

Abstract
The Hippo–YAP signaling pathway plays an essential role in epithelial cells during intestinal regeneration and tumorigenesis. However, the molecular mechanism linking stromal signals to YAP‐mediated intestinal regeneration and tumorigenesis is poorly defined. Here, we report a stroma–epithelium ISLR–YAP signaling axis essential for stromal cells to modulate epithelial cell growth during intestinal regeneration and tumorigenesis. Specifically, upon inflammation and in cancer, an oncogenic transcription factor ETS1 in stromal cells induces expression of a secreted protein ISLR that can inhibit Hippo signaling and activate YAP in epithelial cells. Deletion of Islr in stromal cells in mice markedly impaired intestinal regeneration and suppressed tumorigenesis in the colon. Moreover, the expression of stromal cell‐specific ISLR and ETS1 significantly increased in inflamed mucosa of human IBD patients and in human colorectal adenocarcinoma, accounting for the epithelial YAP hyperactivation. Collectively, our findings provide new insights into the signaling crosstalk between stroma and epithelium during tissue regeneration and tumorigenesis.
Keywords: colorectal cancer, Hippo signaling, intestinal regeneration, ISLR, stroma–epithelia crosstalk
Subject Categories: Cancer, Digestive System, Signal Transduction
Disease‐associated or regenerative YAP activation in the intestinal epithelium is triggered by the mesenchyme‐derived extracellular protein ISLR.
Introduction
Inflammatory bowel disease (IBD), which includes ulcerative colitis (UC) and Crohn's disease (CD), is a type of chronic inflammatory disease associated with increased risk of colorectal carcinoma (CRC). IBD is characterized by submucosal accumulation of inflammatory cells and damage to epithelial cells (Romano et al, 2016; Kim & Cheon, 2017). This damage triggers a complex repair program that activates proliferation of epithelial cells that ultimately repopulate the damaged epithelium. When the damage cannot be repaired, the chronic proliferation caused by constant inflammation can drive the development of CRC (Romano et al, 2016). During tissue repair and CRC development, crosstalk between intestinal epithelium and the underlying mesenchyme is critical for promoting epithelial cell proliferation. Mesenchymal stromal cells form the supportive microenvironment that maintains intestinal epithelial architecture during homeostasis (Powell et al, 2011), become reactive upon inflammation and tissue damage, and further transform into tumor‐associated stromal cells upon CRC (Bussard et al, 2016). Reactive or tumor‐associated stromal cells can contribute to epithelial cell proliferation in a variety of ways, including through the production of cytokines, such as IL‐6 (Zhu et al, 2014; Francescone et al, 2015). Although the importance of mesenchymal stromal cells in promoting epithelial cell proliferation is well established, how stromal signals regulate their proliferation in the context of colitis and CRC remains unclear. A comprehensive understanding of the underlying paracrine mechanisms is of broad interest, because it can uncover new signaling targets for treating these major intestinal diseases.
The Hippo pathway is an evolutionarily conserved signaling cascade, consisting of the core kinase components MST1/2 and LATS1/2, the transcriptional coactivators YAP and TAZ, and the transcriptional factors TEADs (TEAD1‐TEAD4) (Huang et al, 2005; Pan, 2010; Meng et al, 2016). As a tumor suppressor pathway, Hippo signaling is constitutively active under physiological conditions: The MST1/2 kinases phosphorylate and activate LATS1/2 kinases, which in turn phosphorylate YAP/TAZ, leading to cytoplasmic retention and degradation of the latter. However, in the context of tissue regeneration and tumorigenesis, Hippo signaling is frequently dampened, enabling activation of YAP for cell proliferation. Many reports have indicated that YAP/TAZ activity is essential for intestinal epithelial regeneration and tumorigenesis. However, it remains unknown how stromal signals regulate epithelial YAP/TAZ activity, especially in colitis and CRC.
ISLR, also known as Meflin, is a conserved protein containing an immunoglobulin (Ig)‐like domain and five leucine‐rich repeat (LRR) domains (Nagasawa et al, 1997, 1999). Recently, ISLR has been identified as a marker of mesenchymal stromal cells, and it functions as a secreted protein to maintain the undifferentiated state of stromal cells (Maeda et al, 2016). Recently, two reports have shown the importance of ISLR in promoting muscle regeneration and cardiac tissue repair (Zhang et al, 2018; Hara et al, 2019). However, it is unknown whether stromal ISLR regulates intestinal epithelial regeneration and CRC. Given its abundant expression in stromal cells, it is intriguing to postulate that stroma‐secreted ISLR can transduce signals from the microenvironment to epithelial cells, thus mediating stroma–epithelium communication during regeneration and CRC development.
In this study, we utilized stromal cell‐specific conditional knockout (cKO) mouse model to demonstrate that stromal cell‐secreted ISLR promotes intestinal regeneration and concomitantly acts as a pro‐tumorigenic factor in CRC. Mechanistically, extracellular ISLR secreted by stromal cells suppressed Hippo signaling and activated YAP in intestinal epithelial cells. We identified a stroma–epithelium ISLR–YAP signaling axis mediating the regulation of stromal signals to epithelial cell proliferation essential for intestinal regeneration and colorectal cancer development.
Results
Islr/ISLR is increased in stromal cells of the inflamed mucosa in IBD and CRC
To begin elucidating a potential role for ISLR in intestinal homeostasis and in colitis, we first examined its expression patterns in the intestine. Both qRT–PCR and Western blotting assays showed that Islr is abundantly expressed in the intestinal lamina propria, but is nearly undetectable in the intestinal epithelium of mice (Fig 1A and B). Furthermore, we found that Islr was primarily expressed in non‐hematopoietic and non‐epithelial cells (CD45−Epcam−CD90+ and CD45−Epcam−CD90− cells), while it was also mildly expressed in CD45+ immune cells and CD45−Epcam+ epithelial cells (Fig 1C). In situ hybridization also showed primary expression pattern of Islr in stromal cells of both the small intestine and the colon (Fig 1D). Interestingly, Islr levels are the highest at embryonic stage when intestinal epithelium is developing, then decrease to an intermediate level at postnatal day 10, and drop to the lowest level in adulthood (Fig 1D and Appendix Fig S1A), suggesting a potential function in promoting cell proliferation.
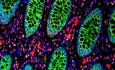
Figure 1. Islr is primarily expressed in stromal cells and upregulated in colitis and colorectal cancer.
-
AqRT–PCR analysis for Islr, Chd1, and Vim in total intestinal tissues, intestinal epithelium, and lamina propria; n = 4.
-
BWestern blotting for Islr and E‐cadherin in total intestinal tissues, intestinal epithelium, and lamina propria. α‐Tubulin was used as a loading control.
-
CqRT–PCR analysis for Islr in CD45+, CD45−Epcam+, CD45−Epcam−CD90+, and CD45−Epcam−CD90− cells sorted from intestinal tissues; n = 3 technical replicates.
-
DIn situ hybridization for Islr with RNAscope probe in intestine and colon from 8‐week‐old mice, showing that Islr is primarily expressed in stromal cells. Negative and positive controls are shown in Appendix Fig S1A. Scale bar: 25 μm.
-
EIn situ hybridization for ISLR with RNAscope probe in human mucosa, and inflamed mucosa from CD and UC patients. Scale bar: 25 μm.
-
FqRT–PCR analysis showing the dynamic changes in Islr in colonic tissues from mice upon DSS treatment and after DSS removal; n = 4 at each timepoints.
-
GIn situ hybridization for Islr with RNAscope probe in mouse colons without or with 3‐day or 5‐day DSS treatments. Scale bar: 50 μm.
-
HThe Cancer Genome Atlas (TCGA) RNA‐Seq analysis showing that ISLR is upregulated in human rectal adenocarcinoma relative to normal rectal tissues. The horizontal lines between the box limits represent the median, the box limits indicate the interquartile ranges, and the whiskers indicate limit superior and limit inferior, respectively; n = 65.
-
IKaplan–Meier survival curve of 270 CRC cases. P = 0.02 (log‐rank test).
-
JISLR level increased with nodal metastasis in colorectal cancer patients. N0, no regional lymph node metastasis; N1, metastases in 1–3 axillary lymph nodes; N2, metastases in 4–9 axillary lymph nodes; and N3, metastases in 10 or more axillary lymph nodes. The solid horizontal lines represent the median, the box limits indicate the interquartile ranges, and the whiskers indicate limit superior and limit inferior, respectively.
-
K, LIn situ hybridization for ISLR/Islr with RNAscope probes in normal human colon, human malignant adenocarcinoma, metastatic adenocarcinoma (K), and mouse colon tumors from AOM–DSS model (L). t indicating tumor; a indicating adjacent tissues of tumor. Scale bar: 50 μm.
Next, we examined Islr/ISLR expression in response to pathological stimuli, including inflammation. In situ hybridization showed that ISLR levels become dramatically elevated in inflamed mucosa from both CD (n = 10) and UC (n = 10) patients, while they were low in unaffected intestinal tissues (Fig 1E). We then induced experimental colitis in mice by administering dextran sulfate sodium (DSS) in drinking water. Consistently, DSS‐induced inflammation progressively resulted in marked upregulation of Islr in the colon, which gradually recovered to basal levels after DSS removal (Fig 1F and G, Appendix Fig S1B).
To further evaluate the relevance of ISLR in inflammation‐induced tumorigenesis, we analyzed clinical samples from human CRC patients. ISLR was dramatically increased in human CRC tissues relative to normal human colon tissues (Fig 1H), and its expression levels were positively correlated with poor survival and nodal metastasis (Fig 1I and J, Appendix Fig S1C). In situ hybridization also showed that ISLR was primarily expressed in the stromal cells of human colorectal tumors (Fig 1K), and in mouse colon tumors from an azoxymethane (AOM)–DSS model mimicking inflammation‐driven colorectal adenocarcinoma (Fig 1L). Taken together, we demonstrate that Islr/ISLR is primarily and specifically expressed in stromal cells in the intestine and becomes markedly upregulated in inflamed mucosa and in CRC.
Ets1 activity induces Islr expression in the context of colitis and CRC
To understand how Islr expression is regulated, we analyzed the potential binding sites of transcription factors in the 2‐kb region upstream from the transcription start site (TSS) of the Islr gene locus using the JASPAR database. The analysis identified two Ets1 binding sites (Fig 2A). ETS1 has been identified as an oncogenic transcriptional factor, and its higher expression is associated with higher grading, increased invasion, and poorer survival in most cancer types (Dittmer, 2015). Interestingly, Ets1/ETS1 is also primarily expressed in the intestinal stromal cells (Fig 2B and C) and is markedly increased in inflamed mucosa from UC and CD patients, experimental colitis, and human colorectal adenocarcinoma (Fig 2C–E). The expression levels of ETS1 were positively correlated with ISLR in CRC tissues (Fig 2F). Together, Ets1/EST1 exhibits an overlapping expression pattern with Islr/ISLR.
