

Abstract
BackgroundEbola virus (EBOV) infection causes a frequently fatal hemorrhagic fever (HF) that is refractory to treatment with currently available antiviral therapeutics. RNA interference represents a powerful, naturally occurring biological strategy for the inhibition of gene expression and has demonstrated utility in the inhibition of viral replication. Here, we describe the development of a potential therapy for EBOV infection that is based on small interfering RNAs (siRNAs)
MethodsFour siRNAs targeting the polymerase (L) gene of the Zaire species of EBOV (ZEBOV) were either complexed with polyethylenimine (PEI) or formulated in stable nucleic acid–lipid particles (SNALPs). Guinea pigs were treated with these siRNAs either before or after lethal ZEBOV challenge
ResultsTreatment of guinea pigs with a pool of the L gene–specific siRNAs delivered by PEI polyplexes reduced plasma viremia levels and partially protected the animals from death when administered shortly before the ZEBOV challenge. Evaluation of the same pool of siRNAs delivered using SNALPs proved that this system was more efficacious, as it completely protected guinea pigs against viremia and death when administered shortly after the ZEBOV challenge. Additional experiments showed that 1 of the 4 siRNAs alone could completely protect guinea pigs from a lethal ZEBOV challenge
ConclusionsFurther development of this technology has the potential to yield effective treatments for EBOV HF as well as for diseases caused by other agents that are considered to be biological threats
Ebola virus (EBOV; family Filoviridae) is a single-stranded, negative-sense RNA virus that is among the best known of the viruses that cause hemorrhagic fever (HF). Although outbreaks have been sporadic and geographically restricted to areas of Central Africa, the HFs caused by these viruses are remarkably severe and are associated with high case fatality rates, which often exceed 80%. In addition to causing human infection, these viruses have decimated populations of wild apes in Central Africa [1]. Although significant advances have been made in the development of vaccines to combat EBOV infection, there are at present no vaccines or effective therapies available for human use. There is clearly a need to develop effective treatments that can be used to respond to outbreaks of EBOV in Africa and to counter acts of bioterrorism that may occur. In addition, the unfortunate death of a Russian scientist after accidental exposure to EBOV [2] underscores the need for medical countermeasures for postexposure prophylaxis
EBOV particles contain an ∼19-kb noninfectious RNA genome that encodes 7 structural proteins and 1 nonstructural protein, with a gene order of 3′ leader, nucleoprotein (NP), virion protein (VP) 35, VP40, glycoprotein, VP30, VP24, polymerase (L) protein, and 5′ trailer [3]. Four of these proteins are associated with the viral genomic RNA in the ribonucleoprotein complex: NP, VP30, VP35, and the L protein. The L and VP35 proteins together comprise the polymerase complex, which is responsible for transcribing and replicating the EBOV genome. The L protein provides the RNA-dependent RNA polymerase activity of the complex and, thus, is an ideal target for antiviral approaches—not only because its suppression should lead to a nearly total loss of all RNA synthesis, but also because of the absence of similar proteins in mammalian cells
RNA interference (RNAi) represents a powerful, naturally occurring biological strategy for inhibiting gene expression. RNAi has been used in cell-culture systems to inhibit the replication of a number of viruses that cause disease in humans, including HIV, hepatitis B virus (HBV), hepatitis C virus, influenza virus, herpesviruses, poliovirus, human papillomavirus, respiratory syncytial virus, and coxsackievirus (reviewed in [4, 5]); more recently, it has been used to inhibit some emerging and reemerging viruses, including Marburg virus [6], lymphocytic choriomeningitis virus [7], and severe acute respiratory syndrome coronavirus [8]. Although these in vitro results have been highly encouraging, the difficulty involved in the effective delivery of small interfering RNAs (siRNAs) in vivo has been the major obstacle to their use as therapeutic agents
Several approaches have been employed in attempts to develop an in vivo siRNA delivery system. Early proof-of-concept studies included the use of mouse models and rapid hydrodynamic intravenous (iv) injection of large volumes of siRNA solution [9–11]. However, this invasive procedure appears to have little utility for human use. Several key breakthroughs have highlighted the feasibility of the use of siRNAs as antiviral therapeutics. Researchers have shown that cationic polymers have the ability to promote the successful delivery of siRNA by iv administration in influenza virus–infected mice [12]. More recently, the efficacy of lipid-encapsulated siRNAs targeted to HBV has been demonstrated in an in vivo mouse model of HBV replication [13]. In that study, siRNA targeted to HBV RNA was incorporated into a specialized liposome to form a stable nucleic acid–lipid particle (SNALP) and administered to HBV-infected mice. Importantly, the reduction in the quantity of HBV DNA observed was specific and lasted for up to 7 days after administration. Here, we used SNALPs as a postexposure treatment in a lethal animal model of EBOV HF
