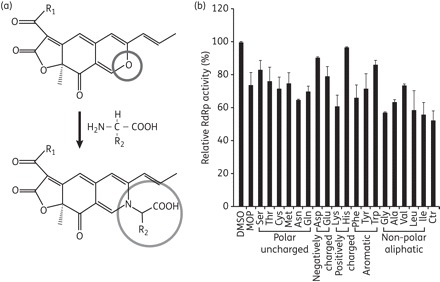
Inhibitory effect of MOP and its AADs on HCV NS5B RdRp activity. (a) Chemical structure of MOP and conversion scheme for the preparation of MOP AADs. R1, C5H11 or C7H15; R2, functional group of amino acids. (b) RdRp inhibitory activity of MOP AADs (10 μM) was evaluated in an in vitro RdRp assay using poly(A) RNA/biotinylated oligo(U)12 as a substrate in a FlashPlate coated with streptavidin. DMSO (0.25%), used as a solvent, and benzothiadiazinylquinolinone (Ctr; 0.2 mM) were used as a negative and positive control, respectively. Incorporated radioactivity was measured in a PerkinElmer TopCount. Three independent experiments were performed in triplicate. Data shown are the means ± SD of three experiments.
Monascus pigment derivatives suppress HCV replication in cell culture
We tested the HCV inhibitory effect of the above-described MOP AADs in R-1 cells that harboured an HCV genotype 1b subgenomic replicon. This replicon consists of the HCV internal ribosome entry site (IRES), which directs expression of a G418 selectable neor marker, and an encephalomyocarditis virus IRES, which directs the expression of the HCV NS proteins (NS3 to NS5B) required for HCV replication (Figure 2a, top panel). R-1 cells were treated with 10 μM of each MOP AAD for 48 h and then the remaining steady-state level of NS5B protein was determined by western blot analysis. As shown in Figure 2(a), among the selected group of MOP AADs that inhibited NS5B RdRp activity in vitro, Lys, Phe, Val, Leu and Ile derivatives also inhibited HCV replication in Huh7 cells, which harbour a HCV subgenomic replicon RNA. Treatment of cells with the positive control, IFN-α (100 IU/mL), or the MOP AAD both reduced the NS5B protein levels to different degrees. IFN-α reduced NS5B by 75% while the reductions for the AADs were: Lys 30%; Phe 39%; Val 38%; Leu 45%; and Ile 35%. In the conditions of this assay, none of the MOP compounds significantly affected Huh7 cell proliferation (at most <10% at 10 μM concentration), as assessed by a colorimetric MTT assay (data not shown). Notably, in contrast to the in vitro RdRp assay results, the parental MOP compound did not significantly reduce HCV protein expression levels in the Huh R-1 cells, which might be due to its lower affinity to the RdRp or inability to pass through the plasma membrane.
Figure 2.
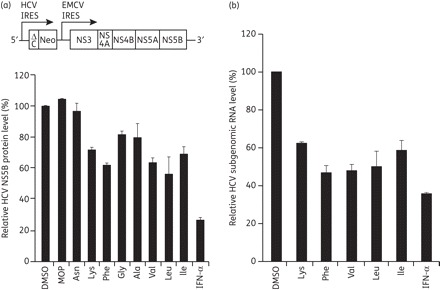
Anti-HCV effect of MOP AADs in HCV subgenomic replicon cells. (a) Schematic diagram of an HCV subgenomic replicon RNA (top panel). The R-1 cells that support constitutive replication of an HCV subgenomic replicon, depicted in (a), were left untreated (DMSO) or treated with the indicated MOP AADs (10 μM) or 100 IU/mL IFN-α for 48 h. Thereafter, whole-cell lysates were prepared and subjected to western blot analysis using an anti-NS5B antibody. Expression of α-tubulin was used to control for equal protein loading. The normalized level of NS5B was determined by densitometric analysis of immunoblots. The relative level of NS5B, expressed as the percentage of the DMSO-treated control, is shown. (b) R-1 cells were treated with 10 μM of the indicated, selected MOP AADs for 48 h. The intracellular HCV genome copy number was quantified by real-time qRT–PCR and expressed as the percentage of the DMSO vehicle-treated control cells.
