

Abstract
Four single-chain variable fragments (scFvs) against protective antigen (PA) and 2 scFvs against lethal factor (LF) of anthrax were isolated from a phage display library generated from immunized chimpanzees. Only 2 scFvs recognizing PA (W1 and W2) neutralized the cytotoxicity of lethal toxin in a macrophage lysis assay. Full-length immunoglobulin G (IgG) of W1 and W2 efficiently protected rats from anthrax toxin challenge. The epitope recognized by W1 and W2 was conformational and was formed by C-terminal amino acids 614–735 of PA. W1 and W2 each bound to PA with an equilibrium dissociation constant of 4×10-11 mol/L to 5 × 10−11 mol/L, which is an affinity that is 20–100-fold higher than that for the interaction of the receptor and PA. W1 and W2 inhibited the binding of PA to the receptor, suggesting that this was the mechanism of protection. These data suggest that W1 and W2 chimpanzee monoclonal antibodies may serve as PA entry inhibitors for use in the emergency prophylaxis against and treatment of anthrax
Anthrax has emerged as a serious bioterrorist threat. Inhalational anthrax is usually fatal if treatment is delayed [1]. The lethality of anthrax is primarily the result of the effects of anthrax toxin, which has 3 components: a receptor-binding protein known as “protective antigen” (PA) and 2 catalytic proteins known as “lethal factor” (LF) and “edema factor” (EF). Entry of the toxins into the cell is initiated by rapid binding of the 83-kDa PA to the cellular receptor, whereupon a furinlike protease cleaves the bound PA into an N-terminal 20-kDa protein (PA20) and a C-terminal 63-kDa protein (PA63) [2]. There is spontaneous oligomerization of PA63 into an antigenically distinct heptameric ring that can no longer be displaced from the cellular receptor. The heptamer binds up to 3 molecules of LF or EF. The resulting complexes enter the cell by endocytosis, and a conformational change induced by low pH results in the release of bound LF and EF into the cytosol. Several steps in this process could be targets for antibodies; for example, antibodies to PA might block receptor binding, oligomerization, or binding of LF and EF, and antibodies to LF and EF might prevent their binding to PA
Passive immunization with polyclonal antibodies protects laboratory animals from the effects of anthrax toxins [3, 4]. Passive immunization of humans with anthrax neutralizing antibodies may provide an effective treatment when vaccination against anthrax is not practical or antibiotic treatment is not effective. Antibody therapy given in conjunction with antibiotics would also be useful when the accumulated level of toxin would be detrimental, even if further bacterial growth was inhibited. Recently, several recombinant monoclonal antibodies (MAbs) against PA were shown to protect animals from challenge with anthrax toxin [5–7 ]. Because chimpanzee immunoglobulins are virtually identical to human immunoglobulins [8–10 ], high-affinity chimpanzee antibodies that neutralize anthrax toxins should have therapeutic value comparable to that of human antibodies. Here, we report the identification and characterization of potent neutralizing chimpanzee MAbs against PA
Materials and Methods
ReagentsRecombinant PA, PA63, LF, and EF were obtained from List Biologicals or were made in our laboratory. Enzymes used in molecular cloning were purchased from New England BioLabs, and oligonucleotides were synthesized by Invitrogen. Anti-His horseradish peroxidase (HRP) conjugate, anti–human Fab HRP conjugate, and anti–human Fc agarose were purchased from Sigma. Nickel-agarose beads were obtained from Invitrogen
AnimalsChimpanzees 1603 and 1609 were immunized 3 times, at 2-week intervals, with 50 μg each of recombinant PA, LF, and EF with alum adjuvant. Eight weeks after the last immunization, bone marrow was aspirated from the iliac crest of these animals. Fischer 344 rats were purchased from Taconic Farms. All experiments involving animals were performed under protocols approved by the Animal Care and Use Committee of the National Institute of Allergy and Infectious Diseases, National Institutes of Health
Library construction and selectionLymphocytes from the bone marrow aspirate were isolated on a Ficoll gradient. mRNA was extracted from 108 lymphocytes by use of an mRNA purification kit (Amersham Biosciences). cDNA was synthesized using a first-strand cDNA synthesis kit (Amersham Biosciences). The heavy-chain (VH) and κ light-chain (Vκ) genes were amplified by polymerase chain reaction (PCR) for 30 cycles (for 1 min at 95°C, for 1 min at 58°C, and for 1 min at 72°C), by use of a mixture of primers for human V genes described elsewhere [11]. The single-chain variable fragment (scFv) was assembled from VH and Vκ via splicing by overlap extension PCR. Gel-purified scFv DNA was digested with SfiI and then was cloned into a pAK100 vector that also had been cut with SfiI [12]
