

Abstract
The main protease of the coronavirus causing severe acute respiratory syndrome performs proteolytic processing of the viral polyproteins. The active form of the enzyme is a homodimer with each subunit consisting of three structural domains. Domains I and II, hosting the complete catalytic machinery, constitute the N-terminal chymotrypsin-like folding scaffold and connect to the extra C-terminal domain III by a long loop. Previously, the domain III-truncated enzyme was demonstrated to fold independently into an intact chymotrypsin-like fold, but it showed no enzyme activity. To further delineate the structure–function relationships of the domain III and the long loop, we generated some truncated and mutated Mpro forms bearing various combinations of the loop with other structural parts of the enzyme. Their conformational and association properties were investigated in detail. Far-ultraviolet circular dichroism (CD) measurements revealed that these fragments could fold independently. The secondary, tertiary and quaternary structures of these mixtures were monitored by CD, fluorescence spectroscopy and analytical ultracentrifugation. However, no enzyme activity was observed for any mutant or mixtures. These observations indicate that the covalent linkage between the chymotrypsin like and the extra domain is essential for enzymatic activity of the main coronavirus protease and for the integrity of its quaternary structure.
Keywords: SARS-CoV, Mpro, folding, protein stability, nicked protein
Coronaviruses (CoVs) are extremely common in animals but usually not pathogens for humans. The severe acute respiratory syndrome (SARS) CoV is an exception in this respect, which caused a board range of diseases and raised a worldwide health hazard in 2003. Maturation of the SARS-CoV involves highly complicated events of proteolytic processing of the polyproteins by the main protease (Mpro) and the papain-like protease (1–5). All Mpro forms, including that from SARS-CoV, have a common homologous structural feature, irrespective of their origin (1). The development of wide-spectrum antiviral drugs against Mpro is thus an attractive therapeutic aim.
SARS-CoV Mpro is a homodimer in the crystalline form and a monomer-dimer equilibrium in solution (2, 6–7). Available evidence indicates that the dimeric form is the enzymatically active form and that the monomer is inactive (8). Each subunit of the dimeric Mpro consists of three structural domains [Fig. 1A, generated with Pymol (9)]. The N-terminal Domains I (residues 8–99) and II (residues 100–183) have an anti-parallel β-barrel structure with a chymotrypsin-like folding scaffold. The C-terminal domain III (residues 200–306) has five α-helices arranged into a globular cluster that is connected to Domain II by a long loop (residues 184–199). The SARS-CoV Mpro has a catalytic Cys–His dyad, instead of the Ser–His–Asp triad of chymotrypsin. The active centre is located in the interface between Domains I and II. The functional role of domain III but not the loop that links domain III and domain (I + II) has been investigated intensively (8, 10, 11).
Fig. 1.
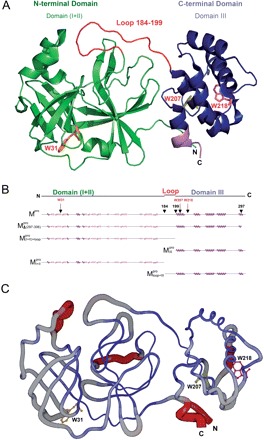
Structural features of the SARS-CoV Mpro. (A) The three structural domains of SARS-CoV Mpro (pdb code: 1uk3) are shown in ribbon representation. The N-terminal domain (residues 1–183) is shown in green, C-terminal domain (residues 200–306) in blue, the loop (residues 184–199) connecting the two domains in red, the deletion region (residues 297–306) of 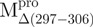 in violet. The tryptophanyl residues Trp31, Trp207 and Trp218 are shown in orange, yellow and pink, respectively, as bond model. Generate with Pymol. (B) Secondary structure features of the enzyme and the truncated regions of the various truncation mutants. (C) The structural flexibility of the enzyme is shown as a variable worm of the polypeptide chain based on the B factor of the pdb file (pdb code: 1uk3). The rigid structure is shown as a thinner worm in blue. The more flexible peripheral regions are shown in red. Only one of the subunits is shown. This figure was generated with SPOCK (http://quorum.tamu.edu).
in violet. The tryptophanyl residues Trp31, Trp207 and Trp218 are shown in orange, yellow and pink, respectively, as bond model. Generate with Pymol. (B) Secondary structure features of the enzyme and the truncated regions of the various truncation mutants. (C) The structural flexibility of the enzyme is shown as a variable worm of the polypeptide chain based on the B factor of the pdb file (pdb code: 1uk3). The rigid structure is shown as a thinner worm in blue. The more flexible peripheral regions are shown in red. Only one of the subunits is shown. This figure was generated with SPOCK (http://quorum.tamu.edu).
We have developed a domain III-truncated mutant to study the functional role of this extra C-terminal domain in SARS-CoV Mpro (10). In chemically induced denaturation experiments, the free energy change of the C-terminal domain-truncated mutant was higher than the wild-type (WT) Mpro, which indicated that domain III might contribute to stabilization of the domain (I + II) (10). To further delineate the structure and function relationship of the domain III and the long loop, we generated more Mpro mutants bearing various combinations of the loop connecting to other structural parts of the enzyme (Fig. 1B). The secondary, tertiary and quaternary structural features were monitored by circular dichroism (CD), fluorescence spectrometry and analytical ultracentrifugation (AUC). Our results indicate that the covalent linkage between the catalytic N-terminal domain and the extra C-terminal domain is essential for both enzyme activity and for the integrity of the quaternary structure of SARS-CoV Mpro.
Materials and Methods
Construction, expression and purification of various SARS-CoV M pro
The genes of the full-length, truncated, 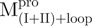 and deletion mutant,
and deletion mutant, 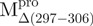 , of SARS-CoV Mpro inserted into the vector pET29(+) (Novagen, Madison, WI, USA) had been described previously (8, 10). The genes of the N-terminal truncated (
, of SARS-CoV Mpro inserted into the vector pET29(+) (Novagen, Madison, WI, USA) had been described previously (8, 10). The genes of the N-terminal truncated (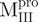 ,
, 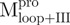 ) and C-terminal truncated (
) and C-terminal truncated (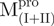 ) mutants of SARS-CoV Mpro were made by polymerase chain reaction (PCR) with appropriate primers using the full-length Mpro as the template. The forward primers for
) mutants of SARS-CoV Mpro were made by polymerase chain reaction (PCR) with appropriate primers using the full-length Mpro as the template. The forward primers for 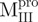 ,
, 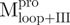 and
and 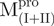 were 5′-GGTGGTCATATGATAACATTAAAT-3′, 5′-GGTGGTCATATGCCATTTGTTGAC-3′ and 5′-GGGCCATGGATAGTGGTTTTAGGAAAATG-3′, respectively; and the reverse primers were 5′-AACTCGAGGGTAACACCAGAG-3′, 5′-AACTCGAGGGTAACACCAGAG-3′ and 5′-GGGCTCGAGACCATAGAATTTACCTTC-3′, respectively. The NdeI, NcoI and XhoI sites are shown underlined and in bold. After digestion with NdeI and XhoI, each PCR product was ligated to the NdeI and XhoI sites of the pET-SARS-CoV Mpro except
were 5′-GGTGGTCATATGATAACATTAAAT-3′, 5′-GGTGGTCATATGCCATTTGTTGAC-3′ and 5′-GGGCCATGGATAGTGGTTTTAGGAAAATG-3′, respectively; and the reverse primers were 5′-AACTCGAGGGTAACACCAGAG-3′, 5′-AACTCGAGGGTAACACCAGAG-3′ and 5′-GGGCTCGAGACCATAGAATTTACCTTC-3′, respectively. The NdeI, NcoI and XhoI sites are shown underlined and in bold. After digestion with NdeI and XhoI, each PCR product was ligated to the NdeI and XhoI sites of the pET-SARS-CoV Mpro except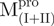 , which was inserted into the NcoI and XhoI sites of the pET-28(a) (Novagen, Madison, WI, USA) after digestion with NcoI and XhoI.
