

Abstract
Rotaxane building blocks bearing 3,5‐bis(trifluoromethyl) benzenesulfonate (BTBS) stoppers have been efficiently prepared from a pillar[5]arene derivative, 3,5‐bis(trifluoromethyl) benzenesulfonyl chloride (BTBSCl) and different diols, namely 1,10‐decanediol and 1,12‐dodecanediol. The BTBS moieties of these compounds are good leaving groups and stopper exchange reactions could be achieved by treatment with different nucleophiles thus affording rotaxanes with ester, thioether or ether stoppers.
Keywords: rotaxanes, pillar[5]arenes, arylsulfonate, stopper exchange, supramolecular chemistry
Rotaxanes, just pop the stoppers: Rotaxane building blocks bearing 3,5‐bis(trifluoromethyl) benzenesulfonate (BTBS) stoppers have been efficiently prepared. The BTBS moieties of these compounds are good leaving groups allowing to perform stopper exchange by treatment with different nucleophiles to afford rotaxanes with ester, ether or thioether stoppers.

1. Introduction
Mechanically interlocked molecules (MIMs) are attractive building blocks for the preparation of molecular machines.1 The synthesis of MIMs such as rotaxanes remains however a challenge as the key supramolecular intermediates needed for their construction are not always stable under classical reaction conditions.2 The active template approach developed by Leigh and co‐workers is an interesting alternative to circumvent this problem but it remains limited to a few specific macrocyclic building blocks.3, 4 On the other hand, the preparation of rotaxanes based on stopper exchange reactions present also clear advantages.5, 6 In this particular case, a rotaxane building block equipped with activated stoppers has to be prepared first. The stopper exchange is then achieved to generate the final product. Importantly, this second step does not rely on the formation of a host‐guest intermediate. It therefore allows for the preparation of rotaxanes difficult or even impossible to prepare by a one‐step synthetic approach based on the direct introduction of stoppers. As part of this research, we have recently shown that a stopper exchange approach is particularly well suited for the synthesis of pillar[5]arene‐containing [2]rotaxanes.6, 7, 8 This methodology is based on the reaction of a [2]rotaxane with two 2,4‐dinitrophenyl ester stoppers with amines to afford the corresponding [2]rotaxane with amide stoppers (Figure 1). In this paper, we now report new building blocks allowing the preparation of pillar[5]arene‐containing [2]rotaxanes with a larger structural diversity. Specifically, the introduction of arylsulfonates as exchangeable stoppers allowed us to perform the stopper exchange with a wide range of nucleophiles (Figure 1). As these reactions occur through a concerted nucleophilic substitution mechanism (SN2 reactions), unthreading of the axle is prevented thus leading to [2]rotaxanes with ester, ether or thioether stoppers.
Figure 1.
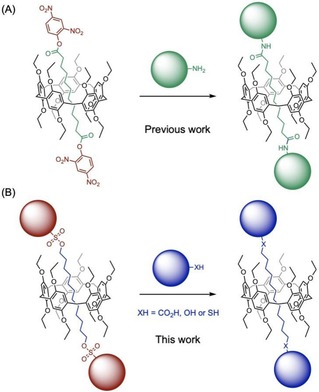
(A) Pillar[5]arene building block with activated ester stoppers and stopper exchange reaction resulting in rotaxanes with amide stoppers (Ref. 6). (B) Proposed building block with arylsulfonate stoppers allowing stopper exchange reactions with various nucleophiles.
2. Results and Discussion
2.1. Preparation of the Rotaxane Building Blocks
Pillar[5]arene‐containing rotaxanes incorporating an axle equipped with bulky leaving groups are in principle good candidates for further chemical transformations by stopper exchange reactions under SN2 conditions. Sulfonates appear as perfectly suited for such a purpose as the reaction conditions used for their preparation from sulfonyl chloride derivatives and alcohols can be in principle used to produce pillar[5]arene‐containing rotaxanes. Classical leaving groups such as mesylates or tosylates are however too small to play the role of stoppers. Amongst all the commercially available sulfonyl chloride reagents, 3,5‐bis(trifluoromethyl) benzenesulfonyl chloride (BTBSCl) attracted our attention. The corresponding sulfonates should be effective stoppers for pillar[5]arene‐containing [2]rotaxane as they are bulky enough to prevent the macrocyclic moiety from escaping the thread. Moreover, the 3,5‐bis(trifluoromethyl) benzenesulfonates (BTBS) are expected to be good leaving groups owing to the electro‐deficient nature of their aromatic units. The preparation of the rotaxane building blocks with BTBS stoppers is shown in Scheme 1. Reaction of BTBSCl (2.2 equiv.) with diol 1 (1 equiv.) in the presence of pillar[5]arene 2 (3 equiv.) gave [2]rotaxane 3. The reaction was performed in CHCl3, a solvent that does not compete with diol 1 for the formation of inclusion complexes with pillar[5]arene 2.9 To further favor the formation of pseudorotaxane intermediates, the reaction was performed at the highest possible concentration (0.49 M for 2) and at the lowest possible temperature (−15 °C).9 Under optimized conditions, compound 3 was obtained in 62 % yield.
Scheme 1.

Preparation of rotaxane building blocks 3 and 6 with BTBS stoppers. Reagents and conditions: (i) Et3N, CHCl3, −15 °C (62 %); (ii) BTBSCl, Et3N, THF, 0 °C (57 %); (iii) 2, BTBSCl, Et3N, CHCl3, −15 °C (64 %).
Molecular modeling studies revealed however that the −(CH2)10− chain in 3 might be slightly too short (Figure S1) and steric hindrance may limit the yields in [2]rotaxane during the stopper exchange reactions. It was thus decided to also prepare the −(CH2)12− analogue from 1,12‐dodecanediol (4) and pillar[5]arene 2 (Scheme 1). Due to the very low solubility of 4 in CHCl3, the direct preparation of rotaxane 6 could not be achieved under the experimental conditions used for the synthesis of 3. For this reason, mono‐BTBS 5 was prepared first from 4 and BTBSCl. Intermediate 5 was highly soluble in CHCl3 thus allowing the preparation of rotaxane 6 under optimal conditions. Treatment of 5 (1 equiv.) with BTBSCl (1.4 equiv.) in the presence of pillar[5]arene 2 (3 equiv.) and Et3N (1.5 equiv.) followed by filtration on SiO2 and gel permeation chromatography (Biobeads SX‐1, CH2Cl2) furnished [2]rotaxane 6 in 64 % yield.