The recombinant plasmids were transformed into Escherichia coli XL1-blue (Stratagene) by electroporation, resulting in a library of 5×107 individual clones. The phagemid production, panning, and screening were essentially the same as those reported by Krebber et al. [12] in 1997. In brief, the phagemids were rescued by superinfection with helper phage VCS M13 (Stratagene) and then were subjected to panning on PA, LF, and EF proteins coated on ELISA wells. Nonspecifically adsorbed phages were removed by extensive washing. Specifically bound phages were eluted with 100 mmol/L triethylamine, neutralized in pH, and amplified. After 3 rounds of panning, randomly chosen single-phage scFv clones were screened for specific binding by phage ELISA [13]. Clones that differentially bound to specific antigens with values of >1.0 for absorbance measured at 450 nm (A450) were considered to have a positive result, whereas A450 values of <0.2 were considered to denote a negative result. For clones that bound specifically to PA, LF, or EF, the variable regions of VH and light (VL)–chain genes were sequenced, and their corresponding amino acid sequences were aligned
Conversion of scFv to Fab and to full-length IgGTo convert scFv to Fab, the following 8 primers were designed: First-strand cDNA of constant chain 1 of heavy chain (CH1) and constant region of κ light chain (Cκ) gene fragments was synthesized from total RNA obtained from human spleen cells (BD Biosciences) and was amplified by PCR performed using Fab H-5′/Fab H-3′ and Fab L-5′/Fab L-3′ primers, respectively. The VH and Vκ gene fragments were amplified by PCR performed using anti-PA scFv DNA as a template and PA H-5′/PA H-3′ and PA L-5′/PA L-3′ as primers. The Fd (VH-CH1) and κ-chain (Vκ-Cκ) segments were produced through splicing by overlap extension PCR of VH and CH1 and of Vκ and Cκ, with use of PA H-5′/Fab H-3′ and PA L-5′/Fab L-3′ as primers. The Fd region was digested with XhoI and NotI, and the κ-chain region was digested with XbaI and SacI. The digested DNA fragments were cloned into pComb3H at the matching restriction sites [14]
Fab H-5′: ACCGTCTCCTCAGCCTCCACCAAGGGCCCATCGGT
Fab H-3′: AATGAGATCTGCGGCCGCTTAAATTAATTAAT
Fab L-5′: GTGGAAATCAAACGAACTGTGGCTGCACCATCTGT
Fab L-3′: AGGTATTTCATTTTAAATTCCTCCT
PA H-5′: GAGGTGCAGCTGCTCGAGACTGGAGGAGGCTT
PA H-3′: CTTGGTGGAGGCTGAGGAGACGGTGACCGTGGTCCCT
PA L-5′: TGGAGGTGGATCCGAGCTCGTAATGACGCAGTCT
PA L-3′: AGCCACAGTTCGTTTGATTTCCACCTTGGTCCCAGG
Fab was converted to full-length IgG by digestion of Fd with XhoI and ApaI and by cloning into a pCDHC68b vector [15] that contains human heavy-chain constant region to yield plasmid pPAH. The κ-chain was digested with XbaI and SacI and was cloned into a pCNHLC vector [15] to yield plasmid pPAκ
Expression and purification of scFv, Fab, and IgGBoth scFv and Fab were expressed in E. coli In brief, bacteria were cultured at 30°C in 2× YT (yeast extract–tryptone–rich) medium containing 2% glucose and appropriate antibiotics, until the OD600 was 0.5–1. The culture was diluted 5-fold in 2× YT without glucose and containing 0.2 mmol/L isopropyl-β-d-thiogalactopyranoside, and it then was incubated for 20 h at 27°C. The expressed proteins were purified by chromatography performed on nickel-charged affinity resins (Invitrogen)
The full-length IgG plasmids pPAH and pPAκ, which contained the VH and VL of anti-anthrax toxin components, were cotransfected into 293T cells for transient expression. The IgG was purified by affinity chromatography performed on anti–human Fc agarose (Sigma)
The purity of each antibody was determined by SDS-PAGE (Novex; Invitrogen), and the protein concentration was determined by bicinchoninic assay (BCA; Pierce Biotechnology) and ELISA performed with a purified human IgG (Jackson ImmunoResearch) as a reference standard
ELISAPA, LF, and unrelated proteins (bovine serum albumin [BSA], thyroglobulin, lysozyme, and phosphorylase b) were coated onto a 96-well plate by placing 100 μL of protein in carbonate buffer (pH 9.5) at a concentration of 5 μg/mL in each well and incubating the plate at room temperature overnight. Serial dilutions of soluble scFv, Fab, IgG, or phage were added to the wells, and the plates were incubated for 2 h at room temperature. The plates were washed, and the secondary antibody conjugate (anti–His-HRP, anti–human Fab-HRP, or anti–M13-HRP) was added and incubated for 1 h at room temperature. The plates were washed, and the color was developed by adding tetramethylbenzidine solution (TMB; Sigma). The plates were read, at an optical density measured at 450 nm, in an ELISA plate reader
Competitive ELISARecombinant PA at a concentration of 5 μg/mL in carbonate buffer (pH 9.5) was coated onto the wells of an ELISA plate. The wells were then incubated with anti-PA W2 Fab at different concentrations for 1 h at room temperature. Anti-PA W2 Fab was removed from the wells, and mouse anti-PA MAbs 14B7 and 2D3 [16], respectively, were added to the wells at a concentration of 0.5 μg/mL. Plates were incubated for 1 h and washed, and bound MAbs were detected by the addition of HRP-conjugated anti–mouse antibody, followed by TMB substrate. Binding to PA by 14B7 and 2D3 was calculated by dividing the optical density value in the absence of W2 by the optical density value in the presence of W2. Competition between W1 Fab and W2 IgG or between W2 Fab and W1 IgG was tested as described above, except that HRP-conjugated anti–human Fc was used as the secondary antibody