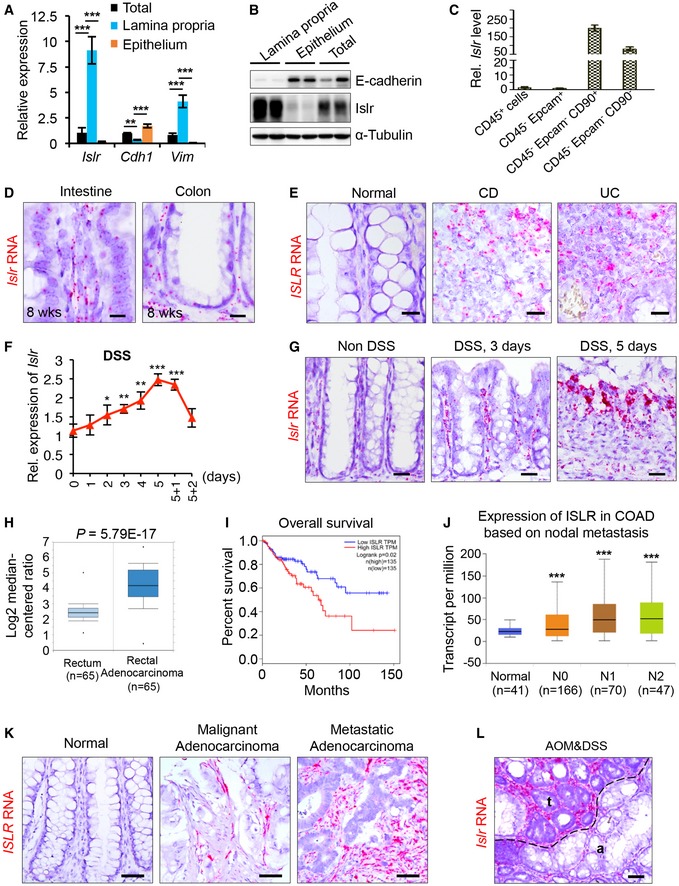
Figure 2. Ets1 activity directly regulated Islr expression.
- The schematic diagram showing two potential Ets1 binding sites in the Islr promoter.
- qRT–PCR analysis for Ets1 in colonic epithelium and lamina propria; n = 3.
- Double immunofluorescence for ETS1 and β‐catenin in intestinal biopsies of healthy controls and inflamed mucosa from UC patients. Scale bar: 50 μm.
- qRT–PCR for Ets1 in colons with or without 5‐day DSS treatment; n = 4.
- Double immunofluorescence for Ets1 and β‐catenin in normal mouse colons without DSS treatment, mouse colons with 5‐day DSS treatments, and AOM–DSS mouse colon tumors. Scale bar: 50 μm.
- Pearson correlation analysis on ETS1 and ISLR in colorectal adenocarcinoma TCGA RNA‐Seq (P < 0.001; R = 0.68).
- Western blotting for ISLR and HA in lysates of UC‐MSCs transfected with pCMV5‐HA or pCMV5‐ETS1. α‐Tubulin was used as a loading control.
- Luciferase activity in lysates of UC‐MSCs transfected with luciferase reporter plasmids of pGL3‐basic empty vector (basic), wild‐type ISLR promoter, or mutant promoter with mutation of ETS1 binding sites under normal and ETS1 overexpression conditions; n = 3.
- Chromatin immunoprecipitation assay was carried out on UC‐MSCs using antibodies against ETS1 and Histone H3. The antibody against Histone H3 was used as a positive control. IgG was used as a negative control. The enrichment of ETS1 binding to ISLR promoter was quantified using qPCR; n = 3 technical replicates.
- Chromatin immunoprecipitation assay was carried out on primary intestinal stromal cells isolated from WT mice without DSS treatment or WT mice after 5‐day DSS treatment using antibodies against ETS1 and Histone H3. The antibody against Histone H3 was used as a positive control. IgG was used as a negative control. The enrichment of Ets1 binding to Islr promoter was quantified using qPCR; n = 3 technical replicates.
To verify the potential regulatory effect of ETS1 on the expression of ISLR, we overexpressed ETS1 in umbilical cord mesenchymal stem cells (UC‐MSCs) and found that ETS1 overexpression could induce ISLR upregulation (Fig 2G). Luciferase reporter assays revealed that ETS1 overexpression was able to activate ISLR promoter activity, while mutation in the ETS1 binding site blocked it (Fig 2H). Chromatin immunoprecipitation (ChIP) assays showed that ETS1 is recruited to its binding site on the ISLR promoter (Fig 2I). The recruitment of Ets1 to the Islr promoter was further corroborated in primary intestinal stromal cells, in which DSS‐induced inflammatory response promoted such recruitment (Fig 2J). Taken together, these results demonstrate that ETS1 activity directly regulates ISLR induction in stromal cells.
Stromal cell‐derived ISLR is essential for intestinal epithelial regeneration
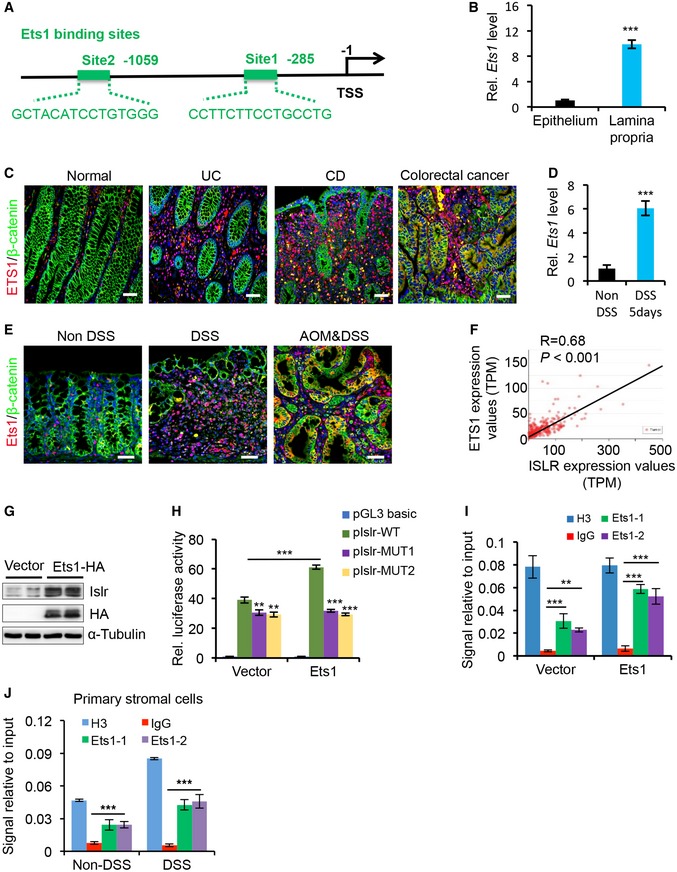
Next, to investigate the in vivo physiological and pathological roles of Islr‐mediated communication between stromal and epithelial cells in the intestine, we generated Twist2‐Cre‐driven Islr cKO mice, in which Islr was specifically deleted in stromal cells (Fig EV1A and B). The expression level of Islr was markedly reduced in the colonic and intestinal tissues of cKO mice (Fig EV1C and D). In situ hybridization showed that Islr was specifically deleted in stromal cells but not in epithelial cells (Fig EV1E). Meanwhile, the Islr cKO mice were viable and fertile with no apparent gross phenotypes. No differences were found in colon microanatomy, in proliferation and apoptotic patterns within the crypt, or in the number of goblet cells between control and cKO mice (Fig EV1F–H). These results suggest that stromal cell‐expressed ISLR is dispensable for intestinal homeostasis under physiological condition.
Figure EV1. No apparent phenotype was observed in Islr cKO mice at homeostasis.
- X‐gal staining showing the Twist2 +/Cre activity in stromal cells using Twist2 +/Cre ;R26R LacZ mice. Scale bar: 50 μm.
- Schematics of Islr genomic locus and strategy for generating loxP‐targeted alleles.
- qRT–PCR for Islr in intestinal and colonic tissues from control and cKO mice; n = 4.
- Western blotting for Islr in colonic tissues from control and cKO mice. α‐Tubulin was used as a loading control.
- In situ hybridization for Islr with RNAscope probe in colons from control and cKO mice, showing that Islr is deleted in stromal cells, while not in the epithelial cells. Dashed lines marked the border of epithelium; n = 3. Scale bar: 25 μm.
- H&E staining of colons from control and cKO mice, which is identical to panels with non‐DSS in Fig 3C. Scale bar: 100 μm.
- Double immunofluorescence for Ki67 and β‐catenin, immunohistochemistry for cleaved caspase3 (Casp3), and PAS staining in colons from control and cKO mice; n = 3. Scale bar: 50 μm.
- Quantification for crypt number, Ki67+ cells, and Casp3+ and PAS+ cells in (G); n = 4.
- Immunohistochemistry for CD45 and immunofluorescence for F4/80 in colons from control and cKO mice after 5‐day DSS treatment; n = 3. Scale bar: 100 μm.
Next, we sought to assess the pathological role of Islr in colitis. To this end, we induced experimental colitis in both control and cKO mice by administering DSS in drinking water for 5 days, followed by 3 days of recovery after DSS removal. Body weight did not significantly differ between control and cKO mice during the DSS treatment (Fig 3A). However, after DSS removal, the body weight of cKO mice continued to decrease, while that of control mice appeared to recover toward the starting level (Fig 3A), indicating impaired recovery in the absence of stromal cell‐expressed Islr. Correspondingly, the inflammatory response was not altered in cKO mice during DSS treatment (Figs 3B and C, and EV1I), but the regenerative response was impaired after DSS removal (Fig 3B and C). In line with this finding, cKO mice exhibited signs of impaired regeneration, with higher clinical scores, shorter colon lengths, and lower percentage of proliferative epithelial cells than the control mice 2 or 3 days after DSS removal (Fig 3D–F). Furthermore, an impaired regenerative response upon Islr deletion in stromal cells was also found in another mouse model of colitis induced by TNBS (Fig EV2). Together, these results indicate that Islr plays a key role in promoting epithelial regeneration in colitis.
Figure 3. Deletion of Islr in stromal cells impaired intestinal regeneration.
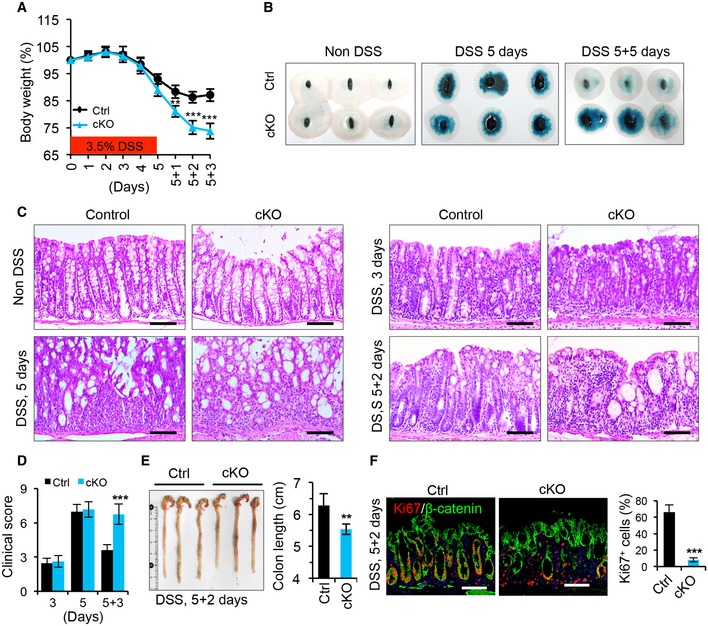
- Quantification of body weight change in control (n = 12) and cKO (n = 13) mice after DSS treatment and DSS removal.
- Fecal occult blood test in control and cKO mice at indicated timepoints; n = 3 at each timepoint.