Rotaxanes 3 and 6 incorporating BTBS stoppers were thus easily prepared on a multigram scale. These compounds were found moisture sensitive and storage under an Ar atmosphere was essential to prevent partial hydrolysis of the BTBS stoppers. For both compounds, the NMR and mass spectra were consistent with the proposed structure. The structure of 6 was further confirmed by X‐ray crystallography. Crystals suitable for X‐ray crystal analysis were obtained by slow diffusion of CH3CN into a solution of 6 in CH2Cl2. As shown in Figure 2, the macrocyclic component of [2]rotaxane 6 is not located at the center of the −(CH2)12− chain of its axel in the solid state. NMR studies revealed however a symmetrical structure in solution. Moreover the signals of all the methylene subunits of the axle are significantly upfield shifted owing to the ring current effect of the pillar[5]arene aromatic subunits. In other words, the different CH2 groups are all located within the pillar[5]arene cavity for some time. All these observations show that gliding motions of the macrocycle along the axle are faster than the NMR timescale in solution. It is thus believed that the peculiar conformation adopted by 6 in the solid state allows for optimized intermolecular interactions in the crystal lattice but it does not necessarily represent an energetically favored conformation of the rotaxane.
Figure 2.

ORTEP plot of the structure of 6 (H: white, F: light green, O: red, S: yellow, C: gray for the dumbbell and dark green for the pillar[5]arene moiety; the disorder of one CF3 unit has been omitted for clarity; thermal ellipsoids are shown at 30 % probability level).
2.2. Stopper Exchange Reactions
With building blocks 3 and 6 in hands, stopper exchange reactions were first carried out by using carboxylates as nucleophiles (Scheme 2). Under optimized conditions, treatment of 3 with carboxylic acids 7 a–c in the presence of K2CO3 and 18‐crown‐6 gave the corresponding [2]rotaxanes with ester stoppers (8 a–c) in 67–73 % yields. Similarly, [2]rotaxanes 9 a–c were obtained in 79–86 % yields under the same conditions starting from 6 and 7 a–c. Rotaxanes 8 d–e and 9 d–e with thioether stoppers were also obtained from the reaction of thiol reagents 7 d–e and BTBS‐stoppered rotaxanes 3 and 6, respectively.
Scheme 2.

Stopper exchange reactions. Reagents and conditions: (i) K2CO3, 18‐crown‐6, Acetone, Δ [from 3: 8 a (67 %), 8 b (73 %), 8 c (72 %), 8 d (44 %), 8 e 87(%); from 6: 9 a (86 %), 9 b (81 %), 9 c (79 %), 9 d (97 %), 9 e (99 %)]; (ii) K2CO3, DMF, 70 °C [from 3: 8 f (62 %), 8 g (63 %); from 6: 9 f (71 %), 9 g (77 %)].
The preparation of rotaxanes with ether stoppers was also attempted by reaction of 3 or 6 with 7 f–g under the same conditions (K2CO3, 18‐crown‐6, acetone, Δ). The yields were however only moderate. Better results were obtained by using a more polar solvent, namely DMF. Compound 8 f–g and 9 f–g were effectively obtained in satisfactory yields when the reaction of 3 or 6 with phenols 7 f–g was performed in DMF at 70 °C in the presence of K2CO3. As anticipated, the nucleophilic substitution reactions are sensitive to steric effects. The stopper exchange reactions performed from rotaxane 3 with the shortest axle were always significantly slower when compared to those carried out from 6. As a result, formation of by‐products originating from the slow decomposition of 3 (mainly hydrolysis) started to compete significantly with the formation of the desired products during the reactions and the final rotaxanes were obtained in lower yields when compared to those prepared from the ‐(CH2)12‐ analogue 6.
Rotaxanes 8 a–g and 9 a–g were all characterized by combination of analytical techniques. In all the cases, the expected molecular ion peak was detected in the MALDI‐TOF mass spectra. It can be noted that all the compounds are rotaxanes since the typical fragmentation pattern of MIMs10 was systematically observed. Specifically, all the MS spectra revealed a peak corresponding to the macrocyclic component ([M‐dumbbell]+). This peak results from the cleavage of the axel followed by the fast unthreading of the linear fragment. Importantly, no further fragments could be detected in the m/z range between the macrocycle and the molecular ion peak. The NMR spectra were also consistent with the proposed structures. Importantly, the interlocked structures of 8 a–g and 9 a–g were unambiguously demonstrated by the chemical shifts of the signals arising from the −(CH2)10− or −(CH2)12− chain of their axle. The methylene groups being located within the cavity of their pillar[5]arene moiety, they appear all dramatically upfield shifted in their 1H NMR spectra. This is illustrated in Figure 3 with the 1H NMR spectra recorded in CD2Cl2 for compounds 8 a and 9 a.
Figure 3.
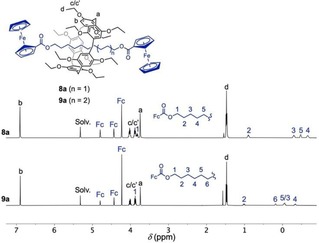
1H NMR spectra (CD2Cl2, 25 °C, 500 MHz) of rotaxanes 8 a and 9 a.
Protons H(2‐5) for 8 a and H(2‐6) for 9 a are observed between δ=−0.7 and 1 ppm. These dramatic shielding is due to the ring current effect of the pillar[5]arene aromatic moieties on the CH2 groups of the axle and represents a diagnostic signature for such rotaxanes. The three expected signals arising from the two equivalent ferrocene (Fc) stoppers are also observed. Finally, the 1H NMR spectra of 8 a and 9 a show all the characteristic features of their pillar[5]arene component. It can be also noted that compounds 8 a and 9 a are D 5‐symmetrical and therefore chiral. As a result, all the methylene groups are diastereotopic. This is particularly clear for H(c/c’) and explains the ABX3 system observed in the spectra of both 8 a and 9 a for the ten equivalent ethyloxy substituents of their pillar[5]arene subunit.
X‐ray quality crystals were obtained for five rotaxanes (8 a, 8 d, 9 d, 9 e and 9 g). In all the cases, the compounds crystallized as racemates. The relative position of the pillar[5]arene moiety onto the alkyl chain of the axle is quite different from one system to the other (Figure 4). Nonetheless, two CH2 moieties of the axle are always located within the cavity of the macrocycle. Careful analysis of the short C−H⋅⋅⋅ plane distances and angles revealed however rather variable orientations for the different rotaxanes thus suggesting that the dispersive forces between the two components must be rather weak. As already mentioned in the case of rotaxane 6, the position of the pillar[5]arene subunit observed for each of the rotaxanes in the solid state is most likely related to optimized intermolecular packing interactions in the crystal lattice rather than the result of an energetically favored conformation of the compounds.
Figure 4.