Affinity measurementSurface plasmon resonance (SPR) biosensor experiments were conducted using a Biacore 3000 instrument (Biacore) with short carboxymethylated dextran sensor surfaces (CM3; Biacore) and standard amine coupling, as described in detail elsewhere [17]. Experiments were conducted in 2 configurations. First, antibodies were immobilized to the surface, and the kinetics of binding and dissociation of PA were recorded for 10 min and 2 h, respectively, at PA concentrations of 1, 10, 100, and 1000 nmol/L. To eliminate the effects of immobilization-induced surface-site heterogeneity and mass transport limitation in the determination of the binding constants [18], the kinetic traces were globally fitted with a model for continuous ligand distributions [19] combined with a 2-compartment approximation of mass transport. Second, we conducted solution competition experiments in which different concentrations of soluble Fab were preincubated with PA for 24 h and the concentration of unbound PA was determined from the initial slope of surface binding noted when the mixture was passed over the antibody-functionalized surface. Standard competition isotherms were used for analysis [17, 18]
Epitope mappingA series of PA fragments that differed in size were amplified from PA-encoding vector pPA26 [20] by means of PCR, as described elsewhere [21], and were inserted into pGEM-T vectors (Promega). PA peptides labeled with 35S-methionine (Amersham Biosciences) were prepared with the TNT T7/SP6–coupled in vitro transcription/translation system (Promega). A radioimmunoprecipitation assay was performed essentially as described elsewhere [21]. In brief, a mixture of a 35S-labeled peptide and anti-PA W2 was incubated at 4°C overnight, and immune complexes were collected with protein G–coupled agarose beads (Amersham Biosciences). The precipitated complex was washed and subjected to SDS-PAGE. The PA peptide was detected by exposing the dried gel to an x-ray film
PA binding assaysRAW264.7 cells [22] were grown, in 6-well plates, to 90% confluence. Ten microliters of PA (10 μg/mL) was mixed with neutralizing antibody 14B7 [23] or with W2 at molar ratios of PA to MAb of 1:1 or 1:10 in a final volume of 200 μL, and it then was incubated for 5 min or 15 min. PBS was mixed with PA in control samples. The mixture was added to the cells and was incubated for 20 min at 37°C. The medium was removed, and cells were washed 5 times with ice-cold PBS, followed by lysis in radioimmunoprecipitation assay buffer (1% Nonidet, 0.5% sodium deoxycholate, and 0.1% SDS in PBS) plus EDTA-free complete protease inhibitor cocktail (Roche Applied Science). Equal amounts of protein (as measured with BCA; Pierce Biotechnology) were loaded onto SDS-PAGE gels. Western blot analysis was performed using anti-PA polyclonal antibody 5308 (dilution, 1:3500), which was developed in our laboratory (Bacterial Toxins and Therapeutics Section, National Institute of Allergy and Infectious Diseases), and HRP-conjugated goat anti–rabbit IgG secondary antibody (dilution, 1:2000) (Santa Cruz Biotech)
In vitro neutralization assayAn established RAW264.7 cell–based assay was used to determine the in vitro neutralization activity of antibodies [22, 24]. Results were plotted, and the effective concentration for 50% neutralization (EC50) was calculated using Prism software (Graphpad Software)
In vivo neutralization assayGroups of 3 Fischer 344 rats (sex, female; body weight, 150–170 g) were injected via the tail vein with PBS or a mixture of antibody and PA+LF (lethal toxin [LT]), at different molar ratios (antibody:LT, 1:3–4), prepared in sterile PBS. Injection volumes were 200 μL/rat. Rats were observed continuously for the first 8 h and then were observed at 16 h and 24 h; twice-daily checks then occurred for 1 week. Rats were monitored for signs of malaise and death. When rats were pretreated with antibody, they were injected intravenously with PBS or antibody at 5 min, 4 h, or 1 week before an intravenous injection with PA+LF (7.5 μg each)
Results
Isolation of anti-PA and anti-LF clonesRecombinant proteins were used as antigens to select antibodies from an scFv library derived from immunized chimpanzees. After 3 rounds of selection with PA, LF, or EF, a total of 192 phagemid clones were screened for specific binding by use of ELISA. We did not detect clones that bound specifically to EF protein. Ninety percent of the clones recognized PA or LF proteins but not BSA control protein. Four unique anti-PA and 2 unique anti-LF scFv phagemid clones were identified (W1, W2, A63-6, W5, F3-6, and F5-1) by sequence analysis (figure 1). Two clones, W1 and W2, had very similar sequences in the VH and Vκ regions (figure 1), with only a 9-aa difference (3 of the amino acids are located within the primer region). The amino acid sequences of the other 4 clones differed greatly. As expected, the greatest divergences in sequence and length were noted in the CDR regions. Because there is extensive homology between chimpanzee and human immunoglobulins [8], similarity searches of all known human immunoglobulin genes in the V-BASE database were conducted [25]. The VH3 family of VH and the Vκ I and II families of Vκ were preferentially used by anti-anthrax antibodies (table 1). As expected, W1 and W2 clones were derived from the same VH and Vκ germline genes
Figure 1.