Materials and Methods
Guinea pigs and virusAn animal model of EBOV infection and pathogenesis has been developed in inbred strain 13 guinea pigs (US Army Medical Research Institute of Infectious Diseases) by serial passage of the Zaire species of EBOV (ZEBOV; Mayinga isolate) 4 times [14]. The resulting guinea pig–adapted strain gives rise to high plasma viremia (typically >1×105 pfu/mL) and is highly lethal to guinea pigs, typically causing death 7–12 days after challenge. Infection experiments were performed under biosafety level (BSL)–4 biocontainment. Research was conducted in compliance with the Animal Welfare Act and other federal statues and regulations relating to animals and to experiments involving animals and adhered to the principles stated in the Guide for the Care and Use of Laboratory Animals [15]. The BSL-4 facility used is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. Noninfectious mouse and guinea pig clearance and biodistribution experiments were conducted at Protiva Biotherapeutics, in accordance with the guidelines of the Canadian Council on Animal Care
siRNAssiRNAs were designed to target individual regions of the ZEBOV L gene, in accordance with “the Tuschl rules” (available at: http://www.rockefeller.edu/labheads/tuschl/sirna.html). The siRNAs duplexes were chemically synthesized by Dharmacon or TriLink Biotechnologies. Sequences used were designated as follows: EK1, 5′-GUACGAAGCUGUAUAUAAAdTdT-3′ (sense) and 5′-UUUAUAUACAGCUUCGUACdTdT-3′ (antisense); EK2, 5′-GGAUCUUGGUACAGUGUUAdTdT-3′ (sense) and 5′-UAACACUGUACCAAGAUCCdTdT-3′ (antisense); EK3, 5′-CAGGCUUAUUCCAGUUAAAdTdT-3′ (sense) and 5′-UUUAACUGGAAUAACCUGdTdT-3′ (antisense); EK4, 5′-GUAAACGGCUGAACAUUAUdTdT-3′ (sense) and 5′-AUAAUGUUCAGCCGUUUACdTdT-3′ (antisense); and EbL-Scram1, 5′-CAAAAAAAUUUUUACGGGGdTdT-3′ (sense) and 5′-CCCCGUAAAAAUUUUUUUGdTdT-3′ (antisense)
Lipid encapsulation of siRNAsiRNA were encapsulated by the process of spontaneous vesicle formation reported by Jeffs et al. [16]. SNALPs were composed of synthetic cholesterol (Sigma), the phospholipid DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine; Avanti Polar Lipids), the PEG lipid PEGC-DMA (3-N-[(ω-methoxy poly(ethylene glycol)2000)carbamoyl]-1,2-dimyrestyloxy-propylamine), and the cationic lipid DLinDMA (1,2-dilinoleyloxy-3-N,N-dimethylaminopropane), at the molar ratio 48:20:2:30. PEG-C-DMA and DLinDMA were synthesized as described elsewhere [17]. The resulting SNALPs were dialyzed in PBS and filter sterilized (0.2-μm filter) before use. Particle sizes ranged from 71 to 84 nm, and typically 90%–95% of the siRNA was found to be encapsulated within these liposomes
Cell-culture experimentssiRNAs (60 pmol) were transfected into Vero cells by use of Oligofectamine (Invitrogen), in accordance with the manufacturer’s instructions. At 0, 24, or 48 h after transfection, Vero cells were infected with ZEBOV at an MOI of 1.0. Culture fluids were collected at 24, 48, and 96 h for determination of the level of infectious ZEBOV, and Vero cells were collected for immunofluorescence staining
In vivo pharmacokinetics and tissue distributionRadiolabeled SNALPs were prepared for plasma clearance and biodistribution experiments by incorporation of 16 μCi of the nonexchangeable lipid label [3H]cholesteryl oleyl ether (CHE) per milligram of total lipid [18]. SNALPs were administered in a single bolus of 0.75 mg of siRNA per kilogram of body weight to 6-week-old female and male Hartley guinea pigs (Charles River Laboratories) via ear vein injection, and blood was collected via contralateral ear nicks over the course of 24 h. At 24 h, guinea pigs were killed by CO2 inhalation, and harvested tissues were homogenized in lysing matrix tubes (MP Biomedicals) containing 500 μL of distilled water. Homogenates were assayed for radioactivity by liquid scintillation counting with Picofluor 40 (tissues) or Picofluor 15 (blood) (PerkinElmer)
siRNA treatment and ZEBOV challenge of guinea pigs siRNAs (30 nmol total) were mixed with polyethylenimine (PEI) (in vivo jetPEI; Qbiogene) at an N/P ratio of 5 at room temperature for 20 min, in accordance with the manufacturer’s instructions. Guinea pigs were treated via intraperitoneal (ip) injection of 300 μL of the PEI polyplexes, corresponding to 8 mg/kg siRNA. Three hours after treatment, guinea pigs were challenged via subcutaneous (sc) injection of 1000 pfu of guinea pig–adapted ZEBOV. The guinea pigs received additional treatments with the PEI polyplexes (prepared as described above immediately before administration) at 24, 48, and 96 h after the ZEBOV challenge
For experiments evaluating the SNALP delivery system, the SNALP-formulated siRNAs were administered ip 1 h after challenge of guinea pigs via sc injection of 1000 pfu of guinea pig–adapted ZEBOV. The guinea pigs received additional treatments with the SNALP-formulated siRNAs at 24, 48, 72, 96, 120, and 144 h after the ZEBOV challenge. The guinea pigs were carefully monitored for signs of disease and survival during the 30-day course of the experiment
Virus titration by plaque assayVirus titration was performed by conventional plaque assay on Vero E6 cells from cell-culture fluids of blood collected from guinea pigs, as described elsewhere [14, 19]