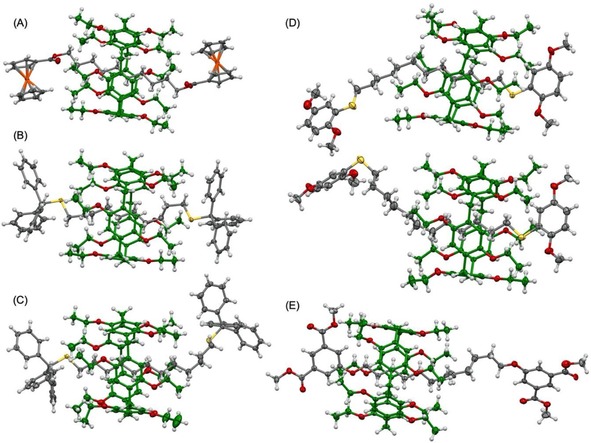
ORTEP plots of the structure of 8 a (A); 8 d (B); 9 d (C); the two conformers of 9 e present in the crystal lattice (D); and 9 g (E). The disorder (in the case of 9 e and 9 g) and the co‐crystalized solvent molecules (in the case of 9 e and 9 g) have been omitted for clarity; thermal ellipsoids are shown at 30 % probability level; colour code: H: white, Fe: orange, O: red, S: yellow, C: gray for the dumbbells and dark green for the pillar[5]arene moieties.
3. Conclusions
In conclusion, we have prepared pillar[5]arene‐containing [2]rotaxanes bearing two BTBS stoppers. These compounds are versatile building blocks and stopper exchange reactions have been efficiently achieved by treatment with various nucleophiles to afford rotaxanes with ester, thioether or ether stoppers. As the nucleophilic substitution occurs via a concerted mechanism (SN2), the rotaxane structure is preserved during these chemical transformations. Given the structural diversity of the final products, this new methodology paves the way towards the preparation of new pillar[5]arene‐containing [2]rotaxanes for various applications. It may be also easily transferred to other families of macrocycles such as cyclodextrins or calixarenes for the preparation of unprecedented rotaxanes.
Experimental Section
General Methods
Reagents were purchased as reagent grade and used without further purification unless specified. Compound 2 was prepared according to a previously reported procedure.11 The carboxylic acid and phenol reagents were dried before use by three successive azeotropic distillations (51 mL of toluene and 35 mL of absolute ethanol) and subsequent drying over P2O5 in a vacuum. All reactions were performed in standard glassware under an inert Ar atmosphere. Evaporation and concentration were done at water aspirator pressure and drying in vacuo at 10−2 Torr. Column chromatography: silica gel 60 (230–400 mesh, 0.040–0.063 mm) was purchased from E. Merck. Thin Layer Chromatography (TLC) was performed on aluminum sheets coated with silica gel 60 F254 purchased from E. Merck. NMR spectra were recorded with a Bruker AC 400 or AC 500 spectrometer with solvent peaks as reference. The 1H signals were assigned by 2D experiments (COSY and NOESY). IR spectra (cm−1) were recorded with a Perkin‐Elmer Spectrum One spectrophotometer. Absorption spectral measurements were carried out with PerkinElmer Lambda 365 spectrophotometer equipped with PCB 1500 water peltier system. MALDI‐TOF mass spectra were recorded by the analytical service of the School of Chemistry (Strasbourg, France). Elemental analyses were performed by the analytical service of the Chemistry Department of the University of Strasbourg (France). X‐ray single crystal structure determinations were performed by the service of radiocristallography of the Fédération de Chimie Le Bel FR (Strasbourg, France).
Compound 3. A mixture of 1 (0.20 g, 1.15 mmol) and 2 (3.07 g, 3.44 mmol) in anhydrous CHCl3 (6.0 mL) was stirred at −15 °C. After 30 min, a solution of BTBSCl (0.86 g, 2.75 mmol) and Et3N (0.35 g, 3.44 mmol) in anhydrous CHCl3 (1 mL) was added dropwise within 2 minutes. The resulting mixture was stirred during 4 h at −15 °C, then filtered through a short plug (SiO2, CH2Cl2) and concentrated. Purification by gel permeation chromatography (Biobeads SX‐1, CH2Cl2) gave compound 3 (1.15 g, 62 %) as a pale‐yellow solid (dec.: 114 °C). 1H NMR (500 MHz, CD2Cl2): δ=8.42 (s, 4H), 8.26 (s, 2H), 6.86 (s, 10H), 3.95–3.88 (m, 10H), 3.87–3.81 (m, 14H), 3.71 (s, 10H), 1.40 (t, J=7 Hz, 30H), 0.87 (m, 4H), −0.41 (m, 8H), −0.71 (broad s, 4H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=149.6, 139.8, 133.1 (q, 2 J C‐F=34 Hz), 128.3, 128.1 (q, 3 J C‐F=3.8 Hz), 127.4 (septet, 3 J C‐F=3.8 Hz), 122.5 (q, 1 J C‐F=272 Hz), 114.0, 72.4, 63.4, 29.6, 29.2, 28.9, 28.5, 24.1, 15.2 ppm; 19F NMR (376 MHz, CD2Cl2): δ=−63.3 ppm. MALDI‐TOF‐MS: m/z=1616.54 (100 %, [M]+, calcd for C81H96F12O16S2: 1616.59), 890.47 (10 %, [M‐dumbbell]+, calcd for C55H70O10: 890.50). Anal. (%) calcd for C81H96F12O16S2.CH2Cl2 (1702.67): C, 57.84; H, 5.80; Found: C, 57.36; H, 5.65.
Compound 5. A mixture of BTBSCl (1.00 g, 3.20 mmol), Et3N (0.49 g, 4.80 mmol) and 4 (2.59 g, 12.80 mmol) in anhydrous THF (22 mL) was stirred at 0 °C. After 4 h, the resulting milky mixture was filtered through a short plug (SiO2, CH2Cl2) and concentrated. Purification by column chromatography (SiO2, CH2Cl2/EtOAc 9 : 1) followed by gel permeation chromatography (Biobeads SX‐1, CH2Cl2) gave compound 5 (0.87 g, 57 %) as a colorless solid (m.p.: 72 °C). IR (neat): 3410 (broad, O−H) cm−1. 1H NMR (500 MHz, CD2Cl2): δ=8.41 (s, 2H), 8.22 (s, 1H), 4.18 (t, J=7 Hz, 2H), 3.61 (t, J=7 Hz, 2H), 1.72 (m, 2H), 1.55 (m, 2H), 1.33–1.27 (m, 16H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=139.2, 133.0 (q, 2 J C‐F=35 Hz), 128.3 (q, 3 J C‐F=3.8 Hz), 127.4 (septet, 3 J C‐F=3.8 Hz), 122.5 (q, 1 J C‐F=272 Hz), 72.7, 62.8, 32.9, 29.6, 29.5, 29.4, 29.3, 28.9, 28.8, 25.7, 25.3 ppm; 19F NMR (376 MHz, CD2Cl2): δ=−63.0 ppm. Anal. (%) calcd for C20H28F6O4S (478.49): C, 50.23; H, 6.00; Found: C, 50.23; H, 5.90.
