

Abstract
Autophagy is a conserved cellular process that acts as a key regulator in maintaining cellular homeostasis. Recent studies implicate an important role for autophagy in infection and immunity by removing invading pathogens and through modulating innate and adaptive immune responses. However, several pathogens, notably some positive-stranded RNA viruses, have subverted autophagy to their own ends. In this review, we summarize the current understanding of how viruses with a positive-stranded RNA genome interact with the host autophagy machinery to control their replication and spread. We review the mechanisms underlying the induction of autophagy and discuss the pro- and anti-viral functions of autophagy and the potential mechanisms involved.
Keywords: autophagy, positive-stranded RNA viruses, autophagosome, p62/SQSTM1, double-membrane vesicle, xenophagy
Introduction
Viruses are intracellular pathogens that depend solely on host cells to replicate their genome and assemble intact viral progeny. Positive-stranded RNA viruses are well known for their ability to induce the remodeling of intracellular membranes to form a scaffold for viral replication complexes [1,2]. However, the exact origins of the membranous structures and the pathways responsible for their formation remain to be fully elucidated. Although endoplasmic reticulum (ER) is utilized by a large number of viruses, other membranous organelles can also be exploited by viruses. Recent studies have shown that the membranous structures contain markers for autophagosomes, suggesting a pivotal role for autophagy in controlling replication of positive-stranded RNA viruses [3,4].
Autophagy is a conserved cellular process responsible for removing damaged organelles and misfolded proteins to maintain cellular homeostasis under both normal and stress conditions [5]. Autophagy was previously regarded to be non-selective. However, increasing evidence has suggested the selectivity of autophagy in recycling unwanted organelles, removing aggregate-prone proteins, and clearing specific viral proteins [6–8]. The importance of selective autophagy in different physiological and pathological states has been increasingly recognized. Recently, autophagy has emerged as a critical player in the control of viral infection and immunity [9–13]. On the one hand, autophagy can serve as a host defense mechanism for some pathogens, such as sindbis virus and herpes simplex virus, by clearing them out of the cells [14–16]. On the other hand, many positive-stranded RNA viruses have been reported to subvert this cellular machinery to favor their own replication and release.
In this review, we discuss the current understanding as to how positive-stranded RNA viruses interact with the host autophagy machinery to control their replication and spread. We review the mechanisms underlying the induction of autophagy. We also discuss the pro- and anti-viral functions of autophagy and the potential mechanisms involved. Understanding how the autophagy pathway is activated and the biological significance of autophagy in the control of viral life cycle is essential for exploration of new anti-viral targets.
Autophagy: Mechanism and Regulation
Among the three types of autophagy (macroautophagy, microautophagy, and chaperon-mediated autophagy), macroautophagy (hereafter referred to as autophagy) is the most extensively studied in both yeast and mammalian cells [6,17,18]. The process of autophagy can be divided into four sequential steps (Fig. 1). Initially, a crescent-shaped double-membrane vesicle (DMV) called isolation membrane or phagophore is formed to sequester misfolded proteins and damaged organelles (induction and nucleation step). Subsequently, the two ends of phagophore fuse to form a complete DMV termed as autophagosome (elongation step). Finally, the outer membrane of the autophagosomes fuses with lysosomes to form autolysosomes while the inner membrane and the cargo enwrapped in autophagosomes are degraded by hydrolyses (fusion step). Autophagosomes can also fuse with early or late endosomes to form amphisomes [19].
Figure 1.
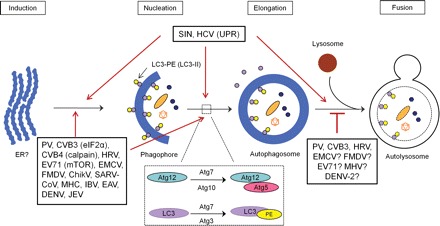
The activation of autophagy pathway by different positive-stranded RNA viruses The process of autophagy consists of four steps: induction, nucleation, elongation, and fusion with lysosomes. Two ubiquitination-like conjugation systems, Atg12–Atg5 and LC3-PE, are essential for the formation of autophagosomes. Positive-stranded RNA viruses induce either complete or incomplete autophagy as indicated. ER stress-induced UPR, eIF2α phosphorylation, and mTOR/p70S6K signaling pathway have been associated with the activation of autophagy in HCV, EV71, and CVB3 infection, respectively. Calpain pathway is required for the activation of autophagy in CVB4 infection. ER, endoplasmic reticulum; UPR, unfolded protein response; eIF2α, eukaryotic initiation factor 2α; mTOR, mammalian target of rapamycin; p70S6K, p70 ribosomal protein S6 kinase; PE, phosphatidylethanolamine; PV, poliovirus; CVB, coxsackievirus; HRV, human rhinovirus; EV71, enterovirus 71; EMCV, encephalomyocarditis virus; FMDV, foot and mouth disease virus; ChikV, chikungunya virus; SARS-CoV, severe acute respiratory syndrome-coronavirus; MHV, mouse hepatitis virus; IBV, infectious bronchitis virus; EAV, equine arteritis virus; DENV, dengue virus; JEV, Japanese encephalitis virus; SIN, sindbis virus; HCV, hepatitis C virus; ?, implicated by indirect experimental evidence but direct evidence is still missing.
More than 30 autophagy-related genes (Atg) have been identified to participate in the autophagic process [5,20,21]. The key proteins involved in the formation of autophagosomes include: (i) uncoordinated (UNC)-51-like kinase (ULK) complex, composed of ULK1, ULK2, Atg13, focal adhesion kinase family interacting protein of 200 kDa (FIP200), and Atg101; (ii) class III phosphatidylinositol 3 (PI3)-kinase complex, comprising of Vps34, p150, beclin-1, Atg14, and Ambra1 (activating molecule in Beclin-1-regulated autophagy protein 1); (iii) two ubiquitination-like conjugation systems, composed of Atg4, Atg12, Atg5, Atg16L1, Atg7, Atg10, Atg3, and microtubule-associated protein light chain (LC3) [5,20,21].
