

Abstract
Viral upper respiratory infections are the most common cause of clinical olfactory dysfunction, but the pathogenesis of dysosmia after viral infection is poorly understood. Biopsies of the olfactory mucosa in patients that complain of dysosmia after viral infection fall into two categories: one in which no olfactory epithelium is seen and another in which the epithelium is disordered and populated mainly by immature neurons. We have used intranasal inoculation with an olfactory bulb line variant of MHV to study the consequences of viral infection on peripheral olfactory structures. MHV OBLV has little direct effect on the olfactory epithelium, but causes extensive spongiotic degeneration and destruction of mitral cells and interneurons in the olfactory bulb such that the axonal projection from the bulb via the lateral olfactory tract is markedly reduced. Moreover, surviving mitral cells apparently remain disconnected from the sensory neuron input to the glomerular layer, judging from retrograde labeling studies using Dil. The damage to the bulb indirectly causes a persistent, long-term increase in the turnover of sensory neurons in the epithelium, i.e. the relative proportion of immature to mature sensory neurons and the rate of basal cell proliferation both increase. The changes that develop after inoculation with MHV OBLV closely resemble the disordering of the olfactory epithelium in some patient biopsies. Thus, damage to the olfactory nerve or bulb may contribute to a form of post-viral olfactory dysfunction and MHV OBLV is a useful model for studying the pathogenesis of this form of dysosmia.
Introduction
The olfactory system has a remarkable capacity to recover after injury, which is based on the capacity of the olfactory epithelium (OE) to generate new sensory neurons throughout life (Monti Graziadei et al., 1979; Graziadei et al., 1980; Costanzo and Graziadei, 1983; Schwob et al., 1995). Nonetheless, anosmia and hyposmia are not uncommon clinical complaints. Data accumulated from the various clinical olfactory centers indicate that viral upper respiratory infection (URI) is one of, if not the, most common causes of a disordered sense of smell (Henkin et al., 1975; Deems et al., 1991). In addition, axoplasmic transport along the primary olfactory pathway, composed of OE, olfactory nerve (ON) and olfactory bulb (OB) is a route of entry to the CNS for many neurotropic viruses after intranasal inoculation (Tomlinson and Esiri, 1983; Stroop et al., 1984; Perlman et al., 1990; Lafay et al., 1991; Barnett et al., 1993) and, in some cases, hematogenous spread (Lafay et al., 1991).
The histopathological correlates of post-viral olfactory disease (PVOD) have been investigated to a limited extent (Douek et al., 1975; Yamagishi et al., 1988, 1990, 1994; Moran et al., 1992; Akerlund et al., 1995). In some patients with PVOD biopsies of the mucosa in the olfactory area include only respiratory epithelium, which suggests that a very large proportion of the mucosa has lost its character as olfactory (Douek et al., 1975). Presumably, the viral infection and/or the host's response to the infection have also destroyed the progenitor cells required for reconstitution of sensory neurons. In these cases virally mediated damage and its sequelae have effects analogous to the severe, widespread destruction of the OE caused by exposure to methylbromide (MeBr) or 3-methylindole (3-MI); some olfactotoxin-damaged areas of the OE are reconstituted as respiratory epithelium as an apparent consequence of obliteration of the neuroepithelial stem cells (Schwob et al., 1994, 1995).
In other patients classified as dysosmic due to PVOD biopsies of the mucosa include epithelium that was still recognizably olfactory, but was disordered and composed mainly of immature sensory neurons (Yamagishi et al., 1988, 1990, 1994; Moran et al., 1992). In this other form of PVOD progenitor cells have been spared, but neuronal maturation is blocked. In animals and in humans newly formed neurons that are unable to connect with the OB, either because of bulbectomy (Schwob et al., 1992), avulsion of the olfactory nerve (Schwob et al., 1994) or congenital absence of the OB (Schwob et al., 1993), have an abbreviated lifespan due to absence of the trophic support that is normally supplied by the OB. As a result, newly generated neurons in these settings lack the time to make the transition to a mature phenotype before dying or die shortly thereafter (Schwob et al., 1992, 1993, 1994). This accelerated neuronal turnover has predictable consequences for the cellular composition of the OE: the rate of neurogenesis increases in an attempt to compensate for the accentuated loss of neurons, and immature neurons are more numerous and mature neurons are sparse in the absence of contact with the bulb, as compared with normal (Schwob et al., 1992, 1993, 1994). Thus, the histopathological findings in cases of PVOD in which OE is present but abnormal are not inconsistent with the notion that viral infection causes a form of damage to the olfactory nerve or bulb and that the predominance of immature neurons in the biopsies of the mucosa of some patients with PVOD is a consequence of the accelerated neuronal turnover that accompanies damage to the bulb or nerve.
In general, the agent that incites PVOD has not been identified in afflicted individuals, but viral URIs in adult humans are primarily caused by coronaviruses, adenoviruses, rhinoviruses, influenza viruses and enteroviruses (Larson et al., 1980). Coronaviruses, which are enveloped, positive-strand RNA viruses, are the second most common cause of influenza-like symptoms and are a not infrequent cause of cold symptoms, which are the two syndromes associated with viral URI (Larson et al., 1980; Sugiura et al., 1998). Coronaviruses are known to mutate rapidly in the host and exhibit a high frequency of recombination; some strains are neurotropic (Holland et al., 1982; Holmes and Lai, 1996). In particular, intranasal inoculation with many strains of mouse hepatitis virus (MHV) produces widespread dissemination and damage in the CNS, including the OB and more central parts of the olfactory system (Barthold, 1988; Barnett and Perlman, 1993). MHV is classified in antigenic group II of the coronaviruses, as are some of the human coronaviruses which cause colds (Holmes and Lai, 1996). In this context, it is also interesting to note that coronavirus persistence in the human CNS has been demonstrated at autopsy by RT-PCR in both neurologically normal individuals and patients with neurological disease (Arbour et al., 2000).
We report here studies using intranasal inoculation of the olfactory bulb line variant (OBLV) of MHV strain JHM (Gallagher et al., 1991) as a model of virus-induced, centrally mediated olfactory dysfunction. The cell line (OBL21A) used to host and propagate the virus was originally generated by avian myc transformation of primary cultures of neonatal OB, harvested at a time when progenitor cells in the bulb give rise primarily to granule and periglomerular neurons (Ryder et al., 1990). MHV OBLV was isolated during persistent infection of the host cells with MHV JHM when the cytopathic effect of infection shifted from syncytium formation to plaque formation at day 60 of passage (Ryder et al., 1990). We describe here the extent and severity of damage to olfactory structures after intranasal inoculation with the neurotropic coronavirus MHV OBLV. After inoculation MHV OBLV causes minimal disruption of the epithelium directly, but causes very extensive damage to the OB, like other MHV JHM variants. In contrast to other strains, however, damage induced by MHV OBLV is mostly limited to the olfactory system, with few if any animals lost due to encephalitis or hepatitis. In addition, we note that changes indicative of accelerated neuronal turnover occur in the OE as an apparent reflection of the damage to the bulb. Thus, the paradigm that we report here mimics the histopathological correlates of one form of PVOD in humans and may be a useful model for understanding the pathogenesis of that form of PVOD.
Materials and methods
Virus and cells
A viral stock of MHV OBLV was obtained as a gift from Dr Michael Buchmeier (Scripps Research Institute, La Jolla, CA) (Gallagher et al., 1991) and was propagated in OBL21A cells, which is a cell line derived from the neonatal olfactory bulb (a gift of Dr Connie Cepko) (Ryder et al., 1990), resulting in a final titer of 2.56 × 106 p.f.u./ml. Viral titers were plaque assayed on DBT cells (ATCC, Rockville, MD).
Animals
Twelve-week-old male BALB/c mice, weighing 20-25 g, were obtained from a commercial supplier (Taconic Farms, Germantown, NY). Some animals were maintained on ad libitum chow and later inoculated with MHV OBLV; others were food-restricted and maintained at 80% of body weight. During the course of the experiment the animals were maintained in a biohazard P2 facility within a heat and humidity controlled environment until they were killed.