Immunofluorescence assayCells were fixed with 10% neutral-buffered formalin for 24 h, to inactivate infectious ZEBOV. After fixation, cells were washed with copious amounts of PBS and processed for immunofluorescence staining for viral proteins. Briefly, cells were incubated in ready-to-use Proteinase K (Dako) for 10 min at room temperature. Cells were then washed with PBS and blocked in normal goat serum (KPL Laboratories) for 20 min at room temperature. Viral antigen was detected by incubating cells with a mouse monoclonal antibody against the guinea pig–adapted ZEBOV for 20 min at room temperature, rinsing in PBS, and incubating with an anti-mouse–AlexaFluor 488 conjugate (Invitrogen) for 20 min at room temperature. Cells were counterstained with 4′,6′-diamidino-2-phenylindole and Evans blue, to aid visualization. The percentage of antigen-positive cells was determined by examining random fields for fluorescence
In vivo cytokine inductionSNALPs were administered in 0.2 mL of PBS to 6–8-week-old CD1 ICR mice (Harlan) by standard iv injection in the lateral tail vein. Blood was collected by cardiac puncture 6 h after administration and then processed as plasma for cytokine analysis. Levels of the mouse cytokines interferon (IFN)–α and IFN-β were measured by use of sandwich ELISA kits (PBL Biomedical), in accordance with the manufacturer’s instructions
Results
To determine whether siRNAs can inhibit the replication of ZEBOV in vitro, we first transfected Vero cells with either a pool of 4 different siRNAs (EK1–EK4) specific for the ZEBOV L gene or an equivalent dose of an irrelevant scrambled sequence (EbL-Scram1). We used Vero cells because they lack the structural genes for IFN-α and IFN-β [20–22], thus precluding any confounding effects of these cytokines. At various time points after transfection (0, 24, and 48 h), the transfected cells were infected with ZEBOV. Cells and culture fluids were harvested 24 h later, to determine the level of virus production. The results showed that the siRNA pool inhibited the production of infectious ZEBOV 2–10-fold, depending on when the transfected cells were infected (figure 1). By immunofluorescence antibody staining, we were able to demonstrate a mean ± SD reduction of 83%±14% (P=.006, Student’s t test) in the numbers of cells expressing EBOV protein (figure 2). Individual testing of the 4 L gene–specific siRNAs yielded similar results (data not shown). The irrelevant scrambled sequence did not inhibit the production of infectious ZEBOV and failed to reduce the numbers of cells expressing EBOV protein (figures 1 and 2)
Figure 1.
Inhibition of the replication of the Zaire species of Ebola virus (ZEBOV) in Vero cells by a pool of 4 different small interfering RNAs (siRNAs) targeting individual regions of the ZEBOV polymerase (L) gene. Cells were transfected with either the siRNA pool or an equivalent dose of EbL-Scram1, an irrelevant scrambled sequence (SCR), as a control. At various time points after transfection (0, 24, and 48 h), the transfected cells were infected with ZEBOV, and cells and culture fluids were harvested 24 h later, for determination of the level of infectious virus by plaque assay
Figure 2.
Inhibition of the replication of the Zaire species of Ebola virus (ZEBOV) in Vero cells, as demonstrated by immunofluorescence staining. Cells were transfected with either a pool of 4 different small interfering RNAs (siRNAs) targeting individual regions of the ZEBOV polymerase (L) gene or an equivalent dose of EbL-Scram1, an irrelevant scrambled sequence (SCR), as a control. Cells were counterstained with 4′,6′-diamidino-2-phenylindole and Evans blue, to aid visualization. ZEBOV-positive cells are identified by green fluorescence
We then tested whether the pool of 4 siRNAs could protect guinea pigs from a lethal ZEBOV challenge. Although both mouse and guinea pig models are available for ZEBOV infection [14, 23, 24], we chose to use guinea pigs for these experiments because (1) they appear (on the basis of evaluation of coagulation changes [25, 26]) to reproduce human filoviral infection slightly better than mice and (2) there are no mouse models available for other filoviruses, whereas guinea pigs have been used to study the Sudan species of EBOV [27] and as models for infection with several strains of the EBOV-related Marburg virus [28, 29]. In the first in vivo experiment, guinea pigs were treated with either the L gene–specific siRNA pool (n=5) or the irrelevant scrambled sequence (n=5), which were mixed with PEI and injected retroorbitally 3 h before the ZEBOV challenge; the guinea pigs then received equivalent doses of the siRNAs (8 mg/kg) at 24, 48, and 96 h after the ZEBOV challenge. A significant reduction in plasma viremia (P=.02) was demonstrated on day 4 after challenge in the guinea pigs receiving the L gene–specific siRNA pool, compared with that in the guinea pigs receiving the irrelevant scrambled sequence (figure 3). All of the control guinea pigs treated with the irrelevant scrambled sequence died or were euthanized by day 12 after challenge, whereas 1 of the guinea pigs treated with the siRNA pool survived and the other 4 guinea pigs either died or were euthanized on day 10, 13, 13, and 14, respectively
Figure 3.