We next confirmed the anti-HCV activity of the five selected MOP derivatives (Lys, Phe, Val, Leu and Ile derivatives) by measuring HCV subgenomic RNA levels by real-time qRT–PCR. The HCV RNA levels were decreased by 38%–53% by treatment with 10 μM of each of the MOP derivatives (Figure 2b). We finally selected MOP Phe, Val and Leu derivatives to further characterize the mechanism of HCV inhibition in HCV-infected cells. We used the HCV infection system established with the genotype 2a HCV clone JFH1, which yields infectious HCV virus from Huh7 or Huh7-derived cell lines.17 Because combination therapy with pegylated IFN-α and ribavirin is the current standard therapy for the treatment of HCV infection,26 we also sought to evaluate the potential of the MOP AADs to act in combination with IFN-α. HCV-infected Huh7 cells were treated with 10 μM of each MOP AAD alone or in combination with IFN-α (100 IU/mL) for 72 h and the HCV genome level was analysed by real-time qRT–PCR. As shown in Figure 3(a), HCV RNA abundance was decreased by 52%–58% when the virus-infected cells were treated with 10 μM of the Phe, Val or Leu MOP derivatives. Combination of IFN-α (100 IU/mL) with the Phe, Val or Leu MOP AADs (10 μM) reduced the HCV RNA levels by 77%, 82% or 88%, respectively. The MOP-Leu derivative showed the greatest reduction of HCV RNA when combined with IFN-α and inhibited HCV replication in a dose-dependent manner, with an IC50 value of 8.9 μM (Figure 3b). We also monitored viral NS5A and NS5B protein expression by western blot analysis. As shown in the Figure 3(b) inset, the MOP-Leu derivative reduced NS5A and NS5B expression in a dose-dependent manner. Lastly, the compound displayed no significant cytotoxicity at concentrations up to 50 μM (<20% decrease of cell viability at 50 μM), as assessed by the MTT cell viability assay (Figure 3c). Together, these results demonstrate that MOP AADs had antiviral activity against both genotype 2a HCV (from the Huh7 infection model) and genotype 1b HCV (from the subgenomic replicon system in Figure 2), and suggest their potential as novel HCV antiviral drugs when used in combination with IFN-α.
Figure 3.
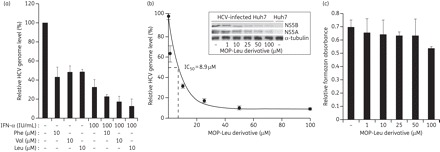
Anti-HCV activity of the selected MOP derivatives alone or in combination with interferon in HCV (genotype 2a)-infected cells. (a) Huh7 cells grown to ∼60% confluency in 6 cm plates were infected with genotype 2a HCV (JFH1) for 4 h in serum-free medium. Then, cells were washed and complete medium was added. Cells were treated with 10 μM MOP AADs alone or in combination with IFN-α (100 IU/mL) for 72 h. The intracellular HCV genome copy number was quantified by real-time qRT–PCR as in Figure 2(b). (b) Huh7 cells were treated with the indicated concentrations of the MOP-Leu derivative for 72 h before measurement of HCV genome copy number to determine the IC50 value. The inset shows the steady-state expression levels of NS5A and NS5B analysed by western blot of HCV-infected cell lysates treated with MOP-Leu. The NS5A and NS5B levels were determined by densitometric analysis of immunoblots that were normalized to α-tubulin levels. (c) Cytotoxicity of MOP derivatives against Huh7 cells was measured by the MTT assay. Error bars represent standard deviations of triplicates per condition.