Animal inoculation and observations
Fifteen minutes prior to anesthesia each mouse was injected with glycopyrrolate at a dose of 0.02 ml/g body wt in order to minimize bronchial secretions. Animals were placed in a Plexiglas box and anesthetized by gas inhalation with Fluothane. Following induction, the mice were placed on their backs and rapidly intubated, using a 20 gauge i.v. catheter. Throughout the course of the inoculation the animals were maintained in a surgical plane of anesthesia, using gas inhalation delivered to an open loop system attached to the i.v. catheter. One 22 gauge i.v. catheter was then inserted into each nostril through which the viral inoculum was delivered. Each animal received 300 μl of the MHV OBLV virus stock per nostril over 20 min for a total of 1.5 × 106 p.f.u. in 600 μl. Following recovery from anesthesia each mouse was returned to its home cage. One set of animals was observed for disease frequency for 45 days post-inoculation.
Histology and immunohistochemistry
Mice inoculated with MHV OBLV and age-matched control mice were killed 1, 2 and 3 weeks and 1 and 5 months after infection by CO2 asphyxiation and perfused with Bouin's fluid. Soft tissues and teeth were discarded and the bone was decalcified using RDO (Apex Engineering Products, Napierville, IL). The skull and its contents were embedded in paraffin, sectioned at 5-15 μm and stained with hematoxylin and eosin (H&E). Sections were immunostained with antiserum directed against olfactory marker protein (OMP) or with antiserum directed against 43 kDa growth-associated protein (GAP-43) following published protocols, using diaminobenzidine as the chromogen (Schwob et al., 1992, 1994, 1995). In addition, some of the material was stained with mouse monoclonal antibody J3.3, which is directed against MHV JHM (a kind gift of Dr John Fleming) (Fleming et al., 1983), using similar protocols. Leakage of endogenous mouse immunoglobulins into brain tissue as a consequence of inflammation and massive neuronal destruction interfered with our use of indirect immunostaining to map spread of the virus; i.e. background staining in the olfactory bulb and piriform cortex was very dense in sections incubated in the absence of primary antibody as a control. Thus, the antibody was only useful in identifying those regions of the CNS in which virus concentration during the acute phase of infection was very high.
Dil labeling of lateral olfactory tract (LOT) axons and centrifugal projections to the bulb
Two ad libitum fed, virally infected animals and three age-matched controls were perfused with 4% paraformaldehyde/0.5% glutaraldehyde in phosphate buffer 5 months following inoculation. After washing in phosphate-buffered saline for 1 day the bone overlying the region of the LOT and adjacent anterior piriform cortex was removed and an insect pin was used to transfer crystalline DiI (Molecular Probes, Eugene, OR) into the tissue. After removing the pin the hole was sealed with 1% agar solution. Tissues were promptly returned to fixative and maintained at 37°C for 6 months. At that time the bulb and brain were embedded in 0.3% albumin/0.03% gelatin medium and sectioned at 50 μm in the coronal plane with a Vibratome. The sections were immediately mounted, coverslipped with a glycerol mixture containing antifade (Vectashield, Vector Laboratories, CA) and photographed.
Autoradiography
Three virus-inoculated animals and three age-matched controls were injected with 2 μCi/g body wt [3H]thymidine i.p. 5 months after infection and perfusion fixed exactly 2 h after tracer injection. The olfactory epithelium was sectioned at 5 μm and sections were dipped in NTB-2 emulsion for autoradiography as described (Schwob et al., 1992). Thymidine-labeled basal cells were counted on five coronal sections equally spaced through the rostro-caudal extent of each animal's epithelium and the length of the epithelium was measured in order to determine the number of labeled cells per millimeter (labeling index).
Measurement of the external plexiform layer (EPL) of the OB
The area of the EPL was measured on captured images of six evenly spaced sections through the rostro-caudal extent of both OBs in the three control and three MHV OBLV-inoculated mice that were injected with [3H]thymidine using the image analysis program IPLab (Scanalytics, Vienna, VA). The summed area of the EPL across the six sections was plotted in relation to [3H]thymidine labeling index for each of the animals.
Counts of granule cells and mitral cells
The numbers of granule cells were determined by sampling five evenly spaced levels through the anteroposterior extent of the bulbs of animals killed 1 and 5 months after inoculation with OBLV while food-restricted. Images of 10 μm thick H&E stained sections were captured with a CCD camera using a 20× objective and assembled into mosaics. After establishing the internal granule layer (IGL) as the region of interest, granule cells were identified by segmenting the images according to size and intensity of hematoxylin staining. The limits for segmentation were chosen to exclude glial cells in the IGL and the effectiveness of the chosen parameters was established by direct visual inspection. The segmented area was converted to a cell count for each case by dividing the total segmented area by the average size of an individual cell; that average was determined by segmenting the image to highlight only those objects that were visually confirmed to be individual cells, summing their area and dividing by the number of objects. While the procedure for converting the segmented area to cell number may be an underestimate of the total number of granule cells due to the potential for two overlapping cells to occupy less area than the sum of two isolated cells, any discrepancy would be more pronounced as the density of granule cells increases. Thus, the discrepancy, should it exist, has the effect of reducing the magnitude of the recovery of granule cell number with time after lesion. In other words, any underestimate of granule cell number works against our hypothesis by opposing the difference that we were trying to establish as significant.
Mitral cells were counted in a set of five hematoxylin stained 10 μm thick sections, evenly spaced along the anteroposterior axis of the bulbs of infected, food-restricted mice that survived for 2 months after inoculation and normal, age-matched controls. Mitral cells were identified by their large size, open chromatin, prominent nucleoli and location at the interface between the EPL and IGL. Any shrinkage of the surviving mitral cells was insufficient to render them difficult to discriminate from the surrounding interneurons. Thus, size remained an important criterion for mitral cell identification in the infected animals. Although the regular arrangement of layers and cell types in the bulb is partially disrupted in infected animals, mitral cells were consistently located just superficial to the mass of granule cells. Mitral cell profiles that included a nucleolus were manually counted in both bulbs and summed across the sections.
Results
Clinical disease after intranasal inoculation with MHV OBLV
Two groups of mice were inoculated with the virus intranasally. In one the animals were fed ad libitum throughout the experiment. In the other the animals were maintained on food-restriction, as described in Materials and methods, in order to determine the effects of a chronically reduced body weight on the extent and consequences of viral infection; this comparison was motivated by the well-known impact of nutritional status on immunological function (Chandra, 1981) and the need to food-restrict animals for purposes of operant evaluation of olfactory function in virus-inoculated mice (Youngentob et al., 2001). A non-significant increase in mortality was observed among the group of food-restricted mice (6%, five out of a total of 80 inoculated mice), as compared with ad libitum fed mice (4%, one out of 25), over the 45 day observation period post-inoculation. Symptoms of systemic disease (lethargy, inanition, weight loss and other behavioral manifestations) were noted in nearly all food-restricted mice, while minimal signs of disease were noted in five of 80 (6%) of the inoculated ad libitum fed animals. Viral replication and clearance studies show little or no difference between the two groups. Virus was undetectable in lung and brain by 15 days post-intranasal inoculation and the liver was not infected (Youngentob et al., 2001). With regard to the anatomical changes described here, food restriction prior to inoculation has the effect of accentuating the damage to the olfactory bulb slightly by comparison with ad libitum fed mice. However, the damage to central olfactory structures is widespread and qualitatively similar in both groups. For the sake of clarity photomicrographs that derive from the food-restricted group are indicated in both the legend and the figure. If not otherwise specified, illustrations are taken from the group of ad libitum fed, infected mice.
Acute effects of MHV OBLV on the OE
Intranasal inoculation with MHV OBLV has minimal effects on the olfactory epithelium during the acute period after infection (Figure 1), especially by comparison with the widespread destruction occasioned by exposure to olfactotoxins like MeBr and 3-MI (Schwob et al., 1994, 1995). Either of these compounds causes the death of all neurons, all supporting cells and many basal cells in >90% of the epithelium (Schwob et al., 1994, 1995). In contrast, only scattered epithelial foci, totalling <10% of the extent of the OE, were badly damaged here. The damaged areas are characterized by a substantial decline in the number of OMP+, i.e. mature, neurons and an increase in the number of GAP-43+, i.e. immature, neurons, in comparison with control OE (Figure 2). The preponderance of immature neurons demonstrates that neurons are being actively replaced there. However, it should be emphasized that the initial damage to the epithelium is scant in extent. For example, the epithelium lining the dorsal vault of the nasal cavity at the level illustrated in Figure 2 is indistinguishable from normal controls. Furthermore, the damage is relatively modest in severity even in the affected areas (Figure 2), in comparison with MeBr exposure, in which the epithelium is very badly disorganized 1 week after lesion and lacks OMP+ neurons completely (Schwob et al., 1995).