Plasma viremia levels of inbred strain 13 guinea pigs 4 days after challenge with the Zaire species of Ebola virus (ZEBOV). Guinea pigs were treated 3 h before the ZEBOV challenge and 1, 2, and 4 days afterward with either a pool of 4 different small interfering RNAs (siRNAs) targeting individual regions of the ZEBOV polymerase (L) gene (n=5) or an equivalent dose of EbL-Scram1, an irrelevant scrambled sequence (SCR) (n=5). Data are means±SDs
To improve the potency of the 4 L gene–specific siRNAs, they were encapsulated in lipid particles previously shown to have antiviral efficacy in a mouse model of HBV infection [13]. The SNALP method yields particles with uniform, reproducible performance specifications regardless of the siRNA payload. The mean±SD particle size of the 7 SNALP preparations used in the present study was 81±3.0 nm, and the mean±SD polydispersity was 0.11±0.024. The mean±SD encapsulation efficiency was 92%±1.5%, with a mean±SD nucleic acid:lipid ratio of 49±2.7 μg of siRNA/μmol of lipid
To assess plasma clearance and biodistribution, SNALPs were prepared containing the nonexchangable lipid label 3H-CHE [18], as described elsewhere [30]. The plasma clearance of SNALPs in guinea pigs was determined after a single iv administration. We have previously shown that iv administration of unstabilized, unformulated siRNA in mice results in rapid elimination from the plasma compartment, with an elimination half-life of ∼2 min [13]. The calculated serum half-life for the SNALPs used in the present study was 39.3 min after administration in guinea pigs (figure 4A)
Figure 4.
In vivo clearance and biodistribution of stable nucleic acid–lipid particles (SNALPs). Shown are plasma clearance (A) and biodistribution (B and C) of [3H]cholesteryl oleyl ether–labeled SNALPs. Each guinea pig received a single intravenous injection of 0.75 mg/kg small interfering RNAs (siRNA) formulated as SNALPs. Biodistribution data were collected 24 h after injection. Data are means±SDs (for all tissues, n=5 guinea pigs, except for testes [n=3] and ovaries [n=2])
Because the liver is known to be one of the early and primary sites of ZEBOV replication in rodents and nonhuman primates [14, 31], it was also of interest to determine the pattern of biodistribution after SNALP administration. As is shown in figure 4B and 4C, substantial quantities of SNALPs accumulated in the liver (mean±SD, 83.4%±6.5%) within 24 h after injection. Substantially fewer SNALPs accumulated in the spleen (mean±SD, 2.2%±0.27%) and the lungs (mean±SD, 2.29% ± 0.28%). The brain (mean±SD, 0.068%±0.016%), gonadal tissues (mean±SD, 0.016%±0.002% and 0.08% ± 0.012% for ovaries and testes, respectively), and thymus (mean ± SD, 0.0044%±0.0006%) remained relatively inaccessible, accumulating very few SNALPs. This rapid and selective accumulation of SNALPs in the liver compares favorably with the results of targeted delivery technologies that use receptor-ligand interactions as well as with the results of other targeting technologies [32–35]
We evaluated the pool of the 4 L gene–specific siRNAs using the SNALP delivery system in a rodent model of EBOV HF. As with the previous challenge experiment, 5 guinea pigs received the siRNA pool (1.0 mg/kg), and 5 control guinea pigs received the irrelevant scrambled sequence (1.0 mg/kg). However, in this experiment, the guinea pigs were not treated with siRNA before the ZEBOV challenge. Rather, treatment was initiated 1 h after the challenge, and additional treatments were administered daily on days 1–6 after infection. Plasma viremia levels, which peak on day 7 in this guinea pig model [14], were not detected on day 7 in any of the guinea pigs treated with the siRNA pool but ranged from ∼3.5 to 4.5 log10 pfu/mL in the control guinea pigs (figure 5A). One guinea pig treated with the siRNA pool died on day 6, most likely because of toxicity, given that we were unable to demonstrate the presence of infectious ZEBOV in this animal. Another guinea pig from this treatment group had to be euthanized on day 26; this death could not be attributed to viral replication either. The remaining 3 guinea pigs treated with the siRNA pool did not show any evidence of illness and survived the ZEBOV challenge, whereas all 5 of the control guinea pigs died or were euthanized by day 13 (figure 5B)
Figure 5.