Molecular docking analysis of selected MOP AADs on the HCV NS5B atomic structure
To understand the molecular mechanism of RdRp inhibition by the selected MOP AADs, we performed molecular docking of the inhibitors to the crystal structures of HCV NS5B–inhibitor complexes.27,28 MOP and its AADs (the Phe, Ile and Leu derivatives; Figure 4a) were successfully docked to the thumb subdomain of NS5B, which is ∼35 Å away from the polymerase active site in the palm domain (Figure 4b). As shown in Figure 4(c), similar to the docking model built with the reference compound, compound 1 [(S,Z)-2-{5-[(5-ethylfuran-2-yl)methylene]-4-oxo-4,5-dihydrothiazol-2-yl}-2-(4-fluorophenyl) acetic acid],28 the MOP AADs (compounds 2–4) displayed binding interactions on the pocket consisting of two hydrophobic sites defined by amino acids Trp528/Leu497/Tyr477 and Leu489/Ile482/Met423, and a hydrogen bond interaction site defined by amino acids Arg501 and Lys533. The propenyl moiety of MOP AADs is bound to the surface of one of the hydrophobic pockets defined by Trp528/Leu497/Tyr477. The predicted binding mode of this moiety is similar to that of the ethylfuran moiety of the reference compound. The hexanoyl group of the MOP AADs, like the 4-fluorophenyl ring of the reference compound, is buried into a narrow deep hydrophobic pocket defined by Leu489, Ile482 and Met423. Lastly, two hydrogen bonds between oxygen atoms of a carboxylate group on the inhibitors and Arg501 and Lys533 side chains on NS5B were also observed, as with the carboxylic acid moiety of the reference compound. The location of this inhibitor-binding site suggests that the binding of these inhibitors could interfere with a conformational change essential for HCV NS5B polymerase activity.29
Figure 4.
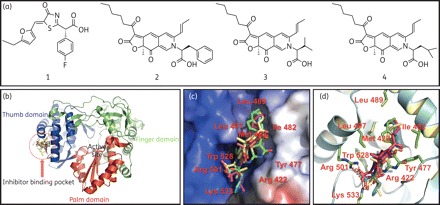
Model for docking of MOP AADs on HCV NS5B 3D structure. (a) The chemical structure of a reference NS5B inhibitor [(S,Z)-2-{5-[(5-ethylfuran-2-yl)methylene]-4-oxo-4,5-dihydrothiazol-2-yl}-2-(4-fluorophenyl) acetic acid (compound 1)] and MOP AADs (compounds 2, 3 and 4 are MOP with Phe, Val and Leu conjugate, respectively). (b) A ribbon diagram of HCV NS5B complexed with MOP AADs at the thumb subdomain. HCV NS5B domains are coloured as follows: thumb, blue; fingers, green; and palm, red. (c) A surface representation of the allosteric site of NS5B to which reference compound 1 (in green) and MOP AADs [2, Phe (in cyan); 3, Val (in pink); and 4, Leu (in yellow)] bind. (d) Overlay of the binding modes of MOP AADs in the allosteric site of HCV NS5B. The dashed red lines indicate hydrogen bonds.
Interference with mevalonate pathway by the MOP AADs
Lipid metabolism and cholesterol synthesis play a critical role in HCV replication. Because the HMG-CoA reductase, which catalyses the conversion of HMG-CoA to mevalonate, is the rate-limiting step in cholesterol biosynthesis, various inhibitors of this enzyme were previously tested for anti-HCV activity.30 In particular, lovastatin, one of the Monascus-produced secondary metabolites, was shown to effectively suppress HCV replication by inhibiting the interaction between HCV NS5A and the host protein FBL2.31,32 We also observed that lovastatin treatment (10 μM) for 48 h reduced viral genome abundance by ∼70% in HCV-replicating R-1 cells (data not shown), confirming previous reports.30,31,33 Currently, it is not known whether MOP, like lovastatin, also interferes with cholesterol biosynthesis. To assess the possibility that MOP AADs may inhibit HCV replication by inhibiting cholesterol biosynthesis, we first performed in vitro HMG-CoA reductase assays in HCV replication-supporting Huh7 cells. As shown in Figure 5(b), MOP derivatives (the Phe, Val and Leu derivatives) did not have any observable HMG-CoA reductase inhibitory activity, even at a high concentration (10 μM), while pravastatin, a HMG-CoA reductase inhibitor, significantly decreased the enzyme activity at a concentration of 1 μM.