Compound 6. A mixture of 5 (0.55 g, 1.14 mmol) and 2 (3.05 g, 3.42 mmol) in anhydrous CHCl3 (10.0 mL) was stirred at −15 °C. After 30 min, a solution of BTBSCl (0.43 g, 1.67 mmol) and Et3N (0.17 g, 1.71 mmol) in anhydrous CHCl3 (1 ml) was added dropwise within 2 min. The mixture was stirred during 4 h at −15 °C, then filtered through a short plug (SiO2, CH2Cl2) and concentrated. Purification by gel permeation chromatography (Biobeads SX‐1, CH2Cl2) gave compound 6 (1.20 g, 64 %) as a pale yellow solid (dec.: 118 °C). 1H NMR (500 MHz, CD2Cl2): δ=8.39 (s, 4H), 8.23 (s, 2H), 6.84 (s, 10H), 3.94–3.88 (m, 10H), 3.85–3.79 (m, 14H), 3.69 (s, 10H), 1.39 (t, J=7 Hz, 30H), 0.88 (m, 4H), 0.44 (broad s, 4H), 0.06 (broad s, 4H), −0.36 (m, 4H), −0.50 (m, 4H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=149.6, 139.8, 133.1 (q, 2 J C‐F=35 Hz), 128.2, 128.1 (q, 3 J C‐F=3.8 Hz), 127.4 (septet, 3 J C‐F=3.8 Hz), 122.5 (q, 1 J C‐F=271 Hz), 113.9, 72.5, 63.3, 30.6, 30.1, 29.0, 28.9, 28.4, 23.9, 15.2 ppm; 19F NMR (376 MHz, CD2Cl2): δ=−63.3 ppm. MALDI‐TOF‐MS: m/z=1644.59 (100 %, [M]+, calcd for C83H100F12O16S2: 1644.63), 890.45 (6 %, [M‐dumbbell]+, calcd for C55H70O10: 890.50). Anal. (%) calcd for C83H100F12O16S2 (1645.79): C, 60.57; H, 6.13; Found: C, 60.53; H, 6.18.
General procedure for the preparation of [2]rotaxanes with ester stoppers (8 a–c and 9 a–c). A mixture of 3 (or 6) (1 equiv.), the appropriate carboxylic acid (7 a–c, 2.5 equiv.), K2CO3 (2.4 equiv.) and 18‐crown‐6 (0.8 equiv.) in acetone (5 mL/160 mg of 3 (or 6)) was heated under reflux. After 24 h (from 6) or 40 h (from 3), the resulting mixture was filtered through a short plug (SiO2, CH2Cl2 containing 1 % of MeOH) and concentrated. The [2]rotaxanes were then purified by gel permeation chromatography (Biobeads SX‐1, CH2Cl2).
General procedure for the preparation of [2]rotaxanes with thioether stoppers (8 d–e and 9 d–e). A mixture of 3 (or 6) (1 equiv.), the appropriate thiol (7 d–e, 2.5 equiv.), K2CO3 (2.4 equiv.) and 18‐crown‐6 (0.8 equiv.) in acetone (5 mL/160 mg of 3 (or 6)) was heated under reflux. After 24 h (from 6) or 40 h (from 3), the reaction mixture was filtered through a short plug (SiO2, CH2Cl2 containing 1 % of MeOH) and concentrated. The [2]rotaxanes were then purified by gel permeation chromatography (Biobeads SX‐1, CH2Cl2).
General procedure for the preparation of [2]rotaxanes with ether stoppers (8 f–g and 9 f–g). A mixture of 3 (or 6) (1 equiv.), the appropriate phenol (7 f–g, 2.5 equiv.), and K2CO3 (2.4 equiv.) in anhydrous DMF (4 mL/160 mg of 3 (or 6) was heated at 70 °C. After 24 h (from 6) or 40 h (from 3), the solvent was removed in vacuum. The resulting solid was diluted with H2O (50 ml) and the product was extracted with Et2O (4 x 40 mL). The combined organic layers were washed with H2O (2×45 ml), dried (MgSO4), filtered and concentrated. The [2]rotaxanes were then purified by gel permeation chromatography (Biobeads SX‐1, CH2Cl2).
Compound 8 a. Orange solid (0.099 g, 67 %, m.p.: 233 °C). UV/Vis (CH2Cl2) λmax (ϵ): 291 (27000), 344 (sh, 660), 440 (broad, 450 M−1cm−1) nm. IR (neat): 1710 (C=O) cm−1. 1H NMR (500 MHz, CD2Cl2): δ=6.91 (s, 10H), 4.80 (broad s, 4H), 4.44 (broad s, 4H), 4.23 (s, 10H), 4.05–3.99 (m, 10H), 3.91–3.85 (m, 10H), 3.82 (t, J=8 Hz, 4H), 3.74 (s, 10H), 1.47 (t, J=7 Hz, 30H), 0.90 (m, 4H), −0.29 (m, 4H), −0.48 (broad s, 4H), −0.67 (broad s, 4H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=170.9, 149.6, 128.0, 113.8, 72.1, 71.1, 70.0, 69.9 (2 peaks), 69.7, 64.8, 63.2, 29.9, 29.1, 29.0, 28.9, 25.0, 15.4 ppm. MALDI‐TOF‐MS: m/z=1488.57 (100 %, [M]+ calcd for C87H108Fe2O14: 1488.64), 891.38 (3 %, [M‐dumbbell+H]+, calcd for C55H71O10 891.50). Anal. (%) calcd for C87H108Fe2O14 (1489.47): C, 70.15, H, 7.31; Found: C, 70.24, H, 7.40.
Compound 9 a. Orange solid (0.127 g, 86 %, m.p.: 200 °C). UV/Vis (CH2Cl2) λmax (ϵ): 295 (31030), 344 (sh, 666), 444 (broad, 470 M−1cm−1) nm. IR (neat): 1710 (C=O) cm−1. 1H NMR (500 MHz, CD2Cl2): δ=6.92 (s, 10H), 4.81 (m, 4H), 4.45 (t, J=1.9 Hz, 4H), 4.25 (s, 10H), 4.06–4.00 (m, 10H), 3.92–3.86 (m, 14H), 3.76 (s, 10H), 1.49 (t, J=7 Hz, 30H), 1.03 (m, 4H), 0.20 (m, 4H), −0.03 (m, 8H), −0.32 (m, 4H) ppm. 13C NMR (125 MHz, CDCl3): δ=171.3, 149.6, 128.1, 113.9, 72.2, 71.6, 70.4, 70.0, 64.9, 63.2, 30.5, 30.2, 29.2, 29.0, 28.9, 25.1, 15.6 ppm. MALDI‐TOF‐MS: m/z=1516.68 (100 %, [M]+ calcd for C89H112Fe2O14: 1516.68), 891.19 (5 %, [M‐dumbbell+H]+, calcd for C55H71O10 891.50). Anal. (%) calcd for C89H112Fe2O14 (1517.53): C, 70.44, H, 7.44; Found: C, 70.37, H, 7.44.
