

Abstract
Kawasaki disease causes systemic vasculitis. The development of skin lesions at the vaccination site with Bacillus Calmette‐Guérin (BCG) is an important diagnostic symptom. We hypothesized that infection with ubiquitous microorganisms immunogenically related to BCG might induce an immunopathologic reaction leading to the development of Kawasaki disease. Mice were first inoculated with BCG, and then secondarily inoculated 4 weeks later with crude extract from Mycobacterium intracellulare (cMI), an abundant atypical mycobacterium. Animals inoculated with BCG followed by cMI developed coronary arteritis with infiltration of inflammatory cells, whereas control animals inoculated with only cMI or BCG did not, suggesting that the immune response to the mycobacteria induced autoimmunity to the vascular wall. Intravenous injection with antibodies to peroxiredoxin II, a modulator of vascular remodeling and a suggested target for autoimmune vasculitis, also resulted in coronary arteritis, but only after prior inoculation with BCG. Tumor necrosis factor‐α, MCP1 and interferon‐γ production were significantly higher in the animals inoculated with BCG than in the control groups (P<0.05). BCG immunization was required for the development of coronary arteritis, suggesting that these cytokines might play important roles. The results indicate that BCG induces primary autoimmunity and stimulates cytokine induction, and that atypical mycobacterial infection boosts the autoimmunity resulting in coronary arteritis.
Keywords: Kawasaki disease, BCG, atypical mycobacterium, mouse model, peroxiredoxin
Introduction
Kawasaki disease with subsequent coronary aneurism was first reported by the Japanese pediatrician Tomisaku Kawasaki in 1967 (Kawasaki, 1967, 1974). To date, many cases have been reported from over 60 countries (Burns & Glode, 2004). Several investigations have revealed that Kawasaki disease consists of systemic vasculitis with hypercytokinemia (2000, 1995, 1988, 1990, 1988, 1989). A number of reports indicate that the development of Kawasaki disease might be associated with bacterial or viral infections such as those caused by Staphylococcus aureus, hemolytic Streptococcus, Yersinia, Epstein–Barr virus, human herpesvirus‐6, or human coronavirus (1997, 1993, 2005, 2002, 1988, 1993). However, the etiology of Kawasaki disease is still controversial.
Several investigations have demonstrated coronary arteritis in experimental animals, and in some of these, dilatation of the coronary artery developed (1983, 1988, 1979, 2004). However, these models were not directly applicable to the clinical phenomenon, as the methods of induction were not based on clinical observations.
Flare and crust formation at Bacillus Calmette‐Guérin (BCG) inoculation sites is one of the typical symptoms of Kawasaki disease listed in the diagnostic criteria, and is probably an immunopathologic response (2005, 1997, 1993). Although not a principal symptom of Kawasaki disease, it allows for early diagnosis of most cases. This response indicates that the immune response to BCG antigens, which reside intradermally in vaccine recipients, might be boosted when the systemic vasculitis occurs.
In patients with Kawasaki disease, antibodies to the mycobacterial heat shock protein (HSP65) increase and recognize the conserved peptide of the human homolog, suggesting that a cross‐reactive immune response to BCG and human antigens might cause immunopathogenesis that leads to systemic vasculitis (Sireci et al., 2000; Yokota et al., 2006).
Autoantibodies, such as antineutrophil antibody (ANCA) and antiendothelial cell antibody (AECA), are known to be associated with autoimmune vasculitis (1997, 2006). It has been reported elsewhere that AECA is found in patients with Kawasaki disease (Kaneko et al., 1994). Recently, Karasawa et al. have reported that autoantibodies to peroxiredoxin II are found predominantly in the sera of patients with autoimmune diseases who have systemic vasculitis (2003, 2004). A homolog of peroxiredoxin II is also conserved in many microorganisms, and this homolog is also expressed in BCG at a higher level than in Mycobacterium tuberculosis (Springer et al., 2001). Thus, cross‐reactive immune responses, which might be triggered by a microorganism that possesses cross‐reactive antigens, could be associated with the development of Kawasaki's disease.
As described above, many reports indicate that Kawasaki disease is associated with infection. Therefore, we hypothesized that infectious agents that have antigenic proteins homologous to those of BCG and the vascular wall might induce an immunopathologic reaction to both the lesion at the BCG vaccination site and the vascular wall.
In this study, we examined whether coronary arteritis develops from the combination of a primary immunization with BCG and a secondary immunization with a ubiquitous microorganism, Mycobacterium intracellurare (cMI), which should share cross‐reactive antigens. In this model, we demonstrate coronary arteritis with cytokinemia, similar to that found in Kawasaki disease. Furthermore, we show that administration of an antibody to a component of the vascular endotherium, peroxiredoxin II, after BCG priming also induces coronary arteritis, indicating that an immunopathologic reaction causes the coronary arteritis and requires prior BCG immunization.
Materials and methods
Bacteria
The BCG (Tokyo strain) was purchased from Nihon BCG Inc. (Tokyo, Japan). Mycobacterium intracellulare (MI, JCM number 6384) was obtained from the Japanese Collection of Microorganisms (RIKEN BioResource Center, Saitama, Japan).
Antibody
We purchased antiperoxiredoxin II polyclonal antibody from Lab Frontier (Seoul, Korea.). This antibody was obtained from rabbits immunized with recombinant human peroxyredoxin II purified from Escherichia coli. The antibody also recognizes the mouse and rat homologs.
Preparation of crude M. intracellulare lysate
We followed the procedure of Lehman et al. to obtain crude lysate for injection (Lehman et al., 1983). MI was cultured in Middlebrook 7H9 broth (Difco, Detroit, MI) for 24 h at 37°C, and then harvested by centrifugation (1000 g, 10 min). After being washed three times with phosphate‐buffered saline (PBS) (pH 7.4, 1000 g, 10 min), the bacteria were lysed by overnight incubation at room temperature with a 10‐fold volume of 4% sodium dodecyl sulfate (SDS) (EM Science, Gibbstown, NJ), and then washed 10 times with PBS to remove the supernatant and SDS. The bacterial lysate was incubated sequentially with 250 μg mL−1 RNAse, DNAse I, and trypsin (Sigma, St Louis, MO) for 4 h. After centrifugation, the pellets were washed four times with PBS. Crude lysate was then obtained by sonication of the pellet (5 g wet weight in 20 mL of PBS) on ice for 2 h (5‐s pulse, then a 1‐s pause, at a fixed frequency of 20 kHz). The sonicated preparation was then centrifuged for 1 h at 40 000 g. at 4°C, and the supernatant was harvested for injection. The wet volume of the ultracentrifugation pellet was then adjusted to a final concentration of 1 mg mL−1 in PBS.
Animal experiments
Female C57BL/6J mice 3 weeks of age were purchased from the Sankyo Laboratory (Tokyo, Japan). BCG (0.4 mg in 0.05 mL of saline) or saline (as a control) was inoculated intradermally into the left flank. After observation of a nodular formation at the inoculation site, 0.1 mL of the purified protein derivative (PPD) was injected intradermally for the skin test to ascertain immunization. A cutaneous reaction was observed 48 h after the injection. Four weeks after the BCG immunization, the mice were intradermally injected with either 0.5 mg of crude MI lysates (cMI) in 0.5 mL of PBS or with PBS as a control, in either one or four daily dose(s). In other experimental groups, the mice were administered rabbit antiserum to mice peroxiredoxin II, or PBS as a control, through the tail vein 4 weeks after the BCG immunization. Ten days after cMI injection or the administration of peroxiredoxin II antibody, sera were collected from the jugular vein. Specimens for histologic analysis were prepared as described previously (Nakamura et al., 2000). Briefly, after the animals had been sacrificed under anesthesia, the heart was excised and embedded in OCT compound (Sakura Finetechnical Co., Ltd, Tokyo, Japan), and then stored at −30oC. Serial sections 7 μm in width were made using a cryostat.
During experiments, the body weight and rectal temperature of the mice were monitored at the time of BCG inoculation and then every day upon and after the secondary immunization or antibody administration.
Histologic examination
The serial sections from base to apex of the heart were stained with hematoxylin and eosin to evaluate them for coronary arteritis. Eight sections from each animal were evaluated, and five eye‐fields were observed for each section. The criteria for coronary arteritis were as follows: ‘mild’ was defined as infiltration of inflammatory cells surrounding the arterial walls; and ‘severe’ was defined as the infiltration of inflammatory cells within the layers of the arterial walls.
Cytokine analysis
The amounts of tumor necrosis factor‐α (TNF‐α), interferon‐γ (IFN‐γ), interleukin (IL)‐6, IL‐10 and monocyte chemoattractant protein‐1 (MCP‐1) in animal sera were evaluated by the sandwich enzyme‐linked imunosorbent assay (Mouse inflammation kit, BD Pharmingen, San Diego, CA).
Statistical analysis
To compare groups, the Wilcoxon test, χ2 test and Fisher's exact values were calculated using the jmp application (SAS Institute Inc., Cary, NC).
Results
Primary BCG immunization followed by secondary cMI immunization induced coronary arteritis
Three‐week‐old female mice were inoculated with BCG or saline. Four days after BCG immunization, nodular formations were observed in all the mice inoculated with BCG, but not in those inoculated with saline (Table 1). The PPD tests resulted in erythema formation with increased venular appearance in all the animals inoculated with BCG, but not in the saline‐inoculated animals (Table 1). Four weeks after being immunized with BCG or saline, animals were intraperitoneally inoculated with cMI or PBS.
Table 1.
Nodal change and tuberculin reaction of injection site of mice (C57BL/6J) inoculated with BCG or saline
| n | Nodal change on day 4 after BCG administration | Erythema formation by PPD test | |
|---|---|---|---|
| One booster | |||
| B+I+ | 3 | 3/3 (100%) | 3/3 (100%) |
| B+I− | 3 | 3/3 (100%) | 3/3 (100%) |
| B−I+ | 3 | 0/3 (0%) | 0/3 (0%) |
| Four boosters | |||
| B+I+ | 5 | 5/5 (100%) a | 5/5 (100%) a |
| B+I− | 5 | 5/5 (100%) a | 5/5 (100%) a |
| B−I+ | 5 | 0/5 (0%) | 0/5 (0%) |
| B−I− | 5 | 0/5 (0%) | 0/5 (0%) |
| Anti‐PrxII | |||
| B+P+ | 5 | 5/5 (100%) a | 5/5 (100%) a |
| B+P− | 5 | 5/5 (100%) a | 5/5 (100%) a |
| B−P+ | 5 | 0/5 (0%) | 0/5 (0%) |
| B−P− | 5 | 0/5 (0%) | 0/5 (0%) |
One booster: an experimental primary inoculation followed by one cMI inoculation or its substitute.
Four boosters: an experimental primary inoculation followed by four cMI inoculations or their substitutes.
Anti‐PrxII: an experimental primary inoculation followed by administration of antiperoxiredoxin II antibody or its substitute.
B+, BCG administered; B−, saline administered as a substitute for BCG; I+, cMI administered; I−, saline administered as a substitute for cMI; P+, antiperoxiredoxin II antibody administered; P−, saline administered as a substitute for antiperoxiredoxin II antibody.
Significant difference from the control (B−P− or B−I−).
The histologic analysis was performed 4 weeks after the secondary inoculation. Mild coronary arteritis with mononuclear cell infiltration around the arterial wall was demonstrated in six of 120 sections (5%) from animals immunized with both BCG and cMI (B+I+, Fig. 1a, Table 2), but in only one of the 120 sections (0.8%) from animals immunized with BCG alone (B+I−, Table 2). Sections from animals immunized with cMI alone without BCG (B−I+) did not show coronary arteritis.
Figure 1.
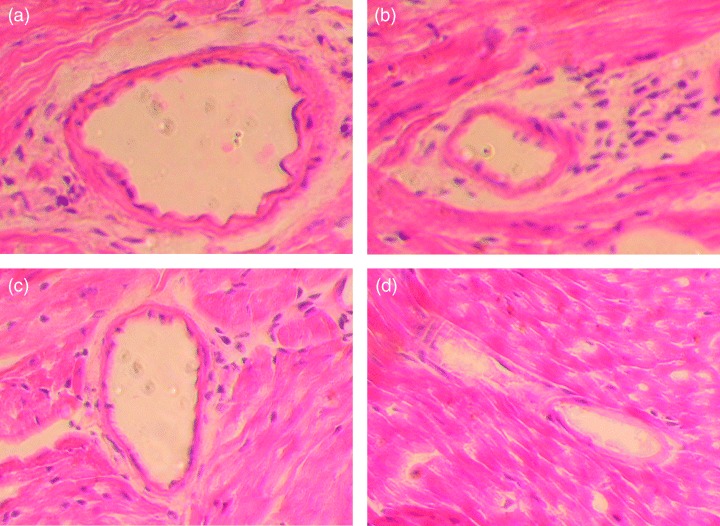
Hematoxylin and eosin staining of coronary arteries 10 days after the secondary inoculation with cMI or antiperoxiredoxin II antibodies. (a) Mild coronary arteritis with inflammatory cell infiltration around the arterial wall in an animal inoculated with one dose of cMI following BCG inoculation. (b) Severe coronary arteritis with inflammatory cell infiltration within the arterial wall in an animal inoculated with four doses of cMI following BCG inoculation. (c) Coronary arteritis in an animal inoculated with anti‐peroxiredoxin II antibody following BCG administration. (d) An artery without cellular infiltration in a control animal inoculated with saline followed by PBS.
Table 2.
Histologic evaluation of heart tissue from the mice (C57BL/6J)
| Coronary arteritis | ||||
|---|---|---|---|---|
| n | Mild | Severe | Total | |
| One booster | ||||
| B+I+ | 3 | 6/120 (5%) | 0/120 (0%) | 6/120 (5%) |
| B+I− | 3 | 1/120 (0.8%) | 0/120 (0%) | 1/120 (0.8%) |
| B−I+ | 3 | 0/120 (0%) | 0/120 (0%) | 0/120 (0%) |
| Four boosters | ||||
| B+I+ | 5 | 19/200 (9.5%) a | 1/200 (0.5%) | 20/200 (10%) a |
| B+I− | 5 | 2/200 (1%) | 0/200 (0%) | 2/200 (1%) |
| B−I+ | 5 | 0/200 (0%) | 0/200 (0%) | 0/200 (0%) |
| B−I− | 5 | 0/200 (0%) | 0/200 (0%) | 0/200 (0%) |
| Anti‐PrxII | ||||
| B+P+ | 5 | 16/200 (8%) a | 0/200 (0%) | 16/200 (8%) a |
| B+P− | 5 | 0/200 (0%) | 0/200 (0%) | 0/200 (0%) |
| B−P+ | 5 | 1/200 (0.5%) | 0/200 (0%) | 1/200 (0.5%) |
| B−P− | 5 | 0/200 (0%) | 0/200 (0%) | 0/200 (0%) |
Data are expressed as the number of positive cases of arteritis per 200 fields.
One booster: an experimental primary inoculation followed by one cMI inoculation or its substitute.
Four boosters: an experimental primary inoculation followed by four cMI inoculations or their substitutes.
Anti‐PrxII: an experimental primary inoculation followed by administration of antiperoxiredoxin II antibody or its substitute.
B+, BCG administered; B−, saline administered as a substitute for BCG; I+, cMI administered; I−, saline administered as a substitute for cMI; P+, antiperoxiredoxin II antibody administered; P−, saline administered as a substitute for antiperoxiredoxin II antibody.
Significant difference from other groups.
In another set of experimental groups (‘Four boosters’ in tables), animals were inoculated four times daily with cMI or PBS following primary inoculation with BCG or saline. As a result, coronary arteritis, including one severe case (infiltration into the arterial wall), was demonstrated in significantly more heart sections (20/200, 10%) in animals that had both BCG and cMI immunization (B+I+) than in those in other groups (Fig. 1b; Table 2, P<0.05 vs. 1% in B+I−, 0% in B−I+, and 0% in B−I−). When BCG or cMI immunizations were performed independently of each other, coronary arteritis was not observed or barely observed in mice treated with either BCG or cMI only. Thus, BCG immunization followed by multiple doses of cMI immunization significantly induced coronary arteritis. Body weight and body temperature were not affected by these immunizations (data not shown).
Development of coronary arteritis as a result of anti‐peroxiredoxin II antibody administration requires BCG immunization
We examined whether the administration of antibodies to peroxiredoxin II induces coronary arteritis. Administration of rabbit antiserum against peroxiredoxin II alone resulted in coronary arteritis in only one of 200 sections examined (0.5%, Table 2). However, when mice were inoculated with BCG prior to antibody administration, coronary arteritis developed in 16 of 200 (8%) sections and at a significantly higher frequency than in the other control groups (P<0.05, Fig. 1c, Table 2), similar to that observed with BCG and cMI immunization. Thus, antibody against peroxiredoxin II induced coronary arteritis, but only when administered after BCG immunization.
Cytokine profiles in animals with vasculitis
Ten days after the cMI or PBS inoculation following BCG or saline inoculation, sera were obtained from the mice to determine cytokine levels. Levels of TNF‐α, IFN‐γ, and MCP‐1, but not IL‐10 or IL‐6, were significantly higher in both the BCG‐inoculated groups (B+I+ and B+I−) than in those mice treated with saline instead of BCG (Table 3, P<0.05). None of these cytokines was elevated in animals inoculated with only cMI (without primary BCG inoculation; B−I+) compared with levels in control animals (Table 3, B−I−).
Table 3.
Serum levels of inflammatory cytokines
| n | TNF‐α | IFN‐γ | MCP‐1 | IL‐10 | IL‐6 | |
|---|---|---|---|---|---|---|
| One booster | ||||||
| B+I+ | 3 | 15.9 ± 3.8 | 4.9 ± 0.8 | 31.5 ± 2.4 | 2.8 ± 4.8 | 2.9 ± 1.9 |
| B+I− | 3 | 14.7 ± 2.8 | 4.2 ± 5.1 | 43.3 ± 9.3 | 0.0 ± 0.0 | 2.3 ± 0.4 |
| B−I+ | 3 | 9.8 ± 1.4 | 3.1 ± 3.0 | 29.1 ± 4.4 | 0.0 ± 0.0 | 1.8 ± 1.7 |
| Four boosters | ||||||
| B+I+ | 5 | 27.4 ± 22.4 a | 10.7 ± 7.6 a | 61.3 ± 27.7 a | 2.7 ± 6.0 | 6.6 ± 2.9 |
| B+I− | 5 | 17.3 ± 3.0 a | 8.4 ± 3.9 a | 51.0 ± 9.1 a | 1.3 ± 1.8 | 6.8 ± 1.8 |
| B−I+ | 5 | 11.4 ± 4.9 | 2.7 ± 1.1 | 42.4 ± 4.6 | 2.2 ± 3.0 | 6.2 ± 2.9 |
| B−I− | 5 | 8.1 ± 1.3 | 0.7 ± 0.9 | 38.1 ± 5.8 | 2.7 ± 4.0 | 5.8 ± 1.4 |
| Anti‐PrxII | ||||||
| B+P+ | 5 | 25.5 ± 13.5 a | 9.5 ± 2.2 b | 83.0 ± 9.8 a | 0.0 ± 0.0 | 5.7 ± 1.2 |
| B+P− | 5 | 18.2 ± 3.7 a | 5.9 ± 1.7 a | 67.8 ± 11.0 a | 0.9 ± 2.0 | 5.5 ± 1.2 |
| B−P+ | 5 | 9.1 ± 2.2 | 1.0 ± 1.1 | 48.1 ± 9.9 | 0.4 ± 0.8 | 4.8 ± 0.9 |
| B−P− | 5 | 7.3 ± 0.6 | 1.3 ± 0.8 | 38.1 ± 5.7 | 0.0 ± 0.0 | 3.9 ± 1.4 |
Data are expressed as mean ± SD (pg mL−1).
One booster: an experimental primary inoculation followed by one cMI inoculation or its substitute.
Four boosters: an experimental primary inoculation followed by four cMI inoculations or their substitutes.
Anti‐PrxII: an experimental primary inoculation followed by administration of antiperoxiredoxin II antibody or its substitute.
B+, BCG administered; B−, saline administered as a substitute for BCG; I+, cMI administered; I−, saline administered as a substitute for cMI; P+, anti‐peroxiredoxin II antibody administered; P−, saline administered as a substitute for antiperoxiredoxin II antibody.
Significant difference from control (B−P−).
Significant difference from control and B+I− group.
Similarly, the administration of antibodies to peroxiredoxin II without prior BCG immunization (B−P+) also did not result in elevated cytokine levels (Table 3).
The IFN‐γ level was significantly higher in animals given antibody to peroxiredoxin II following BCG immunization than it was in animals immunized with BCG alone (Table 3, P<0.05).
The average values of TNF‐α, IFN‐γ and MCP1 were higher in the B+I+ and B+P+ groups than in the B+I− and B+P− groups, although these differences were not statistically significant.
Discussion
As flare and crust formation is observed in patients with Kawasaki disease, the immune response to BCG might be associated with the development of systemic vasculitis, a hallmark of this disease. As described above, autoantibodies to peroxiredoxin II and the human homolog of mycobaterial HSP65 are found in patients with Kawasaki disease (Sireci et al., 2000; 2003, 2006). Many reports also indicate that Kawasaki disease is associated with infection. This suggests that an immunopathologic reaction triggered by unknown microbial agents might be associated with the development of Kawasaki disease.
We hypothesized that a ubiquitous microorganism that shares a cross‐reactive antigen with BCG might induce an immunopathologic reaction to both BCG and vascular antigens. Any microorganism could be a candidate, as long as it shares an antigenically related component with BCG and the endothelium. Under this assumption, we did not inoculate animals with BCG twice. Instead, we used an extract from an atypical mycobacterium as a booster immunogen to demonstrate this phenomenon experimentally in mice, as this microorganism has antigens that are closely related to those of BCG. We used the crude extract of M. intracellulare, one of the most abundant atypical mycobacteria. We demonstrated that immunization with atypical mycobacterium components following BCG immunization resulted in significant coronary arteritis. The development of coronary arteritis required both immunization with BCG and a secondary immunization with atypical mycobacterium components.
In this model, four booster doses resulted in an effect superior to that of one booster. As nonreplicating immunogens tend to be rapidly degraded by phagocytes in the absence of an adjuvant (Harlow & Lane, 1988), multiple immunizations might be required for more apparent booster immunity.
TNF‐α, IFN‐γ, and MCP‐1, cytokines that are elevated in Kawasaki disease (1988, 1990, 1988, 1997), were induced after BCG immunization. When the mice were inoculated with atypical mycobacterial antigen alone, coronary arteritis did not develop. This indicated that BCG immunization may induce cytokine production and provide the cytokine levels necessary for the development of coronary arteritis, and this idea was also supported by the experiment involving the use of antibody to one of the endothelial components.
Administration of antibody to peroxiredoxin II alone barely induced coronary arteritis. When animals were first primed with BCG, the administration of the antibody resulted in significant coronary arteritis, similar to that observed after multiple cMI inoculations following BCG immunization.
Seventy per cent of Kawasaki disease is observed in patients who are less than 3 years of age and presents shortly after BCG immunization. Several reports indicate that BCG may reside in the host for a few years after inoculation (Gasior‐Chrzan, 2001). This clinical observation might support the importance of BCG as the first immunogen and as the immune modulator that provides the circumstances for the development of Kawasaki disease.
TNF‐α, IFN‐γ and MCP1 levels tended to be higher in the B+I+ and B+P+ groups than in the B+I− and B+P− groups, although, except for the IFN‐γ level in the B+P+ group compared with the B+P− groups, these differences were not statistically significant. Immunopathologic inflammation may further enhance cytokine production.
Peroxiredoxin II has antioxidant activity is distributed in many types of cell, including those of the endothelium, and is located in both the cytoplasm and the cell membrane (2005, 2003, 2001). Peroxiredoxin II may be a regulator of platelet‐derived growth factor and remodeling (Choi et al., 2005). In patients with autoimmune diseases who have systemic vasculitis, antibodies to peroxiredoxin II have been demonstrated (Karasawa et al., 2003). Thus, peroxiredoxin II might be an important target of systemic vasculitis. This molecule is conserved in mammals and microorganisms, including mycobacteria (Hofmann et al., 2002). Antibodies that are cross‐reactive to this molecule after a microbial infection might induce destruction of the endothelium and also inhibit its function during the healing process.
Another report indicated that antibodies against HSP may cause cross‐linking of HSP molecules, resulting in the subsequent production of IL‐8 and TNF‐α (Yokota et al., 2006). Thus, autoantibodies against molecules that might hamper endothelial repair and stimulate the cytokine network might be associated with the development of Kawasaki disease. As these molecules (peroxiredoxin II and HSP) are conserved between many microorganisms and humans, microorganisms that possess these homologs might also induce similar immunopathogenic reactions.
In our study, none of the animals developed coronary aneurysm. Blood pressure in mice might be too low to allow development of the aneurysm. Flare and crust formation was not observed at the site of BCG inoculation after cMI immunization. As several studies have indicated that Kawasaki disease might be associated with genetic background (2004, 2004, 2003, 2003, 2001, 1997), we may need to examine the host's genetic background associated with Kawasaki disease in order to produce stronger reactions. This might also explain why none of the animals was febrile. These approaches, investigations into infectious agents/immunogens and host factors, might be critical for clarifying the etiology of Kawasaki disease in detail.
In conclusion, BCG inoculation followed by booster immunizations with an atypical mycobacterium antigen resulted in coronary arteritis in mice. This model may lead to a better understanding of Kawasaki disease.
Acknowledgements
We thank Yasuko Nunoura for technical assistance.
Editor: Patrick Brennan
References
- Abe J, Onimaru M, Matsumoto S, Noma S, Baba K, Ito Y, Kohsaka T & Takeda T (1997) Clinical role for a superantigen in Yersinia pseudotuberculosis infection. J Clin Invest 99: 1823–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama T & Yashiro K (1993) Probable role of Streptococcus pyogenes in Kawasaki disease. Eur J Pediatr 152: 82–92. [DOI] [PubMed] [Google Scholar]
- Asano T & Ogawa S (2000) Expression of IL‐8 in Kawasaki disease. Clin Exp Immunol 122: 514–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayusawa M, Sonobe T, Uemura S, Ogawa S, Nakamura Y, Kiyosawa N, Ishii M & Harada K (2005) Kawasaki Disease Research Committee. Revision of diagnostic guidelines for Kawasaki disease (the 5th revised edition). Pediatr Int 47: 232–234. [DOI] [PubMed] [Google Scholar]
- Belizna C & Tervaert JW (1997) Specificity, pathogenecity, and clinical value of antiendothelial cell antibodies. Semin Arthritis Rheum 27: 98–109. [DOI] [PubMed] [Google Scholar]
- Bosch X, Guilabert A & Font J (2006) Antineutrophil cytoplasmic antibodies. Lancet 368: 404–418. [DOI] [PubMed] [Google Scholar]
- Burns JC & Glode MP (2004) Kawasaki syndrome. Lancet 364: 533–544. [DOI] [PubMed] [Google Scholar]
- Choi MH, Lee IK, Kim GW et al (2005) Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature 435: 347–353. [DOI] [PubMed] [Google Scholar]
- Eberhard BA, Andersson U, Laxer RM, Rose R & Silverman ED (1995) Evaluation of the cytokine response in Kawasaki disease. Pediatr Infect Dis J 14: 199–203. [DOI] [PubMed] [Google Scholar]
- Esper F, Shapiro ED, Weibel C, Ferguson D, Landry ML & Kahn JS (2005) Association between a novel human coronavirus and Kawasaki disease. J Infect Dis 191: 499–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukazawa R, Sonobe T, Hamamoto K et al (2004) Possible synergic effect of angiotensin‐I converting enzyme gene insertion/deletion polymorphism and angiotensin‐II type‐1 receptor 1166A/C gene polymorphism on ischemic heart disease in patients with Kawasaki disease. Pediatr Res 56: 597–601. [DOI] [PubMed] [Google Scholar]
- Furukawa S, Matsubara T, Jujoh K, Yone K, Sugawara T, Sasai K, Kato H & Yabuta K (1988) Peripheral blood monocyte/macrophages and serum tumor necrosis factor in Kawasaki disease. Clin Immunol Immunopathol 48: 247–251. [DOI] [PubMed] [Google Scholar]
- Gasior‐Chrzan B (2001) Delayed granulomatous lesion at the bacillus Calmette‐Guerin vaccination site. Acta Derm Venereol 81: 302–304. [DOI] [PubMed] [Google Scholar]
- Harlow E & Lane D (1988) Antibodies: a Laboratory Manual. Cold Spring Harbor Laboratory Press, NY. [Google Scholar]
- Hofmann B, Hecht HJ & Flohe L (2002) Peroxiredoxins. Biol Chem 383: 347–364. [DOI] [PubMed] [Google Scholar]
- Ihira M, Yoshikawa T, Ishii J et al (2002) Serological examination of human herpesvirus 6 and 7 in patients with coronary artery disease. J Med Virol 67: 534–537. [DOI] [PubMed] [Google Scholar]
- Kaneko K, Savage CO, Pottinger BE, Shah V, Pearson JD & Dillon MJ (1994) Antiendothelial cell antibodies can be cytotoxic to endothelial cells without cytokine pre‐stimulation and correlate with ELISA antibody measurement in Kawasaki disease. Clin Exp Immunol 98: 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasawa R, Sekine T, Oota S et al (2003) Targets of anti‐endothelial cell antibodies in patients with systemic vasculitis: identification by the proteomic approach. Arthritis Rheum 48 (Suppl), 391. [Google Scholar]
- Kariyazono H, Ohno T, Khajoee V, Ihara K, Kusuhara K, Kinukawa N, Mizuno Y & Hara T (2004) Association of vascular endothelial growth factor (VEGF) and VEGF receptor gene polymorphisms with coronary artery lesions of Kawasaki disease. Pediatr Res 56: 953–959. [DOI] [PubMed] [Google Scholar]
- Kawasaki T (1967) Pediatric acute mucocutaneous lymph node syndrome: clinical observation of 50 cases. Arerugi (Jpn J Allergy) 16: 178–222. [Google Scholar]
- Kawasaki T, Kosaki F, Okawa S, Shigematsu I & Yanagawa H (1974) A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan. Pediatrics 54: 271–276. [PubMed] [Google Scholar]
- Kikuta H, Taguchi Y, Tomizawa K et al (1988) Epstein–Barr virus genome‐positive T lymphocytes in a boy with chronic active EBV infection associated with Kawasaki‐like disease. Nature 333: 455–457. [DOI] [PubMed] [Google Scholar]
- Kuniyuki S & Asada M (1997) An ulcerated lesion at the BCG vaccination site during the course of Kawasaki disease. J Am Acad Dermatol 37: 303–304. [PubMed] [Google Scholar]
- Lee SC, Na YP & Lee JB (2003) Expression of peroxiredoxin II in vascular tumors of the skin: a novel vascular marker of endothelial cells. J Am Acad Dermatol 49: 487–491. [DOI] [PubMed] [Google Scholar]
- Lehman TJ, Allen JB, Plotz PH & Wilder RL (1983) Polyarthritis in rats following the systemic injection of Lactobacillus casei cell walls in aqueous suspension. Arthritis Rheum 26: 1259–1265. [DOI] [PubMed] [Google Scholar]
- Lehman TJ, Warren R, Gietl D, Mahnovski V & Prescott M (1988) Variable expression of Lactobacillus casei cell wall‐induced coronary arteritis: an animal model of Kawasaki's disease in selected inbred mouse strains. Clin Immunol Immunopathol 48: 108–118. [DOI] [PubMed] [Google Scholar]
- Leung DY, Meissner HC, Fulton DR, Murray DL, Kotzin BL & Schlievert PM (1993) Toxic shock syndrome toxin‐secreting Staphylococcus aureus in Kawasaki syndrome. Lancet 342: 1385–1388. [DOI] [PubMed] [Google Scholar]
- Matsubara T, Furukawa S & Yabuta K (1990) Serum levels of tumor necrosis factor, interleukin 2 receptor, and interferon gamma in Kawasaki disease involved coronary artery lesions. Clin Immunol Immunopathol 56: 29–36. [DOI] [PubMed] [Google Scholar]
- Maury CPJ, Salo E & Pelkonen P (1988) Circulating interleukin‐1 beta in patients with Kawasaki disease. N Engl J Med 319: 1670–1671. [DOI] [PubMed] [Google Scholar]
- Murata H (1979) Experimental candida‐induced arteritis in mice. Relation to arteritis in the mucocutaneous lymph node syndrome. Microbiol Immunol 23: 825–831. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Ueda Y, Juan Y, Katsuda S, Takahashi H & Koh E (2000) Fas‐mediated apoptosis in adriamycin‐induced cardiomyopathy in rats: in vivo study. Circulation 102: 572–578. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Zaitsu M, Hara M, Yokota G, Watanabe M, Ueda Y, Imayoshi M, Ishii E, Tasaki H & Hamasaki Y (2003) A polymorphism in the promoter of the CD14 gene (CD14/−159) is associated with the development of coronary artery lesions in patients with Kawasaki disease. J Pediatr 143: 357–362. [DOI] [PubMed] [Google Scholar]
- Ouchi K, Suzuki Y, Shirakawa T & Kishi F (2003) Polymorphism of SLC11A1 (formerly NRAMP1) gene confers susceptibility to Kawasaki disease. J Infect Dis 187: 326–329. [DOI] [PubMed] [Google Scholar]
- Ozaki S (2004) Molecular mechanism of vasculitis. Molecular Medicine. (Japanese) 41: 191–198. [Google Scholar]
- Philip S, Lee WC, Liu SK, Wu MH & Lue HC (2004) A swine model of horse serum‐induced coronary vasculitis: an implication for Kawasaki disease. Pediatr Res 55: 211–219. [DOI] [PubMed] [Google Scholar]
- Quasney MW, Bronstein DE, Cantor RM et al (2001) Increased frequency of alleles associated with elevated tumor necrosis factor‐alpha levels in children with Kawasaki disease. Pediatr Res 49: 686–690. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Kang SW, Chang TS, Jeong W & Kim K (2001) Peroxiredoxin, a novel family of peroxidases. IUBMB Life 52: 35–41. [DOI] [PubMed] [Google Scholar]
- Sato N, Sagawa K, Sasaguri Y, Inoue O & Kato H (1993) Immunopathology and cytokine detection in the skin lesions of patients with Kawasaki disease. J Pediatr 122: 198–203. [DOI] [PubMed] [Google Scholar]
- Sireci G, Dieli F & Salerno A (2000) T cells recognize an immunodominant epitope of heat shock protein 65 in Kawasaki disease. Mol Med 6: 581–590. [PMC free article] [PubMed] [Google Scholar]
- Springer B, Master S, Sander P, Zahrt T, McFalone M, Song J, Papavinasasundaram KG, Colston MJ, Boettger E & Deretic V (2001) Silencing of oxidative stress response in Mycobacterium tuberculosis : expression patterns of ahpC in virulent and avirulent strains and effect of ahpC inactivation. Infect Immunol 69: 5967–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Yamamoto K, Kataoka S, Kakihara T, Tanaka A, Sato S & Uchiyama M (1997) High incidence of angiotensin I converting enzyme genotype II in Kawasaki disease patients with coronary aneurysm. Eur J Pediatr 156: 266–268. [DOI] [PubMed] [Google Scholar]
- Ueno Y, Takano N, Kanegane H, Yokoi T, Yachie A, Miyawaki T & Taniguchi N (1989) The acute phase nature of interleukin 6 studies in Kawasaki disease and other febrile illnesses. Clin Exp Immunol 76: 337–342. [PMC free article] [PubMed] [Google Scholar]
- Wong M, Silverman ED & Fish EN (1997) Evidence for RANTES, monocyte chemotactic protein‐1, and macrophage inflammatory protein‐1 beta expression in Kawasaki disease. J Rheumatol 24: 1179–1185. [PubMed] [Google Scholar]
- Yokota S, Minota S & Fujii N (2006) Anti‐HSP auto‐antibodies enhance HSP‐induced pro‐inflammatory cytokine production in human monocytic cells via Toll‐like receptors. Int Immunol 18: 573–580. [DOI] [PubMed] [Google Scholar]