, which was inserted into the NcoI and XhoI sites of the pET-28(a) (Novagen, Madison, WI, USA) after digestion with NcoI and XhoI.
Mutagenesis of full-length SARS-CoV proteases was performed using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA, USA). For the tryptophan mutants, the forward primers for 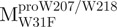 ,
, 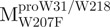 and
and 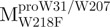 were 5′-CTCTTAATGGATTGTTCTTGGATGACACAGT-3′, 5′-CCATAACA TTAAATGTTTTGGCATTCCTGTATGCTGC-3′ and 5′-CAATGGTGATAGGTTCTT TCTTAATAGATTCACC-3′, respectively. The reverse primers were 5′-ACTGTGTC ATCCAAGAACAATCCATTAAGAG-3′, 5′-GCAGCATACAGGAATGCCAAAACA TTTAATGTTATGG-3′ and 5′-GGTGAATCTATTAAGAAAGAACCTATCACCATTG-3′, respectively. The primers as described above were used to mutate the Trp31, Trp207 and Trp218 codons to phenylalanine codon by PCR. The mutational sites are shown underlined and in bold. The pET-Mpro expression vector was used as template to construct pET-
were 5′-CTCTTAATGGATTGTTCTTGGATGACACAGT-3′, 5′-CCATAACA TTAAATGTTTTGGCATTCCTGTATGCTGC-3′ and 5′-CAATGGTGATAGGTTCTT TCTTAATAGATTCACC-3′, respectively. The reverse primers were 5′-ACTGTGTC ATCCAAGAACAATCCATTAAGAG-3′, 5′-GCAGCATACAGGAATGCCAAAACA TTTAATGTTATGG-3′ and 5′-GGTGAATCTATTAAGAAAGAACCTATCACCATTG-3′, respectively. The primers as described above were used to mutate the Trp31, Trp207 and Trp218 codons to phenylalanine codon by PCR. The mutational sites are shown underlined and in bold. The pET-Mpro expression vector was used as template to construct pET-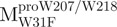 , pET-
, pET-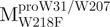 . For pET-
. For pET-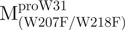 double mutants, pET-
double mutants, pET-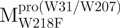 was used as the template.
was used as the template.
All nucleotide sequences were confirmed by autosequencing analysis. All genes coding for the enzymes were successfully expressed and the corresponding enzymes purified to apparent homogeneity by protocols described previously (8).
CD analysis
Far-UV CD spectra were measured at 30°C with a Jasco J-815 spectropolarimeter under constant N2 flush. The temperature was controlled by air-controlling Peltier device (PTC-423S/15, JASCO, Tokyo, Japan). The protein solution was scanned from 250 to 190 nm in cuvettes with a 0.1 mm path length at 0.1-nm intervals with a bandwidth of 1.0 nm. The photomultiplier absorbance was always below 600 V in the analysed region. Each scanning was repeated 10× and an average was reported. Data were corrected for the absorbance of buffer, and the results were expressed as molar ellipticity [Θ] in the units of degrees cm2 decimol−1.
Molar ellipticity [Θ] was calculated according to equation 1:
| (1) |
Where l represents the light path length (cm); C, the molar concentration of protein (mol/l) and Θ, the observed ellipticity (mdeg).
The secondary structural contents were estimated by the online DICHROWEB server (web site, http://www.cryst.bbk.ac.uk/cdweb/html/home.html) (12, 13) using the implemented CDSSTR programme (14–16) (reference set 7). Spectral analysis was taken over the wavelength range from 190 to 240 nm.
Thermal denaturation experiments were performed by monitoring the ellipticity at 222 nm. The temperature was increased with a heating rate of 1°C/min from 30 to 90°C and the temperature at which half of the protein molecules were unfolded was recorded (Tm).
Spectrofluorimetric analysis
Fluorescence spectra of the enzyme were monitored in a Perkin-Elmer LS-50B luminescence spectrometer at 30°C. All spectra were corrected for buffer absorption. The excitation wavelength was set at 295 nm and the fluorescence emission spectrum was scanned from 300 to 400 nm. When excited with 295 nm UV light, the protein intrinsic emission fluorescence was originated from tryptophanyl residue only. The maximal peak of the fluorescence spectrum and the change in fluorescence intensity were used in monitoring the unfolding processes of the enzyme. Both the red shift and the changes in fluorescence intensity were analysed together using the average emission wavelength (AEW) (<λ>) according to equation 2 (17):
| (2) |
in which Fi is the fluorescence intensity at the specific emission wavelength (λi).
Enzyme activity assay of SARS-CoV M pro using fluorogenic substrate
The enzyme activity measurements were performed in 30 mM Tris–HCl buffer (pH 7.7) at 30°C. Enhanced fluorescence due to cleavage of the internal quenched fluorogenic substrate peptide (ortho-aminobenzoic acid-TSAVLQSGFRK-2,4-dinitrophenyl amide) was monitored at 420 nm with excitation at 362 nm using a Perkin-Elmer LS-50B luminescence spectrometer.
AUC
The molar mass and sedimentation coefficient of the proteases were analysed by a sedimentation velocity experiment. Sedimentation velocity was performed in a Beckman-Coulter XL-A analytical ultracentrifuge (Fullerton, CA, USA) with an An50Ti rotor at 20°C and 42,000 rpm in 12-mm double-sector Epon charcoal-filled centerpieces. Prior to the experiments, the sample was diluted to various protein concentrations with 30 mM Tris–HCl. The UV absorption of the cells at 280 nm was scanned in a continuous mode with time interval of 8 min and a step size of 0.003 cm. The partial specific volume of the enzyme, solvent density and viscosity were calculated by the software SEDNTERP (http://www.jphilo.mailway.com/). All samples were visually checked for clarity after ultracentrifugation to make sure that there was no indication of precipitation. The recorded scans at different time points were collected and fitted to a continuous size distribution model by the SEDFIT programme (18, 19). The observed sedimentation profiles of a continuous size distribution [c(s)] can be calculated from equation 3.
| (3) |
where a(r,t) denotes the experimentally observed signal, L(s,D,r,t) denotes the solution of the Lamm equation for a single species, and ε is the noise component.
Reversible unfolding/refolding of the enzyme in GdmCl
The folding experiments of all the full-length WT, truncated and mutated SARS-CoV Mpro were examined in different concentrations of GdmCl in Tris–HCl buffer (30 mM, pH 7.7) at 30°C. The unfolding of the enzyme was monitored by protein fluorescence change. The unfolding was found to reach equilibrium in transient. A 10-min incubation, both unfolding and refolding, was used in the experiments in the present study.