Compound 8 b. Colorless solid (0.107 g, 73 %, m.p.: 193 °C). IR (neat): 1714 (C=O) cm−1. 1H NMR (500 MHz, CD2Cl2): δ=7.90 (d, J=1.9 Hz, 4H), 7.68 (t, J=1.9 Hz, 2H), 6.90 (s, 10H), 4.04–3.98 (m, 14H), 3.89–3.83 (m, 10H), 3.73 (s, 10H), 1.43 (t, J=7 Hz, 30H), 1.39 (s, 36H), 1.11 (m, 4H), −0.11 (m, 4H), −0.76 (m, 4H), −0.83 (m, 4H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=167.0, 151.0, 149.6, 130.3, 128.0, 126.9, 123.5, 113.8, 65.6, 63.2, 34.9, 31.2, 29.5, 29.2, 29.0, 28.8, 25.5, 15.3 ppm. MALDI‐TOF‐MS: m/z=1497.01 (100 %, [M]+, calcd for C95H132O14: 1496.96), 891.42 (15 %, [M‐dumbbell+H]+, calcd for C55H71O10 : 891.50). Anal. (%) calcd for C95H132O14 (1498.06): C, 76.17, H, 8.88; Found: C, 75.97, H, 8.87.
Compound 9 b. Colorless solid (0.120 g, 81 %, m.p.: 138 °C). IR (neat): 1715 (C=O) cm−1. 1H NMR (500 MHz, CD2Cl2): δ=7.91 (d, J=1.9 Hz, 4H), 7.69 (t, J=1.9 Hz, 2H), 6.90 (s, 10H), 4.08 (t, J=2 Hz, 4H), 4.04–3.98 (m, 10H), 3.89–3.84 (m, 10H) 3.74 (s, 10H), 1.44 (t, J=7 Hz, 30H), 1.39 (s, 36H), 1.24 (m, 4H), 0.18 (m, 4H), −0.10 (m, 4H), −0.30 (m, 8H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=167.0, 151.1, 149.6, 130.3, 128.0, 126.9, 123.5, 113.8, 65.4, 63.2, 34.9, 31.2, 30.3, 30.0, 29.0, 28.9, 28.8, 25.5, 15.3 ppm. MALDI‐TOF‐MS: m/z=1525.04 (100 %, [M]+, calcd for C97H136O14: 1524.99), 891.41 (5 %, [M‐dumbbell+H]+, calcd for C55H71O10 : 891.50). Anal. (%) calcd for C97H136O14 (1526.11): C, 76.34, H, 8.98; Found: C, 75.96, H, 8.97.
Compound 8 c. Colorless solid (0.106 g, 72 %, m.p.: 183 °C). IR (neat): 1734 (C=O) cm−1. 1H NMR (500 MHz, CD2Cl2): δ=7.34–7.27 (m, 16H), 7.24–7.21 (m, 4H), 6.87 (s, 10H), 4.59 (t, J=8 Hz, 2H), 3.95‐3.89 (m, 10H), 3.87‐3.81 (m, 10H), 3.71 (s, 10H), 3.49 (t, J=8 Hz, 4H), 3.05 (d, J=8 Hz, 4H), 1.41 (t, J=7 Hz, 30H), 0.58 (m, 4H), −0.26 (m, 4H), −0.51 (m, 4H), −0.58 (m, 4H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=171.1, 149.5, 144.07, 144.02, 144.0, 128.6, 127.9, 127.6 (2 peaks), 126.5 (2 peaks), 113.6, 65.2, 63.1, 46.7, 40.3, 30.0, 29.1, 28.9, 28.4, 24.5, 15.2 ppm. MALDI‐TOF‐MS: m/z=1480.89 (100 %, [M]+, calcd for C95H116O14: 1480.84), 891.46 (5 %, [M‐dumbbell+H]+, calcd for C55H71O10: 891.50). Anal. (%) calcd for C95H116O14 (1481.93): C, 77.00, H, 7.89; Found: C, 77.31, H, 8.18.
Compound 9 c. Colorless solid (0.116 g, 79 %, m.p.: 115 °C). IR (neat): 1733 (C=O) cm−1. 1H NMR (500 MHz, CD2Cl2): δ=7.34‐7.28 (m, 16H), 7.24‐7.20 (m, 4H), 6.87 (s, 10H), 4.59 (t, J=7.7 Hz, 2H), 3.97–3.91 (m, 10H), 3.88–3.82 (m, 10H), 3.72 (s, 10H), 3.54 (t, J=7.7 Hz, 4H), 3.06 (d, J=8 Hz, 4H), 1.42 (t, J=7 Hz, 30H), 0.66 (m, 4H), 0.40 (m, 4H), 0.11 (m, 4H), −0.34 (m, 8H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=171.1, 149.5, 144.1 (2 C), 128.6, 127.9, 127.6, 126.5, 113.6, 65.2, 63.0, 46.8, 40.3, 30.7, 30.2, 28.9 (2 C), 28.3, 24.4, 15.3 ppm. MALDI‐TOF‐MS: m/z=1508.93 (100 %, [M]+, calcd for C97H120O14: 1508.87), 891.36 (3 %, [M‐dumbbell+H]+, calcd for C55H71O10: 891.50). Anal. (%) calcd for C97H120O14 (1509.99): C, 77.16, H, 8.01; Found: C, 77.15, H, 7.90.
Compound 8 d. Colorless solid (0.069 g, 44 %, m.p.: 248 °C). 1H NMR (500 MHz, CD2Cl2): δ=7.47–7.45 (m, 12H), 7.35–7.32 (m, 12H), 7.28–7.25 (m, 6H), 6.81 (s, 10H), 3.90–3.78 (m, 20H), 3.69 (s, 10H), 2.01 (t, J=8 Hz, 4H), 1.37 (t, J=7 Hz, 30H), 1.14 (m, 4H), 0.25 (m, 4H), −0.95 (m, 4H), −2.05 (m, 4H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=149.5, 145.3, 129.5, 127.9, 127.8, 126.6, 113.7, 65.9, 63.2, 32.5, 30.5, 29.5, 29.3, 28.9, 27.9, 15.4 ppm. MALDI‐TOF‐MS: m/z=1580.73 (100 %, [M]+, calcd for C103H120O10S2: 1580.83), 890.42 (39 %, [M‐dumbbell]+, calcd for C55H70O10: 890.50). Anal. (%) calcd for C103H120O10S2 (1582.18): C, 78.19, H, 7.65; Found: C, 78.15, H, 7.38.
