

Abstract
Structural maintenance of chromosomes flexible hinge domain‐containing protein 1 (SMCHD1) has been implicated in X‐chromosome inactivation, imprinting, and DNA damage repair, and mutations in SMCHD1 can cause facioscapulohumeral muscular dystrophy. More recently, SMCHD1 has also been identified as a component of telomeric chromatin. Here, we report that SMCHD1 is required for DNA damage signaling and non‐homologous end joining (NHEJ) at unprotected telomeres. Co‐depletion of SMCHD1 and the shelterin subunit TRF2 reduced telomeric 3′‐overhang removal in time‐course experiments, as well as the number of chromosome end fusions. SMCHD1‐deficient cells displayed reduced ATM S1981 phosphorylation and diminished formation of γH2AX foci and of 53BP1‐containing telomere dysfunction‐induced foci (TIFs), indicating defects in DNA damage checkpoint signaling. Removal of TPP1 and subsequent activation of ATR signaling rescued telomere fusion events in TRF2‐depleted SMCHD1 knockout cells. Together, these data indicate that SMCHD1 depletion reduces telomere fusions in TRF2‐depleted cells due to defects in ATM‐dependent checkpoint signaling and that SMCHD1 mediates DNA damage response activation upstream of ATM phosphorylation at uncapped telomeres.
Keywords: ATM, DNA damage response, non‐homologous end joining, SMCHD1, telomeres
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; DNA Replication, Repair & Recombination
Fusion of telomeres lacking shelterin protein TRF2 requires the recently‐identified telomeric chromatin component SMCHD1 for induction of ATM‐mediated damage responses, or alternative ATR activation.

Introduction
Arguably, the most fundamental function of telomeres is to suppress at chromosome ends DNA damage signaling and DNA end repair (Muller, 1938; McClintock, 1941). This is achieved through the recruitment of specialized proteins that bind directly or indirectly to telomeric repeat DNA, which consists of hundreds to thousands of 5′‐TTAGGG‐3′/5′‐CCCTAA‐3′ repeats in vertebrates. Most abundant at telomeres are the shelterin proteins comprising TRF1, TRF2, RAP1, TIN2, TPP1, and POT1 (Lazzerini‐Denchi & Sfeir, 2016; de Lange, 2018). TRF1 and TRF2 bind as homodimers to the double‐stranded telomeric DNA repeats. Depletion of TRF2 from chromosome ends occurs naturally upon telomere shortening in senescent cells (Karlseder et al, 2002; Cesare et al, 2013). TRF2 depletion leads to ATM kinase activation and a long‐lasting DNA damage response (DDR) promoting cellular senescence (Denchi & de Lange, 2007) or apoptosis (Karlseder et al, 1999). Inactivation of the DDR in senescent cells occurs during tumorigenesis (Shay & Wright, 2011; Maciejowski & de Lange, 2017). The ensuing cell proliferation leads to further telomere shortening and further TRF2 depletion culminating in telomere crisis in which chromosome ends are fused to one another by alternative non‐homologous DNA end joining (alt‐NHEJ), which relies on DNA ligase 3 and poly(ADP‐ribose) polymerase 1 (PARP1) (Jones et al, 2014). Experimental depletion of TRF2 in cells with normal telomere length also leads to ATM‐dependent DDR activation and telomere end joining, which in this case is mediated by the classical NHEJ pathway involving DNA ligase 4 and the KU70/80 heterodimer (Celli & de Lange, 2005). Significantly, classical NHEJ at TRF2‐depleted telomeres requires DDR activation (Denchi & de Lange, 2007).
The DDR promotes genome stability regulating DNA repair, chromatin remodeling, transcription, cell cycle arrest, senescence, and apoptosis (Ciccia & Elledge, 2010; Panier & Durocher, 2013). DDR activation at DNA double‐strand breaks and uncapped telomeres involves ATM recruitment to chromatin by the MRE11/RAD50/NBS1 (MRN) complex. In addition, the MRN complex promotes conformational changes stimulating ATM kinase activity (Paull, 2015). Apart from the interaction with NBS1 in the MRN complex, ATM activation also depends on Tip60/KAT5‐dependent acetylation of K3016 in ATM (Sun et al, 2009). Active ATM leads to autophosphorylation at S1981 and phosphorylation and activation of hundreds of downstream DDR substrates (Matsuoka et al, 2007), such as the CHK2 kinase, p53, NBS1, 53BP1, and H2AX.
SMCHD1 is a non‐canonical member of the structural maintenance of chromosomes (SMC) protein family (Blewitt et al, 2008), which includes among others the SMC1/3 cohesion and SMC2/4 condensin complex components, the SMC5/6 complex which is involved in homologous recombination and RAD50. As other SMC proteins, SMCHD1 contains a hinge domain flanked by coiled‐coil domains. However, unlike SMC1‐6, SMCHD1 forms homodimers (Brideau et al, 2015). Furthermore, it contains a GHKL (gyrase, Hsp90, histidine kinase, MutL)‐type ATPase rather than the bipartite ABC‐type ATPase domain typically seen in SMC proteins (Brideau et al, 2015). Several studies implicate roles of SMCHD1 in modulating chromosome architecture at the inactive X chromosome and at Hox clusters (Nozawa et al, 2013; Jansz et al, 2018; Wang et al, 2018; Gdula et al, 2019). SMCHD1 mediates the compaction of the inactive X chromosome in females linking the H3K9me3 and the XIST‐H3K27me3 domains (Nozawa et al, 2013). Through mediating chromatin interactions on the inactive X chromosome, SMCHD1 may also promote chromatin mixing and drive attenuation of chromosomal compartments and topologically associated domains (TADs) (Wang et al, 2018). SMCHD1 localization to the inactive X chromosome in mouse cells is dependent on a pathway involving the Xist long non‐coding RNA, Hnrnp K, and PRC1 (Jansz et al, 2018). Alternative mechanisms of SMCHD1 recruitment to chromatin may involve H3K9me3, HP1γ, and affinity for nucleic acids (Nozawa et al, 2013). Apart from binding the inactive X chromosome, SMCHD1 is also recruited to sites of DNA damage induced by laser micro‐irradiation (Coker & Brockdorff, 2014) or zeocin drug treatment and it has been implicated in promoting DNA repair by NHEJ over homologous recombination (Tang et al, 2014). Finally, SMCHD1 has been detected in proteomic analyses of telomeric chromatin (Déjardin & Kingston, 2009; Grolimund et al, 2013; Bartocci et al, 2014). Specifically, SMCHD1 was enriched at telomeres that were overly long and showed a lower density of TRF2 (Grolimund et al, 2013). However, the roles of SMCHD1 at telomeres remained enigmatic.
Here, we discover critical functions of SMCHD1 at telomeres that were deprived of TRF2. Significantly, depletion of SMCHD1 prevents efficient ATM‐dependent DNA damage signaling at TRF2‐depleted telomeres. At the same time, telomere end fusions were diminished indicating crucial roles of SMCHD1 in DNA damage signaling or repair. Experimental activation of the ATR checkpoint at TRF2‐depleted telomeres reinstigated chromosome end fusions in the absence of SMCHD1 unraveling a requirement of SMCHD1 for checkpoint activation but not directly the DNA repair reaction. Our data indicate that SMCHD1 is required for promoting ATM‐dependent DDR activation.
Results
SMCHD1 is required for efficient telomere end‐to‐end fusions at TRF2‐depleted telomeres
In previous work, we observed in HeLa cells enrichment of SMCHD1 at long telomeres with an average length of 30 kb over telomeres with an average length of 10 kb (Grolimund et al, 2013). In addition, over‐elongated telomeres showed a lower density of TRF2. We therefore tested if shRNA‐mediated TRF2 depletion in HeLa cells is sufficient to enhance association of SMCHD1 with telomeres of normal length. SMCHD1 association with telomeric DNA was assessed by chromatin immunoprecipitation upon which co‐precipitated telomeric DNA was detected by hybridization. Indeed, immunoprecipitated SMCHD1 was associated with more telomeric DNA in TRF2‐depleted cells (Fig EV1). A probe for Alu‐repeat DNA served as a negative control.
Figure EV1. SMCHD1 association with telomeres is increased upon TRF2 removal.
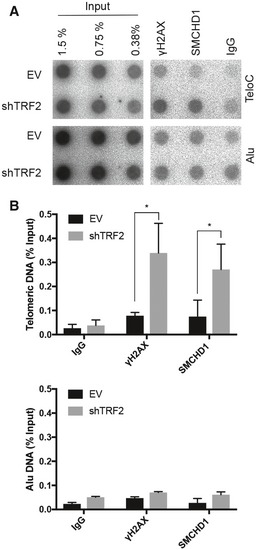
- Telomeric DNA ChlP with antibodies against γH2AX, SMCHD1, and rabbit lgG. Representative dot blot images of precipitated DNA detected with a (CCCTAA)n or Alu probe. ChlPs were performed in HeLa cells transfected with shTRF2 or empty vector (EV) control.
- Bar graph for quantification of γH2AX, SMCHD1, and rabbit lgG binding to telomeric or Alu DNA as assessed by ChlP. The bars represent average values from three independent experiments for telomeric DNA and two independent experiments for Alu DNA. Error bars represent the standard deviation. P‐values were calculated by unpaired two‐tailed Student's t‐test *P < 0.05.
TRF2‐depleted telomeres trigger an ATM‐dependent DNA damage response, and they undergo NHEJ‐mediated telomere end‐to‐end fusions (Denchi & de Lange, 2007). In order to assess potential roles of SMCHD1 for these processes, we used CRISPR/Cas9 technology to disrupt the SMCHD1 gene and we developed shRNA vectors for the depletion of SMCHD1 (Fig 1). Three different guide RNAs were used for generating SMCHD1 knockout clones in HeLa cells and in a HeLa cell clone in which TRF2 could be depleted using an inducible shRNA (Grolimund et al, 2013). Individual clones were screened for loss of SMCHD1 protein expression on Western blots using antibodies recognizing SMCHD1 peptides near the N‐ and C‐termini (Figs 1A and EV2A). This analysis suggested complete loss of SMCHD1 protein expression in all three clones (Fig EV2A). Analysis of the knockout clones by PCR amplification of the targeted loci and DNA sequencing revealed introduction of frameshift mutations near the N‐terminus of SMCHD1 leading to premature stop codons (Fig EV2B), which can explain the loss of SMCHD1 protein expression. In addition, two shRNAs mediated efficient depletion of SMCHD1 protein (Fig EV3 and further below). shRNA‐mediated TRF2 depletion during 5 days (Fig 1A) triggered end‐to‐end fusions at 20% of the chromosome ends as assessed by the analysis of metaphase chromosome spreads (Fig 1B and C). Strikingly, the telomere fusions were reduced to roughly 3–4% when TRF2 was depleted in two different SMCHD1 knockout clones. Similar results were obtained upon shRNA‐mediated co‐depletion of SMCHD1 with TRF2 (Fig EV3) confirming critical roles of SMCHD1 for efficient telomere end‐to‐end fusions upon TRF2 loss. The effects of SMCHD1 loss on telomere end‐to‐end fusions were not due defects in cell cycle progression as the cell cycle profile was not strongly affected in SMCHD1 knockout cells versus wild‐type (WT) (Fig 1D).