Protein unfolding data analysis
Since unfolding/refolding of our sample is a reversible process, the unfolding data were treated with the following thermodynamic models, the two-state (Scheme I) and three-state unfolding model (Scheme II), by global fitting of the data to equations described previously (10, 20):
Results
Engineered variants of SARS-CoV M pro for probing linker function
We used molecular biology techniques to create SARS-CoV Mpro variants, which allowed us to study the structural role of the linker between domain (I+II) and domain III in Mpro. We created four linker variants: 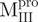 ,
, 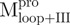 ,
, 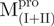 and
and 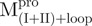 . Among them,
. Among them, 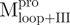 and
and 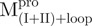 have the loop covalently linked to other parts of the enzyme, but
have the loop covalently linked to other parts of the enzyme, but 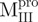 and
and 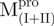 lack the linker. The yields of the purified protein were usually in the range of 5–15 mg/ml from 200 ml of cell culture medium. The purity of the enzymes was determined to be >97% by SDS-PAGE (Supplementary Fig. S1).
lack the linker. The yields of the purified protein were usually in the range of 5–15 mg/ml from 200 ml of cell culture medium. The purity of the enzymes was determined to be >97% by SDS-PAGE (Supplementary Fig. S1).
AUC analysis verified the expected decreases in molar mass of 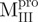 (13.2 kDa),
(13.2 kDa), 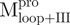 (16.98 kDa),
(16.98 kDa), 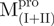 (22.13 kDa) and
(22.13 kDa) and 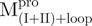 (23.23 kDa) versus 35 kDa for the monomeric mutant,
(23.23 kDa) versus 35 kDa for the monomeric mutant, 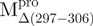 and 64.26 kDa for the dimeric full-length WT Mpro and mutants. All these experimentally determined molar mass values were in agreement with the theoretical values calculated from amino acid sequences.
and 64.26 kDa for the dimeric full-length WT Mpro and mutants. All these experimentally determined molar mass values were in agreement with the theoretical values calculated from amino acid sequences.
Essential role of the covalent linker between domain (I+II) and domain III
We prepared three kinds of solutions comprising the entire enzyme (nicked Mpro): 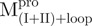 /
/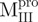 (Mixture 1),
(Mixture 1), 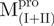 /
/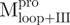 (Mixture 2) and
(Mixture 2) and 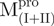 /
/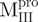 (Mixture 3), all at a molar ratio of 1:1 (146 µM). The conformational feature of this nicked Mpro and its constituent fragments were evaluated by CD spectroscopy as shown in Fig. 2. Results indicated that the enzymes have a well-defined secondary structure, except for that of
(Mixture 3), all at a molar ratio of 1:1 (146 µM). The conformational feature of this nicked Mpro and its constituent fragments were evaluated by CD spectroscopy as shown in Fig. 2. Results indicated that the enzymes have a well-defined secondary structure, except for that of 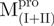 , which had a low CD signal, as anticipated. The secondary structure of the enzymes was estimated using the DICHROWEB server (12, 13). To estimate the secondary structure of
, which had a low CD signal, as anticipated. The secondary structure of the enzymes was estimated using the DICHROWEB server (12, 13). To estimate the secondary structure of 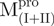 , a 1.5-fold excess of enzyme was employed. The CDSSTR analysis is shown in Table I. The normalized root mean square deviation (NRMSD) values of the data fitting for WT, mutants and various truncations were all <0.05 showing excellent goodness-of-fit. The calculated helical and β-strand contents of the recombinant WT were 0.23 and 0.24, respectively, consistent with the data derived from the crystal structure (Table I).
, a 1.5-fold excess of enzyme was employed. The CDSSTR analysis is shown in Table I. The normalized root mean square deviation (NRMSD) values of the data fitting for WT, mutants and various truncations were all <0.05 showing excellent goodness-of-fit. The calculated helical and β-strand contents of the recombinant WT were 0.23 and 0.24, respectively, consistent with the data derived from the crystal structure (Table I).
Fig. 2.
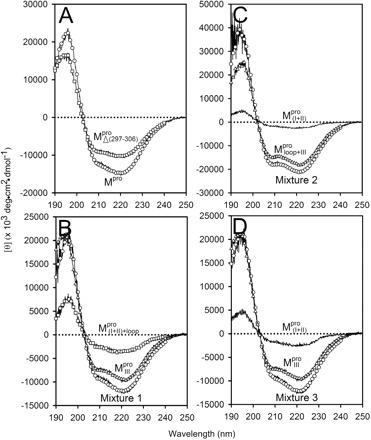
Far-UV CD spectra of Mpro, mixtures and each constituting fragments. Spectra were recorded at 30°C in 30 mM Tris–HCl buffer (pH 7.7) at protein or fragment concentration of 146 μM except for WT (95.6 μM) and 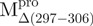 (106 μM). (A)
(106 μM). (A) 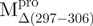 and Mpro; (B)
and Mpro; (B) 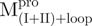 ,
, 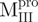 and Mixture 1 (
and Mixture 1 (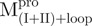 /
/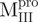 ); (C)
); (C) 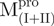 ,
, 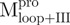 and Mixture 2 (
and Mixture 2 (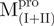 /
/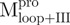 ); (D)
); (D) 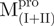 ,
, 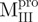 and Mixture 3 (
and Mixture 3 (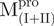 /
/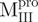 ).
).
Table I.
Structural characteristic of SARS-CoV full-length, N- and C-terminal-truncated Mpro and complex in crystal and solution.
| Protease | Quaternary structure | α-Helix (%) | β-strand (%) | Random coil (%) | Normalized NRMSDa | T m b | |
|---|---|---|---|---|---|---|---|
| 1uk3c | Dimer | 24 | 29 | 47 | |||
| 2qcyc | Monomer | 26 | 28 | 46 | |||
| Mpro | Dimer | 23d | 24 | 53 | 0.032 | 53.9 | |
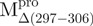
|
Monomer | 17 | 29 | 54 | 0.027 | 68.2 | |
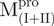 e
e
|
Monomer | 8 | 33 | 59 | 0.022 | 50.2 | |
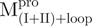
|
Monomer | 5 | 37 | 58 | 0.045 | 52.5 | |
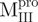
|
Monomer | 48 | 15 | 37 | 0.021 | 57.7 | |
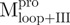
|
Monomer | 71 | 8 | 21 | 0.009 | 61.2 | |
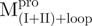
|
19 | 26 | 55 | 0.035 | 49.2 | ||
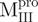 (Mixture1) (Mixture1) |
59.2 | ||||||
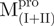
|
41 | 15 | 44 | 0.039 | 49.5 | ||
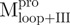 (Mixture2) (Mixture2) |
62.8 | ||||||
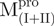
|
19 | 27 | 54 | 0.031 | 46.5 | ||
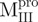 (Mixture3) (Mixture3) |
58.6 |
aNormalized root mean SD of the secondary structure fitting results.
bMelting temperature determined by the CD spectropolarimeter.
cCrystal structure determined by X-ray crystallography.
dPercentage calculated by CDSSTR programme.
eThe data is from 171.36 μM 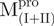 , which is more than the other enzyme, CD analysis is only reliable under this condition.
, which is more than the other enzyme, CD analysis is only reliable under this condition.