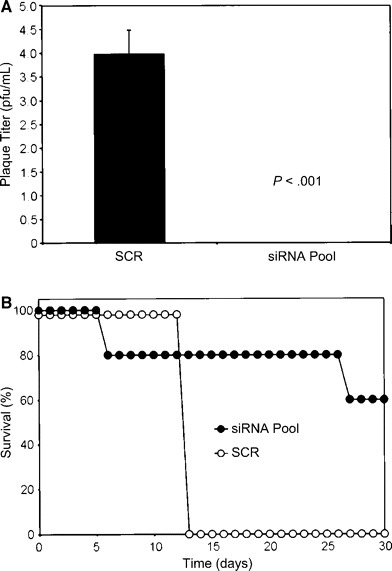
Antiviral efficacy of a pool of 4 different small interfering RNAs (siRNAs) targeting individual regions of the Zaire species of Ebola virus (ZEBOV) polymerase (L) gene and encapsulated in stable nucleic acid–lipid particles (SNALPs). Shown are plasma viremia levels (A) and survival rates (B) for inbred strain 13 guinea pigs after ZEBOV challenge. One hour after challenge and daily on days 1–6 thereafter, guinea pigs were treated, via the SNALP delivery system, with the siRNA pool (1.0 mg/kg) or an equivalent dose of EbL-Scram1, an irrelevant scrambled sequence (SCR). Plasma viremia levels were determined on day 7. Viremia data are means±SDs (n=5)
A further experiment was performed to assess the efficacy of the siRNA pool at a lower dose and to individually evaluate the 4 L gene–specific siRNAs (EK1–EK4). ZEBOV-infected guinea pigs that received a lower dose (0.75 mg/kg) of the siRNA pool were completely protected from viremia and death (figure 6). Although varying levels of viremia and mortality were observed among the groups treated with individual siRNAs (figure 6), the lowest levels of mortality were associated with the EK1- and EK4-treated guinea pigs
Figure 6.
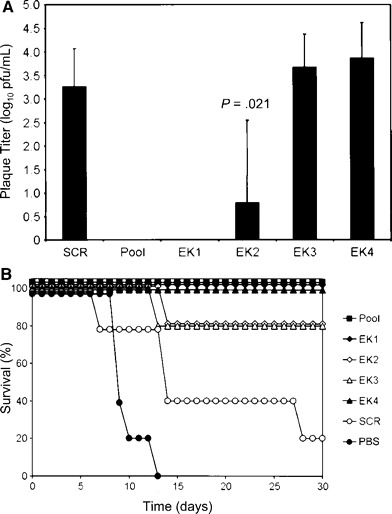
Antiviral efficacy of small interfering RNAs (siRNAs) targeting individual regions of the Zaire species of Ebola virus (ZEBOV) polymerase (L) gene and encapsulated in stable nucleic acid–lipid particles (SNALPs). Shown are plasma viremia levels (A) and survival rates (B) for inbred strain 13 guinea pigs after ZEBOV challenge. One hour after the challenge and daily on days 1–6 thereafter, guinea pigs were treated, via the SNALP delivery system, with a pool of 4 siRNA (0.75 mg/kg); an equivalent dose of EbL-Scram1, an irrelevant scrambled sequence (SCR); or 1 of the 4 siRNAs alone (EK1–EK4). Plasma viremia levels were determined on day 7. Viremia data are means±SDs (n=5). The P value is for the comparison between EK2 and SCR
Recent studies have demonstrated that synthetic siRNA can induce a high level of type I IFN and inflammatory cytokines in mammalian cells [36, 37] and that this immune response could contribute to the antiviral efficacy or toxicities associated with systemic administration of formulated siRNA [13, 36]. The immunostimulatory properties of the siRNA-containing SNALPs were examined directly after iv administration in mice. Strikingly, all of the SNALP-formulated siRNAs, including the irrelevant scrambled sequence, induced IFN-α and IFN-β in the serum of the injected mice (figure 7). Both the guinea pigs and mice that were treated with the immunostimulatory SNALPs showed symptoms of systemic toxicity, including a transient decrease in body weight and piloerection (data not shown), that have previously been shown to be associated with siRNA-mediated stimulation of the mammalian innate immune system [13, 36]. These adverse effects were not evident in the control animals treated with PBS or in the animals injected with empty liposomes or naked siRNA. Of note, EK1, which conferred the greatest benefit when used to treat ZEBOV-infected animals, was the least immunostimulatory of the siRNAs examined. This provides further evidence supporting an RNAi-specific antiviral effect in our infection experiments
Figure 7.