The two conjugation systems are essential for the formation of autophagosomes (Fig. 1). For Atg5–Atg12 conjugation, Atg12 is first activated by Atg7 and then transferred to Atg10. Atg12 finally forms a conjugate with Atg5 to activate the formation of autophagosomes [22]. For LC3–phosphatidylethanolamine (PE) conjugation, nascent LC3 is first cleaved by Atg4 to become LC3-I, which is subsequently activated by Atg7 and then transferred to Atg3. Finally, LC3 is conjugated to PE to form a LC3–PE complex (LC3-II), which participates in the formation of autophagosomes [22]. Recruitment of LC3 protein to autophagic vesicles has been considered a common trait of autophagosome formation. In addition, conversion from LC3-I to LC3-II has been widely accepted as a marker of autophagic signaling [23].
Autophagosome formation is also tightly controlled by multiple signaling pathways. Autophagic protein beclin-1 forms the class III PI3-kinase complex with Vps34, a class III PI3-kinase, to facilitate autophagosome formation by providing phosphatidyliositol 3-phosphates to isolation membrane [24,25]. The mammalian target of rapamycin (mTOR) and the eukaryotic initiation factor 2α (eIF2α) kinases also participate in autophagy process by negatively and positively regulating the formation of autophagosomes, respectively [16,26]. The adaptor protein p62/SQSTM1 has been revealed to be essential in mediating selective autophagy [7,8]. It binds to both ubiquitin and LC3 and targets ubiquitinated proteins to autophagosomes for degradation [27]. p62/SQSTM1 can also be selectively degraded by autophagy [28].
Autophagy functions as a protein quality control system. Thus, defects in autophagy have been associated with several pathological conditions, such as neurodegenerative diseases, myopathy, cancer, and aging [29–31]. Autophagy has also been implicated in the modulation of infection and immunity. Autophagy serves as a critical component of innate immune response by removing bacteria, viruses, and protozoans from the host cells through xenophagy, whose targets are foreign bodies rather than self-molecules [32,33]. The antigens are then presented through MHC class II molecules, initiating adaptive immune response [34]. In this process, autophagy participates and assists both innate and adaptive immunity to clear the pathogens out of the body. However, as opposed to the anti-viral activity, many positive-stranded RNA viruses have successfully developed strategies to hijack autophagy to foster their replication.
Activation of Host Autophagy
Since the early reports that coronavirus and poliovirus subvert the host autophagy machinery to support their replication [3,35], growing numbers of studies indicate that the interaction between autophagy and positive-stranded RNA viruses is widely present. In addition to poliovirus, the other members in picornaviridae family, including coxsackievirus group B (CVB) [36–39], enterovirus 71 (EV71) [40,41], foot and mouth disease virus (FMDV) [42], encephalomyocarditis virus (EMCV) [43], and human rhinovirus (HRV) [3,44], also activate the cellular autophagy pathway. Moreover, sindbis virus (SIN) [15,45] and chikungunya virus (ChikV) [46] in the family of togaviridae, severe acute respiratory syndrome-coronavirus (SARS-CoV), mouse hepatitis virus (MHV), and infectious bronchitis virus (IBV) in the family of coronaviridae [35,47–50], equine arteritis virus (EAV) in the family of arteriviridae [51,52], as well as dengue virus (DENV) [53–60], hepatitis C virus (HCV) [54,61–67], and Japanese encephalitis virus (JEV) [68] in the family of flaviviridae have also been found to induce the activation of autophagy (Fig. 1).
Incomplete and complete autophagy activation
The aforementioned viruses have been demonstrated to activate the autophagic process as measured by increased accumulation of autophagosomes or autophagosome-like vesicles, augmented conversion from LC3-I to LC3-II, and elevated number of punctate LC3-expressing cells, which are widely accepted criteria for monitoring autophagy [69,70]. However, since autophagy is a dynamic process composed of autophagosome formation and degradation, the increased autophagosomes can be a result of increased formation, decreased fusion with lysosomes, or both. Autophagic flux is a measurement of the balance between the rate of autophagosome formation and degradation. Thus, it is more meaningful to determine the rate of autophagic flux to have an integrated view of the complete process [69,70].
Recent study has shown that infection with sindbis virus induces autophagosome formation with an increased autophagic flux [15]. Sindbis virus promotes the formation of autophagosomes, as evidenced by increases in the percentage of cells with GFP-LC3 dots and LC3-II conversion and by visualization of DMVs in virus-infected cells [15]. It was further found that the protein levels of p62, a marker for autophagic flux, are reduced without the changes of its mRNA expression [15]. These results suggest that sindbis virus triggers a complete autophagic response.
Unlike sindbis virus, poliovirus in picornaviridae family has been proposed to block the fusion of autophagosomes with lysosomes [3]. In consistent with this proposition, it was demonstrated that protein expression of p62 is unaltered [37] or increased [36] during CVB3 infection, suggesting that CVB3 infection triggers increased autophagosome formation, but with a reduced autophagic flux. These studies suggest that blockage of autophagosome–lysosome fusion may be a viral strategy to ensure the existence of optimal number of autophagosomes for viral benefits. Further study is needed to clarify the mechanisms by which virus blocks the maturation of autophagosomes into autolysosomes.
Incomplete autophagic flux has also been reported in HCV infection [65,71]. Although autophagosome formation is induced during HCV infection, it was shown that the degradation rate of long-lived proteins and autophagic substrate protein p62 is not significantly changed [65,71]. However, these studies are conflicting with the report by Ke and Chen [54], which showed that the process of autophagy is complete during HCV infection. Using a tandem reporter construct (mRFP-GFP-LC3) [72] and by immunogold-electron microscopy analysis of initial- and late-stage autophagic vacuoles, it was demonstrated that HCV-induced autophagosomes fuse with lysosomes [54]. This observation was further supported by the evidence that disruption of autolysosome maturation inhibits HCV RNA replication and protein expression [54]. Further work is required to clarify this apparent discrepancy.