Figure 1.
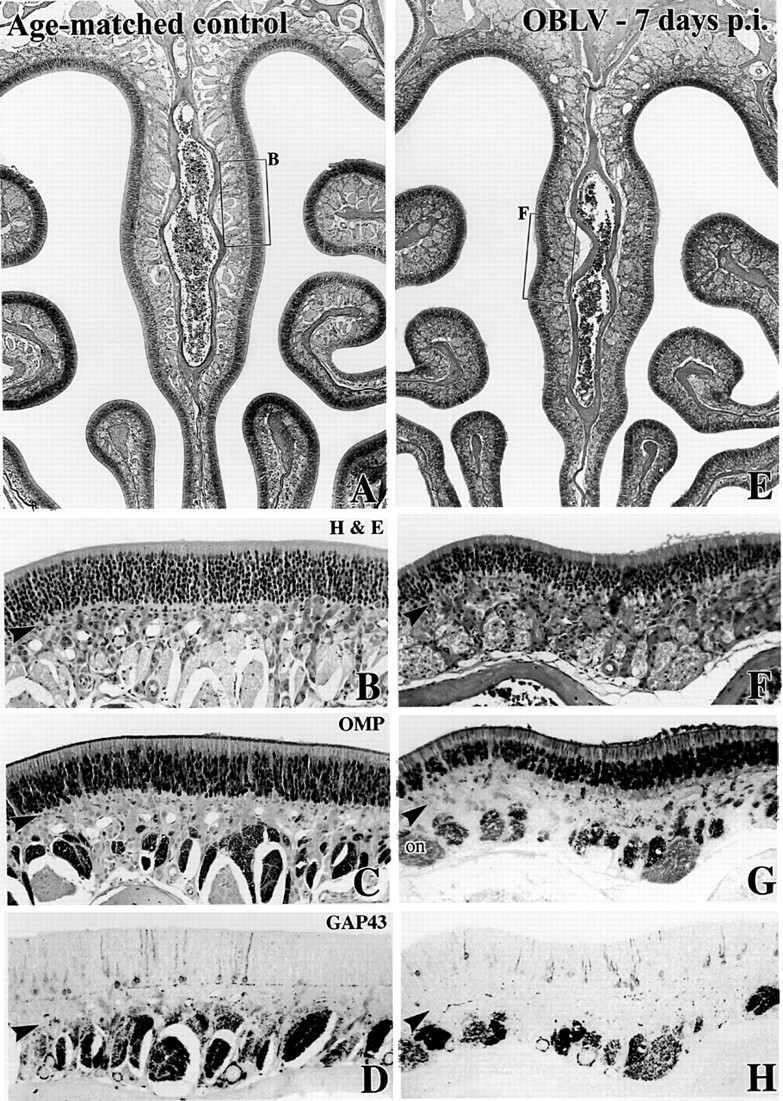
Intranasal inoculation with MHV OBLV causes little damage to the epithelium during the acute phase of infection. (A—D) Age-matched control animal. The boxed area of the hematoxylin and eosin (H&E) stained section in (A) is shown in higher power in (B). (C, D) Comparable location in sections adjacent to (A) stained with anti-OMP and anti-GAP-43, respectively. (E—H)Ad libitum fed mouse inoculated intranasally with MHV OBLV 7 days prior to perfusion. Conventions as in (A)—(D). There are slightly fewer OMP+ neurons in the infected animal, but no increase in GAP-43+ cells, indicating that the epithelium is undamaged and unreactive (cf. Figure. 2). Arrowheads mark the basal lamina. Magnifications: (A, E), 40×; (B—D, F—H), 180×.
Figure 2.
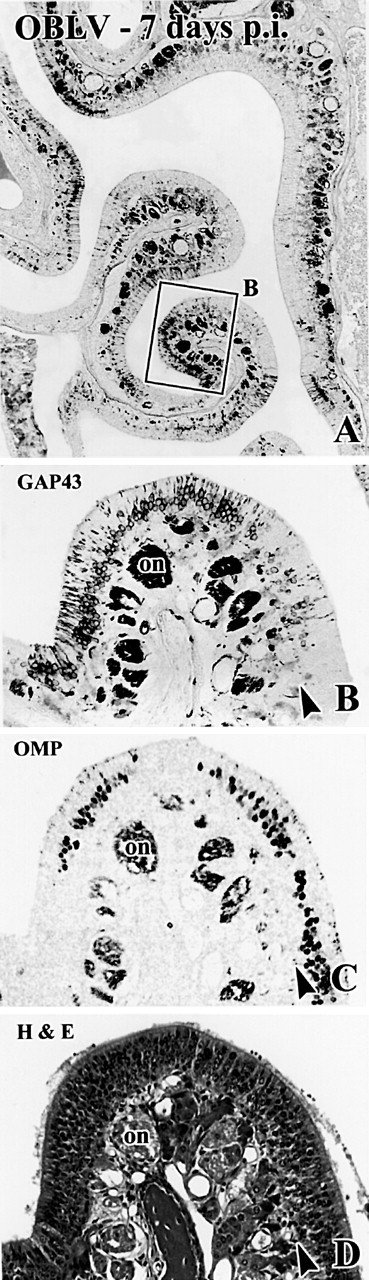
Occasional foci in the epithelium are more severely damaged after acute infection with MHV OBLV. The illustrated case was ad libitum fed at time of inoculation. (A, B) Anti-GAP-43 staining. Boxed area in (A) shown at higher power in (B). (C, D) Comparable location in sections adjacent to (A) that are stained with anti-OMP and H&E, respectively. Note that the epithelium is grossly intact judging from H&E stained material, but that mature, OMP+ sensory neurons have been lost and immature, GAP-43+ sensory neurons are increased as compared with the area of the epithelium illustrated in Figure 1. Arrowheads mark the basal lamina. Magnifications: (A), 38×; (B-D), 143×.
Acute effects on the OB and more central structures
In contrast to the limited damage to the epithelium, the OB was severely disrupted 1 week following inoculation (Figure 3A,D). Spongiosis was widespread and associated with infiltration of lymphocytes and neutrophils. The damage is particularly pronounced in the outer part of the EPL. It is worth noting that trans-synaptic transport of horseradish peroxidase-conjugated wheatgerm agglutinin into the EPL is also most prominent in its most superficial stratum (Shipley, 1985), suggesting a possible means by which virus is preferentially concentrated there rather than more deeply in the EPL. By 2 weeks after infection spongiosis had progressed to the extent that there was a complete or near complete split between the superficial layers of the bulb [the glomerular layer (GL) and olfactory nerve layer (ONL)] and the deeper layers of the bulb (the mitral layer and IGL) around the full circumference of the bulb, which has the effect of more or less obliterating the EPL (Figure 3B,E). By this time many granule cells have died, as shown by the marked decline in their number relative to the normal OB (cf. Figure 3F versus H); it is likely that death of granule cells contributes to spongiotic degeneration of the EPL.
Figure 3.