Compound 9 d. Colorless solid (0.152 g, 97 %, m.p.: 191 °C). 1H NMR (500 MHz, CD2Cl2): δ=7.46–7.44 (m, 12H), 7.35–7.31 (m, 12H), 7.28–7.24 (m, 6H), 6.81 (s, 10H), 3.90–3.77 (m, 20H), 3.69 (s, 10H), 2.07 (t, J=8 Hz, 4H), 1.38 (t, J=7 Hz, 30H), 1.26 (m, 4H), 0.64 (m, 4H), −0.15 (m, 4H), −0.95 (broad s, 4H), −1.28 (broad s, 4H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=149.5, 145.2, 129.5, 127.9, 127.8, 126.6, 113.7, 66.1, 63.1, 32.3, 30.4, 30.2, 29.3, 29.2, 29.1, 28.9, 15.3 ppm. MALDI‐TOF‐MS: m/z=1608.68 (100 %, [M]+, calcd for C105H124O10S2: 1608.86), 890.39 (51 %, [M‐dumbbell]+, calcd for C55H70O10: 890.50). Anal. (%) calcd for C105H124O10S2 (1610.24): C, 78.32, H, 7.76; Found: C, 78.22, H, 7.85.
Compound 8 e. Colorless glassy product (0.118 g, 87 %). 1H NMR (500 MHz, CD2Cl2): δ=6.90 (s, 10H), 6.80–6.75 (m, 4H), 6.65–6.63 (m, 2H), 4.06–4.00 (m, 10H), 3.87‐3.81 (m, 16H), 3.79 (s, 6H), 3.73 (s, 10H), 2.37 (m, 4H), 1.41 (t, J=7 Hz, 30H), 0.58 (m, 4H), −0.24 (m, 8H), −0.41 (broad s, 4H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=154.2, 150.6, 149.6, 129.0, 128.1, 114.4, 113.9, 110.6, 107.8, 63.4, 56.2, 55.4, 31.3, 30.1, 29.5, 29.0, 28.9, 28.4, 15.2 ppm. MALDI‐TOF‐MS: m/z=1368.62 (100 %, [M]+, calcd for C81H108O14S2: 1368.72), 891.38 (3 %, [M‐dumbbell+H]+, calcd for C55H71O10: 891.51). Anal. (%) calcd for C81H108O14S2 (1369.85): C, 71.02, H, 7.95; Found: C, 70.83, H, 7.99.
Compound 9 e. Colorless solid (0.135 g, 99 %, m.p.: 91 °C). 1H NMR (500 MHz, CD2Cl2): δ=6.90 (s, 10H), 6.80–6.77 (m, 4H), 6.65–6.63 (m, 2H), 4.05–3.99 (m, 10H), 3.87–3.81 (m, 16H), 3.79 (s, 6H), 3.73 (s, 10H), 2.42 (m, 4H), 1.42 (t, J=7 Hz, 30H), 0.70 (m, 4H), 0.30 (broad s, 4H), 0.11 (broad s, 4H), −0.03 (m, 4H), −0.16 (m, 4H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=154.2, 150.7, 149.6, 128.8, 128.1, 114.4, 113.9, 110.7, 108.0, 63.4, 56.2, 55.5, 31.3, 30.6, 30.1, 29.4, 28.9, 28.7, 28.4, 15.2 ppm. MALDI‐TOF‐MS: m/z=1396.74 (100 %, [M]+, calcd for C83H112O14S2: 1396.75), 891.50 (4 %, [M‐dumbbell+H]+, calcd for C55H71O10: 891.51). Anal. (%) calcd for C83H112O14S2 (1397.90): C, 71.31, H, 8.08; Found: C, 71.28, H, 8.16.
Compound 8 f. Colorless glassy product (0.104 g, 62 %). 1H NMR (500 MHz, CD2Cl2): δ=7.30–7.25 (m, 24H), 7.23–7.20 (m, 6H), 7.14 (d, J=7 Hz, 4H), 6.89 (s, 10H), 6.76 (d, J=7 Hz, 4H), 3.94‐3.88 (m, 10H), 3.85‐3.80 (m, 10H), 3.72 (s, 10H), 3.22‐3.13 (m, 4H), 1.34 (t, J=7 Hz, 30H), 0.41 (m, 4H), 0.33 (m, 4H), −0.08 (m, 4H), −0.43 (m, 4H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=157.5, 149.7, 147.3, 138.3, 131.9, 131.0, 128.1, 127.4, 125.8, 114.0, 113.0, 68.2, 64.4, 63.4, 30.7, 30.0, 29.0, 28.7, 24.6, 15.3 ppm. MALDI‐TOF‐MS: m/z=1700.84 (100 %, [M]+, calcd for C115H128O12: 1700.94), 890.40 (4 %, [M‐dumbbell]+, calcd for C55H70O10: 890.50). Anal. (%) calcd for C115H128O12 (1702.24): C, 81.14, H, 7.58; Found: C, 81.16, H, 7.53.
Compound 9 f. Colorless glassy product (0.120 g, 71 %). 1H NMR (500 MHz, CD2Cl2): δ=7.29–7.25 (m, 24H), 7.23–7.19 (m, 6H), 7.14 (d, J=7 Hz, 4H), 6.89 (s, 10H), 6.76 (d, J=7 Hz, 4H), 3.95‐3.89 (m, 10H), 3.86‐3.80 (m, 10H), 3.72 (s, 10H), 3.24‐3.15 (m, 4H), 1.36 (t, J=7 Hz, 30H), 0.86 (m, 4H), 0.58 (m, 4H), 0.45 (m, 4H), 0.04 (m, 4H), −0.36 (m, 4H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=157.5, 149.6, 147.3, 138.3, 131.9, 131.0, 128.1, 127.4, 125.8, 113.9, 113.0, 68.2, 64.4, 63.3, 31.0, 30.5, 29.4, 29.0, 28.6, 24.3, 15.2 ppm. MALDI‐TOF‐MS: m/z=1728.89 (100 %, [M]+, calcd for C117H132O12: 1728.97), 891.35 (10 %, [M‐dumbbell+H]+, calcd for C55H71O10: 891.50). Anal. (%) calcd for C117H132O12 (1730.30): C, 81.21, H, 7.69; Found: C, 81.11, H, 7.67.
Compound 8 g. Colorless solid (0.09 g, 63 %, m.p.: 157 °C). IR (neat): 1728 (C=O) cm−1. 1H NMR (500 MHz, CD2Cl2): δ=8.27 (t, J=1.5 Hz, 2H), 7.74 (d, J=1.5 Hz, 4H), 6.93 (s, 10H), 4.01–3.95 (m, 22H), 3.91–3.85 (m, 10H), 3.74 (s, 10H), 3.57–3.48 (m, 4H), 1.40 (t, J=7 Hz, 30H), 0.72 (m, 4H), 0.03 broad s, 4H), −0.36 (m, 8H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=166.0, 159.5, 149.6, 131.9, 128.2, 122.3, 119.3, 114.1, 69.0, 63.4, 52.3, 30.3, 29.5, 29.2, 29.0, 24.4, 15.2 ppm. MALDI‐TOF‐MS: m/z=1448.77 (100 %, [M]+, calcd for C85H108O20: 1448.74), 891.44 (3 %, [M‐dumbbell+H]+, calcd for C55H71O10: 891.50). Anal. (%) calcd for C85H108O20 (1449.76): C, 70.42, H, 7.51; Found: C, 70.19, H, 7.51.