The helical content of the monomeric 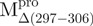 was 0.17, a value significantly different from the 0.26 of the crystal structure of a monomeric Mpro (PDB code: 2QCY) (21). However, this is predictable because a seventh helix was deleted in
was 0.17, a value significantly different from the 0.26 of the crystal structure of a monomeric Mpro (PDB code: 2QCY) (21). However, this is predictable because a seventh helix was deleted in 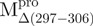 (Fig. 1B).
(Fig. 1B).
Both 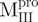 and
and 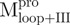 displayed far-ultraviolet (Far-UV) CD spectra with the features of α-helical polypeptides, with minimal values of negative ellipticity at around 208 and 222 nm. In contrast,
displayed far-ultraviolet (Far-UV) CD spectra with the features of α-helical polypeptides, with minimal values of negative ellipticity at around 208 and 222 nm. In contrast, 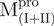 and
and 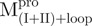 were characteristic for low α-helix and high β-strand contents, as expected. In aqueous solution and at neutral pH, the
were characteristic for low α-helix and high β-strand contents, as expected. In aqueous solution and at neutral pH, the 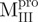 and
and 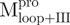 adopted ∼48 and 71% helicity, respectively. On the other hand,
adopted ∼48 and 71% helicity, respectively. On the other hand, 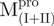 and
and 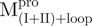 assumed mainly β-strand structures of 33 and 37%, respectively. These results suggest that the overall structure was not destroyed by truncations. This result was also observed in Shi’s study, they found that each fragment (I + II and III) can fold independently (3). As shown in Table I, the secondary structural characteristics of Mixtures 1 or 3 are more similar to
assumed mainly β-strand structures of 33 and 37%, respectively. These results suggest that the overall structure was not destroyed by truncations. This result was also observed in Shi’s study, they found that each fragment (I + II and III) can fold independently (3). As shown in Table I, the secondary structural characteristics of Mixtures 1 or 3 are more similar to 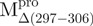 than to the WT. In contrast, a considerable difference in the far-UV CD spectra was observed in Mixture 2 (Fig. 2). These CD data indicate that there are secondary structure differences between the nicked and intact proteins.
than to the WT. In contrast, a considerable difference in the far-UV CD spectra was observed in Mixture 2 (Fig. 2). These CD data indicate that there are secondary structure differences between the nicked and intact proteins.
The thermal stabilities of the recombinant SARS-CoV Mpro and the mixtures were also examined by monitoring the continuous changes in the ellipticity at 222 nm during the temperature range from 30°C to 90°C (Table I). The enzyme precipitated at high temperature and the thermal denaturation was irreversible. The melting profiles are shown in Supplementary Fig. 2S. While two distinct transitions were observed around 49.2 and 59.2°C in the melting profile of the Mixture 1; 49.5 and 62.8°C in the Mixture 2, 46.5 and 58.6°C in the Mixture 3, only one large transition was observed around 53.9°C in the profile of WT. Nevertheless, it can be concluded that the transition proceeds in a three-state fashion with indication of independent behaviour by either of the domains in the mixtures. Among the truncated SARS-CoV Mpro, the C terminally truncated protein had a significantly lower Tm (50.2 for 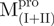 and 52.2 for
and 52.2 for 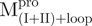 ) than did the N terminally truncated one (57.7°C and 79.2°C for
) than did the N terminally truncated one (57.7°C and 79.2°C for 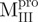 and 61.2°C for
and 61.2°C for 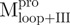 ). The observation is an indication that the catalytic domain is intrinsically less stable and that it derives a significant fraction of its stabilization energy from its interactions with domain III through the linker. The Tm value for WT was 53.9°C (∼54), which is a little higher than that of
). The observation is an indication that the catalytic domain is intrinsically less stable and that it derives a significant fraction of its stabilization energy from its interactions with domain III through the linker. The Tm value for WT was 53.9°C (∼54), which is a little higher than that of 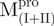 (50.2°C) and
(50.2°C) and 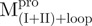 (52.2°C). These results are consistent with the previous report that the C-terminal domain is involved in stabilizing the N-terminal catalytic domain of SARS-CoV Mpro (10). Two transitions with the midpoint temperatures at 57.7 and 79.2°C upon thermal unfolding were also demonstrated in
(52.2°C). These results are consistent with the previous report that the C-terminal domain is involved in stabilizing the N-terminal catalytic domain of SARS-CoV Mpro (10). Two transitions with the midpoint temperatures at 57.7 and 79.2°C upon thermal unfolding were also demonstrated in 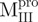 . Moreover, one transition temperature for
. Moreover, one transition temperature for 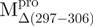 was determined to be 68.2°C, which is higher than that of WT (53.9°C). Further characterization of these transitions is needed to address what structure is attributed to the unfolding, when narrowed down to the area of a single α-helix in
was determined to be 68.2°C, which is higher than that of WT (53.9°C). Further characterization of these transitions is needed to address what structure is attributed to the unfolding, when narrowed down to the area of a single α-helix in 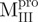 and
and 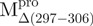 .
.
Reversible unfolding of N- and C-terminal-truncated SARS-CoV M pro in guanidinium chloride
Previously, we found that the guanidinium chloride (GdmCl)-induced unfolding of WT Mpro in 30 mM Tris–HCl was completely reversible (10). Similar results were obtained in all engineered SARS-CoV Mpro used in the present study and representative data are shown in Fig. 3. The C-terminal-truncated Mpro (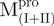 ,
, 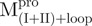 ), with only one tryptophanyl residue, remained intact and showed less fluorescence in the folded state (Fig. 3A and B) than the N-terminal truncated Mpro (
), with only one tryptophanyl residue, remained intact and showed less fluorescence in the folded state (Fig. 3A and B) than the N-terminal truncated Mpro (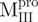 ,
, 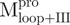 ), which has two tryptophanyl residues (Fig. 3C and D). Domains (I+II) and III were found to fold independently. GdmCl-induced unfolding was thus very suitable for conformational stability studies of the enzyme. We therefore attempted to probe the structural role of the loop of Mpro.
), which has two tryptophanyl residues (Fig. 3C and D). Domains (I+II) and III were found to fold independently. GdmCl-induced unfolding was thus very suitable for conformational stability studies of the enzyme. We therefore attempted to probe the structural role of the loop of Mpro.
Fig. 3.
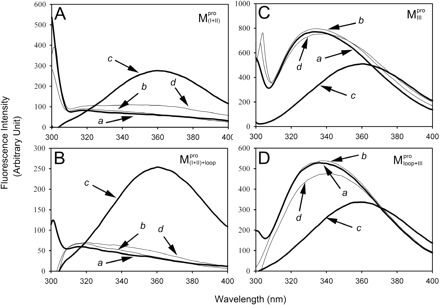
Equilibrium unfolding/refolding of the N- and C-terminal-truncated SARS-CoV Mpro in GdmCl. The enzyme (50.6 µg/ml for 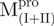 , 48.7 µg/ml for
, 48.7 µg/ml for 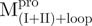 , 2.7 µg/ml for
, 2.7 µg/ml for 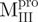 and 6.8 µg/ml for
and 6.8 µg/ml for 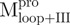 ) in 30 mM Tris–HCl buffer (pH 7.7) was excited with 295 nm UV light and the protein intrinsic fluorescence emission spectra were monitored at 30°C. (A)
) in 30 mM Tris–HCl buffer (pH 7.7) was excited with 295 nm UV light and the protein intrinsic fluorescence emission spectra were monitored at 30°C. (A) 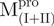 ; (B)
; (B) 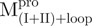 ; (C)
; (C) 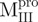 ; (D)
; (D) 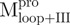 . Curves a, Native enzyme in 30 mM Tris–HCl buffer; b, Native enzyme in the same buffer containing 0.6 M GdmCl; c, Unfolded enzyme in 6 M GdmCl buffer and d, Refolded enzyme in 0.6 M GdmCl buffer. In d, the protein was unfolded in 6 M GdmCl buffer for 10 min and then diluted 10-fold with the buffer. All samples were treated with GdmCl buffer or buffer-only simultaneously for the same duration. The final enzyme concentrations were the same for all spectra.