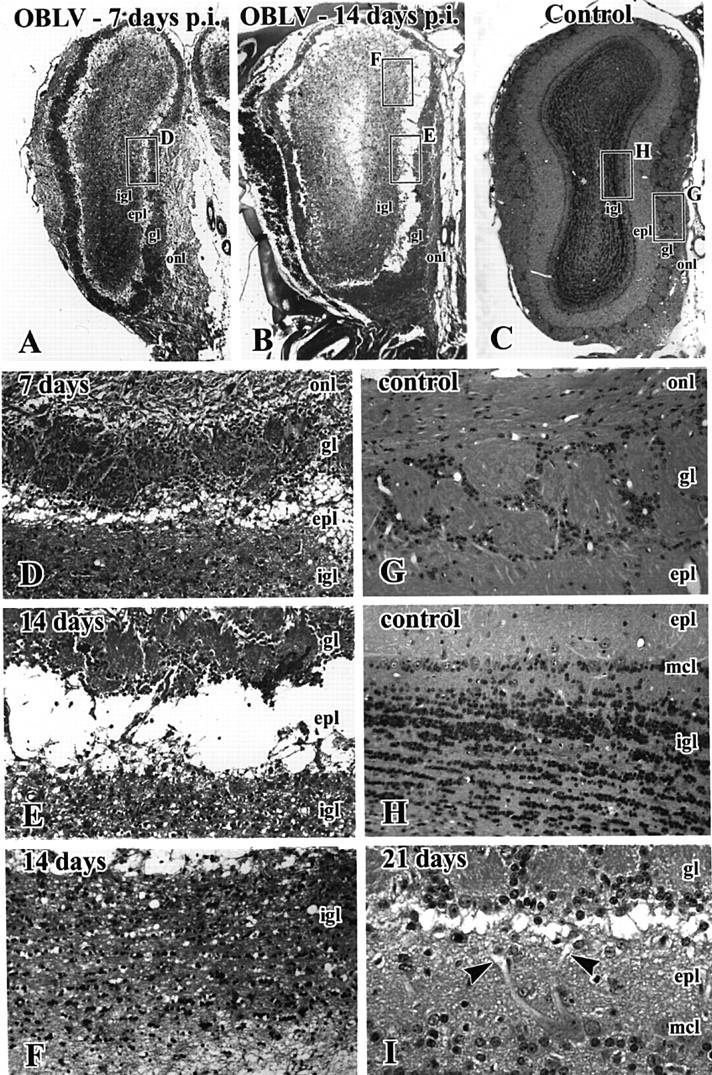
Intranasal inoculation with MHV OBLV causes widespread spongiotic degeneration in the OB during the acute phase of infection that reaches a maximum at 14 days after inoculation. (A-C) H&E stained sections of the bulb from mice surviving 7 and 14 days and an uninoculated control, respectively. All mice were ad libitum fed. Boxed areas in (A)-(C) are shown at higher magnification in (D-H), as indicated. (A, D) At 7 days note the spongiotic change in the outer EPL (epl), immediately deep to the GL (gl). (B, E, F) By 14 days there is frank separation of the deeper layers of the bulb from the GL and ONL due to obliteration of the EPL. There has also been some loss of granule cells as compared with controls (cf. H). (I) Photomicrograph from a comparable level of the bulb from a mouse that survived for 21 days after inoculation. The spongiotic change is resolving, but primary mitral cell dendrites are truncated and form swollen pale endings (arrowheads). igl, internal granule layer; mcl, mitral cell layer. Magnifications: (A-C), 16×; (D-H), 120×; (I), 260×.
By 3 weeks after lesion there has been a partial resolution in the spongiotic change (Figure 3I). Nonetheless, the disconnection between the deeper layers of the bulb and the GL is well demonstrated by the large number of mitral cell primary dendrites that end in a pale club-like expansion, which resembles the reactive end bulb formed after transection of an axon (arrows in Figure 3I). The truncation of mitral cell dendrites may be a consequence of edema and destruction of the surrounding neuropil in the EPL. Alternatively, viral infection of the mitral cells may result in dendritic truncation via some other mechanism.
Virus spreads further centralward from the bulb within the first week after infection. The lateral olfactory tract (LOT) and the underlying piriform cortex show evidence of spongiotic change and an inflammatory infiltrate (Figure 4). In addition, a monoclonal antibody directed against the capsid of MHV JHM (J.3.3) stains many pyramidal neurons in layer II of the piriform cortex, particularly in the areas deep to the LOT (Figure 5A), which receive the densest projection from the mitral and tufted cells (Schwob and Price, 1984). Virus may reach cortical neurons by traveling retrogradely along centrifugal axonal projections from cortex back to bulb; alternatively, spread may occur via anterograde transport down the axons of the LOT and trans-synaptic spread from them to their target neurons in the piriform cortex. In addition, there are foci of spongiosis and inflammation in areas of the ventral forebrain that do not receive a direct projection from the OB, but do receive a projection from cells in the deeper layers of the piriform cortex (Figure 5B).
Figure 4.
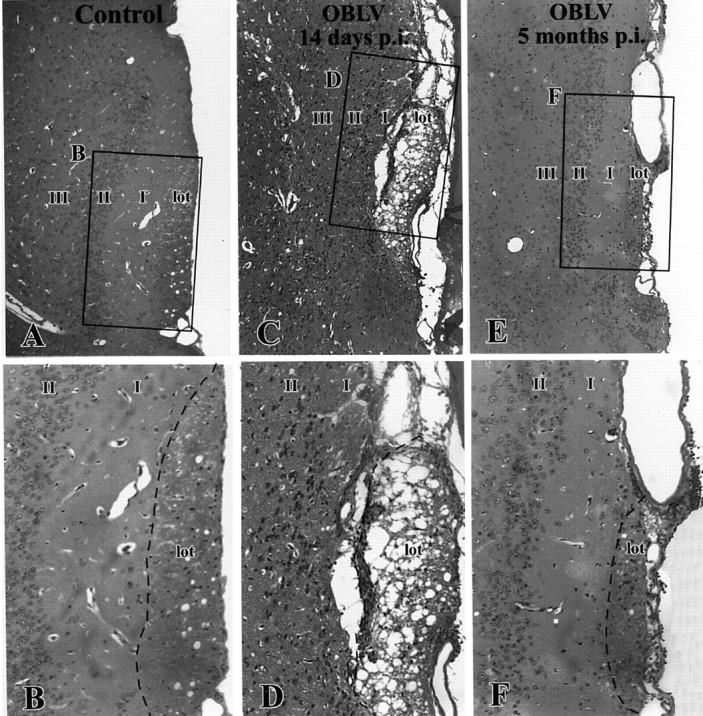
The lateral olfactory tract undergoes spongiotic degeneration during the acute phase of infection and is reduced in cross-sectional area in the long-term, which suggests that mitral cell axons are lost from the tract as a consequence of infection. (A, B) Age-matched control. (C, D) Virally infected, ad libitum fed, 14 day post-infection. (E, F) 5 months post-infection. Boxed areas are shown at higher magnification, as indicated. lot, LOT; I, layer I of piriform cortex; II, layer II of piriform cortex; III, layer III of piriform cortex. Magnifications: (A, C, E), 85×; (B, D, F), 170×.
Figure 5.
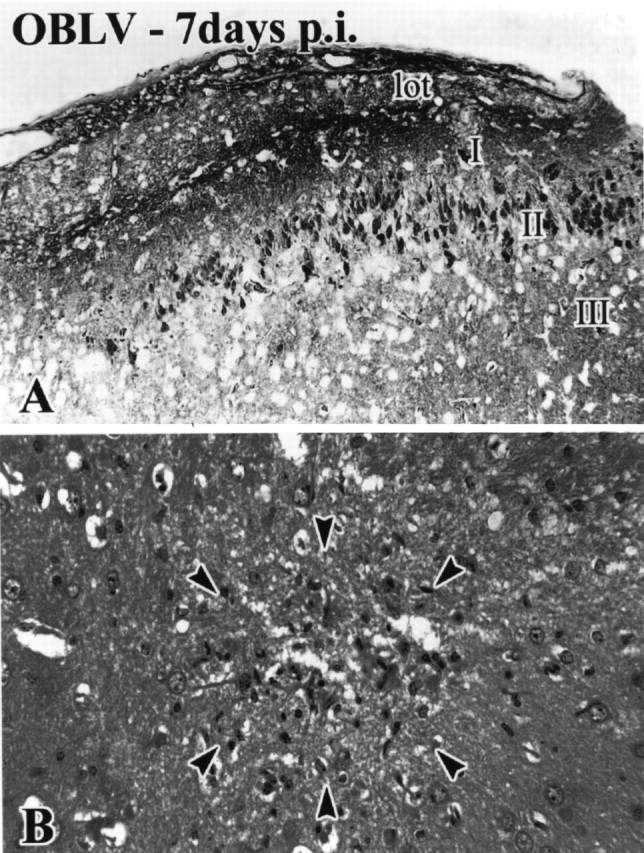
Spread of MHV OBLV to more central structures. (A) anti-MHV-immunoreactive pyramidal neurons in layer II of piriform cortex. (B) Focus of degeneration and mononuclear infiltrate in the caudate nucleus (arrowheads). Ad libitum fed mouse. Magnifications: (A), 180× ; (B), 270 ×.