Figure 1. SMCHD1 promotes c‐NHEJ at dysfunctional telomeres.
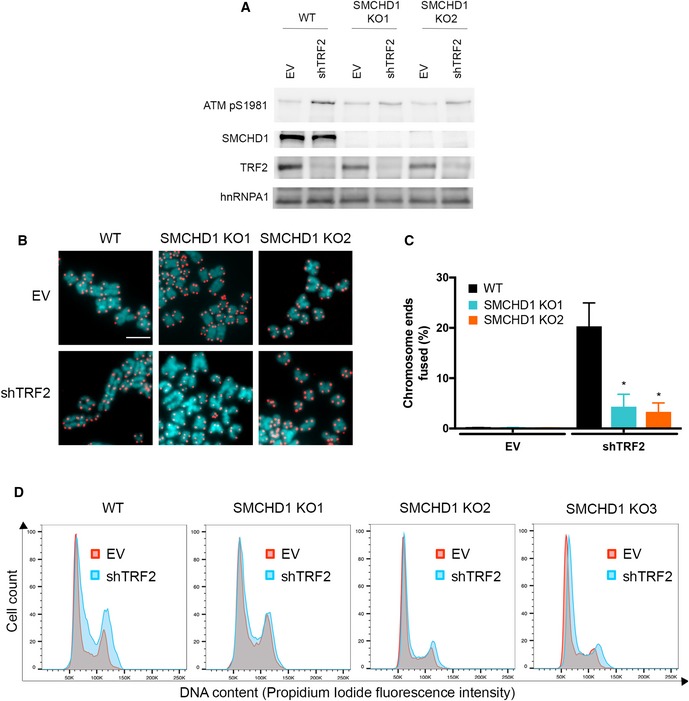
- Western Blot detection of ATM pS1981, SMCHD1, TRF2, and hnRNPA1 loading control in wild‐type and two SMCHD1 knockout (KO1 and KO2) HeLa cells transfected with shTRF2 plasmid or EV control.
- Representative metaphase spreads from wild‐type and SMCHD1 knockout HeLa cells transfected with shTRF2 plasmid or EV control. Telomeric signals were detected with Cy3‐OO‐(CCCTAA)3 and are false‐colored in red; DNA is stained with DAPI and is false‐colored in cyan. Scale bar: 5 µm.
- Quantification of telomere fusions from experiment shown in (B). Bars represent average number of fused chromosome ends. SDs were obtained from three independent experiments (> 3,000 telomeres counted/condition/experiment). *P < 0.05; unpaired two‐tailed Student's t‐test.
- Representative cell cycle profiles from two biological replicates of WT and SMCHD1 KO HeLa cells transfected with shTRF2 plasmid or EV control. Cells were stained with propidium iodide and analyzed by flow cytometry.
Source data are available online for this figure.
Figure EV2. Generation of SMCHD1 knockout cells using CRISPR‐Cas9 technology.
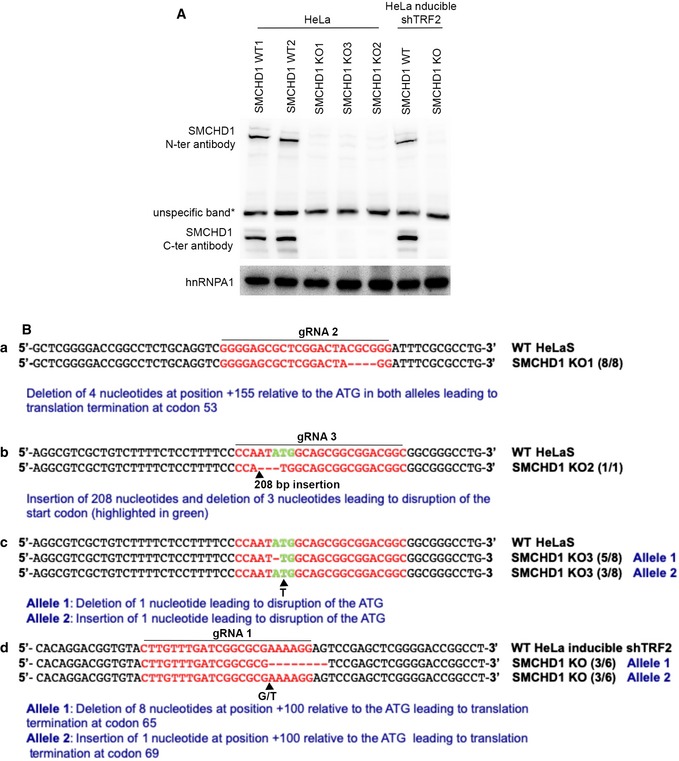
- Western Blot detection of SMCHD1 with an antibody raised against the N‐terminus (aa213–aa300) and the C‐terminus of the protein (aa1955–2005) in SMCHD1 knockout single‐cell clones of HeLa and HeLa inducible shTRF2 cell lines.
- Sequence analysis of edited alleles in SMCHD1 knockout single‐cell clones.
Source data are available online for this figure.
Figure EV3. SMCHD1 stimulates c‐NHEJ and promotes DNA damage signaling at TRF2‐depleted telomeres.
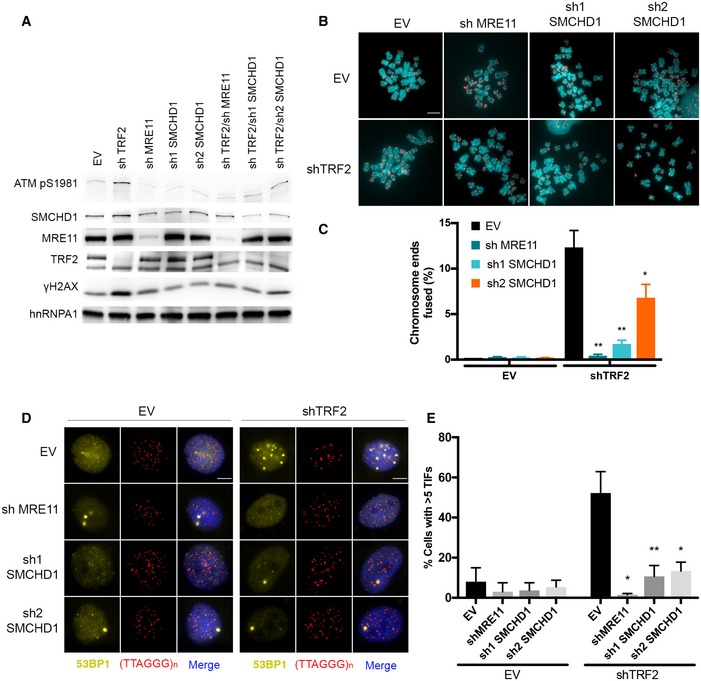
- Western Blot detection of ATM pS1981, SMCHD1, MRE11, TRF2, and γH2AX, in HeLa cells transfected with the indicated shRNA plasmids (shTRF2, shMRE11, sh1 SMCHD1, sh2 SMCHD1, shTRF2/shMRE11, shTRF2/sh1 SMCHD1, shTRF2/sh2 SMCHD1) and empty vector (EV) control.
- Metaphase spreads from HeLa cells transfected with the indicated shRNA plasmids (shTRF2, shMRE11, sh1 SMCHD1, sh2 SMCHD1, shTRF2/shMRE11, shTRF2/sh1 SMCHD1, and shTRF2/sh2 SMCHD1) and empty vector (EV) control. Telomeric signals were detected with Cy3‐(CCCTAA)3 and are false‐colored in red. Scale bar: 5 µm.
- Quantification of telomere fusions in HeLa cells transfected with the indicated shRNAs and EV control. Bars represent average numbers of chromosome ends fused in three independent experiments with SDs. **P < 0.01; *P < 0.05 unpaired two‐tailed Student's t‐test.
- Representative images for detection of 53BP1 at telomeres in HeLa cells transfected with the indicated shRNA plasmids. Immunofluorescence (IF) for 53BP1 (yellow) was combined with telomeric (CCCTAA)3‐FISH (red), and the DNA was stained with DAPI. Scale bars: 5 µm.
- Quantification of the number of cells containing > 5 telomere dysfunction‐induced foci (TIFs) detected as in (A). Data represent mean of three independent experiments ± SD (> 100 cells/condition/experiment). *P < 0.05, **P < 0.01, unpaired two‐tailed Student's t‐test.
Source data are available online for this figure.
SMCHD1 promotes DNA end processing for NHEJ at TRF2‐depleted telomeres
Upon TRF2 depletion, telomeric DNA is first processed to remove the 3′ overhang. The blunt‐end telomeres are then fused by NHEJ (Celli & de Lange, 2005). To better understand the roles of SMCHD1 in these processes, we followed telomeric DNA processing and fusions in time‐course experiments in which TRF2 was depleted using an inducible shRNA in SMCHD1 wild‐type and knockout cells (Fig 2A). Quantification of telomere end‐to‐end fusions showed again strong reduction but not abolishment of fusion events in SMCHD1 knockout cells (Fig 2B). Removal of the telomeric 3′ overhang was assessed by native in‐gel hybridization in which the radiolabeled probe detects only the telomeric 3′ overhang but not the double‐stranded telomeric DNA, which remains base‐paired (Fig 2C, left panel). As expected, the overhang signal was lost upon in vitro treatment of the DNA prior to gel loading with Exonuclease 1 from E. coli which removes the 3′ overhang (left panel, lanes designated with +Exo). Upon denaturation of the same gel, however, single‐ and double‐stranded telomeric DNA are detected with the probe (right panel). Inspection of the native gels (Fig 2C, left panel, see short run) and quantification (Fig 2D) revealed that the SMCHD1 knockout cells lost the telomeric 3′ overhang less efficiently than the SMCHD1 wild‐type cells (Fig 2C and D). Furthermore, the signal for fused telomeres which is fully double‐stranded and therefore can only be detected in the denatured gel (Fig 2C, right panel, see long run) was reduced in the SMCHD1 knockout cells (compare signal of fused to non‐fused telomeres in each lane). These results are consistent with the metaphase chromosome analysis of Figs 1B and C, and 2B. During the time course, we also observed a shift of the telomeric signals over time toward longer telomeres which was expected as TRF2 negatively regulates telomere elongation by telomerase (Smogorzewska et al, 2000). Telomere elongation was also apparent on the native gel, indicating that these telomeres were not fused. When assessing sister telomere exchange by CO‐FISH (Bailey et al, 1996), we did not observe increased frequency of telomere recombination with this assay as seen in alternative lengthening of telomeres (ALT) U2OS cells, which maintain their telomeres by recombination (Fig 3A–C). Altogether, this analysis indicated that the first step of the telomeric DNA end‐fusion reaction, the DNA end processing step, was strongly reduced in the absence of SMCHD1.
Figure 2. SMCHD1 loss slows down overhang processing at TRF2‐depleted telomeres.