Compound 9 g. Colorless solid (0.111 g, 77 %, m.p.: 137 °C). IR (neat): 1725 (C=O) cm−1. 1H NMR (500 MHz, CD2Cl2): δ=8.27 (t, J=1.5 Hz, 2H), 7.74 (d, J=1.5 Hz, 4H), 6.93 (s, 10H), 4.01–3.95 (m, 22H), 3.91–3.85 (m, 10H), 3.74 (s, 10H), 3.57–3.48 (m, 4H), 1.40 (t, J=7 Hz, 30H), 0.72 (m, 4H), 0.03 broad s, 4H), −0.36 (m, 8H) ppm. 13C NMR (125 MHz, CD2Cl2): δ=166.0, 159.5, 149.6, 131.9, 128.2, 122.3, 119.3, 114.1, 69.0, 63.4, 52.3, 30.3, 29.5, 29.2, 29.0, 24.4, 15.2 ppm. MALDI‐TOF‐MS: m/z=1476.77 (100 %, [M]+, calcd for C87H112O20: 1476.77), 891.37 (5 %, [M‐dumbbell+H]+, calcd for C55H71O10: 891.50). Anal. (%) calcd for C87H112O20 (1477.81): C, 70.71, H, 7.64; Found: C, 70.59, H, 7.63.
X‐Ray Crystal Structures
The crystallographic data and the refinement parameters are reported in the supplementary material for all the compounds. The X‐ray crystal structures have been deposited at the Cambridge Structural Database (CCDC deposition numbers: 1980807 for 6, 1980808 for 8 a, 1980817 for 8 d, 1980881 for 9 d, 1980883 for 9 e and 1980886 for 9 g).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the ANR (project FastGiant ANR‐17‐CE07‐0012‐01), the International Center for Frontier Research in Chemistry and the LabEx “Chimie des Systèmes Complexes” is gratefully acknowledged. We further thank L. Karmazin and C. Bailly for the X‐ray crystal structure resolution, E. Wasielewski for high‐field NMR measurements and J.‐M. Strub for the mass spectra.
I. Nierengarten, J.-F. Nierengarten, ChemistryOpen 2020, 9, 393.
Dedicated to Prof. Jean‐Marie Lehn on the occasion of his 80th birthday
References
- 1.
- 1a. Sauvage J.-P., Angew. Chem. Int. Ed. 2017, 56, 11080–11093; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11228–11242; [Google Scholar]
- 1b. Stoddart J. F., Angew. Chem. Int. Ed. 2017, 56, 11228–11242; [Google Scholar]
- 1c. Feringa B. L., Angew. Chem. Int. Ed. 2017, 56, 11060–11078; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11206–11226; [Google Scholar]
- 1d. Balzani V., Credi A., Venturi M., Molecular Devices and Machines – A Journey into the Nano World, Wiley-VCH, 2003; [Google Scholar]
- 1e. Kay E. R., Leigh D. A., Angew. Chem. Int. Ed. 2015, 54, 10080–10088; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10218–10226; [Google Scholar]
- 1f. Erbas-Cakmak S., Leigh D. A., McTernan C. T., Nussbaumer A. L., Chem. Rev. 2015, 115, 10081–10206; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1g. Leigh D. A., Angew. Chem. Int. Ed. 2016, 55, 14506–14508; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14722–14724. [Google Scholar]
- 2.
- 2a.J.-P. Sauvage, C. Dietrich-Buchecker (Eds.), Molecular Catenanes, Rotaxanes and Knots – A Journey Through the World of Molecular Topology, J. Wiley and Sons, 2008;
- 2b. Bruns C. J., Stoddart J. F., The Nature of the Mechanical Bond – From Molecules to Machines, J. Wiley & Sons, 2016. [Google Scholar]
- 3.
- 3a. Crowley J. D., Goldup S. M., Lee A.-L., Leigh D. A., McBurney R. T., Chem. Soc. Rev. 2009, 38, 1530–1541; [DOI] [PubMed] [Google Scholar]
- 3b. Hänni K. D., Leigh D. A., Chem. Soc. Rev. 2010, 39, 1240–1251; [DOI] [PubMed] [Google Scholar]
- 3c. Denis M., Goldup S. M., Nat. Rev. Chem. 2017, 1, 61. [Google Scholar]
- 4.For a recent example, see: Tian C., Fielden S. D. P., Whitehead G. F. S., Victoca-Yrezabal I. J., Leigh D. A., Nat. Commun. 2020, 11, 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The post-modification of a pre-constructed [2]rotaxane scaffold by a stopper exchange reaction has been rarely used and only a few examples have been reported in the literature so far, see:
- 5a. Rowan S. J., Stoddart J. F., J. Am. Chem. Soc. 2000, 122, 164–165; [Google Scholar]; Rowan S. J., Cantrill S. J., Stoddart J. F., White A. J. P., Williams D. J., Org. Lett. 2000, 2, 759–762; [DOI] [PubMed] [Google Scholar]
- 5b. Bordoli R. J., Goldup S. M., J. Am. Chem. Soc. 2014, 136, 4817–4820; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. D. W. Zehnder II , Smithrud D. B., Org. Lett. 2001, 3, 2485–2487; [DOI] [PubMed] [Google Scholar]
- 5d. Kihara N., Motoda S., Yokozawa T., Takata T., Org. Lett. 2005, 7, 1199–1202; [DOI] [PubMed] [Google Scholar]
- 5e. Hannam J. S., Lacy S. M., Leigh D. A., Saiz C. G., Slawin A. M. Z., Stitchell S. G., Angew. Chem. Int. Ed. 2004, 43, 3260–3264; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3322–3326. [Google Scholar]
- 6. Nierengarten I., Meichsner E., Holler M., Pieper P., Deschenaux R., Delavaux-Nicot B., Nierengarten J.-F., Chem. Eur. J. 2018, 24, 169–177. [DOI] [PubMed] [Google Scholar]
- 7.For reviews on pillar[n]arenes, see:
- 7a. Cragg P. J., Sharma K., Chem. Soc. Rev. 2012, 41, 597–607; [DOI] [PubMed] [Google Scholar]