In 17 of 19 infected animals that were perfused 1-2 weeks after inoculation the bulbs were damaged to the degree indicated above. The other two animals showed a lesser extent of disruption. In these the spongiotic degeneration did not fully encircle the bulb. A similar proportion of animals that survived for longer periods showed minimal long-term changes, as shown by preservation of the thickness of the EPL (see below). Invariably, the minimally affected animals also displayed relatively mild clinical symptomatology; there was little or no weight loss, appetite was maintained and activity levels were near normal in the acute post-inoculation period. This clinicopathological constellation was also characteristic of animals that received a deliberately lower dose of virus during the course of pilot studies designed to titrate the optimal viral dose. Thus, the coincidence of minimal clinical signs and limited tissue damage in a few mice that were ostensibly administered a full dose of the virus suggests that those minimally affected mice actually received a lower (and clearly inadequate) effective dose of the virus. The most likely reasons for a reduced dose in these rare cases are technical, i.e. leakage of the inoculum down the pharynx or retrogradely through the nares.
Long-term effects on the OB and its projection via the LOT
By 1 month after inoculation the acute effects of the virus on the OB have resolved and the bulb shows only moderate changes over the ensuing months. As a consequence of infection the OB remains shrunken overall by comparison with normal even 5 months after infection (Figure 6). There are fewer mitral cells overall in the bulbs of MHV OBLV-infected animals (Figure 7A,B versus C,D). Indeed, mitral cell numbers, determined by direct counts at multiple comparable levels of the bulb, are reduced to 30-70% of control in food-restricted, virally inoculated mice (mean reduction 43 ± 9% of normal; n = 4). The loss of mitral cells is further substantiated by a marked reduction in the cross-sectional area of the LOT (Figure 4). Other components of the bulb are also damaged. The EPL is severely and obviously affected and is reduced in volume to less than half in most cases (Figure 6; in addition see Figure 11, in which measures of the EPL are presented for a limited number of OBLV-inoculated animals and controls). Nonetheless, there had been a partial recovery in the thickness of the layer by comparison with its complete obliteration by spongiosis at 2 weeks after lesion (Figure 3 versus 6). Likewise, the IGL also recovered in part and was more densely cellular than at the height of the acute phase (Figure 3 versus 6). Visual comparison of the acute post-lesion animals and others killed at long survival times suggests that granule cell number may recover in part as time passes after lesion. In order to document the partial reconstitution of the granule cell population we compared computer-generated counts of granule cells in the IGL at 1 versus 5 months after lesion (see Materials and methods for details of the analytical procedure) (Figure 8). During that period the number of granule cells increased by 70%, although it still remained below age-matched controls (data not shown). The increase in granule cell number during that period fell just short of statistical significance (t = 2.08, 4 d.f., P = 0.053, one-tailed test).
Figure 6.
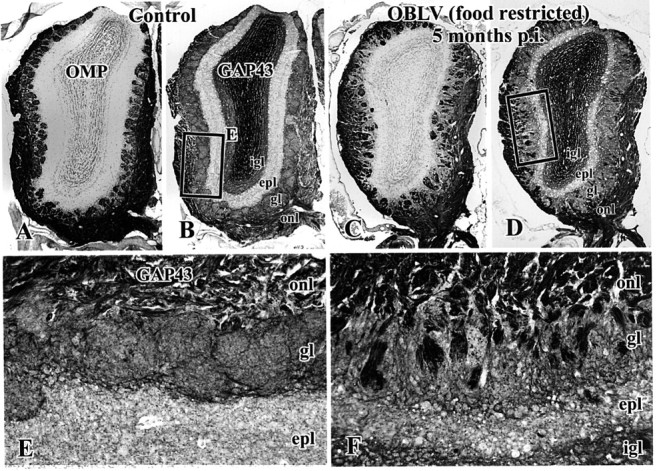
The structure of the OB remains distorted at long survival times after infection. In addition, the projection from the epithelium onto the bulb consists of a larger than normal population of newly innervating axons in infected animals as compared with controls. (A, B, E) Age-matched control. (C, D, F) Mouse inoculated with MHV OBLV and then killed 5 months after infection, food-restricted at time of inoculation. (A, C) Staining with anti-OMP. (B, D, E, F) Staining of adjacent section with anti-GAP-43. Note the marked reduction in thickness of the EPL in the lesioned animals (D, F versus B, E), the presence of OMP-stained sensory axons in the GL of the infected animal (C) and the increase in GAP-43 stained sensory axons in the ONL and GL of the infected animal (D, F versus B, E). Abbreviations as in Figure 3. Magnifications: (A-D), 23×; (E, F), 117×.
Figure 7.

Mitral cells are lost as a consequence of infection. (A, B) Age-matched control. (C, D) Mouse killed 5 months after inoculation, ad libitum fed at time of inoculation. Note the lack of a defined mitral cell layer (A versus C) and loss of mitral cells (arrows in B, D) after lesion. Abbreviations as in Figure 3. Magnifications: (A, C), 108×; (B, D), 315×.
Figure 11.

At long survival times after inoculation the number of proliferating basal cells is increased. Furthermore, the degree of damage to the EPL correlates with the increase in the index of basal cell proliferation in MHV OBLV-infected mice (inoculated when food-restricted) that survived for 5 months after inoculation. (A) Age-matched control, injected i.p with [3H]thymidine 2 h before perfusion. (B) Mouse killed 5 months post-inoculation, ad libitum fed at the time of inoculation, injected i.p. with [3H]thymidine 2 h before perfusion. Arrows indicate [3H]thymidine labeled basal cells. (C) Area of the epithelium illustrated in (A); (B) is indicated by the open arrow. (D) Plot of labeling index versus area of the EPL for each of the experimental (squares) and control (circles) animals. As discussed in the text, one of the three mice in the inoculated group was affected only minimally by the virus in terms of clinical symptomatology and pathological changes to the bulb (curved arrow); presumably the dose of virus was insufficient to produce significant disease in that particular case for technical reasons, most likely leakage of the inoculum. The overall correlation between these two measures is significant across all of the animals. The outlier may be excluded from the experimental data set on the grounds that infusion of virus was clinically ineffective in that case. With this exclusion the difference in labeling index between the two groups is highly significant. Magnifications: (A, B), 270×.
Figure 8.

Granule cell number partially recovers with time after lesion. (A) H&E stained section of olfactory bulb from a representative mouse killed 1 month after inoculation, food-restricted at time of inoculation. Note the paucity of granule cells relative to control (cf. Figure 3C). (B) H&E stained section of olfactory bulb from a representative mouse killed 5 months after inoculation, also food-restricted at time of inoculation. Note the apparent increase in the number of granule cells that has ensued in the prior 4 months. (C) Counts of granule cells confirm the visual impression that granule cell numbers increase between 1 and 5 months after infection. Average number of cells per level, determined bilaterally at five equally spaced levels through the antero-posterior extent of the bulb; comparable levels were counted in each case. Each triangle designates an individual experimental case. The horizontal line indicates the group mean and the error bar represents the SEM. Magnifications: (A, B), 27×.
We further evaluated the relative numbers and status of the mitral and tufted cells remaining after infection by implanting crystalline DiI into the LOT and piriform cortex of perfusion fixed brains of control and inoculated mice (Figure 9). Use of the DiI technique was necessitated by the fragility of the inoculated animals, which prevented the use of tracers, e.g. trans-neuronal transport of wheatgerm agglutinin-horseradish peroxidase, which require surgery. The conventional protocol for using DiI was modified. Deliberately large amounts of DiI were applied and the tissue was incubated for 6 months in order to visualize as extensive a population of surviving mitral and tufted cells as possible.
Figure 9.