- Histological images of colonic tissues from control (n = 10) and cKO (n = 10) mice at indicated timepoints after DSS treatment. The panels with non‐DSS are identical to panels in Fig EV1F. Scale bar: 100 μm.
- Quantification of the clinical scores in control (n = 12) and cKO (n = 13) mice at indicated timepoints.
- Gross images of colons and quantification of colon length from control (n = 6) and cKO (n = 6) mice 2 days after 5‐day DSS treatment.
- Double immunofluorescence for Ki67 and β‐catenin in colons from control (n = 4) and cKO (n = 4) mice 2 days after 5‐day DSS treatment. Quantification of percentage of Ki67+ epithelial cells. Scale bar: 100 m.
Figure EV2. Deletion of Islr in stromal cells impaired intestinal epithelial regeneration in TNBS‐induced colitis.
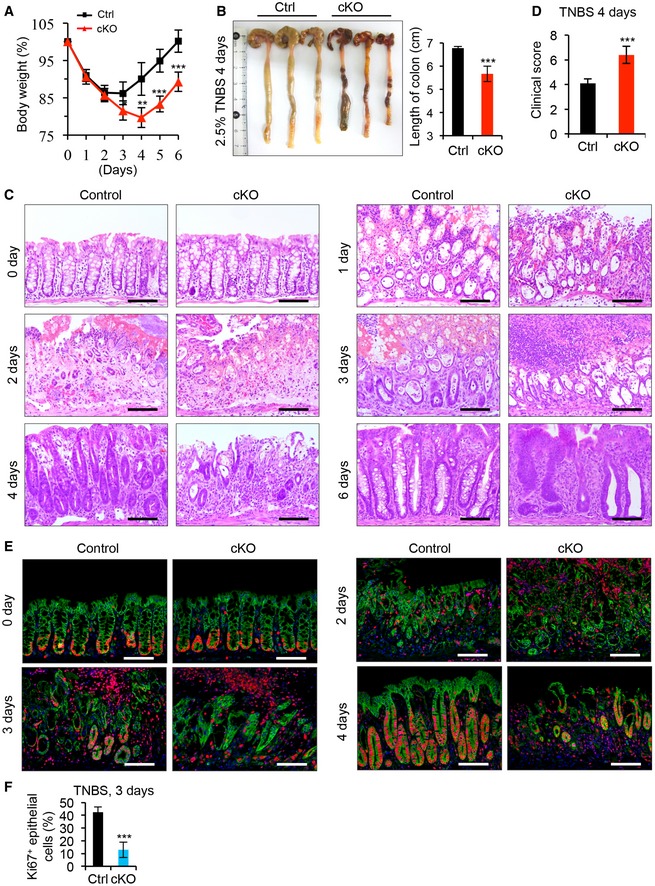
- Quantification of body weight change in control (n = 8) and cKO (n = 8) mice after TNBS treatment.
- Gross images of colons from control (n = 5) and cKO (n = 5) mice 4 days after TNBS treatment, and quantification of colon length from control and cKO mice.
- Histological images of colonic inflamed mucosa from control and cKO mice at indicated timepoints after TNBS treatment; n = 4. Scale bar: 100 μm.
- Quantification of the clinical score of inflamed mucosa in control and cKO mice; n = 5.
- Double immunofluorescence for Ki67 and β‐catenin in colonic inflamed mucosa from control and cKO mice at indicated timepoints after TNBS treatment; n = 4. Scale bar: 100 μm.
- Quantification of the percentage of Ki67+ cells versus epithelial cells at 3 days after TNBS treatment; n = 4.
Stromal cell‐expressed ISLR is required for colorectal tumorigenesis
Given the promoting effect of Islr on epithelial cell proliferation and its high expression in colorectal cancer, we hypothesized that prolonged ISLR expression by stromal cells may exacerbate tumorigenesis. To test this possibility, we examined the in vivo role of Islr in the AOM–DSS‐induced colorectal adenocarcinoma model that recapitulates inflammation‐driven tumorigenesis (De Robertis et al, 2011). We found that deletion of Islr in stromal cells resulted in marked decreases in both tumor size and tumor number (Fig 4A–D), with concomitant reductions in body weight during the process of tumor induction (Fig 4B). The hyperplastic areas in colonic epithelium are markedly reduced in cKO mice compared with control mice (Fig 4E), and cell proliferation levels were significantly lower (Fig 4F–H). Next, we generated xenografted tumors using CT26 CRC cells combined with/without WT or cKO IMCs. WT IMCs markedly promoted tumor growth relative to CT26 CRC cells alone, while ISLR‐deficient cKO IMCs dampened such effect (Appendix Fig S2). In contrast, addition of human recombinant ISLR to the supernatant promoted proliferation of HT29 CRC cells, as compared to non‐ISLR control (Fig 4I). Taken together, these results demonstrate that stromal cell‐secreted ISLR is essential for colorectal tumorigenesis.
Figure 4. Deletion of Islr in stromal cells suppressed tumor growth.
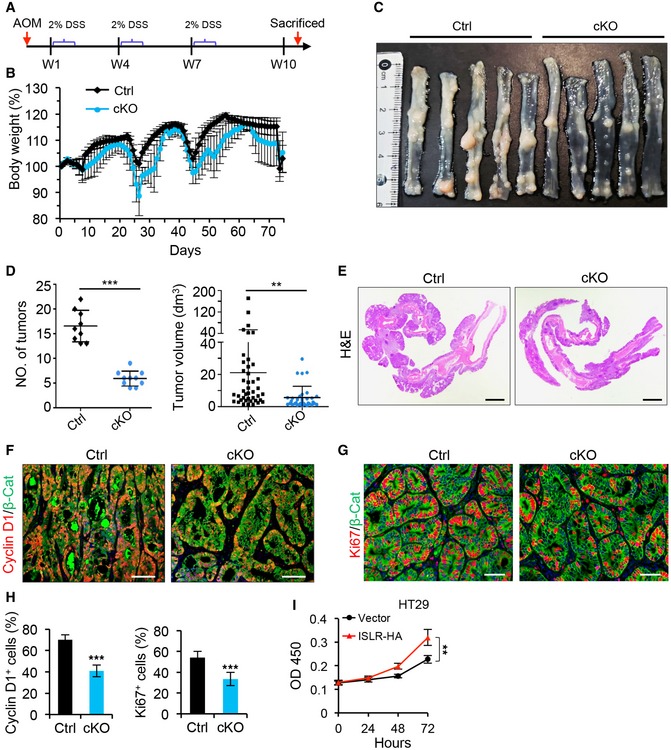
-
ASchematics of generating AOM–DSS mouse colon tumor model.
-
BQuantification of body weight changes in control (n = 9) and cKO (n = 10) mice during tumor development.
-
CGross images of AOM–DSS tumors in control (n = 9) and cKO (n = 10) mice.
-
DQuantification of tumor number per mouse and tumor volume in control (n = 9) and cKO (n = 10) mice.
-
EHistological images of colon tumors from control and cKO mice. Scale bar: 2 mm.
-
F, GDouble immunofluorescence for Cyclin D1 and β‐catenin (F), as well as Ki67 and β‐catenin (G) in colon tumors from control (n = 4) and cKO (n = 4) mice. Scale bar: 100 μm.
-
HQuantification of percentage of Cyclin D1+ and Ki67+ cells in (F and G); n = 4.
-
IThe growth curve of HT29 CRC cells cultured in the supernatant with or without ISLR‐HA. The supernatant was collected for HT29 CRC cells 24 h after transfected with pcDNA3.1 empty vector or pcDNA3.1‐ISLR‐HA vector; n = 5 at each timepoint.
Stromal ISLR dampens epithelial Hippo signaling to activate YAP
To gain insight into the molecular mechanism underlying the regulatory effect of ISLR on epithelial regeneration and tumor growth, we performed genome‐wide transcriptome analysis on colonic epithelial cells from control (n = 3) and cKO (n = 3) mice. We defined the differentially expressed genes (DEGs) as those with P < 0.05 and fold change > 2. This approach yielded 996 downregulated genes and 779 upregulated genes in cKO mice (Fig EV3A). The Hippo signaling pathway was among the enriched KEGG pathway terms (Fig EV3B). We note that Yap1, as a major transcription co‐activator of the Hippo pathway, is essential for promoting intestinal epithelial regeneration and tumorigenesis (Cai et al, 2010; Gregorieff et al, 2015; Taniguchi et al, 2015; Yui et al, 2018; Serra et al, 2019), and that, in particular, Yap1‐null mice with colitis also exhibit impaired regeneration with no alteration in immune response, thus phenocopying Islr cKO mice with colitis (Cai et al, 2010).
Figure EV3. Deletion of Islr in stromal cells suppressed epithelial Hippo signaling.
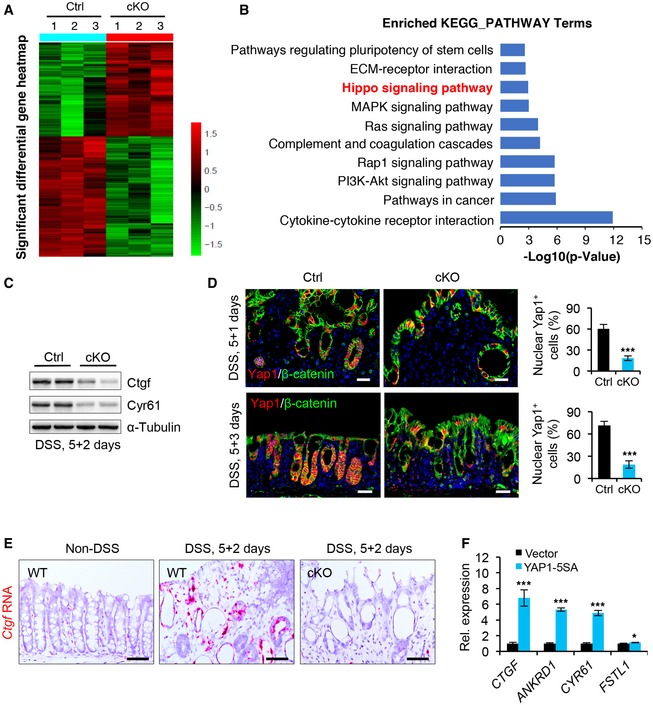
- Heatmaps of differentially expressed genes (DEGs) in colons from control and cKO mice 1 day after DSS removal. Note: The cutoff is P < 0.05 and fold > 2. The parameter of the color key indicated the fold changes converted to log2 of signal value normalized.
- KEGG pathway analysis of DEGs in cKO mice.
- Western blotting for Ctgf and Cyr61 in colon tissues from control and cKO mice 2 days after 5‐day DSS treatment. α‐Tubulin was used as a loading control.
- Double immunofluorescence for Yap1 and β‐catenin in the colons from control and cKO mice 1 or 3 days after 5‐day DSS treatment. Control, n ≥ 3 at each timepoint; cKO, n ≥ 3 at each timepoint. Scale bar: 50 μm.
- RNAscope in situ hybridization assay for Ctgf in colonic epithelium from WT mice or control and cKO mice 2 days after 5‐day DSS treatment. Scale bar: 25 μm.
- qRT–PCR analysis for CTGF, ANKRD1, CYR61, and FSTL1 in NCM460 colon epithelial cells transfected with vector or YAP1‐5SA. *P < 0.05; ***P < 0.001.