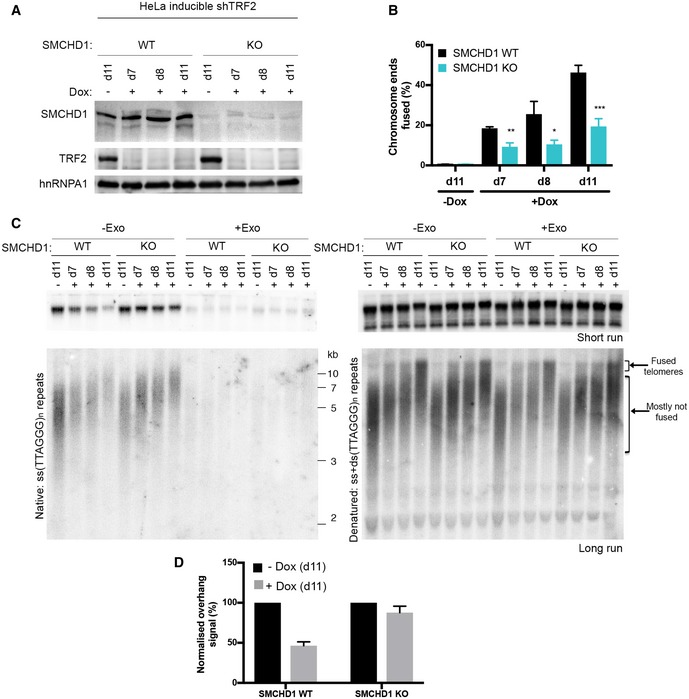
- Western Blot detection of SMCHD1, TRF2, and hnRNPA1 in SMCHD1 wild‐type or SMCHD1 knockout HeLa inducible shTRF2 cells treated with or without doxycycline for the indicated number of days (d7, d8, d11).
- Quantification of telomere fusions in SMCHD1 wild‐type or SMCHD1 knockout HeLa inducible shTRF2 cells treated with or without doxycycline for the indicated number of days (d7, d8, d11). Bars represent average number of fused chromosome ends. SDs were obtained from three independent experiments (> 1,900 telomeres counted/condition/experiment). ***P < 0.001, **P < 0.01; *P < 0.05; unpaired two‐tailed Student's t‐test.
- Terminal restriction fragment (TRF) analysis of telomeric DNA to detect 3′‐overhang processing of genomic DNA isolated from SMCHD1 wild‐type or SMCHD1 knockout HeLa inducible shTRF2 cells treated as in the experiment in (B). (Left) Radiolabeled (CCCTAA)n probe was hybridized with a short run (upper panel) and long run (lower panel) native DNA gel to detect the signal of the telomeric 3′ overhang. Samples used for the short and the long run were from the same digestion split into two. Exo I treatment (+Exo) was used as a control that single‐stranded telomeric signal was terminal. (Right) The total TTAGGG signal in the same lane was detected upon denaturation and hybridization with the same probe.
- Quantification of the telomeric overhang signal at d11 after doxycycline addition to SMCHD1 wild‐type and SMCHD1 knockout HeLa shTRF2 inducible cells. The bar graph represents the average overhang signal intensity from two biological replicates as percentage of the signal in the cells untreated with doxycycline with error bars representing the standard deviation (SD) of the sample.
Source data are available online for this figure.
Figure 3. SMCHD1 loss does not affect recombination frequency at telomeres in TRF2‐depleted cells.
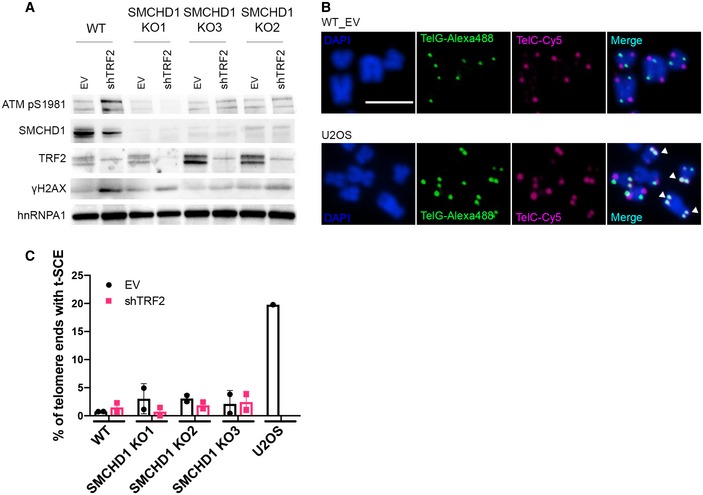
- Western blot detection of ATM pS1981, SMCHD1, TRF2, γH2AX, and hnRNPA1 in WT or SMCHD1 KO HeLa cells transfected with shTRF2 and EV control.
- Representative images for CO‐FISH on metaphase spreads in wild‐type (WT) HeLa and U2OS cells. The leading strand telomeres were labeled by PNA‐FISH with a (TTAGGG)3‐Alexa488 probe (green) and the lagging strand with a (CCCTAA)3‐Cy5 probe (pink), and the DNA was stained with DAPI. Examples of telomere sister chromatid exchange (t‐SCEs) events are marked with white arrows. Scale bar: 5 µm.
- Quantification of chromosome ends with t‐SCEs detected by CO‐FISH. Data represent means of two independent experiments ± SD (> 700 telomeres/condition/experiment) with individual average values indicated.
Source data are available online for this figure.
SMCHD1 promotes ATM activation and DDR at TRF2‐depleted telomeres
NHEJ of TRF2‐depleted telomeres is strictly dependent on activation of the DDR at uncapped telomeres (Denchi & de Lange, 2007). Therefore, we tested if SMCHD1 is required for checkpoint signaling. As expected, TRF2 depletion during 5 days led to induction of TIFs (Takai et al, 2003) in which at S1981 phosphorylated ATM (ATM pS1981), phosphorylated H2AX (γH2AX), and 53BP1 accumulate as foci at telomeres (Fig 4A–E). Strikingly, depletion of TRF2 in the two SMCHD1 knockout clones showed a strong reduction but not abolishment of all TIF markers indicating reduced DDR at TRF2‐depleted telomeres in the absence of SMCHD1. Similarly, we observed reduced TIFs in TRF2‐depleted cells that had been treated with SMCHD1 shRNAs (Fig EV3). Finally, we observed in Western blots that ATM pS1981 was reduced in TRF2‐depleted SMCHD1 knockout cells (Fig 1A) or upon shRNA‐mediated depletion of SMCHD1 (Fig EV3A). Altogether, these results indicate that SMCHD1 is required for efficient ATM activation and the subsequent DDR at TRF2‐depleted telomeres. Notably, however, SMCHD1 is not absolutely essential for the DDR. Thus, SMCHD1 loss has less severe consequences than MRE11 depletion, which completely abolished DDR and NHEJ at TRF2‐depleted telomeres (Fig EV3), reminiscent of results obtained in mouse embryonic fibroblasts (MEFs) in which Mre11 was deleted (Deng et al, 2009).
Figure 4. SMCHD1 promotes TIF formation and stimulates ATM signaling from TRF2‐depleted telomeres.
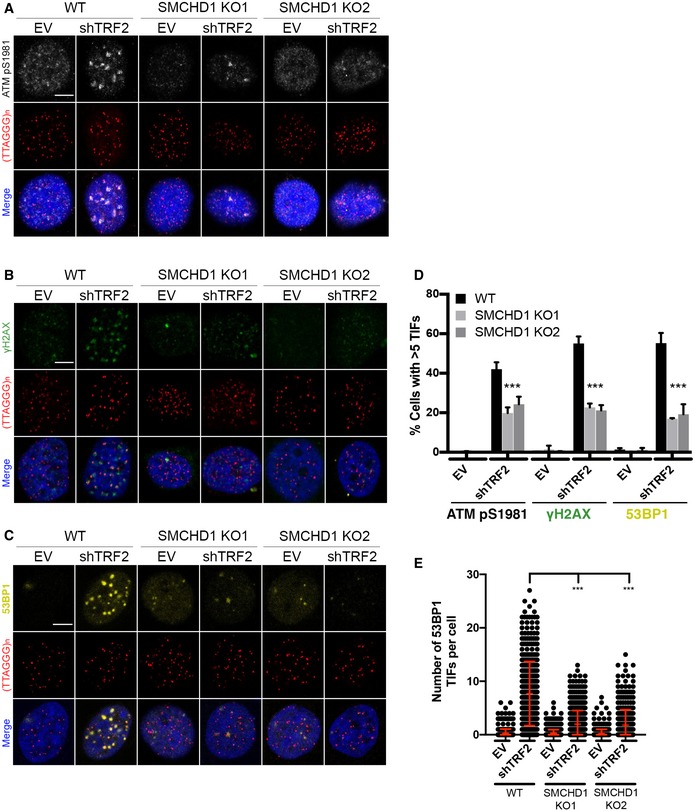
-
A–CRepresentative images for detection of ATM pS9181, γH2AX, and 53BP1 at telomeres in wild‐type (WT) and SMCHD1 knockout HeLa cells transfected with shTRF2 plasmid and empty vector (EV) control. Immunofluorescence (IF) for ATM pS1981 (gray), γH2AX (green), and 53BP1 (yellow) was combined with telomeric (CCCTAA)3‐FISH (red) and DAPI staining total DNA. Scale bars: 5 µm.
-
DQuantification of the number of cells containing > 5 telomere dysfunction‐induced foci (TIFs) detected as in (A‐C). Data represent the mean of four independent experiments ± SD (> 200 cells/condition/experiment) for ATM pS1981 and three independent experiments ± SD (> 200 cells/condition/experiment) for γH2AX and 53BP1. ***P < 0.001; unpaired two‐tailed Student's t‐test.
-
EQuantification of the number of 53BP1 TIFs per cell detected as in (C) from three independent experiments. Data represent individual values per cell with the bar representing mean ± SD. ***P < 0.001; unpaired two‐tailed Student's t‐test.
SMCHD1 is non‐epistatic with TERRA and MDC1
The above analyses indicated that SMCHD1 is required for efficient ATM signaling at uncapped telomeres. TRF2 depletion also leads to upregulation of the long non‐coding RNA TERRA which is thought to activate the nuclease activity of MRE11 promoting the interaction between MRE11 and its activating lysine demethylase LSD1 (Porro et al, 2014a,b). TERRA upregulation occurs independently of ATM, and principally, TERRA could act with SMCHD1 upstream of ATM or it could function in a separate pathway. We therefore tested if SMCHD1 is required for the induction of TERRA expression upon TRF2 depletion quantifying TERRA stemming from different chromosome ends by RT–qPCR (Feretzaki & Lingner, 2017) (Fig EV4A and B). The quantification of TERRA demonstrated that TERRA induction upon TRF2 depletion occurs in SMCHD1 knockout as well as in WT cells, indicating that SMCHD1 is not required for TERRA induction.
Figure EV4. Analysis of epistatic relationship between TERRA and MDC1 with SMCHD1.