Small interfering RNA (siRNA)–mediated cytokine induction in mice. Shown are serum interferon (IFN)–α (A) and IFN-β (B) levels 6 h after intravenous administration of 100 μg (∼5 mg/kg) of stable nucleic acid–lipid particles encapsulating either a pool of 4 different siRNAs that targeted individual regions of the Zaire species of Ebola virus polymerase (L) gene; an equivalent dose of EbL-Scram1, an irrelevant scrambled sequence (SCR); or 1 of the 4 siRNAs alone (EK1–EK4). Injection of PBS alone induced no detectable IFN-α or IFN-β. Note that injection of empty liposomes or naked siRNA alone also failed to induce detectable IFN-α or IFN-β (data not shown). Data are means±SDs (n=4)
Discussion
Currently, there are no known effective pre- or postexposure therapies for human EBOV infection. At this time, treating patients infected with EBOV consists of palliative care directed toward maintaining blood volume and electrolyte balance. A number of therapies—including IFN-α, heparin, convalescent serum, and equine anti-EBOV immunoglobulin—have been used to treat infections in humans and/or nonhuman primates, but the results have been inconsistent and, in general, of little effect [19, 38]. Although a number of postexposure treatments have shown promise in rodent models of EBOV HF [39–41], none has shown any success in nonhuman primates. To date, the only postexposure treatment that has been shown to protect nonhuman primates after challenge with ZEBOV is a strategy designed to modulate the manifestations of disease rather than to block replication of the virus; in a study in rhesus monkeys [42], we demonstrated that a postexposure strategy to mitigate the coagulation disorders that typify EBOV infection improved survival from 0% to 33%. However, improved efficacy is clearly needed
Although there is no assurance that the SNALP-based siRNA strategy described here will protect against EBOV infection in the more-rigorous nonhuman primate models, there is reason for optimism in light of comparative data from historical studies. Specifically, as noted above, several previous studies have shown complete postexposure protection of mice [39, 40] and guinea pigs [41] against a lethal ZEBOV challenge; however, in each of these cases, the rodents were not protected against viremia. Subsequent transition of these strategies to nonhuman primates was uniformly unsuccessful in protecting macaques against a lethal infection [43, 44]. This suggests that the inability to completely inhibit viremia in rodents predicts an unfavorable outcome in nonhuman primates. In the present study, after administration of the EK1 siRNA, we were unable to detect viremia in any of the ZEBOV-challenged guinea pigs. Clearly, this observation does not guarantee success of the SNALP-based siRNA approach in nonhuman primates; on the other hand, it does offer some hope that the prospects for success are at least improved
Here, we have focused on the L gene of ZEBOV, to demonstrate the in vivo utility of the SNALP technology. Future studies will focus on evaluating sequences of other EBOV genes and the possibility of employing cocktails of siRNAs for increased antiviral effect. Furthermore, because of the unique mechanism of RNAi, there may be benefits to combining a siRNA treatment for EBOV infection with complementary antiviral approaches, such as immunoglobulins or coagulation inhibitors
Acknowledgments
We thank Denise Braun and Carlton Rice, for technical assistance and assistance with animal care
Footnotes
Presented in part: 13th International Congress of Virology, San Francisco, 23–28 July 2005 (abstract V-155)
Potential conflicts of interest: T.W.G., L.E.H., E.K., and I.M. claim intellectual property regarding RNA interference for the treatment of filoviral infections; K.M., J.R.P., A.C.H.L., A.J., L.B.J., and I.M. are employees of Protiva Biotherapeutics
Financial support: Defense Threat Reduction Agency and the Medical Chemical/Biological Defense Research Program, US Army Medical Research and Material Command (projects 02-4-4J-081 and 06-4-4J-012)
The opinions, interpretations, conclusions, and recommendations expressed in this article are those of the authors and are not necessarily endorsed by the US Army
References
- 1.Walsh PD, Abernethy KA, Bermejo M, et al. Catastrophic ape decline in western equatorial Africa. Nature. 2003;422:611–4. doi: 10.1038/nature01566. [DOI] [PubMed] [Google Scholar]
- 2.International Society for Infectious Diseases Ebola, lab accident death—Russia (Siberia). Archive no: 20040522.1377. 22 May. 2004 Available at: http://www.promedmail.org. Accessed 1 May 2006.