The experimental data about the effects of positive-stranded RNA viruses on autophagic flux are still sparse. Development of new research technologies will allow for a more reliable and sensitive measurement for monitoring autophagic flow. Understanding the dynamic status of autophagosome formation and degradation after viral infection will not only provide novel insights into the mechanisms by which viruses exploit the autophagic machinery but also assist in the development of anti-viral therapies.
Intracellular signaling pathways trigger autophagy
How does virus infection trigger autophagy? HCV infection has been demonstrated to induce autophagy by activating the unfolded protein response (UPR) [54,65]. The accumulation of misfolded proteins in the ER causes ER stress and activates UPR via three sensors: protein kinase R-like ER kinase (PERK), inositol-requiring kinase 1 (IRE1), and activating transcription factor 6 (ATF6) [73]. It has been shown that HCV infection can induce ER stress to activate all three sensors of UPR [65,71]. Gene silencing of either PERK, IRE1, or ATF6 leads to significant reduction of LC3 lipidation induced by HCV infection, suggesting that all three UPR signaling pathways are required for the induction of autophagy [54,65].
During CVB3 infection, phosphorylation of mTOR remains unchanged whereas the phosphorylation of eIF2α is increased [37], suggesting that eIF2α is a potential signaling pathway responsible for CVB3-induced autophagy. The upstream signaling leading to eIF2α phosphorylation is still unknown. It may be due to the activation of protein kinase R by double-stranded RNA (dsRNA) during CVB3 replication, or it is the downstream effector induced by the activation of PERK as mentioned above. In addition to the eIF2α pathway, it was found that calpain activity is required for CVB4-induced autophagy, as calpain inhibitors were shown to reduce the formation of autophagosomes [39].
The mTOR pathway plays a central role in negatively regulating autophagy activity. The activation of mTOR complex 1 (mTORC1) results in phosphorylation of two effectors, p70 ribosomal protein S6 kinase (p70S6K) and eukaryotic initiation factor 4E binding protein 1 (4E-BP1), which promote protein translation and inhibit autophagy [74]. In EV71-infected SK-N-SH cells, decreased expression of mTOR and p70S6K has been associated with activation of autophagy [40]. But this result was not seen in EV71-infected RD cells, which suggests that EV71 may utilize different signaling pathways in different cell types. The upstream signaling molecules of mTOR in EV71 infection remain unclear. But the activation of mTOR/p70S6K is independent of the activation of class I PI3-kinase/Akt and Erk1/2 signaling pathways [40].
Viral proteins activate autophagy
In addition to cellular components, some viral proteins have been shown to contribute to the biogenesis of autophagosomes. For example, some viral non-structural proteins are able to induce membrane rearrangement. Although expression of either poliovirus non-structural protein 2BC or 3A alone is insufficient to induce autophagosome structure, co-expression of 2BC and 3A has been shown to facilitate the modification of LC3-I to LC3-II and to induce the formation of DMVs that closely resemble the ones induced by poliovirus [3,75]. Further investigation demonstrated that covalent modification of LC3 by expression of polioviral protein 2BC targets LC3 to cellular membranes [76]. The expression of HCV non-structural 4B (NS4B) protein was reported to induce autophagosome-like vehicles similar to those induced in the cells expressing the whole poly-protein or harbor HCV sub-genomic replicons [66]. Similarly, expression of flavivirus non-structural protein NS4A is sufficient to induce autophagy and promote cell survival [57]. It was also reported that co-expression of NSP2 and NSP3 of EAV induces the formation of autophagosome-like DMVs [52].
In summary, despite these positive reports on the activation of autophagy, not all viruses in the family of positive-stranded RNA viruses were shown to activate the cellular autophagy. For example, there was a report showing that autophagy is not induced after HRV2 infection and modulation of the autophagy pathway does not affect viral propagation [77]. In addition, it was demonstrated that infection with HRV-1A, another serotype of HRV, does not induce autophagy and modulation of autophagy has no effects on its replication [44].
Pro-viral Function of Autophagy
Autophagy has been initially identified as a cellular defense mechanism to clear the invading viruses; however, a growing body of research evidence demonstrated that this host machinery can be evolved by numerous positive-stranded RNA viruses, including poliovirus [3], CVB3 [37], CVB4 [39], EV71 [40], HRV [44], FMDV [42], EMCV [43], DENV [53,56], HCV [54,61,62,65,78], MHV and SARS-CoV [35], ChikV [46], and JEV [68] to support their life cycle. Pharmacological inhibition or gene silencing of autophagy pathway in vitro has been demonstrated to inhibit growth and/or spread of these viruses, whereas induction of autophagy results in increased viral yield. Multiple mechanisms have been suggested to be involved in this pro-viral function of autophagy (Fig. 2).
Figure 2.
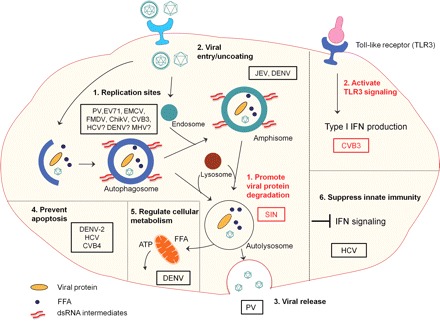
The pro-viral and anti-viral functions of autophagy during positive-stranded RNA viral infection Pro-viral functions of autophagy (viruses are circled in black rectangles): (1) autophagosomes (PV, EV71, CVB3, EMCV, FMDV, ChikV) or amphisomes (DENV) serve as sites for viral replication; (2) amphisomes are also linked to the entry/uncoating of JEV and DENV; (3) the topological structure of autophagosmes is associated with non-lytic egress of PV particles; (4) autophagy prevents premature cell death to maintain favorable cellular environment for viral replication (DENV-2, HCV, and CVB4); (5) autophagy favors DENV replication by selectively degrading lipid droplets to generate ATP for viral replication; (6) suppression of IFN signaling is related to the pro-viral function of autophagy in HCV infection. Anti-viral functions of autophagy (viruses are circled in red rectangles): (1) autophagy inhibits SIN replication by promoting the clearance of viral capsid protein; (2) autophagy is required for TLR3-medicated type-I IFN production during CVB3 infection. PV, poliovirus; CVB, coxsackievirus; EV71, enterovirus 71; FMDV, foot and mouth disease virus; ChikV, chikungunya virus; HCV, hepatitis C virus; HRV, human rhinovirus; EMCV, encephalomyocarditis virus; SIN, sindbis virus; DENV, dengue virus; MHV, mouse hepatitis virus; JEV, Japanese encephalitis virus; FAA, free fatty acid; IFN, interferon; TLR3, toll-like receptor 3; ATP, adenosine triphosphate; ?, implicated by indirect experimental evidence but direct evidence is still missing.