Further analysis of genes related to Hippo signaling revealed that many downstream target genes of YAP/TAZ activity, such as Ctgf and Fstl1, were downregulated, while those upstream regulators of the Hippo pathway, such as Mob1b and Dlg3, were upregulated in cKO mice (Fig 5A). This suggests systematic alteration of Hippo signaling in the intestinal epithelium upon stroma‐specific Islr deletion. These observations were also corroborated by qRT–PCR analysis of individual YAP target genes (Ctgf and Cyr61) in colonic epithelial cells of cKO mice (Fig 5B), as well as by Western blotting results of the corresponding proteins (Fig EV3C).
Figure 5. Deletion of Islr in stromal cells suppressed epithelial Yap activity.
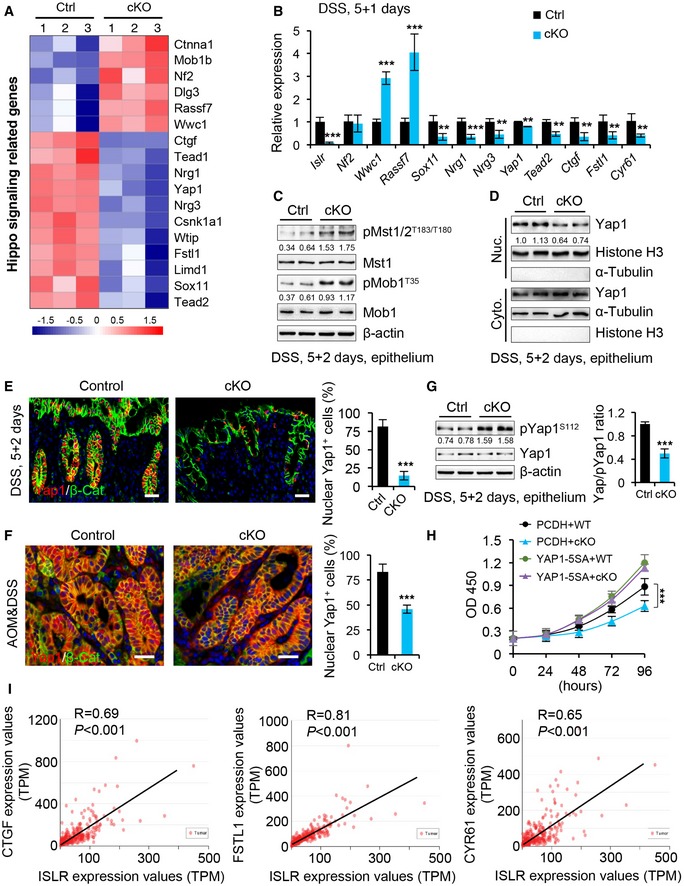
-
AHeatmap of the altered Hippo‐related genes in colons from control and cKO mice 1 day after 5‐day DSS treatment. The parameter of the color key indicated the fold changes converted to log2 of signal value normalized.
-
BqRT–PCR analysis validates the altered Hippo‐related genes in control and cKO mice; n = 4.
-
CWestern blotting for pMst1/2, Mst1, pMob1, and Mob1 in colon epithelium from control and cKO mice 2 days after 5‐day DSS treatment. β‐actin was used as a loading control. The quantification of pMst1/2 versus Mst1 and pMob1 versus Mob1 was shown under the corresponding protein bands.
-
DWestern blotting for Yap1 in nuclear and cytoplasmic proteins isolated from intestinal epithelial cells from control and cKO mice 2 days after 5‐day DSS treatment. Histone H3, a positive control for nuclear proteins. α‐Tubulin, a positive control for cytoplasmic proteins. The quantification of Yap1 versus Histone H3 in nuclear proteins was shown under the corresponding band.
-
E, FDouble immunofluorescence for Yap1 and β‐catenin in the colons from control (n = 4) and cKO (n = 4) mice 2 days after DSS removal (E), and in AOM/DSS tumors from control (n = 4) and cKO (n = 4) mice (F). The percentage of nuclear Yap1+ cells versus epithelial cells was quantified. Scale bar: 50 μm.
-
GWestern blotting for Yap1 and pYap1 in colon epithelial cells from control and cKO mice 2 days after 5‐day DSS treatment. β‐actin was used as a loading control. The ratio of Yap1/pYap1 was quantified; n = 3.
-
HThe growth curve of HCT116 colorectal cancer cells cultured in the supernatant from WT or cKO IMCs, concomitantly transfected with PCDH empty vector or PCDH‐YAP1‐5SA vector; n = 4 technical replicates.
-
IPearson correlation analysis of ISLR and CTGF (P < 0.001; R = 0.69), ISLR and FSTL1 (P < 0.001; R = 0.81), and ISLR and CYR61 (P < 0.001; R = 0.65) in human colorectal cancer based on TCGA RNA‐Seq database.
To clarify the regulatory effect of stromal cell‐secreted ISLR on epithelial Hippo signaling, we examined the activity of Hippo kinases Mst1/2 in the epithelium of DSS‐treated mice 2 days after DSS removal. Our results showed that the phosphorylation of Mst1/2 (pT183/T180 as the marker of kinase activation), as well as that of the substrate Mob1, was markedly increased in the epithelium of cKO mice (Fig 5C), accompanied by the decreased level of nuclear Yap1 (Fig 5D). In line with this, immunofluorescence assay for Yap1 revealed that the proportion of nuclear Yap1+ cells markedly decreased in the inflamed mucosa of cKO mice during the regenerative process (Figs 5E and EV3D) and in AOM–DSS tumors from cKO mice (Fig 5F). In contrast, phosphorylation levels of Yap1 (pS112) were significantly elevated in cKO epithelium upon depletion of stromal Islr (Fig 5G). Consistently, RNAscope in situ hybridization assay showed that the RNA level of Yap1 target gene Ctgf was increased in colonic epithelial cells from mice 2 days after 5‐day DSS treatment, relative to normal WT mice, while it was remarkably reduced in cKO mice (Fig EV3E). Furthermore, we found that the supernatant without recombinant ISLR suppressed cell growth compared with the supernatant with ISLR, while forced activation of YAP (using 5SA mutation) can rescue such inhibitory effect on cell growth caused by ISLR loss (Figs 5H and EV3F), suggesting that YAP activity functionally acts as a downstream effector to mediate ISLR‐induced biological function.
Given the above findings that ISLR is upregulated in CRC and that stromal depletion of Islr results in decreased activity of YAP, we went further to examine the potential correlation between ISLR and YAP activity in clinical samples of human colorectal adenocarcinoma. The transcript levels of ISLR are positively correlated with those of YAP target genes, CGTF, FSTL1, and CYR61 (Fig 5I). Taken together, these results demonstrate that stromal ISLR can suppress epithelial Hippo signaling and activate YAP for tissue regeneration and/or tumorigenesis.
Extracellular ISLR attenuates Hippo signals for YAP activation
To further dissect the mechanism underlying stromal ISLR regulating epithelial Hippo signaling, we asked whether extracellular ISLR secreted by stromal cells could directly suppress the Hippo signaling in epithelial cells. To this end, we first overexpressed HA‐tagged human ISLR (hISLR‐HA) in HEK293FT cells and confirmed the secreted ISLR in the supernatant (Fig 6A). We then treated HEK293FT cells using the supernatant with or without hISLR‐HA and performed transcriptome analysis. Hippo signaling pathway was among the enriched KEGG pathway terms (Fig EV4A). Gene set enrichment assay (GSEA) showed a clear positive association of hISLR‐HA treatment with expression of YAP target genes (Fig 6B and C), suggesting that extracellular hISLR triggers the activation of YAP. In line with this, the levels of pMST1/2, pMOB1, and pYAP1 were reduced in a dose‐dependent manner upon extracellular hISLR‐HA (Fig 6D). Moreover, such ISLR‐induced reduction in pMST1/2, pMOB1, and pYAP upon extracellular hISLR‐HA was further confirmed in NCM460 intestinal epithelial cells (Fig 6E) and in HCT116 CRC cells (Fig EV4B and C). Consistent with the above findings, treatment with the extracellular hISLR‐HA markedly increased the nuclear translocation of YAP in HEK293FT and NCM460 epithelial cells, as evidenced by fluorescent staining and Western blotting assays (Figs 6F and G, and EV4D). Meanwhile, expression of YAP target genes CTGF and CYR61 was also significantly increased in response to the extracellular hISLR‐HA treatment (Fig 6H). Together, these in vitro results clearly demonstrate that extracellular hISLR directly attenuates Hippo signaling in target cells, and consequently triggers YAP activation.
Figure 6. Extracellular Islr attenuates epithelial Hippo signaling to activate YAP signaling in vitro .
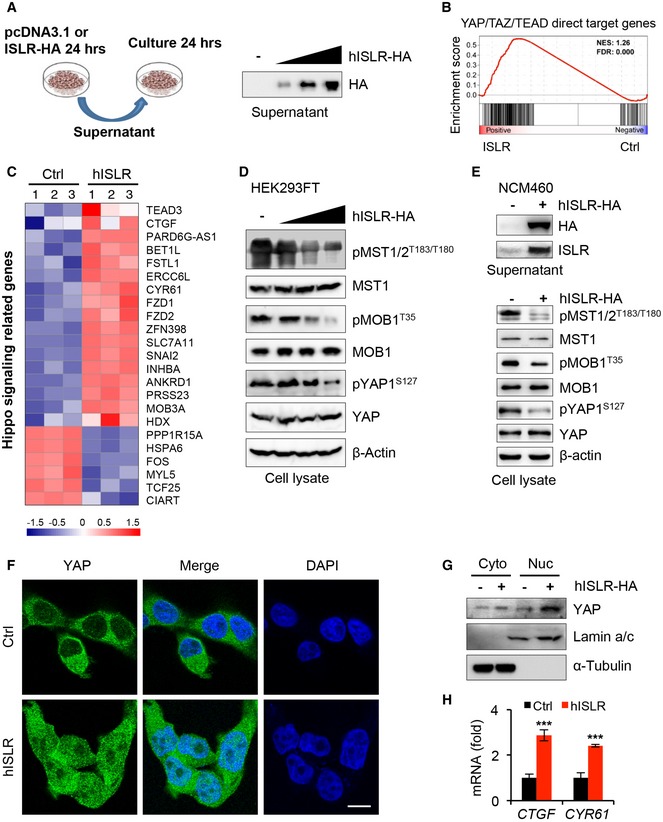
- Schematics of culture epithelial cells with supernatant from HEK293FT cells transfected with pcDNA3.1 empty vector or pcDNA3.1‐hISLR‐HA plasmids. Western blotting for HA in the supernatant from HEK293FT cells transfected with the indicated plasmids.
- Gene set enrichment for YAP/TAZ/TEAD direct target genes in transcriptome profiles for HEK293FT cells treated by hISLR‐HA‐enriched supernatant.
- Heatmap showing the altered Hippo‐related genes in the transcriptome profiles. The parameter of the color key indicated the fold changes converted to log2 of gene fragments per kilobase per million mapped reads (FPKM) values.
- Western blotting for pMST1/2, MST1, pMOB1 and MOB1, pYAP, and YAP in lysates from HEK293FT cells cultured in the supernatants with or without hISLR‐HA. β‐actin was used as a loading control.
- Western blotting for HA and ISLR in the supernatant from NCM460 colonic epithelial cells with pcDNA3.1 empty vector or pcDNA3.1‐hISLR‐HA plasmids. Western blotting for pMST1/2, MST1, pMOB1 and MOB1, pYAP, and YAP in lysates from NCM460 CRC cells cultured in the supernatants with or without hISLR‐HA. β‐actin was used as a loading control.