. Curves a, Native enzyme in 30 mM Tris–HCl buffer; b, Native enzyme in the same buffer containing 0.6 M GdmCl; c, Unfolded enzyme in 6 M GdmCl buffer and d, Refolded enzyme in 0.6 M GdmCl buffer. In d, the protein was unfolded in 6 M GdmCl buffer for 10 min and then diluted 10-fold with the buffer. All samples were treated with GdmCl buffer or buffer-only simultaneously for the same duration. The final enzyme concentrations were the same for all spectra.
The AEW, representing the changes in both fluorescence wavelength and fluorescence intensity, was used to estimate the stability parameters of the unfolding/refolding process (Table II). SARS-CoV Mpro has three Trp residues, Trp31 in domain I and Trp207/Trp218 in domain III. The fluorescence of these Trp mutants showed the conformational changes of individual domains. With both Trp207 and Trp218 at domain III mutated to phenylalanine, 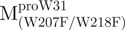 could only report the conformational changes in the N-terminal domain (Fig. 1A).
could only report the conformational changes in the N-terminal domain (Fig. 1A). 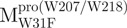 , a single mutant with Trp31 mutated to phenylalanine, revealed structural information of the C-terminal domain (Fig. 1A). All these unfoldings were completely reversible and showed the independent folding of domains (I+II) and III (Fig. 3). Both the N- and C-terminal domains followed monophasic unfolding curves. However these did not coincide, as demonstrated in
, a single mutant with Trp31 mutated to phenylalanine, revealed structural information of the C-terminal domain (Fig. 1A). All these unfoldings were completely reversible and showed the independent folding of domains (I+II) and III (Fig. 3). Both the N- and C-terminal domains followed monophasic unfolding curves. However these did not coincide, as demonstrated in 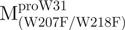 and
and 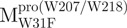 (Fig. 4A). It was obvious that there were differential conformational stabilities for domains (I+II) and III. The unfolding curves of the N-terminal domains, with or without a loop,
(Fig. 4A). It was obvious that there were differential conformational stabilities for domains (I+II) and III. The unfolding curves of the N-terminal domains, with or without a loop, 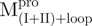 and
and 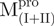 , and the C-terminal domains with or without loops,
, and the C-terminal domains with or without loops, 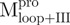 and
and 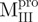 , are shown in Fig. 4B. These data indicate that the conformational changes in the C-terminal domain-truncated enzymes (
, are shown in Fig. 4B. These data indicate that the conformational changes in the C-terminal domain-truncated enzymes (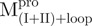 and
and 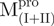 ) and in the N-terminal domain-truncated enzymes (
) and in the N-terminal domain-truncated enzymes (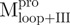 and
and 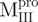 ) were similar to
) were similar to 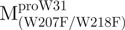 and
and 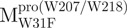 , respectively. It was evident that the N-terminal domain of Mpro underwent unfolding ([GdmCl]0.5 ∼ 1.5 M) earlier than the C-terminal domain ([GdmCl]0.5 ∼ 4 M). According to the stability parameters shown in Table II, the long loop was able to stabilize the truncated protease in GdmCl, especially for the C-terminal domain.
, respectively. It was evident that the N-terminal domain of Mpro underwent unfolding ([GdmCl]0.5 ∼ 1.5 M) earlier than the C-terminal domain ([GdmCl]0.5 ∼ 4 M). According to the stability parameters shown in Table II, the long loop was able to stabilize the truncated protease in GdmCl, especially for the C-terminal domain.
Table II.
Stability parameters of full-length, N- and C-terminal-truncated SARS-CoV Mpro and complex.
| Protease | ΔGN-I (kcal mol−1) | mN-I (kcal mol−1 M−1) | [GdmCl]0.5,N-I (M) | ΔGI-U (kcal mol−1) | mI-U (kcal mol−1 M−1) | [GdmCl]0.5,I-U (M) | |||
|---|---|---|---|---|---|---|---|---|---|
| Mpro | 2.07 ± 0.33 | 1.66 ± 0.23 | 1.3 ± 0.3 | 11.58 ± 0.58 | 2.79 ± 0.14 | 4.2 ± 0.3 | |||
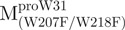
|
2.22 ± 0.43 | 1.62 ± 0.27 | 1.4 ± 0.4 | – | – | – | |||
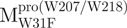
|
– | – | – | 12.08 ± 0.66 | 2.87 ± 0.16 | 4.2 ± 0.2 | |||
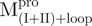
|
2.03 ± 0.52 | 0.98 ± 0.33 | 2.1 ± 0.8 | 8.63 ± 0.60 | 2.13 ± 0.14 | 4.1 ± 0.3 | |||
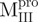 (Mixture 1) (Mixture 1) |
|||||||||
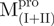
|
3.81 ± 0.95 | 2.62 ± 0.64 | 1.5 ± 0.5 | 9.92 ± 0.33 | 2.36 ± 0.08 | 4.2 ± 0.2 | |||
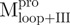 (Mixture 2) (Mixture 2) |
|||||||||

|
4.32 ± 1.74 | 3.19 ± 1.26 | 1.4 ± 0.8 | 6.86 ± 0.45 | 1.80 ± 0.12 | 3.8 ± 0.4 | |||
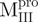 (Mixture 3) (Mixture 3) |
|||||||||
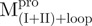
|
3.41 ± 0.78 | 2.29 ± 0.52 | 1.5 ± 0.5 | 9.79 ± 0.41 | 2.34 ± 0.10 | 4.2 ± 0.3 | |||
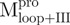 (Mixture 4) (Mixture 4) |
|||||||||
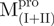
|
5.28 ± 0.35 | 3.87 ± 0.25 | 1.4 ± 0.1 | ||||||
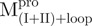
|
5.62 ± 1.04 | 3.97 ± 0.69 | 1.4 ± 0.3 | ||||||
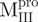
|
5.99 ± 0.15 | 1.71 ± 0.04 | 3.5 ± 0.1 | ||||||
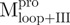
|
9.07 ± 0.30 | 2.23 ± 0.07 | 4.1 ± 0.1 |
Fig. 4.
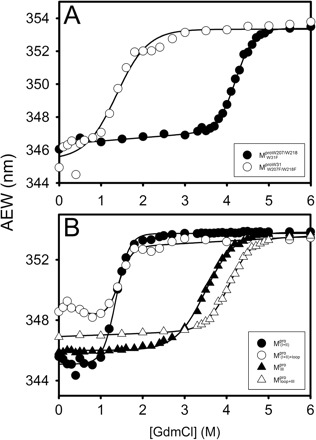
Unfolding curves of the full-length Trp-mutated, N- and C-terminal domain-truncated SARS-CoV Mpro. (A) Unfolding of the full-length Trp-mutated enzyme. 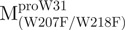 (open circle) in 0.54 μM;
(open circle) in 0.54 μM; 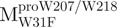 (filled circle) in 0.27 μM.