Serve as viral replication sites
One of the common features shared by positive-stranded RNA viruses is to assemble and replicate on intracellular membranes. The functions of the membranous structures are proposed to provide a scaffold for anchoring and concentrating the replication complexes to prevent the immune response triggered by dsRNA intermediates and to afford certain lipids required by genome synthesis [2]. The replication complexes are usually composed of viral RNA-dependent RNA polymerase, accessory non-structural proteins with helicase and nucleotide triphosphate activity, viral RNA, and host cell factors. As parts of the intracellular membranous structures, autophagosomes or amphisomes have been shown to function as scaffolds required for the replication and assembly of certain positive-stranded RNA viruses.
Replication of several positive-stranded RNA viruses has previously been linked to cellular membranous structures [49,51,79,80]. Recent studies provided the direct evidence of the association between viral replication complexes and autophagosome structures. Confocal microscopy showed the co-localization of polioviral protein 3A, a critical component of the poliovirus RNA replication complex, with the autophagosome marker LC3 [3]. DENV non-structural protein NS1 and dsRNA were reported to co-localize with LC3 and ribosomal protein L28 [55,60]. Immuno-electron microscopy also showed co-localization of EV71 capsid protein VP1 with autophagosomes in virus-infected mouse neurons [40]. Confocal and immune-electron microscopy revealed that both non-structural protein 3A and capsid protein VP1 co-localize with autophagosomes during EMCV infection [43]. Co-localizations of non-structural proteins 2B, 2C, and 3A with LC3 and between structural protein VP1 and Atg5 were also reported in FMDV-infected cells [42]. The ultrastructural analysis showed that the ChikV virions locate in the lumen of autophagomes-like vacuoles [46].
Similar to other positive-stranded RNA viruses, HCV infection induces intracellular membrane redistribution. However, controversy exists as to whether autophagosomes serve as sites for HCV replication. By sucrose gradient analysis, LC3-II was found to co-sediment with HCV RNA and non-structural proteins NS3 and NS5A [63]. However, confocal microscopy showed little evidence of co-localization of LC3 or Atg5 with HCV core, NS3, NS4A/4B, and NS5A proteins [61,62,65,78]. Moreover, it was demonstrated that knockdown of either LAMP2 or Rab7, two critical proteins responsible for the fusion of autophagosomes with lysosomes, inhibits HCV viral replication [54]. These studies suggest that autophagosomes may not be major sites for HCV genome replication.
Although MHV replication complexes were found to be associated with LC3 and Atg12 [35], conflicting results were also reported with regard to the role of autophagy in MHV replication. As opposed to the findings in embryonic stem cell lines that autophagy induced by MHV enhances viral replication, likely through providing a replication site [35], using primary macrophages and murine embryonic fibroblasts it was found that MHV replication does not require the autophagy gene Atg5 [81].
Although direct evidence of the association of viral replication complexes with autophogasomes is lacking for CVB3, blockage of the fusion between autophagosomes and lysosomes using pharmacological inhibitors or knockdown of the genes critical for this fusion increases the accumulation of autophagosomes in virally infected cells and consequently leads to enhanced viral replication [37]. This study provides indirect evidence that autophagosomes are critical components during CVB3 replication, likely by serving as virus anchoring and replication sites. Similar to the observation in CVB3, inhibition of the fusion between autophagosomes or amphisomes and lysosomes was found to increase the viral yield of DENV-2 [60]. These data together with the discoveries that DENV-2 replication complexes co-localize with LC3 and an endosome marker imply that DENV-2 may use the amphisomes as sites for viral replication [60]. However, this effect seems to be viral serotype-specific. It was found that inhibition of lysosome fusion reduces DENV-3 yields and results in an accumulation of viral NS1 [55]. The mechanisms by which autophagy favors DENV-3 replication remain elusive.
Promote viral entry/uncoating and release
Recent evidence has suggested that autophagy may participate in viral life cycle at the early phase of viral infection [59,60,68]. Both DENV and JEV are enveloped RNA viruses in the family of flaviviridae. They enter the host cells primarily via receptor-mediated endocytosis, followed by pH-dependent fusion with endosomes and subsequent release into the cytoplasm. Recent studies demonstrated the co-localization of viral replication complexes or inoculated viral particles with both autophagosome and endosome markers [59,60,68]. These observations imply a potential role for autophagosome–endosome fusion in viral entry/uncoating.
Autophagy has also been suggested to facilitate non-lytic egression of some positive-stranded RNA viruses. Poliovirus, a non-enveloped positive-stranded RNA virus, is often considered as a lytic virus releasing from the cells by cell lysis. However, non-lytic release of poliovirus was also reported [82]. In the study of the biological relevance of autophagy in poliovirus infection, it was found that the decreases in extracellular virus are always more profound than intracellular virus when important autophagic proteins are knocked down [3]. The lower levels of viral particles in extracellular virus in autophagy-suppressing cells have been correlated with reduced non-lytic release of cytoplasmic contents that can be mediated by autophagy [3,83]. This hypothesis is supported by the observation that both LC3 and polioviral VP1 are present in extracellular matrix adjacent to the infected cells [3]. Further investigation is needed to provide direct evidence that autophagy provides a topological mechanism to facilitate viral release.