Figure 5.
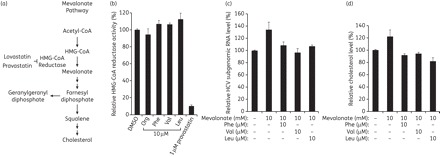
Effect of MOP AADs on the mevalonate pathway. (a) Schematic diagram of the mevalonate pathway. (b) HMG-CoA reductase assays were performed in the presence of the indicated MOP AADs (10 μM) or a known HMG-CoA reductase inhibitor, pravastatin (1 μM). Relative enzyme activity is expressed as the percentage of the control (0.5% DMSO as a solvent). (c) R-1 cells were incubated with 10 mM mevalonate in the absence or presence of 10 μM of the indicated MOP AADs for 48 h. Total cellular RNA was extracted and the HCV RNA level was determined by real-time qRT–PCR as in Figure 2(b). (d) R-1 cells treated as in (c) were subjected to cholesterol assays. The intracellular cholesterol level is expressed as the percentage of the DMSO-treated control.
In the mevalonate pathway, acetyl-CoA is converted into HMG-CoA, mevalonate, farnesyl diphosphate, squalene and cholesterol (Figure 5a).34 The production of geranylgeranyl lipids and cholesterol through the mevalonate pathway is important for HCV RNA replication.31 Consistent with this, we found that mevalonate supplementation to HCV-replicating cells increased HCV subgenomic RNA levels (Figure 5c). This result prompted us to further investigate whether the MOP AADs block HCV replication by inhibiting the cholesterol biosynthetic pathway downstream of mevalonate. To test this possibility, we examined whether the increase in HCV replication by the addition of mevalonate can be suppressed by the MOP AADs. Huh7 R-1 cells, which harbour an HCV subgenomic replicon, were incubated with 10 mM mevalonate in the absence or presence of 10 μM of the Phe, Val or Leu MOP AADs. After 2 days, the cells were harvested and HCV subgenomic RNA levels analysed by real-time qRT–PCR. As shown in Figure 5(c), the HCV RNA level increased by ∼37% with the addition of mevalonate; however, the mevalonate-induced increase was completely blocked by MOP AAD treatment. Moreover, the increase in intracellular cholesterol caused by mevalonate supplementation was also inhibited by MOP AAD treatment and the cholesterol levels were reduced to the level of mock-treated cells (Figure 5d). Together, these results suggest that in addition to a direct inhibition of NS5B RdRp activity, these MOP AAD compounds also inhibit HCV replication by interfering with the cholesterol biosynthetic pathway downstream of the HMG-CoA reductase step.
Discussion
Microbial secondary metabolites have been shown to be effective as anticancer or antimicrobial agents, and for the treatment of metabolic diseases such as hypercholesterolaemia. These natural products are attractive for developing antiviral drugs to treat or control viral infectious diseases, but have previously been overlooked in antiviral research. In this study, we screened an in-house library of microbial secondary metabolites and found a series of MOP AADs that have anti-HCV activity. We have identified two novel activities of these MOP AADs: inhibition of HCV RdRp activity; and interference with the mevalonate pathway. Both of these inhibitory activities were found to contribute to the suppression of HCV replication.