(filled circle) in 0.27 μM. 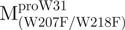 represents the structural change of N-terminal domain.
represents the structural change of N-terminal domain. 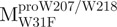 represents the structural change of C-terminal domain. (B) Unfolding of the N- and C-truncated protease bearing loop or not.
represents the structural change of C-terminal domain. (B) Unfolding of the N- and C-truncated protease bearing loop or not. 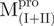 (filled circle) and
(filled circle) and 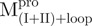 (open circle) in 0.54 μM;
(open circle) in 0.54 μM; 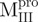 (filled triangle) and
(filled triangle) and 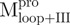 (open triangle) in 0.27 μM. The protein in 30 mM Tris–Cl (pH 7.7) was incubated with various concentrations of GdmCl for 10 min. The protein solution was excited at 295 nm and the emission fluorescence was recorded at 30°C. AEW (Equation 2) of each protein was used to present the unfolding data. Symbols are experimental data and the smooth curves are computer-generated best-fitted lines according to a two-state unfolding model. Truncated mutant had similar curve to the full-length mutated Mpro.
(open triangle) in 0.27 μM. The protein in 30 mM Tris–Cl (pH 7.7) was incubated with various concentrations of GdmCl for 10 min. The protein solution was excited at 295 nm and the emission fluorescence was recorded at 30°C. AEW (Equation 2) of each protein was used to present the unfolding data. Symbols are experimental data and the smooth curves are computer-generated best-fitted lines according to a two-state unfolding model. Truncated mutant had similar curve to the full-length mutated Mpro. 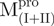 and
and 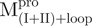 were similar to
were similar to 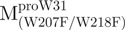 while
while 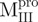 and
and 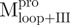 were similar to
were similar to 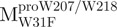 .
.
Unfolding and enzyme activity of the mixtures of the SARS-CoV M pro
To test whether the covalent linkage between domains (I + II) and III would be essential for the structural integrity and enzymatic activity of Mpro, we designed four nicked proteins. In the presence of GdmCl, all mixtures had similar unfolding curves with the WT Mpro, which followed a biphasic-unfolding curve (Fig. 5). All these mixtures had a free energy of the first phase that was lower than the C-terminal domain-truncated enzyme (Table II). These results indicated that there were intimate interactions between N- and C-terminal domains and that the C-terminal domain III could stabilize the N-terminal domain. However, the presence of an N-terminal domain decreased the stability of the C-terminal domain III. The stability parameters of Mixture 3 with no loops were similar to the truncated enzyme. Thus, the absence of the loop weakened the interaction between domains (I+II) and III.
Fig. 5.
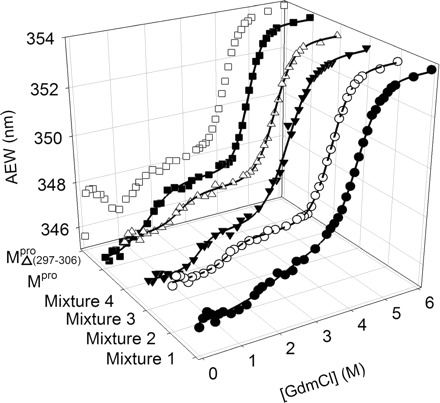
Unfolding curves of the WT, deletion mutants and mixtures of the Mpro. Mixture 1 (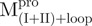 /
/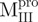 ) (filled circle), Mixture 2 (
) (filled circle), Mixture 2 (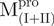 /
/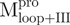 ) (open circle), Mixture 3 (
) (open circle), Mixture 3 (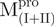 /
/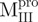 ) (inverted filled triangle), Mixture 4 (
) (inverted filled triangle), Mixture 4 (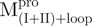 /
/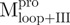 ) (open triangle), WT (filled square) and
) (open triangle), WT (filled square) and 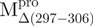 (open square). The enzyme in 30 mM Tris–HCl buffer (pH 7.7) was incubated with various concentrations of GdmCl for 10 min. The enzyme solution was excited at 295 nm and the emission fluorescence was recorded at 30°C. AEW of the enzyme was used to analyse the unfolding data. All mixtures had similar unfolding curve to WT Mpro. For direct comparison purpose, the duplicate presentation of data shown in two-dimensional space is provided (Supplementary Fig. 5S).
(open square). The enzyme in 30 mM Tris–HCl buffer (pH 7.7) was incubated with various concentrations of GdmCl for 10 min. The enzyme solution was excited at 295 nm and the emission fluorescence was recorded at 30°C. AEW of the enzyme was used to analyse the unfolding data. All mixtures had similar unfolding curve to WT Mpro. For direct comparison purpose, the duplicate presentation of data shown in two-dimensional space is provided (Supplementary Fig. 5S).
The enzyme activities of WT Mpro,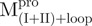 ,
, 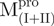 and the mixtures were measured using fluorescence-based peptide cleavage assays. The fluorescence enhancement of WT Mpro was collinear with time (Supplementary Fig. S3). However,
and the mixtures were measured using fluorescence-based peptide cleavage assays. The fluorescence enhancement of WT Mpro was collinear with time (Supplementary Fig. S3). However, 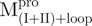 ,
, 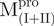 and all nicked enzymes were enzymatically inactive, even at a 10-fold concentration excess of the WT Mpro (Supplementary Fig. S3).
and all nicked enzymes were enzymatically inactive, even at a 10-fold concentration excess of the WT Mpro (Supplementary Fig. S3).
Quaternary structures of the full-length, N- and C-terminal-truncated SARS-CoV M pro and various mixtures
The quaternary structures of SARS-CoV Mpro were examined using AUC and the sedimentation velocity data were analysed using the continuous size distribution model with the SEDFIT programme (Fig. 6A). In experiments, no aggregation was observed at sedimentation coefficients of up to 20 S. The excellent matching of the experimental data points and the curve fitting results (Supplementary Fig. 4SA), the randomly distributed residual values (Supplementary Fig. 4SB), and the homogeneous bitmap picture (Supplementary Fig. 4SC) of the SARS-CoV Mpro all indicated that a highly reliable model for the sedimentation velocity experiments was obtained and the AUC was an excellent biophysical probe for accessing the quaternary structure of the enzyme. The WT Mpro existed as a dimeric form in solution but the 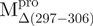 , N- and C-terminal domain-truncated Mpro forms were monomeric (Table I and Fig. 6A). A mixture of N- and C-terminal domain-truncated Mpro forms remained monomeric in solution (Fig. 6A). As dimerization is the prerequisite for enzyme activity of the Mpro, the absence of any proteolytic activity is easily explained.
, N- and C-terminal domain-truncated Mpro forms were monomeric (Table I and Fig. 6A). A mixture of N- and C-terminal domain-truncated Mpro forms remained monomeric in solution (Fig. 6A). As dimerization is the prerequisite for enzyme activity of the Mpro, the absence of any proteolytic activity is easily explained.
Fig. 6.