- 3.Sanchez A, Kiley MP, Holloway BP, Auperin DD. Sequence analysis of the Ebola virus genome: organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res. 1993;29:215–40. doi: 10.1016/0168-1702(93)90063-s. [DOI] [PubMed] [Google Scholar]
- 4.Tan FL, Yin JQ. RNAi, a new therapeutic strategy against viral infection. Cell Res. 2004;14:460–6. doi: 10.1038/sj.cr.7290248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colbere-Garapin F, Blondel B, Saulnier A, Pelletier I, Labadie K. Silencing viruses by RNA interference. Microbes Infect. 2005;7:767–75. doi: 10.1016/j.micinf.2005.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fowler T, Bamberg S, Moller P, et al. Inhibition of Marburg virus protein expression and viral release by RNA interference. J Gen Virol. 2005;86:1181–8. doi: 10.1099/vir.0.80622-0. [DOI] [PubMed] [Google Scholar]
- 7.Sanchez AB, Perez M, Cornu T, de la Torre JC. RNA interference-mediated virus clearance from cells both acutely and chronically infected with the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol. 2005;79:11071–81. doi: 10.1128/JVI.79.17.11071-11081.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu CJ, Huang HW, Liu CY, Hong CF, Chan YL. Inhibition of SARS-CoV replication by siRNA. Antiviral Res. 2005;65:45–8. doi: 10.1016/j.antiviral.2004.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCaffrey AP, Meuse L, Pham TT, Conklin DS, Hannon GJ. RNA interference in adult mice. Nature. 2002;418:38–9. doi: 10.1038/418038a. [DOI] [PubMed] [Google Scholar]
- 10.Giladi H, Ketzinel-Gilad M, Rivkin L, Felig Y, Nussbaum O. Small interfering RNA inhibits hepatitis B virus replication in mice. Mol Ther. 2003;8:769–76. doi: 10.1016/s1525-0016(03)00244-2. [DOI] [PubMed] [Google Scholar]
- 11.Song E, Lee SK, Wang J, et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med. 2003;9:347–51. doi: 10.1038/nm828. [DOI] [PubMed] [Google Scholar]
- 12.Ge Q, Filip L, Bai A, Nguyen T, Eisen HN. Inhibition of influenza virus production in virus-infected mice by RNA interference. Proc Natl Acad Sci USA. 2004;101:8676–81. doi: 10.1073/pnas.0402486101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrissey DV, Lockridge JA, Shaw L, et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat Biotechnol. 2005;23:1002–7. doi: 10.1038/nbt1122. [DOI] [PubMed] [Google Scholar]
- 14.Connolly BM, Steele KE, Davis KJ, et al. Pathogenesis of experimental Ebola virus infection in guinea pigs. J Infect Dis. 1999;179(Suppl 1):S203–17. doi: 10.1086/514305. [DOI] [PubMed] [Google Scholar]
- 15.National Research Council . Guide for the care and use of laboratory animals. Washington, DC: National Academy Press; 1996. [Google Scholar]
- 16.Jeffs LB, Palmer LR, Ambegia EG, Giesbrecht C, Ewanick S. A scalable, extrusion-free method for efficient liposomal encapsulation of plasmid DNA. Pharm Res. 2005;22:362–72. doi: 10.1007/s11095-004-1873-z. [DOI] [PubMed] [Google Scholar]
- 17.Heyes J, Palmer L, Bremner K, MacLachlan I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J Control Release. 2005;107:276–87. doi: 10.1016/j.jconrel.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 18.Stein Y, Halperin G, Stein O. Biological stability of [3H]cholesteryl oleyl ether in cultured fibroblasts and intact rat. FEBS Lett. 1980;111:104–6. doi: 10.1016/0014-5793(80)80771-x. [DOI] [PubMed] [Google Scholar]
- 19.Jahrling PB, Geisbert TW, Geisbert JB, et al. Evaluation of immune globulin and recombinant interferon-α2b for treatment of experimental Ebola virus infections. J Infect Dis. 1999;179(Suppl 1):S224–34. doi: 10.1086/514310. [DOI] [PubMed] [Google Scholar]
- 20.Mosca JD, Pitha PM. Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol Cell Biol. 1986;6:2279–83. doi: 10.1128/mcb.6.6.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spann KM, Tran KC, Collins PL. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-kappaB, and proinflammatory cytokines. J Virol. 2005;79:5353–62. doi: 10.1128/JVI.79.9.5353-5362.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emeny JM, Morgan MJ. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J Gen Virol. 1979;43:247–52. doi: 10.1099/0022-1317-43-1-247. [DOI] [PubMed] [Google Scholar]
- 23.Bray M, Davis K, Geisbert T, Schmaljohn C, Huggins J. A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J Infect Dis. 1998;178:651–61. doi: 10.1086/515386. [DOI] [PubMed] [Google Scholar]
- 24.Ryabchikova EI, Kolesnikova LV, Luchko SV. An analysis of features of pathogenesis in two animal models of Ebola virus infection. J Infect Dis. 1999;179(Suppl 1):S199–202. doi: 10.1086/514293. [DOI] [PubMed] [Google Scholar]