Currently, four different allosteric binding sites for HCV NS5B non-nucleoside inhibitors (NNIs) have been identified: two sites in the thumb domain; and two sites in the palm domain.35 The MOP Leu, Ile and Phe AADs were predicted to bind primarily to the thumb subdomain site II, namely the NNI site II, ∼35 Å away from the active site of the NS5B protein. The MOP AADs failed to dock to the other three NNI sites and did not show any common binding patterns shared with previously reported inhibitors known to bind to NNI-binding sites I, III and IV (data not shown). The pharmacophore of HCV NS5B NNI binding to thumb subdomain site II is composed of the hydrogen-bond interaction sites (Arg501 and Lys533) and a hydrophobic cavity, which appear to be occupied by a carboxylic acid residue and prophenyl residue of MOP AADs, respectively. Among those two predicted interacting parts of the MOP AADs, the carboxylic acid residue seems to be more critical for NS5B inhibition, since the MOP lacking this carboxylic acid residue showed comparably lower NS5B inhibitory activities and no detectable anti-HCV activity in HCV-replicating cells. Further, molecular docking simulations showed that the parental MOP compound did not make hydrogen bonds with Arg501 and Lys533. The NNIs binding to this site, like the inhibitors binding to the NNI-binding site I on the upper section of the HCV NS5B thumb domain, are likely to prevent the polymerase from adopting the closed conformation required for productive polymerization during elongation.29
Genotype-dependent antiviral activity, which is due to the polymorphism of NS5B, is a major obstacle for the development of NNIs against HCV RdRp. Indeed, except for HCV796, an inhibitor that binds to NNI site IV on the palm domain, the activity of NNIs against non-genotype 1 HCV is limited.36,37 The MOP AADs selected in the present study demonstrated similar antiviral potency against HCV genotype 1b-derived subgenomic replicon cells and genotype 2a HCV-infected cells, suggesting the binding mode of these inhibitors was not limited to the genotype 1 NS5B polymerase. However, further studies are needed to determine whether these MOP derivatives provide a novel scaffold binding to the thumb subdomain allosteric sites of NS5B from other genotypes as well. In addition to genotype diversity, the emergence of resistant mutants is another obstacle for the development of NNIs. For instance, treatment of patients with NNI site II inhibitors, such as filibuvir and VCH759, in clinical trials resulted in the selection of resistant mutants with mutations at Leu419, Met423 and Ile482 of NS5B.35,38,39 Our molecular docking simulation predicted that Leu489/Ile482/Met423 is also involved in the binding of the selected MOP AADs to the site II hydrophobic pockets. Thus, it will be of interest to investigate whether the MOP AADs show cross-resistance with other NNI site II inhibitors. If so, MOP AADs would be additional combination choices for NNI cocktail therapy.
Several lines of evidence suggest that cellular lipid and cholesterol metabolism plays either a direct or indirect role in the HCV life cycle. In particular, cholesterol biosynthesis was proposed as an integral part of HCV RNA replication, which occurs on lipid rafts.31,40 In addition, HCV entry through the low-density lipoprotein receptor and viral assembly occurring on the surface of lipid droplets are also linked to cholesterol biosynthesis.41,42 Accordingly, various inhibitors of HMG-CoA reductase have been shown to suppress HCV replication.33,43 Our results suggest that the anti-HCV activity of MOP AADs (the Phe, Val and Leu derivatives) might also be partially due to their ability to interfere with the mevalonate biosynthetic pathway. The key enzyme in the mevalonate–cholesterol pathway, HMG-CoA reductase, was not inhibited by the MOP derivatives. However, the increased HCV RNA levels induced by mevalonate supplementation were inhibited by the MOP AADs. It was previously suggested that geranylgeranylation of the host factor FBL2 is required for targeting of HCV NS5A to the intracellular membranous structure to form the RNA replicase complex.43 This previous study also demonstrated that the addition of cholesterol failed to rescue HCV RNA replication in the presence of lovastatin.43 Thus, it is likely that the MOP AADs prevented HCV replication in part by blocking the generation of geranylgeranyl or its precursors, since the HCV NS5A–FBL2 interaction required for HCV replication depends on geranylgeranylation of FBL2.43 Of note, the increased intracellular cholesterol level in the mevalonate-treated cells decreased upon treatment with the MOP AADs. Therefore, MOP AADs appear to be capable of inhibiting the step(s) involved in converting mevalonate to cholesterol or the enzyme converting farnesyl diphosphate to geranylgeranyl diphosphate.