Alignment of the deduced amino acid sequences of the variable domains of the heavy chain (VH) and κ chain (Vκ). Substitutions relative to W1 are expressed as single letters denoting amino acids. Dashes denote identical residues, and asterisks denote the absence of corresponding residues relative to the longest sequence. Complementarity-determining regions (CDR1–3) and framework regions (FR1–4) are shown above the sequence alignments
Table 1.

Human immunoglobulin germline genes most closely related to the chimpanzee heavy chain (VH) and κ light chain (Vκ) of various anti-anthrax protective antigen (PA) and lethal factor (LF) antibodies
The 6 scFvs were expressed in E. coli purified by affinity chromatography, and tested for binding activity and specificity by ELISA. Clones W1, W2, A63-6, and W5 bound strongly to PA antigen, and clones F3-6 and F5-1 bound strongly to LF antigen (figure 2); none of the clones bound to BSA, thyroglobulin, phosphorylase b, or lysozyme (data not shown)
Figure 2.
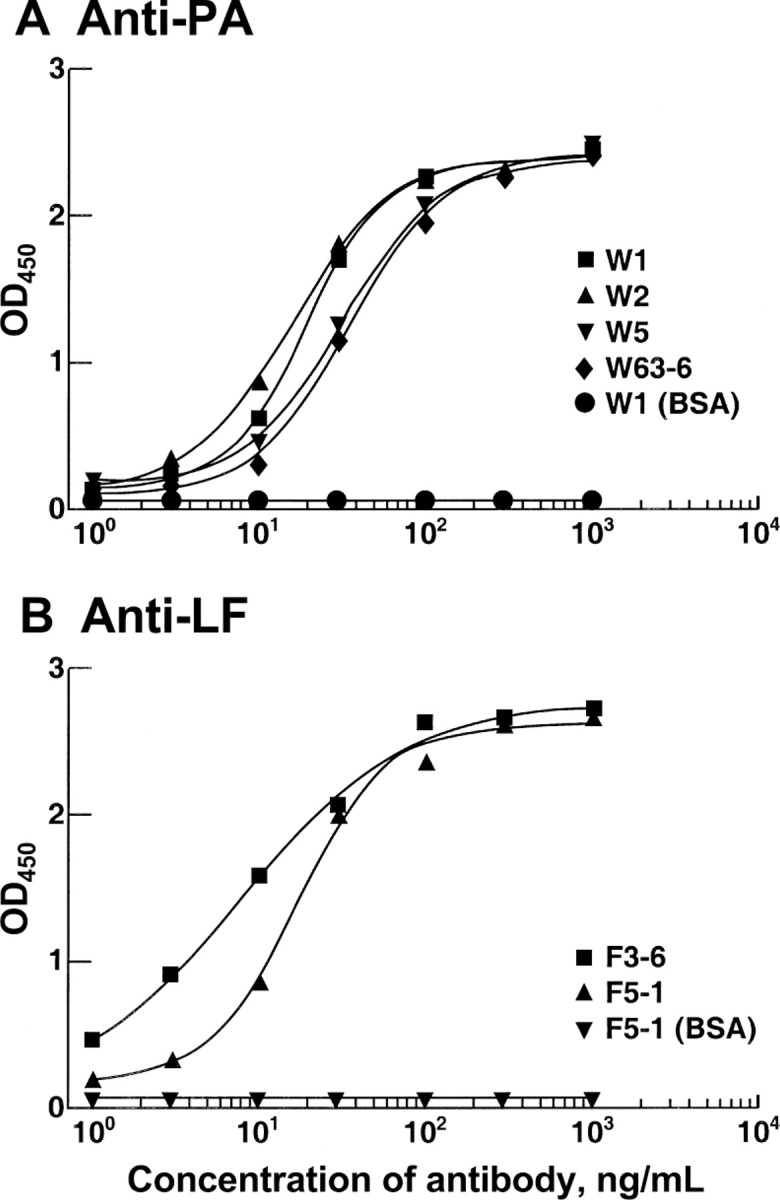
ELISA titration of anti–protective antigen (PA) (A) and anti–lethal factor (LF) (B) single-chain variable fragments (scFvs). Recombinant PA or LF or unrelated proteins, bovine serum albumin (BSA), thyroglobulin, lysozyme, and phosphorylase b were used to coat the wells of an ELISA plate. Wells then were incubated with various dilutions of scFvs. Bound scFv was detected by the addition of peroxidase-conjugated anti-His antibody, followed by tetramethylbenzidine substrate. The anti-PA and anti-LF scFvs did not bind to the unrelated proteins; only BSA is shown as an example. OD, optical density measured at 450 nm
In vitro neutralizing activity and affinity of W1 and W2The effect of anthrax toxin was strongly inhibited by the W1 and W2 scFvs, but not by A63-6, F3-6, F5-1, and W5 scFvs (data not shown). The latter scFvs did not undergo further study
scFv fragments have limited use in passive immunotherapy, because these monovalent fragments are rapidly cleared from the blood. In most cases, bivalent full-length immunoglobulin is more effective than is the corresponding scFv, because of avidity effects, effector functions, and prolonged half-life in the blood. Therefore, W1 and W2 scFvs were converted to bivalent whole IgG1s and were compared for neutralization activity in the RAW264.7 cell-based in vitro assay. Well-characterized mouse anti-PA 14B7 was used as a comparison control [23]
Neutralization by complete IgGs was 5–20-fold better than neutralization by scFvs (data not shown) and was 5-fold higher (for W2) and 15-fold higher (for W1) than that by mouse anti-PA 14B7 (figure 3). The equilibrium dissociation constant (Kd) for W1 and W2 IgG1, respectively, was determined by Biacore analysis. W1 and W2 antibody displayed very high affinity, with a Kd of 4×10-11 and 5×10-11 mol/L, respectively, compared with a Kd of 4×10-9 mol/L for 14B7 (in the present study) (table 2). These affinities compared favorably with those reported for human anti-PA MAbs (table 2), and they were the highest recorded affinities against anthrax PA [6, 7, 26]
Figure 3.