- Immunofluorescence for YAP in HEK293FT cells cultured in the supernatant with or without hISLR‐HA. Scale bar: 10 μm.
- Western blotting for YAP in cytoplasmic or nuclear proteins from HEK293FT cells cultured in the supernatant with or without hISLR‐HA. Lamin a/c, a positive control for nuclear proteins. α‐Tubulin, a positive control for cytoplasmic proteins.
- qRT–PCR for CTGF and CYR61 in HEK293FT cells cultured in the supernatant with or without hISLR‐HA; n = 3.
Figure EV4. Stromal cell‐secreted Islr suppressed epithelial Hippo signaling and activated YAP.
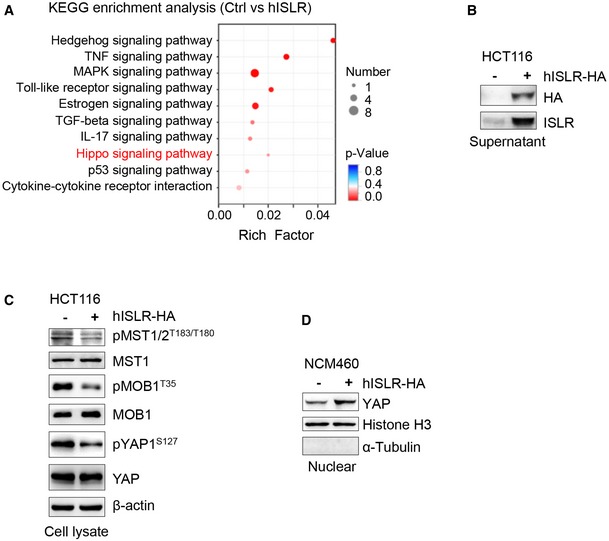
- KEGG enrichment analysis in DEGs of hISLR overexpressing HEK293FT cells.
- Western blotting for HA and ISLR in the supernatant from HCT116 CRC cells transfected with pcDNA3.1 or pcDNA3.1‐hISLR‐HA plasmids.
- Western blotting for pMST1/2, MST1, pMOB1 and MOB1, pYAP, and YAP in lysates from HCT116 CRC cells cultured in the supernatants with or without hISLR‐HA. β‐actin was used as a loading control.
- Western blotting for YAP in nuclear proteins isolated from NCM460 colon epithelial cells cultured in the supernatant with or without hISLR‐HA. Histone H3, a positive control for nuclear proteins. α‐Tubulin, a positive control for cytoplasmic proteins.
Source data are available online for this figure.
To further establish in vivo the promoting effect of extracellular ISLR toward epithelial YAP activity, we first treated primary intestinal epithelial cells with extracellular ISLR. In keeping with the in vitro findings, upon extracellular mouse ISLR (mISLR) treatment, the levels of pMst1/2, pMob1, and pYap1 were reduced in primary intestinal epithelial cells (Fig 7A), while the level of nuclear Yap1 was upregulated (Fig 7B). In agreement, the expression levels of Yap1 target genes, Ctgf, Cyr61, and Fstl1 (Fig 7C), significantly increased upon extracellular mISLR. Subsequent luciferase reporter assay revealed that mISLR significantly increased Yap1‐induced TEAD4 transactivation (Fig 7D). To further test whether this effect is direct, we depleted Yap1 by siRNA in primary intestinal epithelial cells. We found that knockdown of Yap1 abolished ISLR‐mediated promoting effect on cell growth (Figs 7E and EV5A), as well as abrogated the ISLR‐mediated upregulation of Ctgf and Cyr61 (Fig 7F).
Figure 7. Extracellular Islr activates Yap1 signaling in vivo .
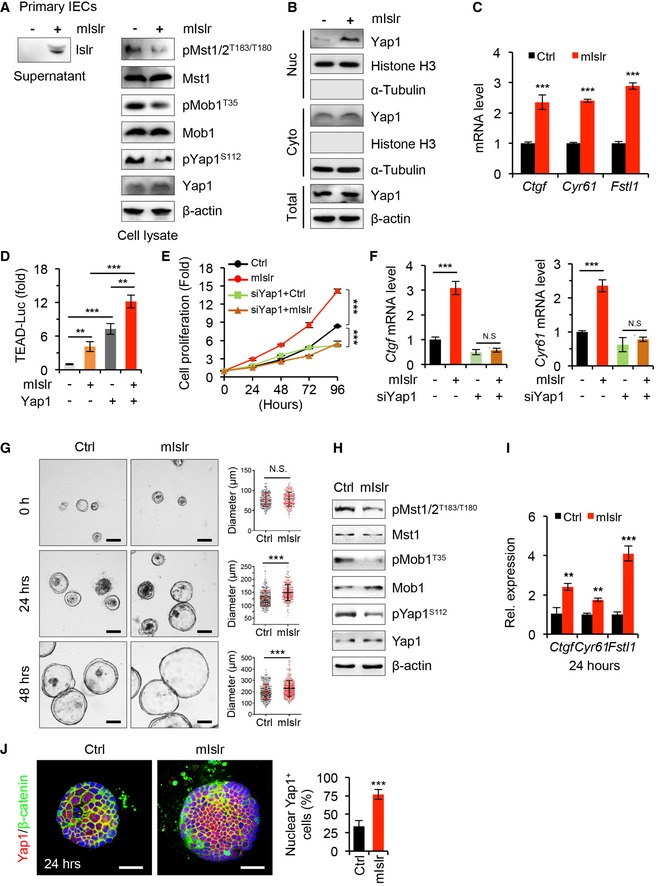
- Western blotting for Islr in the supernatant from primary cultured intestinal epithelial cells (IECs) transfected with pcDNA3.1 or pcDNA3.1‐mIslr‐HA plasmids, and Western blotting for pMst1/2, Mst1, pMob1 and Mob1, pYap1, and Yap1 in lysates from primary IECs cultured in the supernatants with or without mIslr‐HA. β‐actin was used as a loading control.
- Western blotting for Yap1 in nuclear or cytoplasmic proteins from primary intestinal epithelial cells cultured in the supernatant with or without mIslr. Histone H3, a positive control for nuclear proteins. α‐Tubulin, a positive control for cytoplasmic proteins.
- qRT–PCR for Ctgf, Cry61, and Fstl1 in primary intestinal epithelial cells cultured in the supernatant with or without mIslr for 24 h; n = 4.
- Activity of TEAD‐luciferase reporter in primary intestinal epithelial cells upon mIslr, Yap1 or mIslr, and Yap1; n = 4.
- The growth curve of primary intestinal epithelial cells upon mIslr or/and Yap1 siRNA over time; n = 5.
- qRT–PCR for Ctgf and Cry61 in primary intestinal epithelial cells 48 h after treatments of mIslr or/and Yap1 siRNA (siYap1); n = 4.
- Representative images of primary intestinal organoids cultured at indicated conditions and timepoints. Scale bar: 100 μm. The organoids were co‐cultured in a mixed medium of the supernatant with/without mIslr from CT26 cells and regular organoid culture solution at the ratio of 1:4. Quantification of organoids diameter at indicated timepoints; n > 150 organoids at each timepoint.
- Western blotting for pMst1/2, Mst1, pMob1 and Mob1, pYap1, and Yap1 24 h after the treatment. β‐actin was used as a loading control.
- qRT–PCR for Ctgf, Cry61, and Fstl1 in primary intestinal organoids 24 h after the treatments; n = 3.
- Immunofluorescence for Yap1 and β‐catenin in intestinal organoids 24 h after the indicated treatments. The treatments are identical to (G). The percentage of nuclear Yap1+ cells versus epithelial cells was quantified. Scale bar: 50 μm; n = 12.
Figure EV5. Deletion of Yap1 abrogated the mIslr‐mediated promoting effect of tumor growth.
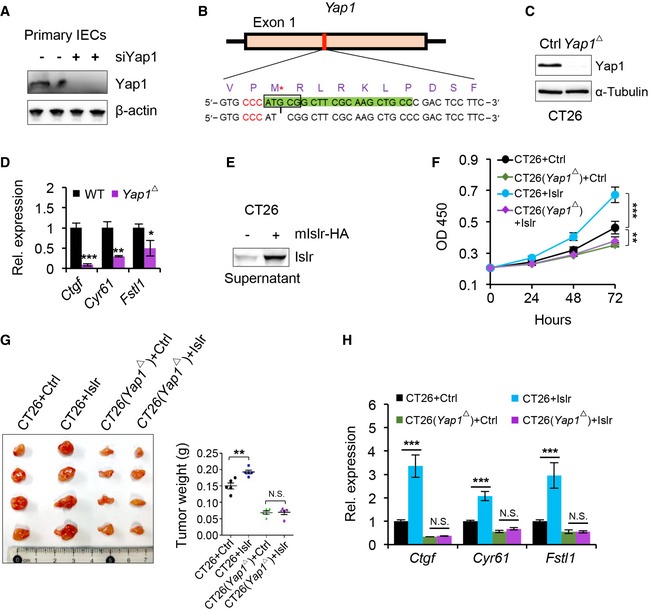
- Western blotting for Yap1 in primary intestinal epithelial cells 24 h after Yap1 siRNA treatment. β‐actin was used as a loading control. The sequences of Yap1 siRNA are in Appendix Table S5.
- Generating Yap1‐deficient CT26 CRC cells using Crispr/Cas9 technique. gRNA marked by green color. Protospacer adjacent motif (PAM) sequence marked by red color. G deletion is indicated by an asterisk.
- Western blotting for Yap1 in normal and Yap1‐deficient (Yap1 Δ) CT26 CRC cells. α‐Tubulin was used as a loading control.
- qRT–PCR for Ctgf, Cry61, and Fstl1 in normal and Yap1‐deficient (Yap1 Δ) CT26 CRC cells; n = 3.
- Western blotting for mIslr in the supernatant from CT26 CRC cells transfected with pcDNA3.1 or pcDNA3.1‐mIslr plasmids.
- The growth curve of CT26 CRC cells under indicated conditions; n = 5.
- Gross images of xenografted tumors 10 days after transplantation. Quantification of tumor weight; n = 5.
- qRT–PCR for Ctgf, Cry61, and Fstl1 in CT26 CRC cells at indicated conditions; n = 3.
Next, we assessed Yap1 activity in intestinal organoids upon extracellular ISLR exposure. Consistent with the above observations, the supernatant with mISLR significantly promoted the growth of organoids (Fig 7G). Similarly, the Hippo signaling was tuned down in these organoids upon ISLR treatment, as shown by reduced levels of pMst1/2, pMob1, and pYap1 (Fig 7H); consequently, Yap1 was activated as shown by increased expression of downstream target genes (Fig 7I) and the increase in nuclear Yap1+ cells (Fig 7J).
Finally, we generated a Yap1‐deficient CT26 CRC cells using Crispr/Cas9 technique (Fig EV5B). We confirmed that Yap1 was completely deleted (Fig EV5C) and the target genes Ctgf, Cyr61, and Fstl1 were indeed downregulated in the Yap1‐deficient CT26 CRC cell line (Fig EV5D). The supernatant containing ISLR promoted the growth of CT26 CRC cells but not the Yap1‐deficient CT26 CRC cells (Fig EV5E and F), clearly indicating that deletion of Yap1 abolished the growth‐promoting effect of ISLR. Consistent with these observations, xenograft tumor assay showed that deletion of Yap1 also suppressed the SLR‐mediated promoting effect on tumor growth (Fig EV5G). Concomitantly, the Yap1 target genes were upregulated upon extracellular SLR, while deletion of Yap1 suppressed such effect (Fig EV5H). Taken together, these findings demonstrated in vitro and in vivo that stromal ISLR activates epithelial YAP for tissue regeneration and/or tumorigenesis.