Quaternary structural analysis of the full-length, N- and C-terminal-truncated SARS-CoV Mpro and non-covalent complexes. Continuous sedimentation of the protein in 30 mM Tris–Cl (pH 7.7) was monitored with analytical ultracentrifuge. The sedimentation velocity data were analysed with SEDFIT. (A) The continuous sedimentation coefficient distribution of the protein [23.85 μM for Mpro,  ,
,  , Mixture 2 (
, Mixture 2 (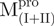 /
/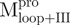 ), Mixture 3 (
), Mixture 3 (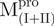 /
/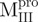 ) and Mixture 4 (
) and Mixture 4 (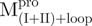 /
/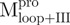 ), 22.8 μM for
), 22.8 μM for 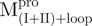 ,
, 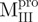 ,
, 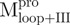 and Mixture 1 (
and Mixture 1 (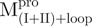 /
/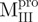 )]. (B) Quaternary structure of the complex in the presence of WT Mpro. All enzyme preparations were in 23.85 μM concentration except for Mixture 1 in 22.8 μM. For direct comparison purpose, the duplicate presentation of data shown in two-dimensional space is provided (Supplementary Fig. 6S).
)]. (B) Quaternary structure of the complex in the presence of WT Mpro. All enzyme preparations were in 23.85 μM concentration except for Mixture 1 in 22.8 μM. For direct comparison purpose, the duplicate presentation of data shown in two-dimensional space is provided (Supplementary Fig. 6S).
We examined the possibility of using WT Mpro as a template to induce the dimerization of mixtures (Fig. 6B). The WT proteases sedimented at 4.4 S with a molar mass of 66 kDa, in agreement with the molecular weight of the dimer (67.6 kDa) calculated from its amino acid sequence. There was a peak between 1.6 S and 2.1 S, corresponding to the truncated proteases. The WT Mpro could not induce dimer formation of the mixture. Thus, the covalent linkage between domains (I + II) and domain III was essential for the structural integrity of SARS-CoV Mpro.
Discussion
Complementation of protein fragments (reconstitution experiments) is an efficient method for inspecting the requirements for protein function and folding pathways (22–23). The function of the protein may be retained in the reconstituted complex depending on the number of cuts and where in the sequence they are made. Fragment complementing systems have been used to probe the molecular features required for fragments to associate and fold into a stable native-like protein complex (24) and to elucidate autonomous folding units. We applied this concept in a broad way to a protein that contains multiple domains. Our previous studies indicated that the C-terminal domain-truncated Mpro could fold independently into an intact chymotrypsin-like fold, but it showed no enzyme activity (10). Our other experimental data showed that the extra C-terminal domain played a pivotal role in controlling dimerization and enzyme activity (25). The present results on Mpro fragments further defined our earlier studies that domain III acts to stabilize the catalytic N-terminal domain (I+II). Furthermore, the different SARS-CoV Mpro-complementing fragment systems described herein were used to probe the structural role of the loop. We confirmed that the covalent linkage between domains (I+II) and (III) is essential, which suggests that the long loop might stabilize the C-terminal domain. It is likely that some critical structural stabilizing interactions are conserved in the loop-retaining fragments with respect to those present in the natural and longer SARS-CoV fragment. Because of this, 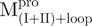 displays a higher content of β-sheets, which is its major secondary structure, than does
displays a higher content of β-sheets, which is its major secondary structure, than does 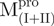 , and
, and 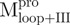 instead displays more helicity than
instead displays more helicity than 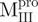 . Examining crystal structures of Mpro reveals that residues around 187 have interactions with residues around 40 and 54 of N-terminal domain, and residues around 198 have interactions with residues around 240 of C-terminal domain. Thus, the truncations we did very likely affected the folding and stability of the domains. This may well explain the differences, we observed by CD and the difference in stability for domains with and without the linker loop.
. Examining crystal structures of Mpro reveals that residues around 187 have interactions with residues around 40 and 54 of N-terminal domain, and residues around 198 have interactions with residues around 240 of C-terminal domain. Thus, the truncations we did very likely affected the folding and stability of the domains. This may well explain the differences, we observed by CD and the difference in stability for domains with and without the linker loop.
Spectroscopic studies by far-UV CD and reversible unfolding analysis in GdmCl indicate that fragments 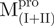 ,
, 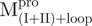 ,
, 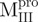 and
and 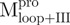 adopted folded states in aqueous solution at neutral pH and therefore appeared to possess correct tertiary structures. The mixtures of various combinations of these fragments (nicked SARS CoV Mpro) revealed structural features similar to those of the intact protein. However, some differences between the nicked and intact species were found from the AUC studies. These negative results seem to base on the mixing of the individual domains which are non-interacting. Little convinced result to show that the component in these mixtures could interact and form the structure like WT Mpro. The nicked proteins attained significant secondary and tertiary structures, as did the native enzyme. However, they seemed to lack specific domain–domain complementation (or interactions), thereby precluding a final folding step to assume the catalytic competent conformation, as in the native Mpro. Therefore, it appears that the individual N- and C-terminal domains can fold independently, but cannot associate to form the enzymatic active dimer. This is consistent with current experimental observations that only the dimeric form of SARS-CoV Mpro possesses biological function and that the dissociated monomer is enzymatically inactive (26–28). Shi et al. (3) have reported that the isolated extra domain III existed as a dimer even at a very low protein concentration and showed no concentration dependence by dynamic light scattering and size-exclusion FPLC analyses. A more recent paper reports that the C-terminal domain of SARS-CoV Mpro can form a 3D domain swapped dimer in the crystalline form (29). However, under our AUC experimental conditions, the
adopted folded states in aqueous solution at neutral pH and therefore appeared to possess correct tertiary structures. The mixtures of various combinations of these fragments (nicked SARS CoV Mpro) revealed structural features similar to those of the intact protein. However, some differences between the nicked and intact species were found from the AUC studies. These negative results seem to base on the mixing of the individual domains which are non-interacting. Little convinced result to show that the component in these mixtures could interact and form the structure like WT Mpro. The nicked proteins attained significant secondary and tertiary structures, as did the native enzyme. However, they seemed to lack specific domain–domain complementation (or interactions), thereby precluding a final folding step to assume the catalytic competent conformation, as in the native Mpro. Therefore, it appears that the individual N- and C-terminal domains can fold independently, but cannot associate to form the enzymatic active dimer. This is consistent with current experimental observations that only the dimeric form of SARS-CoV Mpro possesses biological function and that the dissociated monomer is enzymatically inactive (26–28). Shi et al. (3) have reported that the isolated extra domain III existed as a dimer even at a very low protein concentration and showed no concentration dependence by dynamic light scattering and size-exclusion FPLC analyses. A more recent paper reports that the C-terminal domain of SARS-CoV Mpro can form a 3D domain swapped dimer in the crystalline form (29). However, under our AUC experimental conditions, the 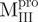 and
and 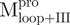 seem to exist majorly as a monomeric form in solution (Fig. 6A).
seem to exist majorly as a monomeric form in solution (Fig. 6A).
Taniuchi et al. (22) developed the concept of permissible sites of cleavage of a protein for producing fragment complementing systems and deduced that cleavages can occur at exposed, flexible loops but not in a typical secondary structure at the level of chain segments (Fig. 1C). In the present case, the cleaved peptide bonds are close to the adjacent essential structural domains. Therefore, the 183–184 and 199–200 peptide bond regions might not be permissible sites of cleavage. It would be intriguing to test chain fission at the middle of this loop.