Suppress innate anti-viral immunity
Recent studies by Ke and Chen [54] and Shrivastava et al. [84] have shed light on how autophagy benefits for HCV infection. They provided evidence that complete autophagic process is required to promote HCV replication and this is largely due to the suppressive effect of autophagy on anti-viral innate immune response [54,84]. Upon HCV infection, the innate immune response is initiated by activation of interferon-β (IFN-β) production mediated by HCV-derived pathogen-associated molecular pattern (PAMP) [54]. Inhibition or activation of UPR-mediated autophagy has been shown to increase or reduce IFN-β production mediated by HCV-derived PAMP, respectively [54]. Similar observation was also made with DENV-derived PAMP, suggesting that both viruses may share the same mechanism to evade the innate immune response [54]. Moreover, Shrivastava et al. [84] also reported that inhibition of autophagy by knocking down beclin-1 or Atg7 reduces HCV replication, which is accompanied by the activation of IFN signaling pathway, as measured by increased levels of IFN-regulated genes, including IFN-β, 2′5′-oligoadenylate synthetase 1, IFN-α, and IFN-α-inducible protein 27 mRNAs. Together, these studies indicate an important mechanism by which HCV avoids the innate immune response through activating the host autophagy pathway.
Regulate cellular metabolism
Autophagy has been demonstrated to be involved in the regulation of cellular metabolism. It regulates lipid metabolism through modulating the degradation of triglycerides stored in lipid droplets, a process called lipophagy [85,86]. Autophagy therefore represents a new cellular process for abnormalities in lipid metabolism and accumulation. Recent study has shown a novel mechanism responsible for autophagy-mediated pro-viral properties toward DENV replication [53]. It was reported that DENV-induced autophagosomes deliver the lipid droplets to lysosomes where triglycerides are depleted and the free fatty acids are released [53]. The released free fatty acids undergo β-oxidation in mitochondria to generate ATP affording the energy for DENV replication [53]. Although the exact signaling pathway contributing to this selective autophagy and the detailed mechanisms responsible for ATP regulation of DENV replication remain unclear, the findings in this research provide the first evidence linking viral infection to autophagy-mediated metabolic regulation. Whether this mechanism also applies to other viruses requiring the host cellular metabolism for their replication warrants further investigation.
Prevent premature cell death
Premature cell death has been considered as an anti-viral host mechanism by providing an unfavorable environment for viral propagation. The cross-talk between autophagy and apoptosis has become evident [87–90]. Induction of autophagy has often been linked to inhibition of apoptosis. DENV-2-induced autophagy has been demonstrated to prevent cells from apoptosis [57]. It was shown that knockdown of autophagy abolishes the protective role of autophagy against cell death and leads to reduced viral replication [57]. In exploring the mechanisms underlying the pro-viral role of autophagy in HCV life cycle, it was found that HCV infection in autophagy-knockdown cells promotes cell death, suggesting a role of autophagy in prolonging cell survival for the establishment of successful viral infection [84]. The cross-talk between autophagy and apoptosis has also been reported in CVB4 infection [38]. Suppression of autophagy by 3-methyladenine (3-MA), a selective class III PI3-kinase inhibitor, triggers caspase activation and inhibition of apoptosis using a pan-caspase inhibitor increases autophagosome formation [38].
Degradation of intracellular or viral proteins
Degradation of intracellular proteins that are against viral replication is a common strategy virus has developed to support its replication [91]. As alluded to earlier, the significance of selective autophagy in many cellular processes has been increasingly appreciated [6,8]. It is therefore conceivable that autophagy-mediated proteolysis of host anti-viral factors may also play a role in promoting viral growth. In addition, appropriate concentrations of viral proteins have been reported to be critical for the optimal replication of viruses and too much viral proteins could be a drawback for positive-stranded RNA viral replication [92–94]. Several lines of evidence have demonstrated that viruses can exploit the host ubiquitin-proteasome system for viral protein degradation to provide the proper stoichiometric ratio of structural and non-structural viral proteins during viral life cycle [95–98]. Thus, it is tempting to postulate that autophagy may also be advantageous for viral replication by degrading excess viral non-structural proteins. Indeed, lysosome-mediated proteolysis has been suggested to be involved in the rapid turnover of HCV NS2 [99]. p62-mediated selective autophagic degradation has also been reported for SIN capsid protein although this role was demonstrated to be anti-viral [15].
In summary, the pro-viral functions of autophagy in positive-stranded RNA viral life cycle apparently involve multiple pathways, either direct effects on viral replication or indirect influences on the host immune and non-immune-related activities. Given the importance of autophagy in regulating diverse cellular functions, it is speculated that other functions of autophagy, for example cell cycle regulation, cell differentiation, and gene transcription, may also contribute to enhanced viral replication and this requires further investigation.
Anti-viral Effects of Autophagy
The autophagy machinery is not always beneficial for positive-stranded RNA viruses. It has been shown that autophagy functions as an anti-viral host defense against SIN infection (Fig. 2) [14,15]. In response to SIN infection, mice with beclin-1 overexpression have improved survival rate, reduced viral loads, and attenuated viral pathogenesis as compared to control mice [14]. Study using neuron-specific Atg5 knockout mice showed that disruption of Atg5 gene leads to enhanced susceptibility of mouse central nervous system to SIN infection [15]. Further investigation demonstrated that loss of Atg5 in SIN-infected neurons results in impaired viral capsid protein clearance, increased p62 accumulation, and accelerated cell death, without affecting viral replication and type I IFN production [15]. In vitro study showed that p62 binds directly to viral capsid protein and transports it to autophagosomes for lysosome-mediated degradation [15]. Electron microscopic analysis provided the direct evidence that SIN virions are captured inside the autophagosomes or autolysosomes [15]. This study suggests that Atg5 plays a crucial role in protecting against SIN infection in mouse central nervous system by promoting p62-mediated clearance of viral proteins, rather than modulating innate immune response or viral replication [15]. Further study is required to elucidate the exact mechanisms by which p62 mediates selective autophagic degradation of SIN capsid protein.