Dil labeling demonstrates that the GL (gl) is largely disconnected from the deeper layers of the bulb in the virally infected mice. (A-D) Age-matched control. (E-H) Mouse killed 5 months after inoculation, ad libitum fed at time of inoculation. Boxed areas shown at higher magnification as indicated. At the level of the anterior olfactory nucleus (AON) retrograde labeling is prominent in the lateral olfactory tract and in the neurons of the AON of the control animal (A) but less so in the MHV OBLV-infected animal (E), despite roughly equivalent injection areas in the piriform cortex in the two animals. (B, C) In the control OB label ends at the boundary between the GL (gl) and the ONL (onl), i.e. the GL is labeled heavily and the ONL is unlabeled. (F, G) In contrast, in the infected mouse label ends at the boundary between the EPL (epl) and GL around much of the circumference of the bulb, i.e. the amount of label in the GL is roughly comparable to that in the ONL. An area of the ventrolateral bulb where some labeling extends into the GL of the inoculated mouse is indicated by the arrows in (F). The short vertical lines mark the boundaries between layers. (D, H) Transmitted light images of sections illustrated in (B), (F), respectively. Abbreviations as in Figure 3. Magnifications: (A, B, D-F, H, 18×; (C, G), 38×.
In control animals under the aforementioned conditions of label application, DiI densely labels mitral and tufted cells (via retrograde spread along the axons of the LOT), neurons in the anterior olfactory nucleus (projecting to the region of piriform cortex deep to the LOT) and fibers centrifugal to the bulb (via anterograde diffusion along axons). As a consequence of the very long incubation, DiI spread beyond the somata of mitral and tufted cells into their apical dendrites and to granule and periglomerular interneurons in the bulb. The labeled fiber pathways sum together to produce dense, nearly homogeneous staining of the IGL and the GL, which prevents visualization of individual cells by either conventional or confocal microscopy. The ONL is left unlabeled, indicating that the dye is not transferred from bulb neurons to primary afferents under these conditions. Thus, there is a sharp demarcation between the GL, which is labeled well by DiI, and the ONL, which is not, in controls.
In the MHV OBLV-lesioned animals the amount of retrograde label in the LOT, anterior olfactory nucleus and OB is reduced despite comparably sized implants of DiI. In addition, the label drops to undetectable at the boundary between the EPL and GL in most areas of the bulb. In other words, there is little label in the GL of the infected, recovered mice, in contrast to controls. The results demonstrate that the GL remains largely disconnected from the deeper layers in most parts of the bulb of the lesioned animals. We interpret this disconnection as an indication that most, if not all, of the remaining mitral cells have lost that part of their dendritic tree that enters the GL and the number, if any, that retain a dendrite in the GL are so few that they are insufficient to produce detectable label. Those parts of the bulb of the infected mice that do have some label in the GL occupy only a small fraction of the overall circumference of the bulb (arrows in Figure 9F).
Despite disruption of the EPL, reduction in mitral cell number and persistent disconnection from the deeper layers of the bulb, the GL and ONL are remarkably intact (Figure 6). Glomeruli are numerous and well-demarcated by the surrounding periglomerular cells. In addition, there is abundant OMP in the ONL and in the glomeruli themselves, indicating that OMP+ olfactory axons reach the bulb and innervate the glomerular neuropil. However, GAP-43+ olfactory axons are markedly more prominent in both the ONL and GL of the OBLV-inoculated animals, as compared with controls (Figure 6B,E versus D,F). Indeed, it is rare for a glomerulus in a control animal to be innervated at a detectable level by GAP-43+ olfactory axons in adult mice (Figure 6B,E), but common for glomeruli in the lesioned animals, even 5 months after infection; by this time any damage incurred by the epithelium as a direct consequence of the MHV OBLV inoculation is repaired (Figure 6D,F). Thus, the results in the OB suggest that the OE is undergoing accelerated turnover of neurons after this form of damage to the bulb, despite the substantial sparing of glomerular structure.
Reflected changes in the epithelium develop as a consequence of damage to the bulb
In keeping with the increased prevalence of GAP-43+ axons in the ONL and GL at long times after infection the OE of lesioned animals contains a higher number of GAP-43+, i.e. immature, sensory neurons than the epithelium of age-matched controls (Figure 10). In addition, the number of OMP+, i.e. mature, sensory neurons is reciprocally reduced as compared with normal. The coincident increase in immature neurons and decrease in mature neurons is an indication that neuronal turnover is accelerated at long survival times after infection and that the average lifespan in the population is reduced (Schwob et al., 1992). That finding is consistently observed and is characteristic of six of the eight inoculated mice allowed to survive for a month or more after infection and stained with anti-OMP and anti-GAP-43 antibodies.
Figure 10.
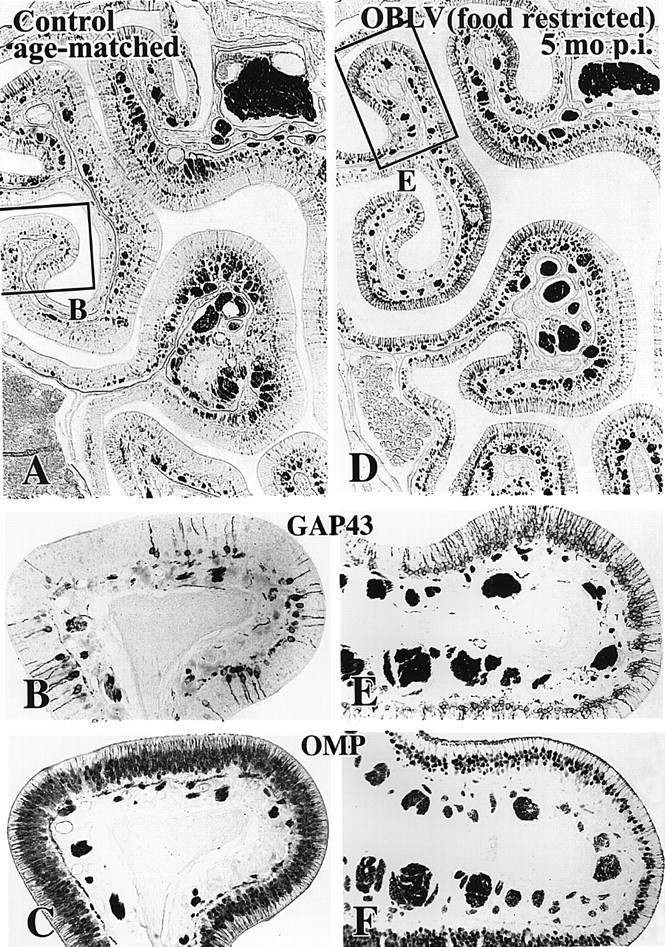
At long survival times after inoculation the number of immature sensory neurons is increased and the number of mature sensory neurons is decreased by comparison with normal. (A-C) Age-matched control. (D-F) Mouse killed 5 months after inoculation, food-restricted at time of inoculation. (A, B, D, E) Anti-GAP-43 immunostaining. Note that the number of GAP-43+ neurons is increased throughout much of the circumference of the epithelium of the lesioned mouse. (C, F) Anti-OMP immunostaining of the same area of the epithelium from sections adjacent to (A and B) and (D and E), respectively. The number of OMP+ neurons is correspondingly reduced in the lesioned animal. Magnifications: (A, D), 57×; (B, C, E, F), 150×.
In keeping with the other indices of accelerated neuronal turnover, the rate of proliferation of basal cells in the olfactory epithelium is also increased in MHV OBLV-infected mice which survived for 5 months after inoculation (Figure 11). The results in the three age-matched normals are highly similar and generate a labeling index of 0.43 ± 0.06 labeled basal cells/mm OE (mean ± SEM). The data in the group of three lesioned animals are more variable; the labeling index was 2.40 ± 1.49 and one of the infected animals showed a much lower labeling index than the other two (Figure 11). Inspection of the OB in that animal and the plot of EPL volume versus labeling index for all control and inoculated mice indicate that the bulb was minimally affected in that inoculated outlier (curved arrow in Figure 11). Likewise, both the density of glomerular labeling with anti-GAP-43 and the number of GAP-43+ neurons in the outlying experimental case were less, i.e. the primary olfactory projection more closely resembles controls (data not shown). It is worth noting that the inoculated mouse in whom the increase in proliferation was minimal was not ill in the period after MHV OBLV infusion. These data, as outlined at the beginning of Results, indicate that the effective dose of inoculation was probably less than in the other two inoculated mice. In contrast, the other two inoculated mice were lethargic and experienced a 15% weight loss due to inanition during the first week after inoculation. Despite this outlier, the difference between the two groups is statistically significant (t = 2.29, 4 d.f., P < 0.05, one-tailed test). If the outlier is eliminated from the OBLV-infected data set either on clinicopathological or statistical criteria (falling greater than three standard deviations beyond the other inoculated animals), the difference between infected and control mice is highly significant (t = 14.69, 3 d.f., P < 0.001, one-tailed test). Alternatively, taking advantage of the minimally affected outlier as a type of negative control for the effect of minimal infection, EPL volume and basal cell labeling index are highly negatively correlated (r2 = 0.90), which serves as an additional indication that basal cell proliferation is increased in infected animals as a consequence of damage to the bulb.