In vitro neutralization assay. Anti–protective antigen (PA) IgG was mixed with anthrax toxin and was incubated for 1 h at 37°C. The mixture was added to RAW264.7 cells in a 96-well plate and was incubated for 4 h at 37°C. After washing, the cells were stained with MTT dye, and then lysis was performed in a solution containing 0.5% SDS in 90% isopropanol and 0.05 mol/L HCl. The plate was read at an optical density measured at 570 nm, and the cell survival was calculated relative to untreated cells. Results were plotted and analyzed using Prism software (version 4; Graphpad Software). EC50, effective concentration for 50% neutralization. ▪, W1; ▾, W2; □, 14B7
Table 2.

Affinity of anti-anthrax protective antigen (PA) W1 and W2 antibodies, compared with other human anti-PA monoclonal antibodies (MAbs)
Characterization of the neutralization epitope and mechanism of neutralizationCompetitive ELISA results indicated that W1 and W2 may recognize the same epitope, because they competed with each other in binding to PA (data not shown), and because the complementarity-determining regions of their VH chains are identical. Therefore, only W2 was used to map the neutralization epitope. In ELISA, competition was also observed between 14B7 and W2 but not between 2D3 and W2 (figure 4). 14B7 binds to the site responsible for binding to the cellular receptor, whereas 2D3 does not compete with 14B7 and binds to a distinct binding site [16]. The binding site for W2 was mapped by radioimmunoprecipitation assay. The smallest peptide that reacted with W2 antibody contained 121 aa, corresponding to aa 614–735 at the C-terminus of PA (figure 5). This result indicated that W2 bound to a conformational epitope that encompassed almost the entire domain 4 of PA [28]. Because domain 4 of PA is responsible for cellular receptor binding [29, 30], the results suggested that W2 neutralizes the toxin by blocking binding of PA to the cellular receptor. This conclusion was confirmed in a binding assay, by use of Western blot analysis, which showed that W2 prevented the binding of PA to RAW264.7 cells (figure 6). The affinity (Kd) of binding of PA to the receptor is 1×10-9 mol/L to 5×10-9 mol/L [31]. Therefore, the affinity of W2 antibody binding to PA is 20–100-fold higher than the affinity of PA for the cellular receptor
Figure 4.

Competitive ELISA. Recombinant protective antigen (PA) was coated onto the wells of an ELISA plate. Wells then were incubated with anti-PA W2 Fab at the concentrations indicated. After incubation for 1 h at room temperature, anti-PA W2 Fab was removed from the wells, and mouse anti-PA monoclonal antibodies (MAbs) 14B7 and 2D3 [16, 23] were added to the wells. Bound MAbs were detected by the addition of peroxidase-conjugated anti–mouse antibody, followed by tetramethylbenzidine substrate. The binding to PA was calculated by dividing the optical density value in the absence of W2 by the optical density value in the presence of W2. ▪, 14B7; ▴, 2D3
Figure 5.

Summary of epitope mapping of anti–protective antigen (PA) W2 antibody by radioimmunoprecipitation assay. 35S-labeled PA peptides, prepared in vitro, were incubated with anti-PA W2. The immune complexes were captured by protein G–coupled agarose beads and were separated by SDS-PAGE. The PA peptide was detected by exposing the dried gel to an x-ray film. Numbers denote the starting and ending amino acids. The peptides that reacted with antibody and, hence, were detected on an x-ray film were considered to be positive (+). Faint intensity of the band on an x-ray film denoted a partial reaction (±)
Figure 6.