In summary, the results presented here demonstrate that MOP AADs can effectively inhibit HCV replication. A double-hit strategy, including inhibition of HCV RdRp activity and interference with the mevalonate synthetic pathway, to inhibit HCV amplification may provide the basis for successful antiviral therapy using the MOP AADs derived from this microbial secondary metabolite. The selected AADs of MOP potentiated the antiviral activity of IFN-α, suggesting that combination therapy with IFN-α or other drugs may offer an alternative strategy for controlling HCV replication.
Funding
This work was supported by a grant from the Korea Research Foundation (KRF 2009-0071221) and in part by a grant from the National Research Foundation of Korea funded by the Korea Government (NRF 2010-0029116). S.-J. K. was the recipient of a postdoctoral fellowship from the BK21 programme of the Korean Ministry of Education, Science and Technology. J.-M. S. and S.-H. L. were supported in part by the BK21 programme.
Transparency declarations
None to declare.
Acknowledgements
We thank Drs Christoph Seeger and Takaji Wakita for providing pZS2 and pJFH1 plasmids, respectively.
References
- 1.Brown RS. Hepatitis C and liver transplantation. Nature. 2005;436:973–8. doi: 10.1038/nature04083. [DOI] [PubMed] [Google Scholar]
- 2.Foster GR. Pegylated interferons for the treatment of chronic hepatitis C: pharmacological and clinical differences between peginterferon-α-2a and peginterferon-α-2b. Drugs. 2010;70:147–65. doi: 10.2165/11531990-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 3.Appel N, Schaller T, Penin F, et al. From structure to function: new insights into hepatitis C virus RNA replication. J Biol Chem. 2006;281:9833–6. doi: 10.1074/jbc.R500026200. [DOI] [PubMed] [Google Scholar]
- 4.Bressanelli S, Tomei L, Roussel A, et al. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc Natl Acad Sci USA. 1999;96:13034–9. doi: 10.1073/pnas.96.23.13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown NA. Progress towards improving antiviral therapy for hepatitis C with hepatitis C virus polymerase inhibitors. Part I: nucleoside analogues. Expert Opin Investig Drugs. 2009;18:709–25. doi: 10.1517/13543780902854194. [DOI] [PubMed] [Google Scholar]
- 6.Demain AL, Sanchez S. Microbial drug discovery: 80 years of progress. J Antibiot (Tokyo) 2009;62:5–16. doi: 10.1038/ja.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong H-C, Koehler PE. Production and isolation of an antibiotic from Monascus purpureus and its relationship to pigment production. J Food Sci. 1981;46:589–92. doi: 10.1111/j.1365-2621.1981.tb04917.x. [DOI] [Google Scholar]
- 8.Lee CL, Tsai TY, Wang JJ, et al. In vivo hypolipidemic effects and safety of low dosage Monascus powder in a hamster model of hyperlipidemia. Appl Microbiol Biotechnol. 2006;70:533–40. doi: 10.1007/s00253-005-0137-0. [DOI] [PubMed] [Google Scholar]
- 9.Endo A. Chemistry, biochemistry, and pharmacology of HMG-CoA reductase inhibitors. Klin Wochenschr. 1988;66:421–7. doi: 10.1007/BF01745510. [DOI] [PubMed] [Google Scholar]
- 10.Carels M, Shepherd D. The effect of different nitrogen sources on pigment production and sporulation of Monascus species in submerged, shaken culture. Can J Microbiol. 1977;23:1360–72. doi: 10.1139/m77-205. [DOI] [PubMed] [Google Scholar]
- 11.Jung H, Kim C, Kim K, et al. Color characteristics of Monascus pigments derived by fermentation with various amino acids. J Agric Food Chem. 2003;51:1302–6. doi: 10.1021/jf0209387. [DOI] [PubMed] [Google Scholar]