Discussion
The results presented here describe infection of the peripheral olfactory system by intranasal inoculation of MHV OBLV, its spread to more central parts of the system and the long-term effects of infection on the structure of the bulb. MHV OBLV produces a more restricted distribution of damage in the CNS, less morbidity and mortality and less systemic disease than does MHV JHM. For example, the parent strain JHM infects not only the brain, and in doing so causes widespread encephalitis, but also spleen, liver and intestine (organs in which MHV OBLV is not found) (Barthold, 1988; Barnett et al., 1993). Moreover, clearance of JHM ranges from 30 days for these latter organs to 60 days for brain (Barthold, 1988). Thus, with respect to clearance from brain tissue, MHV OBLV is cleared five times faster (Youngentob et al., 2001). Therefore, we infer that adaptation of MHV OBLV on an olfactory bulb-related cell line has had the effect of altering its cellular tropism and virulence and results in a restricted focus of damage on the central olfactory pathways. Indeed, the most severely affected CNS structure is the OB.
The adapted virus, like the parent strain, also seems to have a minimal effect on or infect and kill only a small percentage of the sensory neurons in the OE after acute infection. However, the composition of the population of sensory neurons in the epithelium is altered in animals that survive for longer periods after lesion. Given the limited extent of epithelial damage after acute inoculation and the rapid clearance of the virus, the subsequent shifts in the population of sensory neurons must be an indirect effect that reflects disruption of its CNS target rather than persistent infection of the OE. In other words, the shift occurs as a consequence of loss of mitral cells and/or disconnection of the deeper layers of the bulb from the GL and ONL. The coincident increase in immature sensory neurons, decrease in mature sensory neurons and accelerated proliferation of basal cells indicate that neuronal turnover is accelerated in the epithelium and thus that neuronal lifespan is attenuated (Schwob et al., 1992). Based on previous analysis of experimental models of damage to the olfactory system, specifically ablation of the OB and avulsion of the olfactory nerve, the reduction in neuronal lifespan in the epithelium is most likely the consequence of a decrease in the trophic support supplied by the bulb to the sensory neurons, and the most likely cause of that decrease in trophic support is loss of mitral cells and absence of dendrites of the surviving mitral and tufted cells from the GL (Carr and Farbman, 1992, 1993; Schwob et al., 1992).
Despite the severity and persistence of the damage to the bulb caused by MHV OBLV, the changes in the sensory neuronal population are less severe in this setting than in the epithelium of animals that were bulbectomized months prior to study (Schwob et al., 1992). Indeed, the more limited shift of the neuronal population towards immaturity after virus inoculation (in comparison with bulb ablation or nerve transection) resembles the moderate changes observed in the epithelium following manipulations that selectively deplete the mitral cell population. For example, basal cell proliferation and neuronal cell death are increased in parallel after neonatal transection of the LOT, which causes a reduction in the number of mitral cells in the bulb (Weiler and Farbman, 1999). After virus, as after LOT transection, it is likely that the sensory neurons receive some trophic support from the bulb, but at a reduced level. A potential source for that support in the inoculated mice is surviving periglomerular neurons, the dendrites of which apparently remain as a component of the glomerular neuropil. The glial cells of the olfactory nerve layer of the bulb are another potential source of trophic support for the epithelial neurons (Pixley, 1992).
It was striking that the decimation of the population of granule cells that accompanies infection is repaired to a limited extent with long survival times after lesion. That recovery in number most likely reflects ongoing neurogenesis and migration by precursors of bulbar interneurons from the subventricular zone surrounding the anterior end of the lateral ventricle (termed the SVZa), which is known to persist throughout life (Garcia-Verdugo et al., 1998; Luskin, 1998). Further investigation is warranted to determine whether the rate of production of the new neurons is accelerated as a consequence of post-infection depletion of the granule cell population. It also remains to be determined to what extent these deeper cells contribute to the trophic support that the sensory neurons derive from the bulb.
The changes that persist in the OE long after infection with MHV OBLV are highly similar to the histopathology of one set of patients who complain of PVOD (Yamagishi et al., 1988, 1990, 1994; Moran et al., 1992). As noted above, coronaviruses are a prominent cause of URI in humans, are highly mutable, often neurotropic and can persist in the CNS (Larson et al., 1980; Holland et al., 1982; Barthold, 1988; Barnett and Perlman, 1993; Holmes and Lai, 1996; Sugiura et al., 1998; Arbour et al., 2000). Our observations show that a mutated/adapted coronavirus causes destruction of the OB and more central olfactory areas and produces `reflected' changes in the OE that are reminiscent of the histopathology of some patients with PVOD. Despite the extent of the lesion, the infected animals recover the activities of daily living quickly and many are able to perform an odorant detection task and are not anosmic (Youngentob et al., 2001). The distribution of damage with MHV OBLV contrasts with other MHV strains, which are more virulent and spread rapidly and more widely through the CNS. Thus, our findings raise the possibility that a modified/adapted neurotropic virus can spread from the nose via infection of sensory neurons and transport along the olfactory nerve to cause extensive damage that is limited to the OB and cortex, without causing widespread encephalitis, and, in that manner, be responsible for some types of olfactory dysfunction in humans.
Acknowledgments
We thank the following colleagues for their generosity in sharing critical reagents: Dr Michael Buchmeier for his gift of MHV OBLV, Dr John Fleming for his gift of the J.3.3 hybridoma, Dr Frank Margolis for his gift of the anti-OMP antiserum and Dr Karina Meiri for her gift of the monoclonal anti-GAP-43 antibody. This work was supported by Public Health Service grants P01 DC00220 and K04 DC00080 from the National Institute of Deafness and Other Communication Disorders.