Inhibition of the binding of protective antigen (PA) to RAW264.7 cells by preincubation of toxin with monoclonal antibodies. PA at a concentration of 6 nmol/L (500 ng/mL) was incubated with anti-PA 14B7 or W2 antibodies at a molar ratio of 1:1 or 1:10 for 5 min. The mixture was added to RAW264.7 cells and was incubated for 20 min at 37°C. After the cells were washed and lysed, separation by SDS-PAGE was performed. The proteins were transferred to a membrane and were probed with anti-PA polyclonal antibody
In vivo animal protection Because affinity of anti-PA is strongly correlated with its neutralization activity, it is reasonable to assume that anti-PA W1 and W2 are potent neutralizing antibodies. As has been reported for other anti-PA MAbs [6, 7], one remarkable feature of our antibodies is the very slow dissociation rate, which may provide a significant physiological advantage for toxin neutralization in vivo
To evaluate the neutralization of PA by W1 and W2 in vivo, we measured protection against anthrax toxin challenge in the Fischer 344 rat model in 2 ways. First, MAb and toxin were mixed at different molar ratios, and the mixtures were injected into 3 rats each, which were then observed for morbidity and death. Injection of 7.5 μg of LT (7.5 μg of PA + 7.5 μg of LF) alone killed all 3 rats within 100–134 min (table 3). W2 antibody conferred protection at very low concentrations. Addition of W2 antibody at a molar ratio of 1:4 (Ab:PA), the lowest concentration of antibody tested, completely protected the rats from anthrax toxin challenge. In comparison, 14B7 mouse antibody protected only 1 of the 3 rats at this ratio, probably reflecting the difference in affinity between W2 and 14B7
Table 3.

Neutralization of protective antigen (PA) of anthrax toxin by W2 in rats
Second, in a more stringent test, the MAb was injected 5 min, 4 h, and 1 week before injection of LT (7.5 μg of PA + 7.5 μg of LF), to investigate the duration of antibody protection. Single administration of W1 and W2 antibodies at a 2:1 molar ratio (Ab:PA) protected 6 of 6 rats challenged with toxin 5 min, 4 h, or 1 week later (table 4). A lower dose of antibodies (0.5:1) still protected all the rats (n=6) when they were challenged 5 min or 4 h later but not when they were challenged 1 week later (table 4)
Table 4.

Duration of neutralization of protective antigen (PA) of anthrax toxin by W1 and W2 in rats
Discussion
We identified 2 chimpanzee MAbs, W1 and W2, which bind to PA with high affinity. These antibodies neutralized cytotoxicity of anthrax toxin in the picomolar range in vitro, and they efficiently protected animals from anthrax toxin challenge in vivo, most likely by blocking binding to the cell receptor. Our 2 neutralizing MAbs have the highest affinity of any human antibodies for PA reported thus far. Antibody affinity has been shown to correlate well with efficacy [5, 32–36 ]
The neutralization epitope recognized by W1 and W2 MAbs was mapped to a region of PA comprising aa 614–735. So far, 3 neutralization epitopes in PA have been proposed: the site for binding to the cellular receptor, the site for binding to LF [16, 37], and the site for heptamer formation [37]. However, the locations of these putative epitopes have not been precisely defined. For example, the epitope recognized by murine MAb 14B7 was suggested to be between Asp-671 and Ile-721, on the basis of the differential binding of 14B7 to different PA fragments generated through C-terminal deletions [16]. Because N-terminally deleted PA fragments were not tested, the epitope mapping for 14B7 could be incomplete. The previous observation that a PA fragment corresponding to aa 624–735 did not compete with PA for binding to the cell receptor [38] suggests that the region between aa 624 and aa 735 was not sufficient to generate the receptor recognition site. Some of the residues in domain 4 of PA that are critical for binding to the cellular receptor and to anti-PA 14B7 MAb have been determined by alanine-scanning mutations [39]
Anthrax, whether resulting from natural or bioterrorist-associated exposure, is a constant threat to human health. Although production of an efficient anthrax vaccine is an ultimate goal, the benefits of vaccination can be expected only if a large proportion of the population at risk is immunized. The low incidence of anthrax suggests that large-scale vaccination may not be the most efficient means of controlling this disease. In contrast, passive administration of neutralizing human or chimpanzee MAb to a subject at risk for anthrax or exposed to anthrax could provide immediate efficacy for emergency prophylaxis against or treatment of anthrax. This finding is supported by a recent study [27], which indicated that passive immunization with affinity-improved, humanized murine MAb 14B7 against PA (ETI-204) could efficiently protect rabbits before or after challenge with aerosolized Bacillus anthracis spores. However, humanized murine MAbs may retain some antigenic components of the original murine sequences and may elicit antibodies to the MAb in humans. Human- and chimpanzee-derived MAbs would not be expected to have this problem, because the sequences of chimpanzee immunoglobulin genes are virtually identical to those of humans [8–10 , 40]. Furthermore, one would expect that the anti-PA chimpanzee MAbs W1 and W2 will provide better protection than will ETI-204, because these MAbs had higher affinity (Kd) and lower EC50 in in vitro neutralization experiments than did ETI-204