Toll-like receptors (TLRs) play an important role in innate anti-viral immunity against CVB3 infection (Fig. 2) [100]. It was recently reported that autophagy plays a significant role in TLR-mediated type I IFN signaling during CVB3 infection [101]. It was found that blockage of autophagy by either gene-silencing LC3-II, beclin-1 or Atg5, or using pharmacological inhibitor 3-MA inhibits TLR3 signaling in response to dsRNA. Interestingly, in contrast to the earlier observation that incomplete autophagy increases CVB3 replication [37], it was demonstrated that complete autophagy is required for the activation of signaling triggered by TLR3, as inhibition of lysosome activity by bafilomycin A or chloroquine results in decreased type I IFN signaling [101]. The detail mechanisms in relation to a dual function of autophagy in supporting CVB3 replication by providing replication scaffolds and suppressing viral replication by triggering TLR3-mediated innate immune response require further investigation.
Conclusion
The available evidence highlighted in this review points to a crucial role for autophagy in regulating viral infection and/or in manipulating host anti-viral defense. Many positive-stranded RNA viruses interplay with autophagy to optimize their infection. They affect multiple aspects of viral life cycle, through promoting viral entry/uncoating, serving sites for viral replication, preventing cell death, regulating cellular metabolisms, escaping innate immunity, and controlling viral progeny release. Although the reported pro-viral function is dominant for positive-stranded RNA viruses, autophagy also plays an anti-viral role via facilitating the clearance of some viral structural proteins.
Despite significant progress in recent years, many of the details in this field remain to be elucidated, such as the signaling pathways responsible for virus-induced autophagy, the underlying mechanisms by which viruses inhibit the fusion of autophagosomes with lysosomes, and the molecular basis of virus-specific selective autophagy. A better understanding of these questions will be critical for developing novel autophagy-based treatment to control viral infection and viral pathogenesis.
Funding
This work was supported by the Canadian Institutes of Health Research.
References
- 1.den Boon JA, Ahlquist P. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu Rev Microbiol. 2010;64:241–256. doi: 10.1146/annurev.micro.112408.134012. [DOI] [PubMed] [Google Scholar]
- 2.Miller S, Krijnse-Locker J. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol. 2008;6:363–374. doi: 10.1038/nrmicro1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson WT, Giddings TH, Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wileman T. Aggresomes and autophagy generate sites for virus replication. Science. 2006;312:875–878. doi: 10.1126/science.1126766. [DOI] [PubMed] [Google Scholar]
- 5.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 6.Fujita N, Yoshimori T. Ubiquitination-mediated autophagy against invading bacteria. Curr Opin Cell Biol. 2011;23:492–497. doi: 10.1016/j.ceb.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 7.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Komatsu M, Ichimura Y. Selective autophagy regulates various cellular functions. Genes Cells. 2010;15:923–933. doi: 10.1111/j.1365-2443.2010.01433.x. [DOI] [PubMed] [Google Scholar]
- 9.Kirkegaard K. Subversion of the cellular autophagy pathway by viruses. Curr Top Microbiol Immunol. 2009;335:323–333. doi: 10.1007/978-3-642-00302-8_16. [DOI] [PubMed] [Google Scholar]
- 10.Kudchodkar SB, Levine B. Viruses and autophagy. Rev Med Virol. 2009;19:359–378. doi: 10.1002/rmv.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee HK, Iwasaki A. Autophagy and antiviral immunity. Curr Opin Immunol. 2008;20:23–29. doi: 10.1016/j.coi.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orvedahl A, Levine B. Autophagy in Mammalian antiviral immunity. Curr Top Microbiol Immunol. 2009;335:267–285. doi: 10.1007/978-3-642-00302-8_13. [DOI] [PubMed] [Google Scholar]
- 13.Taylor MP, Jackson WT. Viruses and arrested autophagosome development. Autophagy. 2009;5:870–871. doi: 10.4161/auto.9046. [DOI] [PubMed] [Google Scholar]
- 14.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orvedahl A, MacPherson S, Sumpter R, Jr, Talloczy Z, Zou Z, Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7:115–127. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Talloczy Z, Jiang W, Virgin HWt, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA. 2002;99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuervo AM, Dice JF. Lysosomes, a meeting point of proteins, chaperones, and proteases. J Mol Med (Berl) 1998;76:6–12. doi: 10.1007/s001090050185. [DOI] [PubMed] [Google Scholar]
- 18.Dunn WA., Jr Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. 1994;4:139–143. doi: 10.1016/0962-8924(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 19.Fader CM, Colombo MI. Autophagy and multivesicular bodies: two closely related partners. Cell Death Differ. 2009;16:70–78. doi: 10.1038/cdd.2008.168. [DOI] [PubMed] [Google Scholar]
- 20.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–830. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–216. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 23.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001;2:330–335. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeng X, Overmeyer JH, Maltese WA. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci. 2006;119:259–270. doi: 10.1242/jcs.02735. [DOI] [PubMed] [Google Scholar]
- 26.Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12(Suppl. 2):1509–1518. doi: 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- 27.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 28.Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010;584:1374–1378. doi: 10.1016/j.febslet.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 29.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexander DE, Leib DA. Xenophagy in herpes simplex virus replication and pathogenesis. Autophagy. 2008;4:101–103. doi: 10.4161/auto.5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120:159–162. doi: 10.1016/j.cell.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol. 2009;10:461–470. doi: 10.1038/ni.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem. 2004;279:10136–10141. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kemball CC, Alirezaei M, Flynn CT, Wood MR, Harkins S, Kiosses WB, Whitton JL. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J Virol. 2010;84:12110–12124. doi: 10.1128/JVI.01417-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, Luo H. Autophagosome supports coxsackievirus B3 replication in host cells. J Virol. 2008;82:9143–9153. doi: 10.1128/JVI.00641-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoon SY, Ha YE, Choi JE, Ahn J, Lee H, Kim DH. Autophagy in coxsackievirus-infected neurons. Autophagy. 2009;5:388–389. doi: 10.4161/auto.5.3.7723. [DOI] [PubMed] [Google Scholar]
- 39.Yoon SY, Ha YE, Choi JE, Ahn J, Lee H, Kweon HS, Lee JY, et al. Coxsackievirus B4 uses autophagy for replication after calpain activation in rat primary neurons. J Virol. 2008;82:11976–11978. doi: 10.1128/JVI.01028-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang SC, Chang CL, Wang PS, Tsai Y, Liu HS. Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J Med Virol. 2009;81:1241–1252. doi: 10.1002/jmv.21502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang XY, Xi XY, Zhao ZD. Autophagy inhibitor 3-MA decreases the production and release of infectious enterovirus 71 particles. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi. 2011;25:176–178. [PubMed] [Google Scholar]
- 42.O'Donnell V, Pacheco JM, LaRocco M, Burrage T, Jackson W, Rodriguez LL, Borca MV, et al. Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology. 2011;410:142–150. doi: 10.1016/j.virol.2010.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Li Z, Xinna G, Xin G, Yang H. Autophagy promotes the replication of encephalomyocarditis virus in host cells. Autophagy. 2011;7:613–628. doi: 10.4161/auto.7.6.15267. [DOI] [PubMed] [Google Scholar]
- 44.Klein KA, Jackson WT. Human rhinovirus 2 induces the autophagic pathway and replicates more efficiently in autophagic cells. J Virol. 2011;85:9651–9654. doi: 10.1128/JVI.00316-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orvedahl A, Levine B. Autophagy and viral neurovirulence. Cell Microbiol. 2008;10:1747–1756. doi: 10.1111/j.1462-5822.2008.01175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krejbich-Trotot P, Gay B, Li-Pat-Yuen G, Hoarau JJ, Jaffar-Bandjee MC, Briant L, Gasque P, et al. Chikungunya triggers an autophagic process which promotes viral replication. Virol J. 2011;8:432. doi: 10.1186/1743-422X-8-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cottam EM, Maier HJ, Manifava M, Vaux LC, Chandra-Schoenfelder P, Gerner W, Britton P, et al. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy. 2011;7:1335–1347. doi: 10.4161/auto.7.11.16642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Haan CA, Reggiori F. Are nidoviruses hijacking the autophagy machinery? Autophagy. 2008;4:276–279. doi: 10.4161/auto.5241. [DOI] [PubMed] [Google Scholar]
- 49.Gosert R, Kanjanahaluethai A, Egger D, Bienz K, Baker SC. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J Virol. 2002;76:3697–3708. doi: 10.1128/JVI.76.8.3697-3708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reggiori F, Monastyrska I, Verheije MH, Cali T, Ulasli M, Bianchi S, Bernasconi R, et al. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe. 2010;7:500–508. doi: 10.1016/j.chom.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pedersen KW, van der Meer Y, Roos N, Snijder EJ. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J Virol. 1999;73:2016–2026. doi: 10.1128/jvi.73.3.2016-2026.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Snijder EJ, van Tol H, Roos N, Pedersen KW. Non-structural proteins 2 and 3 interact to modify host cell membranes during the formation of the arterivirus replication complex. J Gen Virol. 2001;82:985–994. doi: 10.1099/0022-1317-82-5-985. [DOI] [PubMed] [Google Scholar]
- 53.Heaton NS, Randall G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe. 2010;8:422–432. doi: 10.1016/j.chom.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ke PY, Chen SS. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J Clin Invest. 2011;121:37–56. doi: 10.1172/JCI41474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khakpoor A, Panyasrivanit M, Wikan N, Smith DR. A role for autophagolysosomes in dengue virus 3 production in HepG2 cells. J Gen Virol. 2009;90:1093–1103. doi: 10.1099/vir.0.007914-0. [DOI] [PubMed] [Google Scholar]
- 56.Lee YR, Lei HY, Liu MT, Wang JR, Chen SH, Jiang-Shieh YF, Lin YS, et al. Autophagic machinery activated by dengue virus enhances virus replication. Virology. 2008;374:240–248. doi: 10.1016/j.virol.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McLean JE, Wudzinska A, Datan E, Quaglino D, Zakeri Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J Biol Chem. 2011;286:22147–22159. doi: 10.1074/jbc.M110.192500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Panyasrivanit M, Greenwood MP, Murphy D, Isidoro C, Auewarakul P, Smith DR. Induced autophagy reduces virus output in dengue infected monocytic cells. Virology. 2011;418:74–84. doi: 10.1016/j.virol.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 59.Panyasrivanit M, Khakpoor A, Wikan N, Smith DR. Linking dengue virus entry and translation/replication through amphisomes. Autophagy. 2009;5:434–435. doi: 10.4161/auto.5.3.7925. [DOI] [PubMed] [Google Scholar]
- 60.Panyasrivanit M, Khakpoor A, Wikan N, Smith DR. Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J Gen Virol. 2009;90:448–456. doi: 10.1099/vir.0.005355-0. [DOI] [PubMed] [Google Scholar]
- 61.Ait-Goughoulte M, Kanda T, Meyer K, Ryerse JS, Ray RB, Ray R. Hepatitis C virus genotype 1a growth and induction of autophagy. J Virol. 2008;82:2241–2249. doi: 10.1128/JVI.02093-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci USA. 2009;106:14046–14051. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ferraris P, Blanchard E, Roingeard P. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J Gen Virol. 2010;91:2230–2237. doi: 10.1099/vir.0.022186-0. [DOI] [PubMed] [Google Scholar]
- 64.Mizui T, Yamashina S, Tanida I, Takei Y, Ueno T, Sakamoto N, Ikejima K, et al. Inhibition of hepatitis C virus replication by chloroquine targeting virus-associated autophagy. J Gastroenterol. 2010;45:195–203. doi: 10.1007/s00535-009-0132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology. 2008;48:1054–1061. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Su WC, Chao TC, Huang YL, Weng SC, Jeng KS, Lai MM. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J Virol. 2011;85:10561–10571. doi: 10.1128/JVI.00173-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Taguwa S, Kambara H, Fujita N, Noda T, Yoshimori T, Koike K, Moriishi K, et al. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J Virol. 2011;85:13185–13194. doi: 10.1128/JVI.06099-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li JK, Liang JJ, Liao CL, Lin YL. Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect. 2012;14:159–168. doi: 10.1016/j.micinf.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 69.Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol. 2010;221:117–124. doi: 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sir D, Liang C, Chen WL, Jung JU, Ou JH. Perturbation of autophagic pathway by hepatitis C virus. Autophagy. 2008;4:830–831. doi: 10.4161/auto.6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 73.Lee DY, Lee J, Sugden B. The unfolded protein response and autophagy: herpesviruses rule! J Virol. 2009;83:1168–1172. doi: 10.1128/JVI.01358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 75.Suhy DA, Giddings TH, Jr, Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J Virol. 2000;74:8953–8965. doi: 10.1128/jvi.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taylor MP, Kirkegaard K. Modification of cellular autophagy protein LC3 by poliovirus. J Virol. 2007;81:12543–12553. doi: 10.1128/JVI.00755-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brabec-Zaruba M, Berka U, Blaas D, Fuchs R. Induction of autophagy does not affect human rhinovirus type 2 production. J Virol. 2007;81:10815–10817. doi: 10.1128/JVI.00143-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tanida I, Fukasawa M, Ueno T, Kominami E, Wakita T, Hanada K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy. 2009;5:937–945. doi: 10.4161/auto.5.7.9243. [DOI] [PubMed] [Google Scholar]
- 79.Cho MW, Teterina N, Egger D, Bienz K, Ehrenfeld E. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology. 1994;202:129–145. doi: 10.1006/viro.1994.1329. [DOI] [PubMed] [Google Scholar]
- 80.Goldsmith CS, Tatti KM, Ksiazek TG, Rollin PE, Comer JA, Lee WW, Rota PA, et al. Ultrastructural characterization of SARS coronavirus. Emerg Infect Dis. 2004;10:320–326. doi: 10.3201/eid1002.030913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao Z, Thackray LB, Miller BC, Lynn TM, Becker MM, Ward E, Mizushima NN, et al. Coronavirus replication does not require the autophagy gene ATG5. Autophagy. 2007;3:581–585. doi: 10.4161/auto.4782. [DOI] [PubMed] [Google Scholar]
- 82.Tucker SP, Thornton CL, Wimmer E, Compans RW. Vectorial release of poliovirus from polarized human intestinal epithelial cells. J Virol. 1993;67:4274–4282. doi: 10.1128/jvi.67.7.4274-4282.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kirkegaard K, Jackson WT. Topology of double-membraned vesicles and the opportunity for non-lytic release of cytoplasm. Autophagy. 2005;1:182–184. doi: 10.4161/auto.1.3.2065. [DOI] [PubMed] [Google Scholar]
- 84.Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology. 2011;53:406–414. doi: 10.1002/hep.24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dong H, Czaja MJ. Regulation of lipid droplets by autophagy. Trends Endocrinol Metab. 2011;22:234–240. doi: 10.1016/j.tem.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Djavaheri-Mergny M, Maiuri MC, Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene. 2010;29:1717–1719. doi: 10.1038/onc.2009.519. [DOI] [PubMed] [Google Scholar]
- 88.Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 2005;5:886–897. doi: 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- 89.Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 91.Gao G, Luo H. The ubiquitin-proteasome pathway in viral infections. Can J Physiol Pharmacol. 2006;84:5–14. doi: 10.1139/y05-144. [DOI] [PubMed] [Google Scholar]
- 92.Banerjee R, Weidman MK, Echeverri A, Kundu P, Dasgupta A. Regulation of poliovirus 3C protease by the 2C polypeptide. J Virol. 2004;78:9243–9256. doi: 10.1128/JVI.78.17.9243-9256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Camborde L, Planchais S, Tournier V, Jakubiec A, Drugeon G, Lacassagne E, Pflieger S, et al. The ubiquitin-proteasome system regulates the accumulation of Turnip yellow mosaic virus RNA-dependent RNA polymerase during viral infection. Plant Cell. 2010;22:3142–3152. doi: 10.1105/tpc.109.072090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kerkvliet J, Papke L, Rodriguez M. Antiviral effects of a transgenic RNA-dependent RNA polymerase. J Virol. 2011;85:621–625. doi: 10.1128/JVI.01626-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.de Groot RJ, Rumenapf T, Kuhn RJ, Strauss EG, Strauss JH. Sindbis virus RNA polymerase is degraded by the N-end rule pathway. Proc Natl Acad Sci USA. 1991;88:8967–8971. doi: 10.1073/pnas.88.20.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gladding RL, Haas AL, Gronros DL, Lawson TG. Evaluation of the susceptibility of the 3C proteases of hepatitis A virus and poliovirus to degradation by the ubiquitin-mediated proteolytic system. Biochem Biophys Res Commun. 1997;238:119–125. doi: 10.1006/bbrc.1997.7251. [DOI] [PubMed] [Google Scholar]
- 97.Lawson TG, Gronros DL, Evans PE, Bastien MC, Michalewich KM, Clark JK, Edmonds JH, et al. Identification and characterization of a protein destruction signal in the encephalomyocarditis virus 3C protease. J Biol Chem. 1999;274:9904–9980. [PubMed] [Google Scholar]
- 98.Losick VP, Schlax PE, Emmons RA, Lawson TG. Signals in hepatitis A virus P3 region proteins recognized by the ubiquitin-mediated proteolytic system. Virology. 2003;309:306–319. doi: 10.1016/s0042-6822(03)00071-0. [DOI] [PubMed] [Google Scholar]
- 99.Welbourn S, Jirasko V, Breton V, Reiss S, Penin F, Bartenschlager R, Pause A. Investigation of a role for lysine residues in non-structural proteins 2 and 2/3 of the hepatitis C virus for their degradation and virus assembly. J Gen Virol. 2009;90:1071–1080. doi: 10.1099/vir.0.009944-0. [DOI] [PubMed] [Google Scholar]
- 100.Yajima T, Knowlton KU. Viral myocarditis: from the perspective of the virus. Circulation. 2009;119:2615–2624. doi: 10.1161/CIRCULATIONAHA.108.766022. [DOI] [PubMed] [Google Scholar]
- 101.Gorbea C, Makar KA, Pauschinger M, Pratt G, Bersola JL, Varela J, David RM, et al. A role for Toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J Biol Chem. 2010;285:23208–23223. doi: 10.1074/jbc.M109.047464. [DOI] [PMC free article] [PubMed] [Google Scholar]