- 12.Wang TM, Lin TF. Monascus rice products. Adv Food Nutr Res. 2007;53:123–59. doi: 10.1139/m78-217. [DOI] [PubMed] [Google Scholar]
- 13.Kim C, Jung H, Kim YO, et al. Antimicrobial activities of amino acid derivatives of Monascus pigments. FEMS Microbiol Lett. 2006;264:117–24. doi: 10.1111/j.1574-6968.2006.00451.x. [DOI] [PubMed] [Google Scholar]
- 14.Yasukawa K, Takahashi M, Natori S, et al. Azaphilones inhibit tumor promotion by 12-O-tetradecanoylphorbol-13-acetate in two-stage carcinogenesis in mice. Oncology. 1994;51:108–12. doi: 10.1159/000227320. [DOI] [PubMed] [Google Scholar]
- 15.Kim SJ, Kim JH, Kim YG, et al. Protein kinase C-related kinase 2 regulates hepatitis C virus RNA polymerase function by phosphorylation. J Biol Chem. 2004;279:50031–41. doi: 10.1074/jbc.M408617200. [DOI] [PubMed] [Google Scholar]
- 16.Kim C, Jung H, Kim JH, et al. Effect of Monascus pigment derivatives on the electrophoretic mobility of bacteria, and the cell adsorption and antibacterial activities of pigments. Colloids Surf B Biointerf. 2006;47:153–9. doi: 10.1016/j.colsurfb.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 17.Wakita T, Pietschmann T, Kato T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–6. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim SJ, Kim JH, Sun JM, et al. Suppression of hepatitis C virus replication by protein kinase C-related kinase 2 inhibitors that block phosphorylation of viral RNA polymerase. J Viral Hepat. 2009;16:697–704. doi: 10.1111/j.1365-2893.2009.01108.x. [DOI] [PubMed] [Google Scholar]
- 19.Ahn DG, Lee W, Choi JK, et al. Interference of ribosomal frameshifting by antisense peptide nucleic acids suppresses SARS coronavirus replication. Antiviral Res. 2011;91:1–10. doi: 10.1016/j.antiviral.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung CS, Huang CY, Chang W. Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J Virol. 2005;79:1623–34. doi: 10.1128/JVI.79.3.1623-1634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campos N, Rodriguez-Concepcion M, Sauret-Gueto S, et al. Escherichia coli engineered to synthesize isopentenyl diphosphate and dimethylallyl diphosphate from mevalonate: a novel system for the genetic analysis of the 2-C-methyl-d-erythritol 4-phosphate pathway for isoprenoid biosynthesis. Biochem J. 2001;353:59–67. doi: 10.1042/0264-6021:3530059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang SM, Kim SJ, Kim JH, et al. Interaction of hepatitis C virus core protein with Hsp60 triggers the production of reactive oxygen species and enhances TNF-α-mediated apoptosis. Cancer Lett. 2009;279:230–7. doi: 10.1016/j.canlet.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 23.Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J Comput Chem. 1998;19:1639–62. doi: 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B. [DOI] [Google Scholar]
- 24.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. http://www.pymol.org . [Google Scholar]
- 25.Fitch DM, Evans KA, Chai D, et al. A highly efficient, asymmetric synthesis of benzothiadiazine-substituted tetramic acids: potent inhibitors of hepatitis C virus RNA-dependent RNA polymerase. Org Lett. 2005;7:5521–4. doi: 10.1021/ol052371w. [DOI] [PubMed] [Google Scholar]
- 26.Parfieniuk A, Jaroszewicz J, Flisiak R. Specifically targeted antiviral therapy for hepatitis C virus. World J Gastroenterol. 2007;13:5673–81. doi: 10.3748/wjg.v13.i43.5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang M, Ng KK, Cherney MM, et al. Non-nucleoside analogue inhibitors bind to an allosteric site on HCV NS5B polymerase. Crystal structures and mechanism of inhibition. J Biol Chem. 2003;278:9489–95. doi: 10.1074/jbc.M209397200. [DOI] [PubMed] [Google Scholar]