In summary, because the C- and N-terminal domains of Mpro can fold independently and show a completely reversible unfolding and refolding process in GdmCl, SARS-CoV Mpro provides an ideal model system to delineate complicated protein folding in a feasible stepwise pathway. We demonstrated here that the mixtures of Mpro also showed a reversible biphasic unfolding curve, similar to the intact WT form. Nevertheless, there was no enzyme activity found for any mixture. In addition, no dimer formation between any truncated protease was observed with or without the presence of WT Mpro as a template. These results indicate that the covalent linkage between the chymotrypsin-like domain and the extra domain must be essential for enzyme activity and for the quaternary structural integrity of the SARS-CoV Mpro.
Funding
This work was supported by National Science Council, Republic of China (NSC 96-2113-M-415-010).
Conflict of interest
None decalred.
Supplementary Material
Glossary
Abbreviations
- SARS
severe acute respiratory syndrome
- CoV
coronavirus
- Mpro
main protease
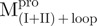
a C-terminal domain-truncated Mpro with loop retained
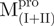
domain (I+II) of Mpro
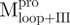
a N-terminal domain-truncated Mpro with loop retained
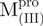
domain III of Mpro
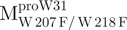
a double mutant of Mpro with the tryptophanyl residues at positions 207 and 218 mutated to phenylalanine, the only unaltered tryptophanyl residue was W31a
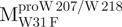
a point mutant of Mpro with tryptophanyl residue at position 31 mutated to phenylalanine
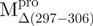
a deletion mutant of Mpro with C-terminal amino acid residus 297–306 deleted
- AEW
average emission wavelength
- CD
circular dichroism
- AUC
analytical ultracentrifugation
- GdmCl
guanidinium chloride
Footnotes
aFor consistency we have adopted a systematic abbreviation nomenclature for all engineered enzymes: superscript font for unaltered residues and subscript for the engineered portion.
References
- 1.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 2.Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H, Gao GF, Anand K, Bartlam M, Hilgenfeld R, Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl Acad. Sci. USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi J, Wei Z, Song J. Dissection study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme: defining the extra domain as a new target for design of highly specific protease inhibitors. J. Biol. Chem. 2004;279:24765–24773. doi: 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liang PH. Characterization and inhibition of SARS-coronavirus main protease. Curr. Top. Med. Chem. 2006;6:361–376. doi: 10.2174/156802606776287090. [DOI] [PubMed] [Google Scholar]
- 5.Ratia K, Saikatendu KS, Santarsiero BD, Barretto N, Baker SC, Stevens RC, Mesecar AD. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl Acad. Sci. USA. 2006;103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartlam M, Yang H, Rao Z. Structural insights into SARS coronavirus proteins. Curr. Opin. Struct. Biol. 2005;15:664–672. doi: 10.1016/j.sbi.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J, Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin PY, Chou CY, Chang HC, Hsu WC, Chang GG. Correlation between dissociation and catalysis of SARS-CoV main protease. Arch. Biochem. Biophys. 2008;472:34–42. doi: 10.1016/j.abb.2008.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeLano WL. The Pymol Molecular Graphic System. San Carlos, CA, USA: DeLano Scientific; 2002. [Google Scholar]
- 10.Chang HP, Chou CY, Chang GG. Reversible unfolding of the severe acute respiratory syndrome coronavirus main protease in guanidinium chloride. Biophys. J. 2007;92:1374–1383. doi: 10.1529/biophysj.106.091736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, Li J, Zhou Z, Ding Y, Zhao Q, Zhang XC, Liao M, Bartlam M, Rao Z. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 2008;82:2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lobley A, Whitmore L, Wallace BA. DICHROWEB: an interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics. 2002;18:211–212. doi: 10.1093/bioinformatics/18.1.211. [DOI] [PubMed] [Google Scholar]
- 13.Whitmore L, Wallace BA. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32:W668–W673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Compton LA, Johnson W.C., Jr Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal. Biochem. 1986;155:155–167. doi: 10.1016/0003-2697(86)90241-1. [DOI] [PubMed] [Google Scholar]
- 15.Manavalan P, Johnson W.C., Jr Variable selection method improves the prediction of protein secondary structure from circular dichroism spectra. Anal. Biochem. 1987;167:76–85. doi: 10.1016/0003-2697(87)90135-7. [DOI] [PubMed] [Google Scholar]
- 16.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 17.Royer CA, Mann CJ, Matthews CR. Resolution of the fluorescence equilibrium unfolding profile of trp aporepressor using single tryptophan mutants. Protein Sci. 1993;2:1844–1852. doi: 10.1002/pro.5560021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown PH, Schuck P. Macromolecular Size-and-Shape Distributions by Sedimentation Velocity Analytical Ultracentrifugation. Biophys. J. 2006;90:4651–4661. doi: 10.1529/biophysj.106.081372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pace CN. Measuring and increasing protein stability. Trends Biotechnol. 1990;8:93–98. doi: 10.1016/0167-7799(90)90146-o. [DOI] [PubMed] [Google Scholar]
- 21.Shi J, Sivaraman J, Song J. Mechanism for controlling the dimer-monomer switch and coupling dimerization to catalysis of the severe acute respiratory syndrome coronavirus 3C-like protease. J. Virol. 2008;82:4620–4629. doi: 10.1128/JVI.02680-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taniuchi H, Parr GR, Juillerat MA. Complementation in folding and fragment exchange. Methods Enzymol. 1986;131:185–217. doi: 10.1016/0076-6879(86)31042-5. [DOI] [PubMed] [Google Scholar]
- 23.de Prat-Gay G. Association of complementary fragments and the elucidation of protein folding pathways. Protein Eng. 1996;9:843–847. doi: 10.1093/protein/9.10.843. [DOI] [PubMed] [Google Scholar]
- 24.Musi V, Spolaore B, Picotti P, Zambonin M, De Filippis V, Fontana A. Nicked apomyoglobin: a noncovalent complex of two polypeptide fragments comprising the entire protein chain. Biochemistry. 2004;43:6230–6240. doi: 10.1021/bi0400103. [DOI] [PubMed] [Google Scholar]
- 25.Hsu WC, Chang HC, Chou CY, Tsai PJ, Lin PI, Chang GG. Critical assessment of important regions in the subunit association and catalytic action of the severe acute respiratory syndrome coronavirus main protease. J. Biol. Chem. 2005;280:22741–22748. doi: 10.1074/jbc.M502556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chou CY, Chang HC, Hsu WC, Lin TZ, Lin CH, Chang GG. Quaternary structure of the severe acute respiratory syndrome (SARS) coronavirus main protease. Biochemistry. 2004;43:14958–14970. doi: 10.1021/bi0490237. [DOI] [PubMed] [Google Scholar]
- 27.Fan K, Wei P, Feng Q, Chen S, Huang C, Ma L, Lai B, Pei J, Liu Y, Chen J, Lai L. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Graziano V, McGrath WJ, Yang L, Mangel WF. SARS CoV main proteinase: The monomer-dimer equilibrium dissociation constant. Biochemistry. 2006;45:14632–14641. doi: 10.1021/bi061746y. [DOI] [PubMed] [Google Scholar]
- 29.Zhong N, Zhang S, Xue F, Kang X, Zou P, Chen J, Liang C, Rao Z, Jin C, Lou Z, Xia B. C-terminal domain of SARS-CoV main protease can form a 3D domain-swapped dimer. Protein Sci. 2009;18:839–844. doi: 10.1002/pro.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.