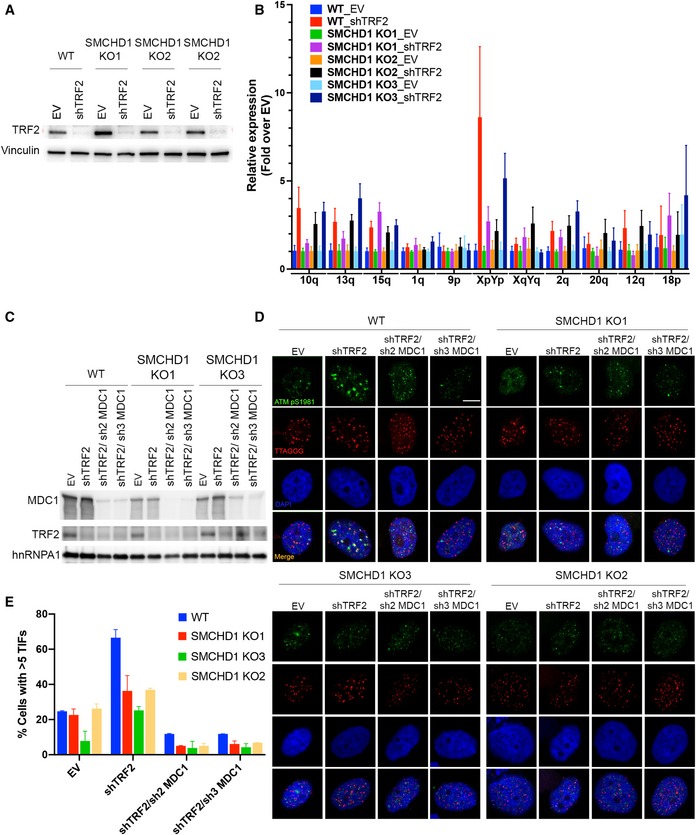
- Western Blot detection of TRF2 and vinculin in WT and SMCHD1 KO HeLa cells transfected with shTRF2 plasmid or EV control.
- TERRA quantification by RT–qPCR using primers specific for the indicated subtelomeric sequences (10q, 13q, 15q, 1 q, 9p, XpYp, XqYq, 2q, 20q, 12q, 18p). Bars represent average expression levels normalized to EV in each indicated cell line. SDs were obtained from three biological and two technical replicates.
- Western Blot detection of MDC1, TRF2, and hnRNPA1 in WT or SMCHD1 KO HeLa cells transfected with the indicated shRNA plasmids (shTRF2, shTRF2/sh2 MDC1, and shTRF2/sh3 MDC1) and EV control.
- Representative images for detection of ATM pS9181 at telomeres in wild‐type (WT) and SMCHD1 KO HeLa cells transfected with indicated shRNA plasmids and empty vector (EV) control. Immunofluorescence (IF) for ATM pS1981 (green) was combined with telomeric (CCCTAA)3‐FISH (red), and the DNA was stained with DAPI.
- Quantification of the number of cells containing > 5 telomere dysfunction‐induced foci (TIFs) detected as in (B). Data represent mean of two independent experiments ± SD (> 50 cells/condition/experiment).
Source data are available online for this figure.
Another factor implicated in TIF formation at uncapped telomeres stimulating chromosome end‐to‐end fusions is MDC1 (Dimitrova & de Lange, 2006). We determined the epistatic relationship of SMCHD1 and MDC1 for TIF formation upon TRF2 loss, depleting MDC1 with shRNAs in WT and SMCHD1 knockout cells (Fig EV4C–E). As expected, TIF formation was reduced upon deletion of SMCHD1 or depletion of MDC1. Significantly, TIF formation was further reduced upon MDC1 depletion in SMCHD1 knockout cells unraveling additive effects. Therefore, the two factors behaved in a non‐epistatic manner, suggesting that SMCHD1 does not directly cooperate with MDC1 for DNA damage signaling. The latter is also consistent with the notion that in contrast to SMCHD1 (Fig 2), MDC1 is not required for efficient removal of the 3′ overhang, indicating that it functions further downstream (Dimitrova & de Lange, 2006).
SMCHD1 is not required for genome‐wide ATM signaling
ATM activation at uncapped chromosome ends may involve distinct steps that do not occur upon induction of chromosome breaks elsewhere in the genome. We tested in time‐course experiments if SMCHD1 is required for ATM activation at chromosome breaks induced by γ‐irradiation and if SMCHD1 influences ATM foci disappearance that occurs upon DNA repair (Fig 5A–C). In addition, we followed accumulation and disappearance of DNA damage markers on Western blots (Figs 5 and EV5). CHK2 which acts downstream of ATM becomes phosphorylated at T68. CHK1 and RPA become phosphorylated by ATR. In the time‐course experiments, no striking difference was observed between WT and SMCHD1 knockout cells with regard to foci accumulation and disappearance and the overall accumulation and disappearance of DNA damage markers (Figs 5 and EV5). As acute RNA interference‐mediated depletion of SMCHD1 did not affect ATM levels (Fig EV5B), we infer that the slight reduction of ATM in the SMCHD1 KO (knockout) clones as compared to WT is due to clonal variation. We conclude that for the bulk of DNA damage sites that were induced by γ‐irradiation, SMCHD1 played no crucial role for DNA damage signaling and DNA repair.
Figure 5. SMCHD1 is not required for genome‐wide ATM signaling.
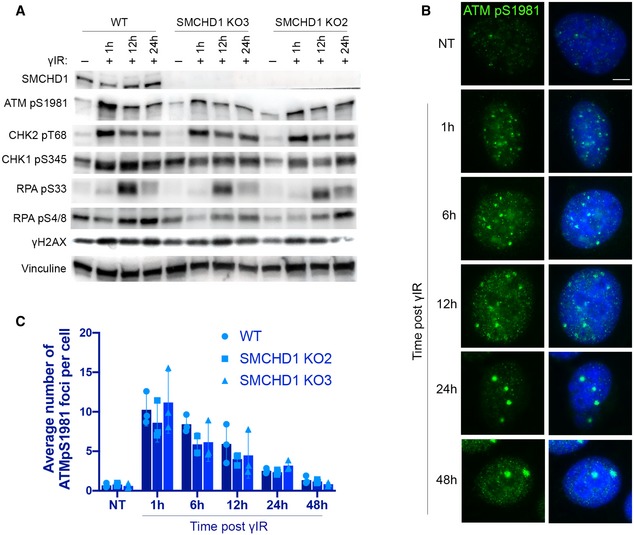
- Western blot detection of SMCHD1, ATMpS1981, CHK2 pT68, CHK1 pS345, RPA pS33, RPA pS4/8, γH2AX, and vinculin in wild‐type or SMCHD1 KO HeLa cells γ‐irradiated with 3 Gy dose and harvested at the indicated times (1, 12, 24 h) after the treatment.
- Representative images for immunofluorescence (IF) detection of ATMpS1981 in wild‐type HeLa cells γ‐irradiated with a dose of 3 Gy and harvested after the indicated times. Scale bar: 5 µm.
- Quantification of the average number of ATMpS1981 foci per cell detected as in (B) in wild‐type and SMCHD1 KO γ‐irradiated HeLa cells. Data represent mean of three independent experiments ± SD (> 200 cells/condition/experiment) with individual average values indicated.
Source data are available online for this figure.
Figure EV5. SMCHD1 loss does not affect genome‐wide DNA damage signaling upon γ‐irradiation, and depletion of alt‐NHEJ repair factor POLθ does not affect repair efficiency after removal of TRF2 and TPP1.
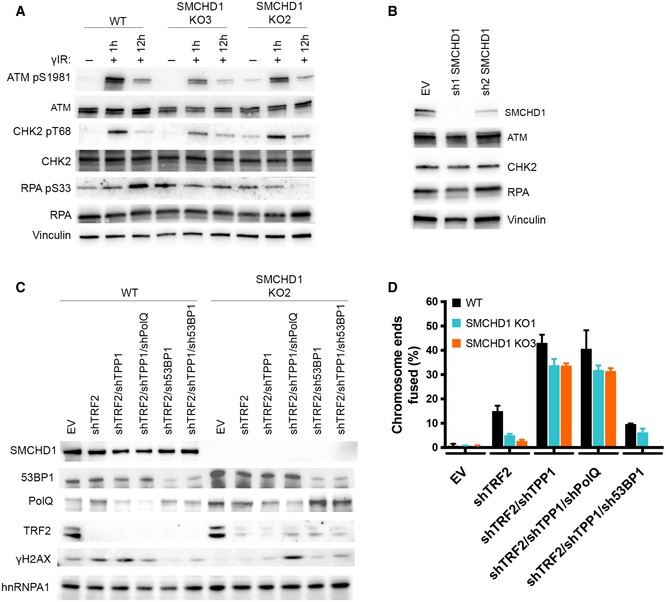
- Western blot detection of ATMpS1981, total ATM, CHK2 pT68, total CHK2, RPA pS33, total RPA, and vinculin in wild‐type or SMCHD1 KO HeLa cells y‐irradiated with a dose of 3Gy and harvested at the indicated times (1, 12 h) after the treatment.
- Western blot detection of SMCHD1, total ATM, total CHK2, total RPA, and vinculin in HeLa cells transfected with the indicated shRNA plasmids (sh1 SMCHD1 and sh2 SMCHD1) and empty vector (EV) control.
- Western blot detection of SMCHD1, 53BP1, POLQ, TRF2, γH2AX, and hnRNPA1 in HeLa cells transfected with the indicated shRNA plasmids (shTRF2, shTRF2, shTRF2/shTPP1, shTRF2/shTPP1/shPOLQ, shTRF2/sh53BP1, and shTRF2/shTPP1/sh53BP1) and empty vector (EV) control.
- Quantification of telomere fusions from experiment shown in (A). Bars represent average number of fused chromosome ends. SDs were obtained from two independent experiments (> 2,000 telomeres counted/condition/experiment).
Source data are available online for this figure.
SMCHD1 activates the DDR at TRF2‐depleted telomeres to promote end joining
The above results unraveled requirements of SMCHD1 for ATM activation and NHEJ of TRF2‐depleted telomeres, but they could not distinguish if the effects on NHEJ were solely due to its involvement in checkpoint activation or if SMCHD1 also played direct roles in the DNA processing or end ligation reactions. For addressing this question, we were inspired by a previous landmark paper (Denchi & de Lange, 2007), which discovered the requirement of ATM for NHEJ of TRF2‐depleted telomeres and which demonstrated that ATM function could be substituted by activated ATR. To activate ATR at telomeres, we depleted TPP1 with shRNAs (Fig 6A), which leads to removal of POT1 from the telomeric 3′ overhang (Frescas & de Lange, 2014). This in turn leads to RPA binding to the single‐stranded 3′ overhang, subsequent ATR/ATRIP recruitment, and checkpoint signaling at chromosome ends (Zou & Elledge, 2003). Significantly, the shRNA‐mediated depletion of TPP1 reinstated efficient chromosome end‐to‐end fusions in SMCHD1 knockout cells that had been depleted for TRF2 (Fig 6B). Concomitant inhibition of the ATR kinase with an inhibitor (VE‐821) (Reaper et al, 2011) again prevented efficient end fusions (Fig 6C), indicating that ATR signaling upon TPP1‐depletion was responsible for triggering chromosome end‐to‐end fusions in the absence of SMCHD1.