- 28.Yan S, Appleby T, Larson G, et al. Structure-based design of a novel thiazolone scaffold as HCV NS5B polymerase allosteric inhibitors. Bioorg Med Chem Lett. 2006;16:5888–91. doi: 10.1016/j.bmcl.2006.08.056. [DOI] [PubMed] [Google Scholar]
- 29.Biswal BK, Cherney MM, Wang M, et al. Crystal structures of the RNA-dependent RNA polymerase genotype 2a of hepatitis C virus reveal two conformations and suggest mechanisms of inhibition by non-nucleoside inhibitors. J Biol Chem. 2005;280:18202–10. doi: 10.1074/jbc.M413410200. [DOI] [PubMed] [Google Scholar]
- 30.Ikeda M, Abe K, Yamada M, et al. Different anti-HCV profiles of statins and their potential for combination therapy with interferon. Hepatology. 2006;44:117–25. doi: 10.1002/hep.21232. [DOI] [PubMed] [Google Scholar]
- 31.Ye J. Reliance of host cholesterol metabolic pathways for the life cycle of hepatitis C virus. PLoS Pathog. 2007;3:e108. doi: 10.1371/journal.ppat.0030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang C, Gale M, Jr, Keller BC, et al. Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication. Mol Cell. 2005;18:425–34. doi: 10.1016/j.molcel.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 33.Kapadia SB, Chisari FV. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc Natl Acad Sci USA. 2005;102:2561–6. doi: 10.1073/pnas.0409834102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–30. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 35.Legrand-Abravanel F, Nicot F, Izopet J. New NS5B polymerase inhibitors for hepatitis C. Expert Opin Investig Drugs. 2010;19:963–75. doi: 10.1517/13543784.2010.500285. [DOI] [PubMed] [Google Scholar]
- 36.Herlihy KJ, Graham JP, Kumpf R, et al. Development of intergenotypic chimeric replicons to determine the broad-spectrum antiviral activities of hepatitis C virus polymerase inhibitors. Antimicrob Agents Chemother. 2008;52:3523–31. doi: 10.1128/AAC.00533-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pauwels F, Mostmans W, Quirynen LM, et al. Binding-site identification and genotypic profiling of hepatitis C virus polymerase inhibitors. J Virol. 2007;81:6909–19. doi: 10.1128/JVI.01543-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooper C, Lawitz EJ, Ghali P, et al. Evaluation of VCH-759 monotherapy in hepatitis C infection. J Hepatol. 2009;51:39–46. doi: 10.1016/j.jhep.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 39.Le Pogam S, Kang H, Harris SF, et al. Selection and characterization of replicon variants dually resistant to thumb- and palm-binding nonnucleoside polymerase inhibitors of the hepatitis C virus. J Virol. 2006;80:6146–54. doi: 10.1128/JVI.02628-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aizaki H, Lee KJ, Sung VM, et al. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology. 2004;324:450–61. doi: 10.1016/j.virol.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 41.Miyanari Y, Atsuzawa K, Usuda N, et al. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol. 2007;9:1089–97. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 42.Alvisi G, Madan V, Bartenschlager R. Hepatitis C virus and host cell lipids: an intimate connection. RNA Biol. 2011;8:258–69. doi: 10.4161/rna.8.2.15011. [DOI] [PubMed] [Google Scholar]
- 43.Ye J, Wang C, Sumpter R, Jr, et al. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc Natl Acad Sci USA. 2003;100:15865–70. doi: 10.1073/pnas.2237238100. [DOI] [PMC free article] [PubMed] [Google Scholar]