References
- Akerlund, A., Bende, M. and Murphy, C. (1995) Olfactory threshold and nasal mucosal changes in experimentally induced common cold Acta Otolaryngol. (Stockh.), 115,88 -92. [DOI] [PubMed] [Google Scholar]
- Arbour, N., Day, R., Newcombe, J. and Talbot, P.J. (2000) Neuroinvasion by human respiratory coronavirusesJ. Virol. , 74,8913 -8921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett, E.M. and Perlman, S. (1993) The olfactory nerve and not the trigeminal nerve is the major site of CNS entry for mouse hepatitis virus, strain JHM Virology,194 , 185-191. [DOI] [PubMed] [Google Scholar]
- Barthold, S.W. (1988) Olfactory neural pathway in mouse hepatitis virus nasoencephalitis Acta Neuropathol., 76,502 -506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr, V.M. and Farbman, A.I. (1992) Ablation of the olfactory bulb up-regulates the rate of neurogenesis and induces precocious cell death in olfactory epithelium Exp. Neurol., 115,55 -59. [DOI] [PubMed] [Google Scholar]
- Carr, V.M. and Farbman, A.I. (1993) The dynamics of cell death in the olfactory epithelium Exp. Neurol., 124,308 -314. [DOI] [PubMed] [Google Scholar]
- Chandra, R.K. (1981) Immunodeficiency in undernutrition and overnutrition Nutr. Rev.,39 , 225-231. [DOI] [PubMed] [Google Scholar]
- Costanzo, R.M. and Graziadei, P.P.C. (1983) A quantitative analysis of changes in the olfactory epithelium following bulbectomy in hamster J. Comp. Neurol., 215,370 -381. [DOI] [PubMed] [Google Scholar]
- Deems, D.A., Doty, R.L., Settle, R.G., Moore-Gillon, V., Shaman, P., Mester, A.F., Kimmelman, C.P., Brightman, V.J. and Snow, J.B. Jr (1991) Smell and taste disorders, a study of 750 patients from the University of Pennsylvania Smell and Taste CenterArch. Otolaryngol. Head Neck Surg. , 117,529 -538. [DOI] [PubMed] [Google Scholar]
- Douek, E., Bannister, L.H. and Dodson, H.C. (1975) Olfaction and its disorders Proc. R. Soc. Med., 68,467 -470. [PMC free article] [PubMed] [Google Scholar]
- Fleming, J.O., Stohlman, S.A., Harmon, R.C., Lai, M.M.C., Frelinger, J.A. and Weiner, L.P. (1983) Antigenic relationships of murine coronavirus: analysis using monoclonal antibodies to JHM (MHV-4) virus Virology, 131,296 -307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher, T.M., Escarmis, C. and Buchmeier, M.J. (1991) Alteration of the pH dependence of coronavirus-induced cell fusion: effect of mutations in the spike glycoprotein J. Virol., 65,1916 -1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Verdugo, J.M., Doetsch, F., Wichterle, H., Lim, D.A. and Alvarez-Buylla, A. (1998) Architecture and cell types of the adult subventricular zone: in search of the stem cellsJ. Neurobiol. , 36,234 -248. [DOI] [PubMed] [Google Scholar]
- Graziadei, P.P.C. and Monti Graziadei, G.A. (1980) Neurogenesis and neuron regeneration in the olfactory system of mammals. III. Deafferentation and reinnervation of the fila olfactoria in rat J. Neurocytol.,9 , 145-162. [DOI] [PubMed] [Google Scholar]
- Henkin, R.I., Larsson, A.L. and Powell, R.D. (1975) Hypogeusia, dysgeusia, hyposmia, and dysosmia following influenza-like infection Ann. Otol.,84 , 672-681. [DOI] [PubMed] [Google Scholar]
- Holland, J.J., Spindle, K., Horodyski, F., Grabau, E., Nichol, S. and Vandepol, S. (1982) Rapid evolution of RNA genomes Science, 215,1577 -1585. [DOI] [PubMed] [Google Scholar]
- Holmes, K.V. and Lai, M.M.C. (1996) Coronaviridae: the viruses and their replication In Fields, B.N., Knipe, D.M. and Howley, P. (eds), Virology, 3rd Edn. Lippincott-Raven, Philadelphia, PA, pp.1075 -1095.
- Lafay, F., Coulon, P., Astic, L., Saucier, D., Riche, D., Holley, A. and Flamand, A. (1991) Spread of the CVS strain of rabies virus and of the avirulent mutant Av01 along the olfactory pathways of the mouse after intranasal inoculationVirology , 183,320 -330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson, H.E., Reed, S.E. and Tyrrell, D.A. (1980) Isolation of rhinoviruses and coronaviruses from 38 colds in adults J. Med. Virol., 5,221 -229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luskin, M.B. (1998) Neuroblasts of the postnatal mammalian forebrain: their phenotype and fate J. Neurobiol., 36,221 -233. [PubMed] [Google Scholar]
- Monti Graziadei, G.A. and Graziadei, P.P.C. (1979) Neurogenesis and neuron regeneration in the olfactory system of mammals. II. Degeneration and reconstitution of the olfactory sensory neurons after axotomy J. Neurocytol. 8 , 197-213. [DOI] [PubMed] [Google Scholar]
- Moran, D.T., Jafek, B.W., Eller, P.M. and Rowley, J.C. (1992) Ultrastructural histopathology of human olfactory dysfunction Microsc. Res. Technol.,23 , 103-110. [DOI] [PubMed] [Google Scholar]
- Perlman, S., Evans, G. and Afifi, A. (1990) Effect of olfactory bulb ablation on spread of a neurotropic coronavirus into the mouse brain J. Exp. Med., 172,1127 -1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pixley, S.K. (1992) CNS glial cells support in vitro survival, division, and differentiation of dissociated olfactory neuronal progenitor cells Neuron,8 , 1191-1204. [DOI] [PubMed] [Google Scholar]
- Ryder, E.F., Snyder, E.Y. and Cepko, C.L. (1990) Establishment and characterization of multipotent neural cell lines using retrovirus vector-mediated oncogene transferJ. Neurobiol. , 21,356 -375. [DOI] [PubMed] [Google Scholar]
- Schwob, J.E. and Price, J.L. (1984) The development of axonal connections in the central olfactory system of rats J. Comp. Neurol., 223,177 -202. [DOI] [PubMed] [Google Scholar]
- Schwob, J.E., Mieleszko Szumowski, K.E. and Stasky, A.A. (1992) Olfactory sensory neurons are trophically dependent on the olfactory bulb for their prolonged survival J. Neurosci., 12,3896 -3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwob, J.E., Mieleszko Szumowski, K.E., Leopold, D.A. and Emko, P.E. (1993) Histopathology of olfactory mucosa in Kallmann's syndrome Ann. Otol. Rhinol. Laryngol.,102 , 117-122. [DOI] [PubMed] [Google Scholar]
- Schwob, J.E., Youngentob, S.L. and Meiri, K.F. (1994) On the formation of neuromata in the peripheral olfactory system J. Comp. Neurol.,340 , 361-380. [DOI] [PubMed] [Google Scholar]
- Schwob, J.E., Youngentob, S.L. and Mezza, R.C. (1995) The reconstitution of the olfactory epithelium after methyl bromide induced lesions J. Comp. Neurol.,359 , 15-37. [DOI] [PubMed] [Google Scholar]
- Shipley, M.T. (1985) Transport of molecules from nose to brain: transneuronal anterograde and retrograde labeling in the rat olfactory system by wheat germ agglutinin-horseradish peroxidase applied to the nasal epithelium Brain Res. Bull.,15 , 129-142. [DOI] [PubMed] [Google Scholar]
- Stroop, W.G., Rock, D.L. and Fraser, N.W. (1984) Localization of herpes simplex virus in the trigeminal and olfactory systems of the mouse central nervous system during acute and latent infections by in situ hybridization Lab. Invest., 51,27 -38. [PubMed] [Google Scholar]
- Sugiura, M., Aiba, T., Mori, J. and Nakai, Y. (1998) An epidemiological study of postviral olfactory disorder Acta Otolaryngol. (Stockh.),538 (suppl.),191 -196. [DOI] [PubMed] [Google Scholar]
- Tomlinson, A.H. and Esiri, M.M. (1983) Herpes simplex encephalitis; Immunohistological demonstration of spread of virus via olfactory pathways in mice J. Neurol. Sci.,60 , 473-484. [DOI] [PubMed] [Google Scholar]
- Weiler, E. and Farbman, A.I. (1999) Mitral cell loss following lateral olfactory tract transection increases proliferation density in rat olfactory epithelium Eur. J. Neurosci., 11,3265 -3275. [DOI] [PubMed] [Google Scholar]
- Yamagishi, M., Hasegawa, S. and Nakano, Y. (1988) Examination and classification of human olfactory mucosa in patients with clinical olfactory disturbances Arch. Otol. Rhinol. Laryngol., 245,316 -320. [DOI] [PubMed] [Google Scholar]
- Yamagishi, M., Nakamura, H., Suzuki, S., Hasegawa, S. and Nakano, Y. (1990) Immunohistochemical examination of olfactory mucosa in patients with olfactory disturbance Ann. Otol, Rhinol. Laryngol., 99,205 -210. [PubMed] [Google Scholar]
- Yamagishi, M., Fujiwara, M. and Nakamura, H. (1994) Olfactory mucosal findings and clinical course in patients with olfactory disorders following upper respiratory viral infection Rhinology, 32,113 -118. [PubMed] [Google Scholar]
- Youngentob, S.L., Schwob, J.E., Saha, S., Manglapus, G. and Jubelt, B. (2001) Functional consequences following infection of the olfactory system by intranasal infusion of the olfactory bulb line variant (OBLV) of mouse hepatitis strain JHM Chem. Senses, 26,953 -963. [DOI] [PMC free article] [PubMed] [Google Scholar]