Figure 6. ATR signaling induction by TPP1 removal rescues the telomere fusion defect in SMCHD1 knockout cells.
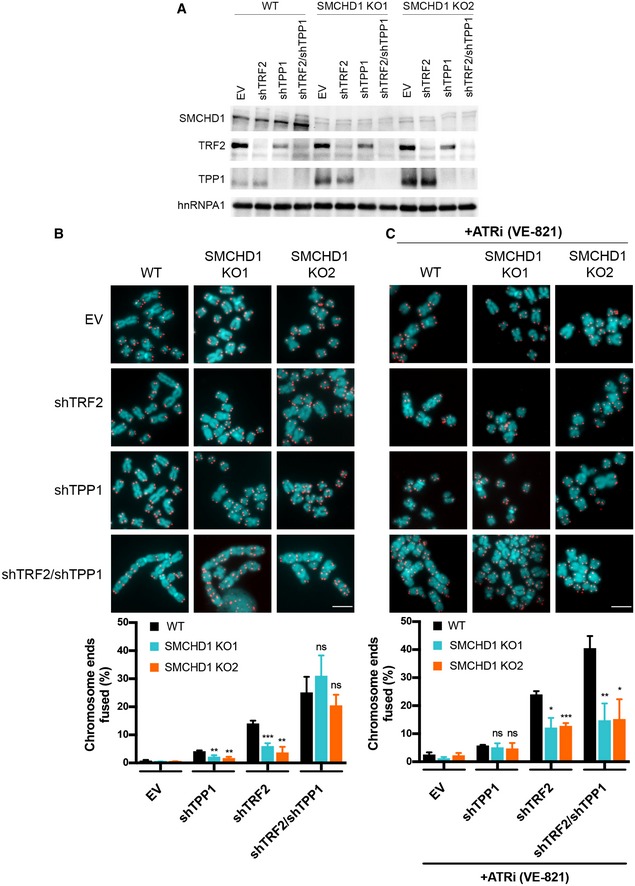
- Western Blot detection of SMCHD1, TRF2, TPP1 and hnRNPA1 in wild‐type or SMCHD1 knockout HeLa cells transfected with the indicated shRNAs (shTRF2, shTPP1, shTRF2/shTPP1) or EV control.
- Representative metaphase spreads (Scale bar: 5 µm) from HeLa cells transfected with indicated shRNAs or EV control and quantification of telomere fusions. Bars represent average number of fused chromosome ends. SDs were obtained from three independent experiments (> 2,500 telomeres counted/condition/experiment). ***P < 0.001; **P < 0.01 unpaired two‐tailed Student's t‐test.
- Representative metaphase spreads (Scale bar: 5 µm) from HeLa cells transfected with indicated shRNAs or EV control treated for 4 days with ATRi (VE‐821) and quantification of telomere fusions. Bars represent average number of fused chromosome ends. SDs were obtained from three independent experiments (> 2,000 telomeres counted/condition/experiment). ***P < 0.001; **P < 0.01; *P < 0.05 unpaired two‐tailed Student's t‐test.
Source data are available online for this figure.
Finally, since in mouse cells removal of Tpp1 and Pot1a/b promoted microhomology‐mediated end joining (MMEJ) of chromosome ends (Rai et al, 2010), we tested if depletion of DNA polymerase ( (POLQ) which is required for MMEJ contributes to the telomere fusions in the human SMCHD1 KO cells that had been depleted for TRF2 and TPP1 (Fig EV5). However, fusions were not reduced upon POLQ depletion whereas they were reduced upon depletion of 53BP1 (Fig EV5), which had been implicated in classical NHEJ of TRF2‐depleted telomeres before (Dimitrova et al, 2008). Therefore, MMEJ did not contribute to telomere fusions in our experimental setting.
Discussion
In this paper, we demonstrate that loss of SMCHD1 abolishes efficient DNA damage signaling and NHEJ at telomeres that are depleted of TRF2. The defects of SMCHD1 knockout cells in signaling and repair can be ascribed to its roles in DDR activation (Fig 7). Indeed, activation of ATR upon depletion of TPP1 was sufficient to suppress the defects of the SMCHD1 knockout for NHEJ at TRF2‐depleted telomeres, suggesting that SMCHD1 is required for checkpoint signaling but it is not directly involved in NHEJ. Since SMCHD1 loss in TRF2‐depleted cells strongly attenuated ATM activation, our data indicate that SMCHD1 functions in the DDR cascade upstream of ATM phosphorylation (Fig 7). During canonical ATM‐dependent DDR at DNA double‐strand breaks, the MRN complex binds and senses DNA ends recruiting and activating ATM, which then initiates the DNA damage signaling cascade (Paull, 2015). At telomeres, the MRN complex is present even when telomeres are intact (Zhu et al, 2000). Indeed, NBS1 of the MRN complex interacts directly with TRF2, but in this context, ATM is not activated (Rai et al, 2017). TRF2 inhibits ATM signaling by several mechanisms involving its TRFH and hinge domains (Okamoto et al, 2013). The TRFH domain of TRF2 promotes formation of t‐loops, which prevents exposure of the chromosome ends to the MRN complex not allowing ATM recruitment or activation (Doksani et al, 2013; Van Ly et al, 2018). In addition, the TRFH domain of TRF2 interacts at intact telomeres with a non‐phosphorylated form of NBS1 preventing ATM activation (Rai et al, 2017). Second, through a portion of the hinge domain of TRF2 referred to as iDDR, TRF2 can sever the DDR at the level of the E3 ubiquitin ligase RNF168 which is required for 53BP1 localization to telomeres (Okamoto et al, 2013). Upon TRF2 removal, NBS1 is phosphorylated by CDK2 at Ser432 (Rai et al, 2017). The t‐loops will unwind and MRE11/RAD50 may associate with the uncapped telomeres possibly at their DNA ends in a similar manner as it does with DNA double‐strand breaks (Syed & Tainer, 2018). Phosphorylated NBS1 may bind to uncapped telomeres via MRE11 enabling ATM recruitment and activation (Rai et al, 2017).
Figure 7. Schematic model of the DNA damage response at uncapped telomeres in SMCHD1 wild‐type and knockout cells.
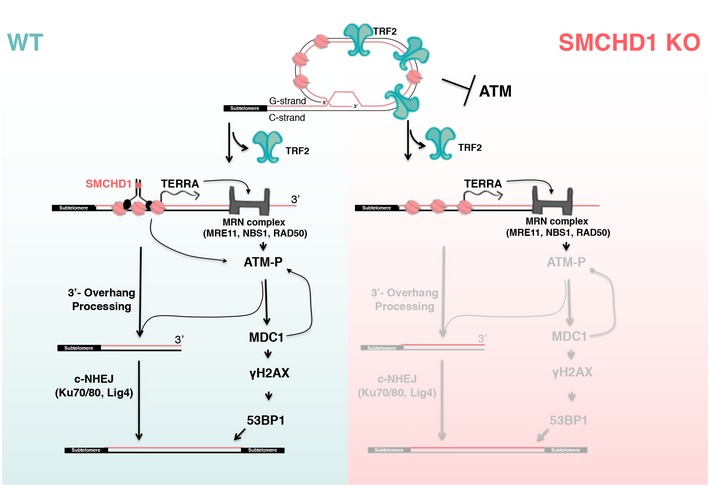
Loss of TRF2 leads to t‐loop unwinding. In wild‐type cells, SMCHD1 may remodel the telomeric chromatin which promotes ATM activation by the MRN complex (arrows). ATM activation is required for NHEJ at TRF2‐depleted telomeres. In SMCHD1 knockout cells, ATM activation and DNA damage signaling are attenuated resulting in inefficient 3′‐overhang processing and impaired telomere end‐to‐end fusions. SMCHD1 loss and lack of ATM activation can be compensated for by ATR (not depicted). Only the subset of events and factors are depicted which are directly relevant to this paper.
Our data implicate SMCHD1 at the onset in the damage signaling cascade. We infer that SMCHD1 is not required for t‐loop unwinding as its function at uncapped telomeres could be reinstated upon ATR activation (Fig 6) and checkpoint kinases have not been implicated in t‐loop unwinding (Van Ly et al, 2018). SMCHD1 promotes ATM activation as in its absence ATM is not efficiently phosphorylated and ATM pS1981 does not accumulate substantially in TIFs (Fig 7). SMCHD1 contains an N‐terminal ATPase domain and a C‐terminal hinge domain mediating homodimerization (Brideau et al, 2015). We speculate that SMCHD1 may promote ATP‐dependent chromatin remodeling at telomeres upon t‐loop unfolding. A role in chromatin architecture and remodeling would be analogous to the functioning of other SMC proteins (van Ruiten & Rowland, 2018) and consistent with the described roles of SMCHD1 in modulating chromosome structure (Nozawa et al, 2013; Jansz et al, 2018; Wang et al, 2018; Gdula et al, 2019). For example, SMCHD1 may modulate the telomere structure at TRF2‐depleted telomeres at the molecular level to expose telomeric DNA ends and favor MRE11 remodeling, thus assisting ATM recruitment and activation (Fig 7). This function of SMCHD1 appears not to involve TERRA induction which occurs upon TRF2 depletion independently of SMCHD1 and ATM. Our epistatic analyses with MDC1 also suggest that SMCHD1 and MDC1 act at separate steps in DNA damage signaling.
At the inactive X chromosome in females, SMCHD1 has been implicated in chromosome compaction linking H3K9me3‐rich with H3K27me3‐rich domains (Nozawa et al, 2013). At telomeres, however, we did not detect notable effects of SMCHD1 depletion on telomere compaction (data not shown). Thus, although H3K9me3 may be important for SMCHD1 binding to uncapped telomeres, SMCHD1 loss does not alter chromatin compaction at telomeres at detectable levels as seen at the inactive X chromosome.
ATM activation is not only required for NHEJ of uncapped telomeres but also for NHEJ of a subset of DNA breaks which occur in heterochromatic regions of the genome (Goodarzi et al, 2008). It has been proposed that ATM signaling at DNA breaks temporarily perturbs heterochromatin to promote processing of otherwise inflexible chromatin (Goodarzi et al, 2008). We tested if SMCHD1 promotes also ATM activation at DNA breaks induced by γ‐irradiation elsewhere in the genome. Our data indicate that SMCHD1 plays no essential role for damage signaling by ATM and DNA repair for the majority of damage sites that were induced by γ‐irradiation. Nevertheless, we do not exclude that SMCHD1 participates in damage signaling at other specialized regions of the genome which possibly adopt a heterochromatic structure similar to telomeres. Consistent with this notion are previous observations, which demonstrated recruitment of SMCHD1 to laser‐beam induced sites of DNA damage (Coker & Brockdorff, 2014; Tang et al, 2014). However, our data do not support a general role of SMCHD1 in ATM activation and DNA repair. They unravel for the first time specific roles of SMCHD1 at uncapped telomeres to trigger damage signaling activation, and we determine that its requirement for NHEJ at uncapped telomeres is solely explained by its contribution to the DDR.