Discussion
Here, we demonstrate that stromal cell‐secreted ISLR promotes intestinal epithelial regeneration by attenuating Hippo signaling to activate YAP signaling in epithelial cells (Fig 8). Consistent with this conclusion, numerous studies showed an essential role for YAP/TAZ in intestinal regeneration upon inflammation or after irradiation (Cai et al, 2010; Gregorieff et al, 2015; Taniguchi et al, 2015; Yui et al, 2018; Serra et al, 2019). Supporting the function of ISLR in regulating Hippo signaling, Yap1 deficiency in the intestine of mice with colitis impairs the regenerative response but does not influence the inflammatory response (Cai et al, 2010). Thus, it results in a phenotype that resembles that of the Islr cKO mice. Given the central importance of YAP/TAZ in tissue repair, it is conceivable that a sophisticated upstream regulatory network is necessary for fine‐tuning YAP/TAZ activity. In this regard, recent reports have indicated that YAP/TAZ signaling can be activated in colitis by the IL‐6/GP130/Src as well as extracellular matrix (ECM)/FAK/Src cascades (Taniguchi et al, 2015; Yui et al, 2018), independent of the classic Hippo pathway. Our current work indicates that epithelial Hippo signaling is directly regulated by secreted ligand of stromal origin upon inflammatory stimuli. Therefore, we propose an inter‐cellular regulatory machinery for the classic Hippo pathway in the epithelial cells that is governed by signals from stromal cells, and provide a parallel mechanism accounting for YAP‐associated cell–cell communications. In addition to highlighting the importance of stromal cells in controlling epithelial YAP activity under pathological conditions, our study also identifies a novel ISLR–YAP signaling axis that links inflammation‐associated stromal signals with epithelial regeneration.
Figure 8.
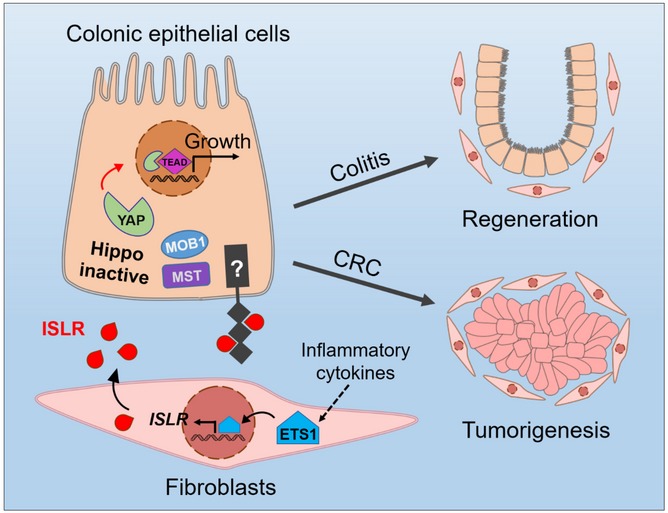
The working model of stromal cell‐secreted Islr regulating intestinal epithelial Hippo signaling.
Finally, our observations that deletion of Islr in stromal cells suppressed tumor growth and that ISLR levels are positively correlated with the poor survival and lymph node metastasis strongly support the idea that ISLR acts as an oncogenic factor during CRC development. Other pro‐tumorigenic factors secreted by tumor‐associated stromal cells, including IL6, VEGF, and MMPs, have been shown to promote tumorigenesis (Bussard et al, 2016). Intriguingly, our results highlight ISLR as a stroma‐secreted oncogenic factor that can enhance YAP activity in CRC. Our finding of the inter‐cellular connection between ISLR and YAP/TAZ explains the molecular basis for YAP/TAZ hyperactivation commonly observed in many types of human cancers (Overholtzer et al, 2006). Consistently, we demonstrate that ETS1, which is an oncogenic transcription factor in most cancer types (Dittmer, 2015), is a direct upstream regulator of ISLR and it increases its expression in response to stress signals. In support of this notion, both ISLR and ETS1 are upregulated in tumor‐associated stromal cells (Wernert et al, 1994), and their expression levels are correlated with tumor progression (Dittmer, 2015). The ETS1‐induced ISLR expression can at least partially account for the oncogenic activity of ETS1. Thus, we posit that ISLR is an important pro‐tumorigenic factor in CRC.
In summary, we discovered a new molecular mechanism of stromal ISLR activating epithelial YAP activity underlying communication between stromal cells and epithelial cells during intestinal tissue regeneration and tumorigenesis. While this type of stroma–epithelium signaling is required for regeneration, its dysregulation exacerbates colorectal cancer—a finding that offers basis for a new therapeutic strategy.
Materials and Methods
Subjects
Colonoscopic biopsies were obtained from inflamed and unaffected sites in the colons of 10 UC and 10 CD patients as well as from the normal colonic mucosa in 10 healthy individuals. All patients were from Department of Gastroenterology, the Shanghai Tenth People's Hospital of Tongji University (Shanghai, China). Each subject provided written informed consent. The diagnosis of UC and CD was based on clinical, radiological, endoscopic, and pathological examinations.
Ethics
All mouse experimental procedures and protocols were evaluated and authorized by the Beijing Laboratory Animal Management and were strictly performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of China Agricultural University (approval number: SKLAB‐2015‐04‐03).
Mouse strains
The Islr‐floxed mouse has been described previously (Zhang et al, 2018). The Twist2‐Cre mice were obtained from the Jackson Laboratory (stock number: 008712).
Xenograft tumor
Five‐week‐old male nude mice (BALB/c nude) were obtained from the Beijing Vital River Laboratory Animal Technology Co., Ltd., and maintained in specific pathogen‐free conditions. Mice were randomly grouped and injected subcutaneously with 5.0 × 105 CT26 cells with/without 2.0 × 105 primary colonic stromal cells in a total volume of 100 μl PBS or supernatant containing mIslr. Mice were sacrificed 10 days after transplantation, and tumor weight was measured. Then, all tumors were fixed in 10% formalin for paraffin embedding.
RNAscope in situ hybridization assay
An RNAscope in situ hybridization assay was performed as described previously (Anderson et al, 2016). The sample sections were processed according to the manufacturer's instructions using an RNAscope Red Kit (ACDBio). Briefly, 5‐μm‐thick sections were deparaffinized and treated with hydrogen peroxide solution for 10 min at room temperature (RT). Target retrieval was performed for 15 min at 100°C, and then, the slides were treated with protease for 15 min at 40°C. Probes were then hybridized for 2 h at 40°C, followed by RNAscope amplification and then Fast Red chromogenic detection RNAscope. Then, the slides were counterstained in hematoxylin. After dehydration, the slides were mounted with EcoMount (EM897L; Biocare). The following RNAscope probes were used in this study: Hs‐ISLR (cat. #455481; ACDBio), Mm‐Islr‐O1 (cat. #453321; ACDBio), Mm‐Ctgf (cat. #314541; ACDBio), Mm‐Ppib (cat. #313911; ACDBio), and DapB (cat. #310043; ACDBio).
DSS treatment
To induce acute colitis with DSS, mice were treated with 3.5% w/v DSS with molecular weight of 36,000–50,000 (MP Biochemicals) in drinking water for 5 days; then, the DSS was withdrawn for another 3 days. The severity of colitis was scored daily on the basis of standard parameters, including body weight and the presence of diarrhea, and/or bloody stools. The mice were sacrificed at different timepoints to obtain colon samples. The clinical score was measured as previously reported with modifications (Tian et al, 2019), and evaluated by an independent investigator who was blinded to the experiment. The parameters of clinical score are included in Appendix Table S1.
TNBS treatment
TNBS was used to induce colitis as described previously (Tian et al, 2019). To induce colitis with TNBS, the mice were weighed and anesthetized with isoflurane‐mixed gas on day 1. Then, a 1.5 × 1.5 cm area of skin was shaved on the back of each mouse between the shoulders, and 150 μl of 1% (w/v) TNBS (Sigma, cat. no. p2297) presensitization solution was applied to the shaved skin. On day 8, after weighing and anesthetizing each mouse, we slowly injected 125 μl of 2.5% (w/v) TNBS solution into the lumen of the colon with 1‐ml syringe and a 3.5 F catheter, and removed the catheter and positioned the mouse head‐down for 5 min. The severity of colitis was scored daily on the basis of standard parameters including body weight and the presence of diarrhea and/or bloody stools. The mice were sacrificed at the indicated timepoints to obtain colon samples.
AOM and DSS treatment
An AOM–DSS mouse model was generated as described previously with modifications (Tian et al, 2017). Briefly, 8‐week‐old control and Islr cKO mice were intraperitoneally injected with AOM at a concentration of 10 mg/kg body weight (Sigma‐Aldrich). Five days after AOM injection, the mice were treated with a so‐called DSS cycle comprising two steps: The mice were first given 2.5% (w/v) DSS (molecular weight: 36,000–50,000, MP Biomedicals) for 7 days and then given normal water for 14 days. The mice were subjected to a total of 3 DSS cycles. After the treatments, the mice were sacrificed, distal colon tissues were collected, and tumor numbers and volumes were evaluated.
qRT–PCR analysis
Total RNA was isolated from sorted cells of colonic tissues, cell lines, and mouse colonic tissues using TRIzol reagent (Life Technologies) according to the manufacturer's instructions. To detect mRNA levels, reverse transcription was carried out using oligo (dT) primers. qRT–PCR was performed using LightCycler 480 SYBR Green I Master Mix on a LightCycler 480 Real‐Time PCR System (Roche, Mannheim, Germany). Relative expression was calculated based on the 2−ΔΔCt method, and Gapdh or Actin was used as the internal control. The primers for qRT–PCR analysis are included in Appendix Table S2.
Flow cytometry and cell sorting
Colonic epithelial cells were collected following vigorous shaking after incubation in 1× DPBS containing 5 mM EDTA and 0.2 mM DTT for 30 min at 37°C on a rotating platform. The remaining tissue was incubated in digestion solution containing 2% FBS, 0.25 mg/ml collagenase XI (Sigma), and 0.1 mg/ml DNase I (Sigma) for 1 h at 37°C. Mesenchymal cells were collected by centrifugation following vigorous shaking. Then, the colonic epithelial cells and mesenchymal fractions were pipetted with prewarmed dispase (2.5 U/ml) and DNase I for 5 min to generate single‐cell suspensions. The single‐cell suspensions from the colon tissues were stained for 30 min on ice using a mix of antibodies (from eBioscience: LIVE/DEAD‐BV421, CD45‐APC, Epcam‐FITC, and CD90‐PE). Viable cells were gated by forward and side scatter; Epcam+, Epcam−CD45+, CD45−Epcam−CD90−, and CD45−Epcam−CD90+ cell subpopulations were gated; and 1 × 105 to 1 × 106 cells were sorted using a BD FACS Aria III Sorter (BD Biosciences) for further RNA isolation.