- 25.Geisbert TW, Pushko P, Anderson K, Smith J, Davis KJ. Evaluation in nonhuman primates of vaccines against Ebola virus. Emerg Infect Dis. 2002;8:503–7. doi: 10.3201/eid0805.010284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bray M, Hatfill S, Hensley L, Huggins JW. Haematological, biochemical and coagulation changes in mice, guinea-pigs and monkeys infected with a mouse-adapted variant of Ebola Zaire virus. J Comp Pathol. 2001;125:243–53. doi: 10.1053/jcpa.2001.0503. [DOI] [PubMed] [Google Scholar]
- 27.Bowen ET, Platt GS, Lloyd G, Raymond RT, Simpson DI. A comparative study of strains of Ebola virus isolated from southern Sudan and northern Zaire in 1976. J Med Virol. 1980;6:129–38. doi: 10.1002/jmv.1890060205. [DOI] [PubMed] [Google Scholar]
- 28.Hevey M, Negley D, Pushko P, Smith J, Schmaljohn A. Marburg virus vaccines based upon alphavirus replicons protect guinea pigs and nonhuman primates. Virology. 1998;251:28–37. doi: 10.1006/viro.1998.9367. [DOI] [PubMed] [Google Scholar]
- 29.Ryabchikova E, Strelets L, Kolesnikova L, Pyankov O, Sergeev A. Respiratory Marburg virus infection in guinea pigs. Arch Virol. 1996;141:2177–90. doi: 10.1007/BF01718224. [DOI] [PubMed] [Google Scholar]
- 30.Fenske DB, MacLachlan I, Cullis PR. Stabilized plasmid-lipid particles: a systemic gene therapy vector. Methods Enzymol. 2002;346:36–71. doi: 10.1016/s0076-6879(02)46048-x. [DOI] [PubMed] [Google Scholar]
- 31.Geisbert TW, Hensley LE, Larsen T, et al. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am J Pathol. 2003;163:2347–70. doi: 10.1016/S0002-9440(10)63591-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishikawa M, Yamauchi M, Morimoto K, Ishida E, Takakura Y. Hepatocyte-targeted in vivo gene expression by intravenous injection of plasmid DNA complexed with synthetic multi-functional gene delivery system. Gene Ther. 2000;7:548–55. doi: 10.1038/sj.gt.3301140. [DOI] [PubMed] [Google Scholar]
- 33.Collard WT, Yang Y, Kwok KY, Park Y, Rice KG. Biodistribution, metabolism, and in vivo gene expression of low molecular weight glycopeptide polyethylene glycol peptide DNA co-condensates. J Pharm Sci. 2000;89:499–512. doi: 10.1002/(SICI)1520-6017(200004)89:4<499::AID-JPS7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 34.Biessen EA, Vietsch H, Rump ET, et al. Targeted delivery of oligodeoxynucleotides to parenchymal liver cells in vivo. Biochem J. 1999;340(Pt 3):783–92. [PMC free article] [PubMed] [Google Scholar]
- 35.Soutschek J, Akinc A, Bramlage B, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–8. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 36.Fang D, McClintock K. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol. 2005;23:457–62. doi: 10.1038/nbt1081. [DOI] [PubMed] [Google Scholar]
- 37.Hornung V, Guenthner-Biller M, Bourquin C, et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. 2005;11:263–70. doi: 10.1038/nm1191. [DOI] [PubMed] [Google Scholar]
- 38.Isaacson M, Sureau P, Courteille G, Pattyn SR. Clinical aspects of Ebola virus disease at the Ngaliema Hospital, Kinshasa, Zaire. In: Pattyn SR, editor. Ebola virus haemorrhagic fever. New York: Elsevier/North-Holland Biomedical Press; 1978. pp. 15–20. [Google Scholar]
- 39.Huggins J, Zhang ZX, Bray M. Antiviral drug therapy of filovirus infections: S-adenosylhomocysteine hydrolase inhibitors inhibit Ebola virus in vitro and in a lethal mouse model. J Infect Dis. 1999;179(Suppl 1):S240–7. doi: 10.1086/514316. [DOI] [PubMed] [Google Scholar]
- 40.Bray M, Raymond JL, Geisbert T, Baker RO. 3-deazaneplanocin A induces massively increased interferon-alpha production in Ebola virus-infected mice. Antiviral Res. 2002;55:151–9. doi: 10.1016/s0166-3542(02)00018-9. [DOI] [PubMed] [Google Scholar]
- 41.Parren PW, Geisbert TW, Maruyama T, Jahrling PB, Burton DR. Pre- and postexposure prophylaxis of Ebola virus infection in an animal model by passive transfer of a neutralizing human antibody. J Virol. 2002;76:6408–12. doi: 10.1128/JVI.76.12.6408-6412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geisbert TW, Hensley LE, Jahrling PB, et al. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet. 2003;362:1953–8. doi: 10.1016/S0140-6736(03)15012-X. [DOI] [PubMed] [Google Scholar]
- 43.Huggins JW, Zhang ZX, Davis K, Coulombe RA. Inhibition of Ebola virus by S-adenosylhomocysteine hydrolase inhibitors. Antiviral Res. 1995;26:A301. [Google Scholar]
- 44.Parren PW, Geisbert TW, Geisbert J, Sullivan NJ, Jahrling PB. Program and abstracts of the VRC Symposium on Viral Hemorrhagic Fevers. Bethesda, MD: National Institute of Allergy and Infectious Diseases; 2003. Antibody activity against Ebola virus in vitro and in vivo. [Google Scholar]