ATM activation upon telomere shortening and TRF2 depletion contributes to the induction of cell cycle arrest and cellular senescence (d'Adda di Fagagna et al, 2003). Our results implicate SMCHD1 in damage signaling from unprotected telomeres. Mutations in SMCHD1 have been linked to several diseases including facioscapulohumeral muscular dystrophy (FSHD) and Bosmia arhinia (Jansz et al, 2017). It will be important to determine whether disease mutations also impact on DNA damage signaling from telomeres and to what extent this may affect disease pathology.
Materials and Methods
Cell culture
HeLa cells harboring 11‐kb‐long telomeres as well as the HeLa cells containing an inducible shTRF2 knockdown cassette cell lines were described previously (Grolimund et al, 2013). They were used for all transient transfection experiments and to derive SMCHD1 knockout clones. Cells were maintained at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% FCS and penicillin/streptomycin. For the experiment in Fig 5, WT and SMCHD1 knockout cells were seeded at 0.5 × 106 cells per dish. They were irradiated 14 h post‐seeding with 3 grays of total γ‐irradiation dosage and harvested after the indicated times for Western blot and indirect immunofluorescence analysis.
Antibodies
The following primary antibodies were used: TRF2 (#05‐521, Millipore, mouse, dilution 1:1,000, used for Western blots (WB)), γH2AX (Millipore, #05‐636, mouse, dilution 1:1,000, used for WB and IF), hnRNPA1 (4B10, #sc‐32301, Santa Cruz Biotechnology, mouse, dilution 1:3,000, used for WB), vinculin (#ab129002, Abcam, rabbit monoclonal [EPR8185], dilution 1:3,000, used for WB), 53BP1 (#NB100‐304, Novus Biologicals, rabbit, dilution 1:2,000, used for IF), phospho‐ATM‐Ser1981 (#ab81292, Abcam, rabbit, dilution 1:1,000, used for WB and IF), ATM (#ab32420, Abcam, rabbit monoclonal [Y170], dilution 1:1,000, used for WB), SMCHD1 (#A302‐871A, Bethyl Laboratories, N‐terminal, rabbit, dilution 1:2,000, used for WB and ChIP), SMCHD1 (#A302‐872A, Bethyl Laboratories, C‐terminal, rabbit, dilution 1:2,000, used for WB and ChIP), POLQ (a generous gift from Dr JS Hoffmann, mouse, dilution 1:1,000), 53BP1 (A300‐272A. Bethyl Laboratories, dilution 1:1,000, used for WB in figure EV9), TPP1 (#H00065057‐M02, Abnova, rabbit, dilution 1:1,000, used for WB), MRE11 (#NB100‐142, Novus Biologicals, rabbit, dilution 1:2,000, used for WB), CHK1 pS345 (#2348S, Cell Signaling, dilution 1:1,000, used for WB), CHK1 (#sc‐8408, Santa Cruz Biotechnologies, mouse, dilution 1:2,000, used for WB), CHK2 pT‐68 (#2661, Cell Signaling, rabbit, dilution 1:1,000, used for WB), CHK2 (#05‐649, Millipore, mouse, dilution 1:2,000, used for WB), RPA pS33 (#A300‐246A, Bethyl Laboratories, rabbit, dilution 1:1,000, used for WB), RPA pS4/8 (#A300‐245A, Bethyl Laboratories, rabbit, dilution 1:1,000, used for WB), RPA (#A300‐244A, Bethyl Laboratories, rabbit, dilution 1:1,000, used for WB), MDC1 (#ab11171, Abcam, rabbit, dilution 1:1,000, used for WB), and normal rabbit IgG (#sc‐2027, rabbit, used for ChIP).
Plasmids
Plasmids containing shRNAs used in this study were prepared by restriction cloning of annealed oligonucleotides into pSUPERpuro or pSUPERblast plasmid backbones (Oligoengine™). The target sequences of the shRNAs were as follows: MRE11 5′‐TGAGAACTCTTGGTTTAAC‐3′ cloned into pSUPERblast plasmid (Porro et al, 2014a); TRF2 5′‐GCGCATGACAATAAGCAGA‐3′ cloned into pSUPERblast and pSUPERpuro plasmid (Porro et al, 2014b); sh1_SMCHD1 5′‐ATTGGATAGCGGGTGATATTA‐3′ cloned into pSUPERpuro plasmid; sh2_SMCHD1 5′‐TTATTCGAGTGCAACTAATTT‐3′ cloned into pSUPERpuro plasmid; shTPP1 5′‐GACTTAGATGTTCAGAAAA‐3′ cloned into pSUPERblast plasmid (Abreu et al 2010); sh2_MDC1 5′‐AGAGGGACAATGATACAAA‐3′ cloned into pSUPERblast plasmid; sh3_MDC1 5′‐GTCTCCCAGAAGACAGTGA‐3′ cloned into pSUPERblast plasmid (Stewart et al, 2003); sh3 53BP1 GGGAGAAGAAGAGTTTGAT cloned into pSUPERpuro plasmid; and shPOLQ 5′‐GGGCCTCTTTAGATATAAA‐3′ cloned into pSuperPURO plasmid. The pSpCas9(BB)‐2A‐puro plasmid (a generous gift from Dr. Feng Zhang, Addgene plasmid #62988) was used for CRISPR/Cas9‐mediated knockout of SMCHD1.
Transfection protocols
For depletion experiments, HeLa cells were transfected in 6‐well plates at 60‐80% confluency using Lipofectamine 2000 according to the manufacturer's protocol (Thermo Fisher, #11668019). Puromycin (conc. 1 μg/ml, #ant‐pr‐1, Invivogen) and blasticidin (conc. 5 μg/ml, #ant‐bl‐1, Invivogen) were added to the media 20–24 h after transfection, and the cells were expanded on 10‐cm dishes. Selection with the antibiotics was maintained for 3–5 days. Empty pSUPERpuro and pSUPERblast plasmids were used as control in all the experiments. For the experiment in Fig 4, ATRi (VE‐821, SelleckChem, #S8007) was added to the cells 24 h after addition of the selection antibiotics at 10 μM concentration and the cells were maintained with the inhibitor for 4 days.
Immunoblotting
After harvesting, cells were counted using CASY Cell Counter and Analyzer, and cell pellets with equal cell numbers were resuspended in 2× Laemmli buffer (20% glycerol, 4% sodium dodecyl sulfate, 10 mM Tris–Cl pH 6.8, 200 mM dithiothreitol, 0.05% bromophenol blue) at final concentration of 10,000 cells/μl and boiled for 5 min at 95°C. Protein extracts were fractionated on 4–20% Mini‐PROTEAN® TGX™ Precast Protein Gels (Bio‐Rad), transferred to a nitrocellulose blotting membrane (Amersham™ Protran™, 0.2 μm NC, GE Healthcare Life Sciences, #10600001), blocked in 3% BSA/1xPBS/0.1% Tween‐20 for 30 min, and incubated with primary antibody overnight at 4°C. Membranes were then washed 3 × 5 min in 1xPBS/0.1% Tween‐20, incubated with anti‐mouse or anti‐rabbit HRP‐conjugated secondary antibody for 30 min (anti‐mouse IgG HRP conjugate Promega #W402B, anti‐rabbit IgG HRP conjugate Promega #W4011, 1:3,000), and chemiluminescence was detected using WesternBright ECL Spray (Advansta, #K‐12049‐D50). Detection of TPP1 was performed using a renaturation protocol as described (Loayza & De Lange, 2003).
Telomere restriction fragment length analysis for detection of single‐stranded and double‐stranded telomeric DNA
Genomic DNA was isolated using the Wizard® Genomic DNA Purification Kit (Promega, #A1120). Isolated DNA (5 μg) was subjected to digestion with 40 U ExoI (New England Biolabs, #M0293S) as control or non‐digested and incubated for 8 h at 37°C in CutSmart® Buffer in a final volume of 80 μl. The samples were then heated at 80°C for 20 min to inactivate the ExoI enzyme. Following the inactivation, 20 μl of digestion mix containing 125U HinfI (New England Biolabs, #R0155M) and 25U RsaI (New England Biolabs, #R0167L) was added to all the samples (Exo+ and Exo−), and the digestion mix was incubated overnight at 37°C. Digested DNA was loaded on a 1% agarose gel (35 μl of the digestion mix was loaded for the short run and 55 μl for the long run in Fig 2C) and separated by regular gel electrophoresis in 1× TBE at 3 V/cm for 1 h (short run) and at 1.5 V/cm for 16 h (long run). Gels were dried for 3 h at 50°C, prehybridized at 50°C in Church buffer (1% BSA, 1 mM EDTA, 0.5 M phosphate buffer pH 7.5, 7% SDS), and hybridized at 50°C overnight to a [32P]‐labeled (CCCTAA)n probe (Grolimund et al, 2013) for detection of single‐stranded (ss) telomeric DNA. After hybridization, the gel was rinsed in 4× SSC and followed by successive 1‐h washes at 50°C in 4 × SSC, 4 × SSC/0.5% SDS, and 2 × SSC/0.5% SDS and exposed to a sensitive phosphoimager screen overnight. After the image was acquired, the gel was denatured with 0.8 M NaOH and 150 mM NaCl, neutralized with 1.5 M NaCl, 0.5 M Tris–HCl pH 7.0, prehybridized in Church buffer at 50° for 1 h, and incubated with the same probe overnight at 50°C. The gel was again washed and exposed as above, and the image was acquired using Amersham™ Typhoon™ Biomolecular Imager (GE Healthcare). The images were quantified using Aida Image Analysis software. The single‐stranded DNA signal was divided by the total denatured DNA signal in each lane and further normalized to −Dox samples.
CRISPR/Cas9 gene editing
The CRISPR/Cas9 gene editing system was used to create SMCHD1 knockout cell lines. To target the SMCHD1 gene locus (NC_000018.10; gene ID 23347), a region of 200 bp encompassing the ATG in Exon 1 was submitted to the Optimal CRISPR design tool (http://crispr.mit.edu). Three gRNAs with scores higher than 93 were chosen for further experiments (gRNA 1: 5′‐CTTGTTTGATCGGCGCGAAA‐3′, gRNA2: 5′‐GGGGAGCGCTCGGACTACGC‐3′, gRNA 3: 5′‐GCCGTCCGCCGCTGCCATAT‐3′). Complementary oligonucleotides harboring the guide RNA sequence and BpiI compatible overhangs were synthetized by Microsynth AG. The oligonucleotides were annealed and ligated into a BpiI (Thermo Fisher Scientific, ER1011)‐digested and BpiI‐dephosphorylated pSpCas9(BB)‐2A‐puro vector (Addgene, 62988). The resulting constructs were transfected into HeLa cells using Lipofectamine™ 2000 (Thermo Fisher Scientific, #11668019). Transfected cells were selected with 1 μg/ml of puromycin for 4 days. Single‐cell clones were obtained by limiting dilution and were screened for the absence of SMCHD1 by Western blotting using the N‐terminal anti‐SMCHD1 antibody. To verify the gene editing in positive clones, the PCR products obtained with two primers (AV48_SMCHD1_gPCR_F: 5′‐AGGAGCGCGTTTGAATCGG‐3′, AV47_SMCHD1_gPCR_R 5′‐CTTCGCGTACCTGACACACAC‐3′) were TOPO‐cloned (Thermo Fisher, #450071) and sent for sequencing.