Isolation and culture of primary IMCs
IMCs were isolated as described previously with modifications (Khalil et al, 2013). Briefly, colonic tissues were removed and placed in cold HBSS to remove debris. Each colon was opened longitudinally and cut into 0.5‐cm pieces, which were then incubated in HBSS containing 5 mM EDTA for 30 min at 37°C on a rotating platform to remove primary intestinal epithelial cells. After isolating the epithelial cells by vigorous shaking, the remaining tissue was rinsed several times with cold HBSS and incubated in digestion solution containing 2% FBS, 0.25 mg/ml collagenase XI (Sigma), 0.08 U/ml dispase II, and 0.1 mg/ml DNase 1 (Sigma) for 1 h at 37°C on a rotating platform. After vigorous shaking, single mesenchymal cells were pelleted at 500 × g in a tabletop and centrifuged for 5 min. The supernatant was discarded, and each pellet was resuspended with modified DMEM (Gibco) containing 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma), 1× l‐glutamine, and 1× nonessential amino acids (NEAAs). Then, the cells from each mouse colon were seeded in a 6‐cm dish and cultured in a 5% CO2 incubator at 37°C. After 2.5 h, the floating debris and nonadherent cells were washed off with PBS. The adherent cells were IMCs consisting of fibroblasts and myofibroblasts. The IMCs were passaged 1 week post‐seeding with a 1:3 ratio. After 3 passages, the supernatant was collected from each dish for follow‐up experiments.
Intestinal organoid culture and supernatant treatment
Intestinal crypts were isolated from small intestine samples by incubation for 10 min at 4°C in HBSS containing 5 mM EDTA as described previously (Sato et al, 2009). Briefly, the isolated crypts were counted and pelleted. A total of 100 isolated crypts from C57 BL/B6 mice were mixed with 30 μl of Matrigel (BD Bioscience) and plated in 48‐well plates. After Matrigel polymerization, 300 μl of crypt culture medium (advanced DMEM/F12 [Gibco], 100 U/ml penicillin, 100 μg/ml streptomycin [Sigma], B27 supplement [Invitrogen], N2 supplement [Invitrogen], 2 mM GlutaMAX [Invitrogen], 1 mM N‐acetyl cysteine [Sigma], 50 ng/ml mouse EGF [PeproTech], 100 ng/ml mouse Noggin [PeproTech], and 10% human R‐spondin‐1 [PeproTech]) was added to each well. The medium was changed every 2 days, and the organoids were passaged every 4 days.
Briefly, isolated crypts were counted and pelleted. A total of 100 isolated crypts from C57 BL/B6 mice were mixed with 30 μl of Matrigel (BD Bioscience) and plated in 48‐well plates. After Matrigel polymerization, 300 μl of crypt culture medium [advanced DMEM/F12 (Gibco), 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma), B27 supplement (Invitrogen), N2 supplement (Invitrogen), 2 mM GlutaMAX (Invitrogen), 1 mM N‐acetyl cysteine (Sigma), 50 ng/ml mouse EGF (PeproTech), 100 ng/ml mouse Noggin (PeproTech), and 10% human R‐spondin‐1 (PeproTech)] was added to each well. Medium was changed every 2 days, and organoids were passaged every 4 days.
For treating organoids with cell supernatant, a total of 100 primary intestinal crypts were mixed with 30 μl of Matrigel and plated in the center of a 48‐well plate. After Matrigel polymerization, 200 μl of crypt organoid culture medium was added to each well. Twelve hours post‐seeding, the medium was changed with 150 μl of regular crypt organoid culture medium and 50 μl of cell supernatant with/without mIslr. Then, the mixed medium was changed daily. The growth of organoids was observed, and the diameters of organoids were quantified at 0, 24, and 48 h, respectively.
Cell lines, culture conditions, transfections, and supernatant collection
HEK293FT and HT29 cell lines were purchased from the American Type Culture Collection (ATCC) (Manassas, VA) and maintained in DMEM supplemented with 10% FBS. CT26 and NCM460 cell lines were purchased from ATCC (Manassas, VA) and cultured in RPMI 1640 supplemented with 10% FBS. HCT116 cell line was purchased from ATCC (Manassas, VA) and cultured in IMDM supplemented with 10% FBS. UC‐MSCs were cultured in DMEM supplemented with 10% FBS, 1× l‐glutamine, and 1× nonessential amino acids (NEAAs). All cell lines were tested and confirmed to be free of mycoplasma infection. The cells were transfected using Lipofectamine 2000 reagent (Invitrogen) with 2 μg of vector or negative control vector for a well of 6‐well plate according to the manufacturer's protocol. Mediums were changed 6 h post‐transfection, and the supernatant was collected after 24 or 36 h.
Histology, immunofluorescence, and immunohistochemistry
For histological analysis, the colonic tissues were rinsed with 1× DPBS, fixed in 10% formalin, paraffin‐embedded, and sectioned at 5 μm. The sections were deparaffinized with xylene followed by treatment with serial dilutions of ethanol. The sections were stained with hematoxylin (Sigma) for 6 min and then washed in running water for 5 min. The sections were stained with eosin (Sigma) for 10 s, dehydrated with serial dilutions of ethanol, and then mounted with coverslips in neutral gum mounting medium.
For immunostaining, antigen retrieval was performed by heating the slides in 0.01 M citrate buffer (pH 6.0) in a microwave. The sections were rinsed three times with ddH2O after natural cooling, immersed in 3% H2O2 for 10 min or PBS‐T (PBS, 1% Triton X‐100) for 20 min, washed twice with PBS, and then blocked for 1 h at RT with blocking solution (10% normal goat serum in TBS‐T). The sections were incubated with primary antibodies overnight at 4°C. Then, the slides were washed three times with PBS and incubated for 1 h at RT with Alexa Fluor 488 and 594 goat anti‐mouse or anti‐rabbit IgG (H+L). The slides were then washed 3 times with PBS and stained with DAPI for 5 min, and the sections were covered with anti‐quenching reagent. The following antibodies were used: anti‐Ki67 (1:500, Thermo Fisher), anti‐ISLR (1:200, Sigma), anti‐Ets1 (1:800, Cell Signaling Technology), anti‐CD45 (1:200, Santa Cruz), anti‐F4/80 (1:200, Santa Cruz), anti‐β‐catenin (1:1,000, Sigma), anti‐cleaved caspase3 (1:1,000, Cell Signaling Technology), anti‐Cyclin D1 (1:1,000, Cell Signaling Technology), anti‐Yap1 (1:500, Cell Signaling Technology), and anti‐Mucin2 (1:100, Santa Cruz).
Confocal imaging
HEK293FT cells were grown on glass bottom cell culture dishes (NEST, 801001), washed once with filtered PBS, and fixed in 4% paraformaldehyde for 30 min at room temperature. After permeabilization, cells were blocked with PBS with 5% BSA and then incubated with primary antibodies. After three separate washes, cells were incubated with Alexa Fluor‐conjugated secondary antibodies and then stained with DAPI. Images were captured using a Leica laser scanning confocal microscope (Leica TCS SP8).
Nucleoprotein extraction
Nucleoprotein was isolated from cells using a nucleoprotein extraction kit (AR0106, BosterBio, USA) according to the manufacturer's instructions. Briefly, HEK293FT, NCM460, and primary IEC were washed once with cold PBS. The cells were pelleted at 600 × g for 5 min, resuspended in reagent A, and incubated on ice for 10 min to lyse the cell membrane. Intact nuclei were pelleted at 16,000 × g for 5 min, and soluble cytoplasmic proteins were removed. The nuclear pellet was resuspended in nucleoprotein extraction reagent and incubated on ice for 40 min. Chromatin was pelleted at 20,000 × g for 5 min, and the soluble nucleoproteins were saved. The protein concentration was determined with a BCA Kit (Beyotime). Lamin a/c or Histone H3 was used as a nuclear internal control.
Western blotting
Cell lysates were subjected to Western blotting according to standard procedures. Fresh tissues were homogenized using RIPA buffer (Beyotime Biotechnology, Shanghai, China) in the presence of protease and phosphatase inhibitor cocktails (Roche), followed by treatment with homogenizer work center (T10 basic, IKA). Proteins were measured by BCA protein assay kit (Beyotime) and denatured, then 30 μg of total protein was separated on a 6–12% SDS–PAGE gel, and transferred to PVDF membranes (GE Healthcare). The PVDF membranes were blocked with 5% nonfat dry milk for 1 h at RT and incubated with primary antibodies overnight at 4°C. Results were collected by chemiluminescence imaging system (Sagecreation, Beijing). The relative protein band intensity was quantified using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA). Antibodies were applied as following: anti‐GAPDH (Sigma), anti‐α‐Tubulin (Sigma), anti‐β‐actin (Sigma), anti‐ISLR (Sigma), anti‐Histone H3 (Santa Cruz), anti‐E‐cadherin (Abcam), anti‐Lamin a/c (Cell Signaling), anti‐HA (Santa Cruz), anti‐CYR61 (Santa Cruz), anti‐CTGF(Santa Cruz), anti‐MOB1(Cell Signaling), anti‐pMOB1T35 (Cell Signaling), anti‐YAP (Cell Signaling), anti‐pYAP1S127 (Cell Signaling), anti‐pMST1/2T183/T180 (Cell Signaling), and anti‐MST1(Cell Signaling).
Plasmid construction
Constructs of full length or truncated human ISLR and mouse Islr were cloned into a pMlink vector and a pcDNA3.1 vector with a C‐terminal 3 × HA tag. A full‐length mouse Ets1 construct was cloned into a modified pCMV5 vector with a C‐terminal 3 × HA tag. YAP (5SA) (S61/S109/S127/S164/S381A) plasmids were constructed in a pCDH1‐MCS‐coGFP vector. For luciferase assays, a construct including 2 Kb Islr or TEAD4 promoter sequence was cloned into a pGL3‐Basic vector. All mutants were generated through site‐directed mutagenesis. All constructs were verified by performing DNA sequencing.
Microarray analysis
Total RNA was isolated from the colons of three littermate controls and three cKO mice using TRIzol reagent (Life Technologies) according to the manufacturer's instructions. RNA was purified with an RNase‐free DNase Kit (Qiagen) and then submitted to CapitalBio Technology, where the samples were labeled and hybridized to Agilent Mouse Gene arrays. The array data were analyzed for data summarization, normalization, and quality control using the GeneSpring software V13 (Agilent). The q value, a measure of the false discovery rate (FDR), was computed using the Significance Analysis of Microarrays software for each probe set by running an unpaired t‐test. The data were log2‐transformed, and the median was centered by genes using the Adjust Data function of Cluster 3.0 software; the data were then further analyzed by hierarchical clustering with average linkages. Finally, tree visualization was performed using Java TreeView (Stanford University School of Medicine, Stanford, CA, USA). The results were visualized as an intensity heatmap with R software. The microarray data have been submitted to the GEO repository: GSE135259.
RNA‐Seq analysis
HEK293FT cells were treated with pcDNA3.1‐hISLR‐HA or empty vector for 24 h. RNA was extracted with a standard procedure. RNA quality was assessed on a 2100 Expert Bioanalyzer (Agilent). RNA samples were sent for library preparation and sequencing on the Illumina NovaSeq 6000 platform by Shanghai Majorbio Bio‐Pharm Technology. The data were analyzed on the free online platform of Majorbio I‐Sanger Cloud Platform (http://www.i-sanger.com) or using R software. The RNA‐Seq data have been submitted to the GEO repository: GSE135280.