Acknowledgments
We thank John Robbins and Rachel Schneerson, for their assistance and helpful discussions, and Charlene Shaver and Jason Wiggins, for excellent technical support
Footnotes
Potential conflicts of interest: none reported
Financial support: Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH); NIH (contract no. N01-AO-02733)
References
- 1.Jernigan JA, Stephens DS, Ashford DA, et al. Bioterrorism‐related inhalational anthrax: the first 10 cases reported in the United States. Emerg Infect Dis. 2001;7:933–44. doi: 10.3201/eid0706.010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collier RJ, Young JA. Anthrax toxin. Annu Rev Cell Dev Biol. 2003;19:45–70. doi: 10.1146/annurev.cellbio.19.111301.140655. [DOI] [PubMed] [Google Scholar]
- 3.Little SF, Ivins BE, Fellows PF, Friedlander AM. Passive protection by polyclonal antibodies against Bacillus anthracis infection in guinea pigs. Infect Immun. 1997;65:5171–5. doi: 10.1128/iai.65.12.5171-5175.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobiler D, Gozes Y, Rosenberg H, Marcus D, Reuveny S, Altboum Z. Efficiency of protection of guinea pigs against infection with Bacillus anthracis spores by passive immunization. Infect Immun. 2002;70:544–60. doi: 10.1128/IAI.70.2.544-550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maynard JA, Maassen CB, Leppla SH, et al. Protection against anthrax toxin by recombinant antibody fragments correlates with antigen affinity. Nat Biotechnol. 2002;20:597–601. doi: 10.1038/nbt0602-597. [DOI] [PubMed] [Google Scholar]
- 6.Wild MA, Xin H, Maruyama T, et al. Human antibodies from immunized donors are protective against anthrax toxin in vivo. Nat Biotechnol. 2003;21:1305–6. doi: 10.1038/nbt891. [DOI] [PubMed] [Google Scholar]
- 7.Sawada‐Hirai R, Jiang I, Wang F, et al. Human anti‐anthrax protective antigen neutralizing monoclonal antibodies derived from donors vaccinated with anthrax vaccine adsorbed. J Immune Based Ther Vaccines. 2004;2:5. doi: 10.1186/1476-8518-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schofield DJ, Satterfield W, Emerson SU, Purcell RH. Four chimpanzee monoclonal antibodies isolated by phage display neutralize hepatitis a virus. Virology. 2002;292:127–36. doi: 10.1006/viro.2001.1252. [DOI] [PubMed] [Google Scholar]
- 9.Ehrlich PH, Moustafa ZA, Justice JC, et al. Human and primate monoclonal antibodies for in vivo therapy. Clin Chem. 1988;34:1681–8. [PubMed] [Google Scholar]
- 10.Ehrlich PH, Moustafa ZA, Harfeldt KE, Isaacson C, Ostberg L. Potential of primate monoclonal antibodies to substitute for human antibodies: nucleotide sequence of chimpanzee Fab fragments. Hum Antibodies Hybridomas. 1990;1:23–6. [PubMed] [Google Scholar]
- 11.Sblattero D, Bradbury A. A definitive set of oligonucleotide primers for amplifying human V regions. Immunotechnology. 1998;3:271–8. doi: 10.1016/s1380-2933(97)10004-5. [DOI] [PubMed] [Google Scholar]
- 12.Krebber A, Bornhauser S, Burmester J, et al. Reliable cloning of functional antibody variable domains from hybridomas and spleen cell repertoires employing a reengineered phage display system. J Immunol Methods. 1997;201:35–55. doi: 10.1016/s0022-1759(96)00208-6. [DOI] [PubMed] [Google Scholar]
- 13.Harrison JL, Williams SC, Winter G, Nissim A. Screening of phage antibody libraries. Methods Enzymol. 1996;267:83–109. doi: 10.1016/s0076-6879(96)67007-4. [DOI] [PubMed] [Google Scholar]
- 14.Glamann J, Burton DR, Parren PW, et al. Simian immunodeficiency virus (SIV) envelope‐specific Fabs with high‐level homologous neutralizing activity: recovery from a long‐term‐nonprogressor SIV‐infected macaque. J Virol. 1998;72:585–92. doi: 10.1128/jvi.72.1.585-592.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trill JJ, Shatzman AR, Ganguly S. Production of monoclonal antibodies in COS and CHO cells. Curr Opin Biotechnol. 1995;6:553–60. doi: 10.1016/0958-1669(95)80092-1. [DOI] [PubMed] [Google Scholar]
- 16.Little SF, Novak JM, Lowe JR, et al. Characterization of lethal factor binding and cell receptor binding domains of protective antigen of Bacillus anthracis using monoclonal antibodies. Microbiology. 1996;142:707–15. doi: 10.1099/13500872-142-3-707. [DOI] [PubMed] [Google Scholar]
- 17.Schuck P, Boyd LF, Andersen PS. Measuring protein interactions by optical biosensors. In: Coligan JE, Dunn BM, Ploegh HL, Speicher DW, Wingfield PT, editors. Current protocols in protein science. Vol 2. New York: John Wiley & Sons; 1999. pp. 20.2.1–20.2.21. [Google Scholar]
- 18.Schuck P. Use of surface plasmon resonance to probe the equilibrium and dynamic aspects of interactions between biological macromolecules. Annu Rev Biophys Biomol Struct. 1997;26:541–66. doi: 10.1146/annurev.biophys.26.1.541. [DOI] [PubMed] [Google Scholar]
- 19.Svitel J, Balbo A, Mariuzza RA, Gonzales NR, Schuck P. Combined affinity and rate constant distributions of analyte or ligand populations from experimental surface binding and kinetics and equilibria. Biophys J. 2003;84:4062–77. doi: 10.1016/S0006-3495(03)75132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Welkos SL, Lowe JR, Eden‐McCutchan F, Vodkin M, Leppla SH, Schmidt JJ. Sequence and analysis of the DNA encoding protective antigen of Bacillus anthracis. Gene. 1988;69:287–300. doi: 10.1016/0378-1119(88)90439-8. [DOI] [PubMed] [Google Scholar]
- 21.Schofield DJ, Purcell RH, Nguyen HT, Emerson SU. Monoclonal antibodies that neutralize HEV recognize an antigenic site at the carboxyterminus of an ORF2 protein vaccine. Vaccine. 2003;22:257–67. doi: 10.1016/j.vaccine.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 22.Varughese M, Chi A, Teixeira AV, Nicholls PJ, Keith JM, Leppla SH. Internalization of a Bacillus anthracis protective antigen‐c‐Myc fusion protein mediated by cell surface anti‐c‐Myc antibodies. Mol Med. 1998;4:87–95. [PMC free article] [PubMed] [Google Scholar]
- 23.Little SF, Leppla SH, Cora E. Production and characterization of monoclonal antibodies to the protective antigen component of Bacillus anthracis toxin. Infect Immun. 1988;56:1807–13. doi: 10.1128/iai.56.7.1807-1813.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pitt ML, Little SF, Ivins BE, et al. In vitro correlate of immunity in a rabbit model of inhalational anthrax. Vaccine. 2001;19:4768–73. doi: 10.1016/s0264-410x(01)00234-1. [DOI] [PubMed] [Google Scholar]
- 25.Cook GP, Tomlinson IM. The human immunoglobulin VH repertoire. Immunol Today. 1995;16:237–42. doi: 10.1016/0167-5699(95)80166-9. [DOI] [PubMed] [Google Scholar]
- 26.Cirino NM, Sblattero D, Allen D, et al. Disruption of anthrax toxin binding with the use of human antibodies and competitive inhibitors. Infect Immun. 1999;67:2957–63. doi: 10.1128/iai.67.6.2957-2963.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohamed N, Clagett M, Li J, et al. A high‐affinity monoclonal antibody to anthrax protective antigen passively protects rabbits before and after aerosolized Bacillus anthracis spore challenge. Infect Immun. 2005;73:795–802. doi: 10.1128/IAI.73.2.795-802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Crystal structure of the anthrax toxin protective antigen. Nature. 1997;385:833–8. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- 29.Varughese M, Teixeira AV, Liu S, Leppla SH. Identification of a receptor‐binding region within domain 4 of the protective antigen component of anthrax toxin. Infect Immun. 1999;67:1860–5. doi: 10.1128/iai.67.4.1860-1865.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu S, Leppla SH. Cell surface tumor endothelium marker 8 cytoplasmic tail‐independent anthrax toxin binding, proteolytic processing, oligomer formation, and internalization. J Biol Chem. 2003;278:5227–34. doi: 10.1074/jbc.M210321200. [DOI] [PubMed] [Google Scholar]
- 31.Singh Y, Chaudhary VK, Leppla SH. A deleted variant of Bacillus anthracis protective antigen is non‐toxic and blocks anthrax toxin action in vivo. J Biol Chem. 1989;264:19103–7. [PubMed] [Google Scholar]
- 32.Adams GP, Schier R, Marshall K, et al. Increased affinity leads to improved selective tumor delivery of single‐chain Fv antibodies. Cancer Res. 1998;58:485–90. [PubMed] [Google Scholar]
- 33.Bachmann MF, Kalinke U, Althage A, et al. The role of antibody concentration and avidity in antiviral protection. Science. 1997;276:2024–7. doi: 10.1126/science.276.5321.2024. [DOI] [PubMed] [Google Scholar]
- 34.Jackson H, Bacon L, Pedley RB, et al. Antigen specificity and tumour targeting efficiency of a human carcinoembryonic antigen‐specific scFv and affinity‐matured derivatives. Br J Cancer. 1998;78:181–8. doi: 10.1038/bjc.1998.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamarre A, Lecomte J, Talbot PJ. Antiidiotypic vaccination against murine coronavirus infection. J Immunol. 1991;147:4256–62. [PubMed] [Google Scholar]
- 36.Lamarre A, Talbot PJ. Protection from lethal coronavirus infection by immunoglobulin fragments. J Immunol. 1995;154:3975–84. [PubMed] [Google Scholar]
- 37.Brossier F, Levy M, Landier A, Lafaye P, Mock M. Functional analysis of Bacillus anthracis protective antigen by using neutralizing monoclonal antibodies. Infect Immun. 2004;72:6313–7. doi: 10.1128/IAI.72.11.6313-6317.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh Y, Klimpel KR, Quinn CP, Chaudhary VK, Leppla SH. The carboxyl‐terminal end of protective antigen is required for receptor binding and anthrax toxin activity. J Biol Chem. 1991;266:15493–7. [PubMed] [Google Scholar]
- 39.Rosovitz MJ, Schuck P, Varughese M, et al. Alanine‐scanning mutations in domain 4 of anthrax toxin protective antigen reveal residues important for binding to the cellular receptor and to a neutralizing monoclonal antibody. J Biol Chem. 2003;278:30936–44. doi: 10.1074/jbc.M301154200. [DOI] [PubMed] [Google Scholar]
- 40.Schofield DJ, Glamann J, Emerson SU, Purcell RH. Identification by phage display and characterization of two neutralizing chimpanzee monoclonal antibodies to the hepatitis E virus capsid protein. J Virol. 2000;74:5548–55. doi: 10.1128/jvi.74.12.5548-5555.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]