Telomeric PNA‐FISH on metaphase spreads
On the day of harvesting, cells were treated with 0.1 μg/ml demecolcine (Sigma‐Aldrich Chemie GmbH #D7385‐10MG) for 2 h, cells were collected, resuspended in hypotonic solution (0.056 M KCl), and swollen for 7 min. Swollen cells were fixed in methanol:acetic acid (3:1) and stored overnight at 4°C. The next day, cell suspensions were dropped onto slides to prepare metaphase spreads, incubated 1 min at 70°C in a wet chamber, and dried for 16–24 h before FISH. FISH staining of human telomeric DNA was performed as described (Vancevska et al, 2017). Slides were rehydrated in 1× PBS for 5 min, treated with 4% formaldehyde in PBS for 5 min, washed 3× with 1xPBS, and dehydrated with increasing amounts of ethanol (70, 95, 100%). Dehydrated slides were then placed on coverslips containing 70 μl hybridization mix (10 mM Tris–HCl, 2% blocking reagent (Roche, #11096176001), 70% formamide, and 0.1 μM Cy3‐labeled (CCCTAA)3 PNA probe (PNA Bio, #F1002)) and denatured at 80°C for 3 min in a hybridization oven. Subsequently, the hybridization was allowed to proceed for 3 h in a light‐protected humified chamber at 25°C. The coverslip was then removed from the slide, washed twice for 15 min in buffer containing 70% formamide and 10 mM Tris–HCl pH 7.4 and three times for 15 min with 0.1 M Tris–HCl pH 7.2, 0.15 M NaCl, 0.08% Tween‐20. For DNA staining, DAPI was added to 1 μg/ml in the second wash. After the washes, slides were stored at 4°C in a dark place until imaging.
Indirect immunofluorescence and telomeric FISH (IF‐FISH)
Indirect immunofluorescence detection of human ATM pS981, 53BP1, and γH2AX followed by telomeric FISH staining was performed as described (Vancevska et al, 2017). For detection of ATM pS1981 before cross‐linking, cells were fractionated with an ice‐cold pre‐extraction buffer containing 0.5% Triton X‐100, 20 mM HEPES‐OH pH 7.5, 50 mM NaCl, 3 mM MgCl2, and 300 mM sucrose for 7 min. Subsequently, cells were washed with 1xPBS and the same protocol was applied as for the other stainings.
Chromosome orientation FISH (CO‐FISH)
CO‐FISH was performed as previously described (Celli et al, 2006). 10 μM BrdU was added to the cells 12–16 h before harvesting. On the day of harvesting, cells were treated with 0.1 μg/ml colcemid (Roche, 10295892001) for 2 h, and cells were collected, resuspended in hypotonic solution (0.075 M KCl), and swollen for 15 min at 37°C. Swollen cells were fixed in methanol:acetic acid (3:1) and stored overnight at 4°C. The next day, cell suspensions were dropped onto slides to prepare metaphase spreads and dried for 16–24 h before CO‐FISH. Slides were rehydrated in 1× PBS for 5 min, treated with 0.5 mg/ml RNase A (Qiagen, 1007885) in PBS for 30 min at 37°C, incubated with 10 μg/ml Hoechst 33258 (Sigma) in 2× SSC for 15 min at room temperature, and exposed to 365 nm UV light (Stratalinker 1800 UV irradiator) for 1 h in 2× SSC at room temperature. The slides were then digested twice with 10 U/μl of Exonuclease III (Promega, M1811) for 1 h, washed with 1xPBS, and then dehydrated with an ethanol series of 70, 95, and 100% for 5 min each. Dehydrated slides were then sequentially hybridized with a G‐rich and C‐rich telomeric PNA probes. The slides were placed on coverslips containing 70 μl hybridization mix (10 mM Tris–HCl pH 7.5, 2% blocking reagent (Roche, #11096176001), 70% formamide, and 0.1 μM Cy5‐labeled (CCCTAA)3 PNA probe (PNA Bio, #F1003)) and denatured at 80°C for 3 min in a hybridization oven. Subsequently, the hybridization was allowed to proceed for 2 h in a light‐protected humified chamber at 25°C. The coverslip was then removed from the slide, rinsed in buffer containing 70% formamide and 10 mM Tris–HCl pH 7.4, and hybridized with 0.1 μM Alexa488‐labeled (TTAGGG) PNA probe (PNA Bio, #F1008) for additional 2 h. The slides were subsequently processed in the same way as for the FISH protocol.
Chromatin immuno precipitation (ChIP)
ChIP protocol for SMCHD1 and γH2AX was performed as described previously (Grolimund et al, 2013). Briefly, 10 million cells per condition were harvested and washed with 1× PBS pH 7.4. The cell pellet was then cross‐linked in 1 ml 1% formaldehyde in 1× PBS pH 7.4 for 15 min at RT. Glycine pH 2.5 was added to 125 mM to quench the reaction and incubated for 5 min, and cells were then washed 3× with 1× PBS pH 7.4. Cells were subsequently incubated 5 min in 1 ml lysis buffer (1% SDS, 10 mM EDTA pH 8.0, 50 mM Tris–Cl pH 8.0, EDTA‐free protease inhibitor complex (Roche, #11836170001)), centrifuged for 5 min at 2,000 g and the chromatin‐enriched pellet was again resuspended in 500 μl lysis buffer and subjected to sonication for 30 min (30‐s ON, 30‐s OFF, total sonication time 15 min) using Bioruptor® Twin Diogenode sonicatior (#UCD‐400). The sonicated lysate was centrifugated at 20,000 g for 15 min at 4°C. Per IP 100 μl of the cleared lysate was diluted with nine volumes of IP buffer (1.2 mM EDTA pH 8.0, 1.1% Triton X‐100, 16.7 mM Tris pH 8.0, 300 mM NaCl) and incubated with 5 μg of the corresponding antibody (normal rabbit IgG, SMCHD1 or γH2AX) and 30 μl of preblocked Protein G Sepharose 4 Fast Flow 50% bead slurry (GE Healthcare, #17‐0618‐01) overnight at 4°C. The beads were then washed once with wash buffer 1 (0.1% SDS, 1% Triton X‐100, 2 mM EDTA pH 8.0, 20 mM Tris pH 8.0, 300 mM NaCl), wash buffer 2 (0.1% SDS, 1% Triton X‐100, 2 mM EDTA pH 8.0, 20 mM Tris pH 8.0, 500 mM NaCl), wash buffer 3 (500 mM LiCl, 1% NP‐40, 1% Na‐deoxycholate, 1 mM EDTA, 10 mM Tris pH 8.0), and twice with wash buffer 4 (1 mM EDTA, 10 mM Tris pH 8.0) at room temperature for 5 min. Elution and cross‐link reversal were performed at 65°C overnight in cross‐link reversal buffer (1% SDS, 0.1 M NaHCO3, 0.5 mM EDTA pH 8.0, 20 mM Tris–Cl pH 8.0, 10 μg DNase‐free RNase (Roche #11119915001)). For DNA extraction, the QIAquick PCR Purification Kit (Qiagen, #28106) was used. Telomeric and Alu‐repeat DNA were detected successively using the conditions described before. After the exposure, the image was acquired using Fujifilm Fluorescent Image Analyzer FLA‐3000 and the image quantification was done using AIDA Image Analyzer software v 4.06.
Cell cycle analysis
For analysis of the cell cycle distribution of SMCHD1 KO cells, propidium iodide‐stained cells were analyzed by fluorescence‐activated cell sorting (FACS). For each sample, 2 × 106 cells were pelleted, washed in 1× PBS, fixed by dropwise addition of 1 ml of ice‐cold 70% ethanol, and incubated at least 24 h. Following the fixation, cells were resuspended in 250 μl 1×PBS containing 0.2 μg/ml of RNase A and incubated for 15 min at 37°C. Cells were then stained by addition of 250 μl of 1× PBS containing 80 μg/ml propidium iodide and incubated at 4°C for 10 min. Subsequently, cells were passed through a strainer and analyzed by FACS on Accuri C6 (BD Biosciences). The percentage of cells in each phase of the cell cycle was determined using the Watson Pragmatic computational model in FlowJo software (TreeStar).
RT–qPCR for measuring TERRA transcript levels
TERRA was measured as previously described (Feretzaki & Lingner, 2017) with slight modifications. Total RNA was extracted with NucleoSpin® RNA isolation kit (Macherey‐Nagel, #740955) from 2 × 106 cells following the manufacturer's protocol with 3 DNase treatment steps. cDNA from three biological replicates was synthetized using Invitrogen's SuperScript III Reverse Transcriptase (#18080044) from 2 μg of total RNA. Reaction mixes in total volume of 20 μl were prepared as follows: 2 μg of total RNA, 0.5 mM dNTP mix, 150 ng random primers (Thermo Fisher #48190011), 250 ng oligo (dT)15 primer (Promega, #C1101), 1× First‐Strand Buffer, 5 mM DTT, 20 U SUPERase IN (Ambion #AM2696), and 200 U SuperScript III RT (200 U/μl) or H2O for no RT control. The cDNA was then diluted to 40 μl and stored at −20°C. Quantitative PCR (qPCR) was performed on an Applied Biosystems 7900HT Fast Real‐Time System using Power SYBR Green PCR Master Mix (Applied Biosystems #4368708) in a 384‐well reaction plate (Applied Biosystems MicroAmp Optical 384‐Well Reaction Plate with Barcode #4309849). Each sample was prepared in three biological and two technical replicates. The master mix for each reaction is prepared as follows: 2 μl diluted cDNA, 5 pmol of forward primer, 5 pmol reverse primer, 1× Power SYBR Green PCR Master Mix, and H2O to a total volume of 10 μl. qPCR data were analyzed using the relative ΔCt quantification method, and GAPDH was used for normalization.
Author contributions
JL, AV, and VP designed research, AV, VP, MF, and WA carried out experiments, SJB provided expertise and resources for conducting experiments during the revision phase, and JL and AV wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 6
Source Data for Expanded View
Acknowledgements
We thank Larissa Grolimund for initiating the work on SMCHD1, and Jana Majerska and Galina Glousker from the Lingner Lab for help in establishing the protocol to measure 3′ overhangs. We also thank Paulina Marzec, Timothy Lippert, and Phil Ruis from the Boulton laboratory for helpful discussions and help with establishing the CO‐FISH protocol. Research in J.L.'s laboratory was supported by the Swiss National Science Foundation (SNSF), the SNSF‐funded NCCR RNA and disease network, the Swiss Cancer League, and EPFL.
The EMBO Journal (2020) 39: e102668
References
- Abreu E, Aritonovska E, Reichenbach P, Cristofari G, Culp B, Terns RM, Lingner J , Terns MP (2010) TIN2‐tethered TPP1 recruits human telomerase to telomeres in vivo. Mol Cell Biol 30: 2971–2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Reaper PM, Clay‐Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP (2003) A DNA damage checkpoint response in telomere‐initiated senescence. Nature 426: 194–198 [DOI] [PubMed] [Google Scholar]
- Bailey SM, Goodwin EH, Meyne J, Cornforth MN (1996) CO‐FISH reveals inversions associated with isochromosome formation. Mutagenesis 11: 139–144 [DOI] [PubMed] [Google Scholar]
- Bartocci C, Diedrich JK, Ouzounov I, Li J, Piunti A, Pasini D, Yates JR, Lazzerini Denchi E (2014) Isolation of chromatin from dysfunctional telomeres reveals an important role for Ring1b in NHEJ‐mediated chromosome fusions. Cell Rep 7: 1320–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blewitt ME, Gendrel A‐V, Pang Z, Sparrow DB, Whitelaw N, Craig JM, Apedaile A, Hilton DJ, Dunwoodie SL, Brockdorff N et al (2008) SmcHD1, containing a structural‐maintenance‐of‐chromosomes hinge domain, has a critical role in X inactivation. Nat Genet 40: 663–669 [DOI] [PubMed] [Google Scholar]
- Brideau NJ, Coker H, Gendrel A‐V, Siebert CA, Bezstarosti K, Demmers J, Poot RA, Nesterova TB, Brockdorff N (2015) Independent mechanisms target SMCHD1 to trimethylated histone H3 lysine 9‐modified chromatin and the inactive X chromosome. Mol Cell Biol 35: 4053–4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli GB, de Lange T (2005) DNA processing is not required for ATM‐mediated telomere damage response after TRF2 deletion. Nat Cell Biol 7: 712–718 [DOI] [PubMed] [Google Scholar]
- Celli GB, Denchi EL, de Lange T (2006) Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat Cell Biol 8: 885–890 [DOI] [PubMed] [Google Scholar]
- Cesare AJ, Hayashi MT, Crabbe L, Karlseder J (2013) The telomere deprotection response is functionally distinct from the genomic DNA damage response. Mol Cell 51: 141–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coker H, Brockdorff N (2014) SMCHD1 accumulates at DNA damage sites and facilitates the repair of DNA double‐strand breaks. J Cell Sci 127: 1869–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Déjardin J, Kingston RE (2009) Purification of proteins associated with specific genomic Loci. Cell 136: 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denchi EL, de Lange T (2007) Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448: 1068–1071 [DOI] [PubMed] [Google Scholar]
- Deng Y, Guo X, Ferguson DO, Chang S (2009) Multiple roles for MRE11 at uncapped telomeres. Nature 460: 914–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, de Lange T (2006) MDC1 accelerates nonhomologous end‐joining of dysfunctional telomeres. Genes Dev 20: 3238–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Chen Y‐CM, Spector DL, de Lange T (2008) 53BP1 promotes non‐homologous end joining of telomeres by increasing chromatin mobility. Nature 456: 524–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doksani Y, Wu JY, de Lange T, Zhuang X (2013) Super‐resolution fluorescence imaging of telomeres reveals TRF2‐dependent T‐loop formation. Cell 155: 345–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feretzaki M, Lingner J (2017) A practical qPCR approach to detect TERRA, the elusive telomeric repeat‐containing RNA. Methods 114: 39–45 [DOI] [PubMed] [Google Scholar]
- Frescas D, de Lange T (2014) Binding of TPP1 protein to TIN2 protein is required for POT1a, b protein‐mediated telomere protection. J Biol Chem 289: 24180–24187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gdula MR, Nesterova TB, Pintacuda G, Godwin J, Zhan Y, Ozadam H, McClellan M, Moralli D, Krueger F, Green CM et al (2019) The non‐canonical SMC protein SmcHD1 antagonises TAD formation and compartmentalisation on the inactive X chromosome. Nat Commun 10: 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Löbrich M, Jeggo PA (2008) ATM signaling facilitates repair of DNA double‐strand breaks associated with heterochromatin. Mol Cell 31: 167–177 [DOI] [PubMed] [Google Scholar]
- Grolimund L, Aeby E, Hamelin R, Armand F, Chiappe D, Moniatte M, Lingner J (2013) A quantitative telomeric chromatin isolation protocol identifies different telomeric states. Nat Commun 4: 2848 [DOI] [PubMed] [Google Scholar]
- Jansz N, Chen K, Murphy JM, Blewitt ME (2017) The epigenetic regulator SMCHD1 in development and disease. Trends Genet 33: 233–243 [DOI] [PubMed] [Google Scholar]
- Jansz N, Keniry A, Trussart M, Bildsoe H, Beck T, Tonks ID, Mould AW, Hickey P, Breslin K, Iminitoff M et al (2018) Smchd1 regulates long‐range chromatin interactions on the inactive X chromosome and at Hox clusters. Nat Struct Mol Biol 25: 766–777 [DOI] [PubMed] [Google Scholar]
- Jones RE, Oh S, Grimstead JW, Zimbric J, Roger L, Heppel NH, Ashelford KE, Liddiard K, Hendrickson EA, Baird DM (2014) Escape from telomere‐driven crisis is DNA ligase III dependent. Cell Rep 8: 1063–1076 [DOI] [PubMed] [Google Scholar]
- Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T (1999) p53‐ and ATM‐dependent apoptosis induced by telomeres lacking TRF2. Science 283: 1321–1325 [DOI] [PubMed] [Google Scholar]
- Karlseder J, Smogorzewska A, de Lange T (2002) Senescence induced by altered telomere state, not telomere loss. Science 295: 2446–2449 [DOI] [PubMed] [Google Scholar]
- de Lange T (2018) Shelterin‐mediated telomere protection. Annu Rev Genet 52: 223–247 [DOI] [PubMed] [Google Scholar]
- Lazzerini‐Denchi E, Sfeir A (2016) Stop pulling my strings ‐ what telomeres taught us about the DNA damage response. Nat Rev Mol Cell Biol 17: 364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loayza D, De Lange T (2003) POT1 as a terminal transducer of TRF1 telomere length control. Nature 423: 1013–1018 [DOI] [PubMed] [Google Scholar]
- Maciejowski J, de Lange T (2017) Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol 18: 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y et al (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316: 1160–1166 [DOI] [PubMed] [Google Scholar]
- McClintock B (1941) The stability of broken ends of chromosomes in zea mays. Genetics 26: 234–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ (1938) The remaking of chromosomes. Collect Net 13: 181–198 [Google Scholar]
- Nozawa R‐S, Nagao K, Igami K‐T, Shibata S, Shirai N, Nozaki N, Sado T, Kimura H, Obuse C (2013) Human inactive X chromosome is compacted through a PRC2‐independent SMCHD1‐HBiX1 pathway. Nat Struct Mol Biol 20: 566–573 [DOI] [PubMed] [Google Scholar]
- Okamoto K, Bartocci C, Ouzounov I, Diedrich JK, Yates JR, Denchi EL (2013) A two‐step mechanism for TRF2‐mediated chromosome‐end protection. Nature 494: 502–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panier S, Durocher D (2013) Push back to respond better: regulatory inhibition of the DNA double‐strand break response. Nat Rev Mol Cell Biol 14: 661–672 [DOI] [PubMed] [Google Scholar]
- Paull TT (2015) Mechanisms of ATM activation. Annu Rev Biochem 84: 711–738 [DOI] [PubMed] [Google Scholar]
- Porro A, Feuerhahn S, Delafontaine J, Riethman H, Rougemont J, Lingner J (2014a) Functional characterization of the TERRA transcriptome at damaged telomeres. Nat Commun 5: 5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porro A, Feuerhahn S, Lingner J (2014b) TERRA‐reinforced association of LSD1 with MRE11 promotes processing of uncapped telomeres. Cell Rep 6: 765–776 [DOI] [PubMed] [Google Scholar]
- Rai R, Zheng H, He H, Luo Y, Multani A, Carpenter PB, Chang S (2010) The function of classical and alternative non‐homologous end‐joining pathways in the fusion of dysfunctional telomeres. EMBO J 29: 2598–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R, Hu C, Broton C, Chen Y, Lei M, Chang S (2017) NBS1 phosphorylation status dictates repair choice of dysfunctional telomeres. Mol Cell 65: 801–817.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaper PM, Griffiths MR, Long JM, Charrier J‐D, Maccormick S, Charlton PA, Golec JMC, Pollard JR (2011) Selective killing of ATM‐ or p53‐deficient cancer cells through inhibition of ATR. Nat Chem Biol 7: 428–430 [DOI] [PubMed] [Google Scholar]
- van Ruiten MS, Rowland BD (2018) SMC complexes: universal DNA looping machines with distinct regulators. Trends Genet 34: 477–487 [DOI] [PubMed] [Google Scholar]
- Shay JW, Wright WE (2011) Role of telomeres and telomerase in cancer. Semin Cancer Biol 21: 349–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, van Steensel B, Bianchi A, Oelmann S, Schaefer MR, Schnapp G, de Lange T (2000) Control of human telomere length by TRF1 and TRF2. Mol Cell Biol 20: 1659–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Wang B, Bignell CR, Taylor AMR, Elledge SJ (2003) MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421: 961–966 [DOI] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, Price BD (2009) Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol 11: 1376–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed A, Tainer JA (2018) The MRE11‐RAD50‐NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu Rev Biochem 87: 263–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, de Lange T (2003) DNA damage foci at dysfunctional telomeres. Curr Biol 13: 1549–1556 [DOI] [PubMed] [Google Scholar]
- Tang M, Li Y, Zhang X, Deng T, Zhou Z, Ma W, Songyang Z (2014) Structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1) promotes non‐homologous end joining and inhibits homologous recombination repair upon DNA damage. J Biol Chem 289: 34024–34032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Ly D, Low RRJ, Frölich S, Bartolec TK, Kafer GR, Pickett HA, Gaus K, Cesare AJ (2018) Telomere loop dynamics in chromosome end protection. Mol Cell 71: 510–525.e6 [DOI] [PubMed] [Google Scholar]
- Vancevska A, Douglass KM, Pfeiffer V, Manley S, Lingner J (2017) The telomeric DNA damage response occurs in the absence of chromatin decompaction. Genes Dev 31: 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C‐Y, Jégu T, Chu H‐P, Oh HJ, Lee JT (2018) SMCHD1 merges chromosome compartments and assists formation of super‐structures on the inactive X. Cell 174: 406–421.e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XD, Küster B, Mann M, Petrini JH, de Lange T (2000) Cell‐cycle‐regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat Genet 25: 347–352 [DOI] [PubMed] [Google Scholar]
- Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA‐ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 6
Source Data for Expanded View