Luciferase assay for Islr promoter activity
The sequence for Islr is located on chromosome 9 (NC_000075.6, base pairs 58156246..58159236, complement) in the mouse genome. An approximately 2‐kb region upstream of transcript start site (TSS) was identified as the Islr promoter in this study, which is located at chromosome 9 (NC_000075.6: base pairs C58161222..58159222) and cloned into the pGL3‐Basic reporter constructs. Binding sites 1 and 2 of ETS1 are located at base pairs 58159520‐58159506 and base pairs 58160294‐58160280, respectively. The construct was verified by DNA sequencing. Nucleotides at Ets1 binding sites were mutated by carrying out site‐directed mutagenesis (BGI, Shenzhen, China). The wild type and mutant plasmids were transfected with phRL‐TK control plasmid to UC‐MSCs, respectively. The luciferase activities of firefly and Renilla were measured after 24 h of transfection by using Dual‐Glo luciferase assay kit (Promega) according to the manufacturer's instructions.
Luciferase assay for TEAD promoter activity
The TEAD‐luciferase reporter plasmids were transfected into primary intestinal epithelial cells with NEPA21 electroporator (NEPA GENE) according to the manufacturer's instructions. The luciferase activities were determined after 24 h using the Dual‐luciferase Assay System (Promega).
Cell proliferation assay
Cells were seeded in 96‐well format. An ATP‐based CellTilter‐LumiTM Plus cell viability assay kit (Beyotime) was used for detecting cell proliferation. 100 μl of the reagent was added into each individual well and mixed for 10 min at room temperature, and then, intracellular ATP content was measured using a luminescence detector (Bio‐Tek Synergy NEO).
Chromatin immunoprecipitation (ChIP) assay
A ChIP assay was performed using the SimpleChIP enzymatic chromatin immunoprecipitation kit (Cell Signaling Technology, #9002) according to the manufacturer's protocol, with modifications. Briefly, for primary intestinal stromal cells, after removing epithelial cells with EDTA, tissue was weighted, finely minced, and cross‐linked immediately with 1.5% (v/v) formaldehyde at room temperature for 20 min. Then, the cross‐linked tissues were disaggregated with Dounce Homogenizer for single cells on ice, and the cross‐linked stromal cells were immediately used for nuclei preparation and chromatin digestion according to the manufacturer's protocol.
For UC‐MSCs, harvested UC‐MSCs were first cross‐linked with 1% (v/v) formaldehyde for 10 min. After isolating the nuclei by lysis of the cytoplasmic fraction, the immunoprecipitation preparations were incubated with micrococcal nuclease for 20 min at 37°C with frequent mixing to digest the DNA into fragments of 150–900 bp. The sonicated nuclear fractions were divided for input control and were incubated with anti‐Ets1, anti‐Histone H3 (as a positive control), and anti‐IgG (as a negative control) at 4°C overnight. The recruited genomic DNA obtained from the ChIP assay was quantified by qRT–PCR with primers specific for the Ets1 binding elements of the Islr promoter regions. The primers are used in Appendix Table S3.
Genome engineering
Gene‐specific sgRNA oligos were cloned into the px459v2.0‐eSpCas9 expression vector (Addgene), which bicistronically expressed sgRNA and Cas9 nuclease. Yap1 sgRNA sequences were determined by the CRISPR Design Tool (http://crispr.mit.edu/) (Appendix Table S4). CT26 cells were transfected with 2 μg of vector DNA by Lipofectamine 2000. 10 μg/ml puromycin was added into CT26 cells for 3 days 24 h post‐transfection. Genomic DNA extracted from engineered cells was identified by T7E1 restriction analysis. Then, engineered cells were seeded for monoclonal screening. Finally, the positive monoclone was identified by DNA sequencing.
Statistical analysis
All analyses were performed at least in triplicate, and the means obtained were subjected to independent t‐tests. In the figures, asterisks denote statistical significance (*P < 0.05; **P < 0.01; ***P < 0.001). All data are reported as the mean ± standard deviation (SD). The means and SDs from at least three independent experiments are presented in all graphs.
Author contributions
ZY designed research; JX, YTa, XS, YTi, GL, YP, MD, SD, CL, YS, and PL performed research; JX, YLo, YLi, BZ, YCh, ZL, YCo, MVP, QM, ZZ, and ZY analyzed data; and JX, ZZ, and ZY wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
ZY is supported by the National Natural Science Foundation of China (Nos. 81772984, 81572614); the Major Project for Cultivation Technology (2016ZX08008001); Basic Research Program (2015QC0104, 2015TC041, 2016SY001, 2016QC086); and SKLB Open Grant (2018SKLAB6‐12). ZZ is supported by the National Key R&D Program of China (2017YFA0504504), the National Natural Science Foundation of China (81725014, 31930026), the ‘Strategic Priority Research Program’ (XDB19020202).
The EMBO Journal (2020) 39: e103255
Contributor Information
Zhaocai Zhou, Email: zczhou@sibcb.ac.cn.
Zhengquan Yu, Email: zyu@cau.edu.cn.
Data availability
The datasets and computer code produced in this study are available in the following databases:
RNA‐Seq data: Gene Expression Omnibus GSE135259 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135259)
RNA‐Seq data: Gene Expression Omnibus GSE135280 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135280)
References
- Anderson CM, Zhang B, Miller M, Butko E, Wu X, Laver T, Kernag C, Kim J, Luo Y, Lamparski H et al (2016) Fully automated RNAscope in situ hybridization assays for formalin‐fixed paraffin‐embedded cells and tissues. J Cell Biochem 117: 2201–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussard KM, Mutkus L, Stumpf K, Gomez‐Manzano C, Marini FC (2016) Tumor‐associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res 18: 84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Zhang N, Zheng Y, de Wilde RF, Maitra A, Pan D (2010) The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev 24: 2383–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Robertis M, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, Signori E, Fazio VM (2011) The AOM/DSS murine model for the study of colon carcinogenesis: from pathways to diagnosis and therapy studies. J Carcinog 10: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer J (2015) The role of the transcription factor Ets1 in carcinoma. Semin Cancer Biol 35: 20–38 [DOI] [PubMed] [Google Scholar]
- Francescone R, Hou V, Grivennikov SI (2015) Cytokines, IBD, and colitis‐associated cancer. Inflamm Bowel Dis 21: 409–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, Wrana JL (2015) Yap‐dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 526: 715–718 [DOI] [PubMed] [Google Scholar]
- Hara A, Kobayashi H, Asai N, Shigeyoshi S, Higuchi T, Kato K, Okumura T, Bando YK, Takefuji M, Mizutani Y et al (2019) Roles of the mesenchymal stromal/stem cell marker meflin in cardiac tissue repair and the development of diastolic dysfunction. Circ Res 125: 414–430 [DOI] [PubMed] [Google Scholar]
- Huang J, Wu S, Barrera J, Matthews K, Pan D (2005) The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 122: 421–434 [DOI] [PubMed] [Google Scholar]
- Khalil H, Nie W, Edwards RA, Yoo J (2013) Isolation of primary myofibroblasts from mouse and human colon tissue. J Vis Exp 80: e50611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Cheon JH (2017) Pathogenesis of inflammatory bowel disease and recent advances in biologic therapies. Immune Netw 17: 25–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Enomoto A, Hara A, Asai N, Kobayashi T, Horinouchi A, Maruyama S, Ishikawa Y, Nishiyama T, Kiyoi H et al (2016) Identification of meflin as a potential marker for mesenchymal stromal cells. Sci Rep 6: 22288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Moroishi T, Guan KL (2016) Mechanisms of Hippo pathway regulation. Genes Dev 30: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa A, Kubota R, Imamura Y, Nagamine K, Wang Y, Asakawa S, Kudoh J, Minoshima S, Mashima Y, Oguchi Y et al (1997) Cloning of the cDNA for a new member of the immunoglobulin superfamily (ISLR) containing leucine‐rich repeat (LRR). Genomics 44: 273–279 [DOI] [PubMed] [Google Scholar]
- Nagasawa A, Kudoh J, Noda S, Mashima Y, Wright A, Oguchi Y, Shimizu N (1999) Human and mouse ISLR (immunoglobulin superfamily containing leucine‐rich repeat) genes: genomic structure and tissue expression. Genomics 61: 37–43 [DOI] [PubMed] [Google Scholar]
- Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng CX, Brugge JS, Haber DA (2006) Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci USA 103: 12405–12410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D (2010) The hippo signaling pathway in development and cancer. Dev Cell 19: 491–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell DW, Pinchuk IV, Saada JI, Chen X, Mifflin RC (2011) Mesenchymal cells of the intestinal lamina propria. Annu Rev Physiol 73: 213–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano M, F DEF, Zarantonello L, Ruffolo C, Ferraro GA, Zanus G, Giordano A, Bassi N, Cillo U (2016) From inflammation to cancer in inflammatory bowel disease: molecular perspectives. Anticancer Res 36: 1447–1460 [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ et al (2009) Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature 459: 262–265 [DOI] [PubMed] [Google Scholar]
- Serra D, Mayr U, Boni A, Lukonin I, Rempfler M, Challet Meylan L, Stadler MB, Strnad P, Papasaikas P, Vischi D et al (2019) Self‐organization and symmetry breaking in intestinal organoid development. Nature 569: 66–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, Yu FX, Wang K, Ho SB, Boland BS, Chang JT et al (2015) A gp130‐Src‐YAP module links inflammation to epithelial regeneration. Nature 519: 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Ma X, Lv C, Sheng X, Li X, Zhao R, Song Y, Andl T, Plikus MV, Sun J et al (2017) Stress responsive miR‐31 is a major modulator of mouse intestinal stem cells during regeneration and tumorigenesis. Elife 6: e29538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Xu J, Li Y, Zhao R, Du S, Lv C, Wu W, Liu R, Sheng X, Song Y et al (2019) MicroRNA‐31 reduces inflammatory signaling and promotes regeneration in colon epithelium, and delivery of mimics in microspheres reduces colitis in mice. Gastroenterology 156: 2281–2296 e6 [DOI] [PubMed] [Google Scholar]
- Wernert N, Gilles F, Fafeur V, Bouali F, Raes MB, Pyke C, Dupressoir T, Seitz G, Vandenbunder B, Stehelin D (1994) Stromal expression of c‐Ets1 transcription factor correlates with tumor invasion. Cancer Res 54: 5683–5688 [PubMed] [Google Scholar]
- Yui S, Azzolin L, Maimets M, Pedersen MT, Fordham RP, Hansen SL, Larsen HL, Guiu J, Alves MRP, Rundsten CF et al (2018) YAP/TAZ‐dependent reprogramming of colonic epithelium links ECM remodeling to tissue regeneration. Cell Stem Cell 22: 35–49 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Zhang Y, Gu L, Lan M, Liu C, Wang M, Su Y, Ge M, Wang T, Yu Y et al (2018) Islr regulates canonical Wnt signaling‐mediated skeletal muscle regeneration by stabilizing Dishevelled‐2 and preventing autophagy. Nat Commun 9: 5129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Cheng X, Ding Y, Shi J, Jin H, Wang H, Wu Y, Ye J, Lu Y, Wang TC et al (2014) Bone marrow‐derived myofibroblasts promote colon tumorigenesis through the IL‐6/JAK2/STAT3 pathway. Cancer Lett 343: 80–89 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The datasets and computer code produced in this study are available in the following databases:
RNA‐Seq data: Gene Expression Omnibus GSE135259 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135259)
RNA‐Seq data: Gene Expression Omnibus GSE135280 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135280)