- 7b. Xue M., Yang Y., Chi X., Zhang Z., Huang F., Acc. Chem. Res. 2012, 45, 1294–1308; [DOI] [PubMed] [Google Scholar]
- 7c. Ogoshi T., J. Inclusion Phenom. Macrocyclic Chem. 2012, 72, 247–262; [Google Scholar]
- 7d. Ogoshi T., Yamagishi T. a., Eur. J. Org. Chem. 2013, 2961–2975; [Google Scholar]
- 7e. Cao D., Meier H., Asian J. Org. Chem. 2014, 3, 244–262; [Google Scholar]
- 7f. Ogoshi T., Yamagishi T., Chem. Commun. 2014, 50, 4776–4787; [DOI] [PubMed] [Google Scholar]
- 7g. Strutt N. L., Zhang H., Schneebeli S. T., Stoddart J. F., Acc. Chem. Res. 2014, 47, 2631–2642; [DOI] [PubMed] [Google Scholar]
- 7h. Yang K., Pei Y., Pei Z., Chem. Commun. 2016, 52, 9316–9326; [DOI] [PubMed] [Google Scholar]
- 7i. Ogoshi T., Yamagishi T. a., Nakamoto Y., Chem. Rev. 2016, 116, 7937–8002; [DOI] [PubMed] [Google Scholar]
- 7j. Kakuta T., Yamagashi T., Ogoshi T., Chem. Commun. 2017, 53, 5250–5266. [DOI] [PubMed] [Google Scholar]
- 8.For examples of pillar[5]arene-containing rotaxanes prepared by introduction of stoppers to an inclusion complex, see:
- 8a. Strutt N. L., Forgan R. S., Spruell J. M., Botros Y. Y., Stoddart J. F., J. Am. Chem. Soc. 2011, 133, 5668–5671; [DOI] [PubMed] [Google Scholar]
- 8b. Ogoshi T., Nishida Y., Yamagishi T. a., Nakamoto Y., Macromolecules 2010, 43, 7068–7072; [Google Scholar]
- 8c. Ogoshi T., Yamafuji D., Aoki T., Yamagishi T. a., Chem. Commun. 2012, 48, 6842–6844; [DOI] [PubMed] [Google Scholar]
- 8d. Ogoshi T., Yamafuji D., Aoki T., Kitajima K., Yamagishi T.-a., Hayashi Y., Kawauchi S., Chem. Eur. J. 2012, 18, 7493–7500; [DOI] [PubMed] [Google Scholar]
- 8e. Dong S., Han C., Zheng B., Zhang M., Huang F., Tetrahedron Lett. 2012, 53, 3668–3671; [Google Scholar]
- 8f. Wei P., Yan X., Li J., Ma Y., Yao Y., Huang F., Tetrahedron 2012, 68, 9179–9185; [Google Scholar]
- 8g. Ogoshi T., Aoki T., Shiga R., Iizuka R., Ueda S., Demachi K., Yamafuji D., Kayama H., Yamagishi T. a., J. Am. Chem. Soc. 2012, 134, 20322–20325; [DOI] [PubMed] [Google Scholar]
- 8h. Hu X.-Y., Wu X., Duan Q., Xiao T., Lin C., Wang L., Org. Lett. 2012, 14, 4826–4829; [DOI] [PubMed] [Google Scholar]
- 8i. Ke C., Strutt N. L., Li H., Hou X., Hartlieb K. J., McGonigal P. R., Ma Z., Iehl J., Stern C. L., Cheng C., Zhu Z., Vermeulen N. A., Meade T. J., Botros Y. Y., Stoddart J. F., J. Am. Chem. Soc. 2013, 135, 17019–17030; [DOI] [PubMed] [Google Scholar]
- 8j. Hou X., Ke C., Cheng C., Song N., Blackburn A. K., Sarjeant A. A., Botros Y. Y., Yang Y.-W., Stoddart J. F., Chem. Commun. 2014, 50, 6196–6199; [DOI] [PubMed] [Google Scholar]
- 8k. Dong S., Yuan J., Huang F., Chem. Sci. 2014, 5, 247–252; [Google Scholar]
- 8l. Trinh T. M. N., Nierengarten I., Holler M., Gallani J.-L., Nierengarten J.-F., Chem. Eur. J. 2015, 21, 8019–8022; [DOI] [PubMed] [Google Scholar]
- 8m. Ogoshi T., Iizuka R., Kotera D., Ymagishi T. a., Org. Lett. 2015, 17, 350–353; [DOI] [PubMed] [Google Scholar]
- 8n. Vincent S. P., Buffet K., Nierengarten I., Imberty A., Nierengarten J.-F., Chem. Eur. J. 2016, 22, 88–92; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8o. Yu G., Wu D., Li Y., Zhang Z., Shao L., Zhou J., Hu Q., Tang G., Huang F., Chem. Sci. 2016, 7, 3017–3024; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8p. Delavaux-Nicot B., Ben Aziza H., Nierengarten I., Trinh T. M. N., Meichsner E., Chessé M., Holler M., Abidi R., Maisonhaute E., Nierengarten J.-F., Chem. Eur. J. 2018, 24, 133–140; [DOI] [PubMed] [Google Scholar]
- 8q. Meichsner E., Nierengarten I., Holler M., Chessé M., Nierengarten J.-F., Helv. Chim. Acta 2018, 101, e1800059; [Google Scholar]
- 8r. Steffenhagen M., Latus A., Trinh T. M. N., Nierengarten I., Lucas I. T., Joiret S., Landoulsi J., Delavaux-Nicot B., Nierengarten J.-F., Maisonhaute E., Chem. Eur. J. 2018, 24, 1701–1708; [DOI] [PubMed] [Google Scholar]
- 8s. Holler M., Stoerkler T., Louis A., Fisher F., Nierengarten J.-F., Eur. J. Org. Chem. 2019, 3401–3405. [Google Scholar]
- 9. Milev R., Lopez-Pacheco A., Nierengarten I., Trinh T. M. N., Holler M., Deschenaux R., Nierengarten J.-F., Eur. J. Org. Chem. 2015, 479–485. [Google Scholar]
- 10.
- 10a. Vetter W., Logemann E., Schill G., Org. Mass Spectrom. 1977, 12, 351–369; [Google Scholar]
- 10b. Dietrich-Buchecker C., Leize E., Nierengarten J.-F., Sauvage J.-P., Van Dorsselaer A., Chem. Commun. 1994, 2257–2258; [Google Scholar]
- 10c. Schalley C. A., Ghosh P., Engeser M., Int. J. Mass Spectrom. 2004, 232, 249–258. [Google Scholar]
- 11.
- 11a. Ogoshi T., Kitajima K., Aoki T., Fujinami S., Yamagishi T. a., Nakamoto Y., J. Org. Chem. 2010, 75, 3268–3273; [DOI] [PubMed] [Google Scholar]
- 11b. Holler M., Allenbach N., Sonet J., Nierengarten J.-F., Chem. Commun. 2012, 48, 2576–2578. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary