

Abstract
Matrix proteins are the driving force of assembly of enveloped viruses. Their main function is to interact with and polymerize at cellular membranes and link other viral components to the matrix–membrane complex resulting in individual particle shapes and ensuring the integrity of the viral particle. Although matrix proteins of different virus families show functional analogy, they share no sequence or structural homology. Their diversity is also evident in that they use a variety of late domain motifs to commit the cellular vacuolar protein sorting machinery to virus budding. Here, we discuss the structural and functional aspects of the filovirus matrix protein VP40 and compare them to other known matrix protein structures from vesicular stomatitis virus, influenza virus and retroviral matrix proteins.
Keywords: Matrix protein, Ebola virus, VP40, Influenza virus M1, VSV M, HIV-1 MA, Late domains
1. Overview of general matrix protein functions
Enveloped RNA viruses from the families of Filoviridae, Paramyxoviridae, Rhabdoviridae and Bornaviridae (constituting the order Mononegavirales), Orthomyxoviridae and Retroviridae are a morphologically diverse group of viruses. The viral shape is determined by the matrix protein and the nucleocapsid that are responsible for producing spherical, rod-shaped, bullet-shaped or bacilliform particles. Viral matrix proteins generally constitute a major structural protein of the viral particle and are located underneath the viral membrane. During the assembly process in a host cell, matrix proteins self-assemble into higher-order oligomers and/or polymerise at cellular membranes [1,2]. They display a high tendency to aggregate in vitro, which may reflect their ability to self-assemble in vivo. Matrix proteins interact with membranes and evidence suggests that they provide a link between the cytoplasmic tails of the glycoproteins and the nucleocapsids that contain the RNA genome within the viral particle [2]. Although in some cases matrix protein expression in eukaryotic cells is sufficient to induce virus-like particles (VLPs) [3–5], the structural requirements for such ordered membrane-associated polymerisation reactions are not well understood and often depend on other viral components such as the glycoprotein [2]. In addition, cellular factors modulating membrane structures might play a role. Consequently, no virus-like particles have yet been produced from soluble matrix proteins and artificial lipid bilayers in vitro.
In contrast to segmented and non-segmented negative strand viruses, retroviral matrix proteins are initially part of the Gag poly-protein. Expression of Gag is largely sufficient to perform virus assembly and budding at the plasma membrane which leads to the release of virus-like particles (VLPs). In immature viral particles, proteolytic processing generates several distinct protein products, including MA (matrix protein), CA (capsid protein) and NC (nucleocapsid protein) thus producing mature infectious virions. Like the matrix protein from negative strand RNA viruses MA is associated with the viral membrane and thus performs essentially the same structural function [6].
2. Lipid rafts are microdomains for enveloped virus assembly
Lipid rafts are enriched in cholesterol and sphingolipids that can selectively incorporate or exclude proteins and have been first implicated in influenza virus budding [7]. Subsequent studies found the same principle for measles virus, HIV and Ebola virus. These studies show that lipid rafts concentrate glycoproteins and matrix proteins thus establishing platforms for efficient assembly and budding [2].
3. The cellular machinery for budding
The first evidence for the involvement of specific Gag domains in viral budding came from the work of Göttlinger and colleagues who reported that a deletion of the C-terminal region of HIV Gag (p6 protein) caused a significant defect in virus particle release [8]. Electron microscopy studies revealed that these particles failed to pinch off the plasma membrane. Subsequent studies then identified a highly conserved Pro-Thr-Ala-Pro sequence motif, termed late domain, as playing a crucial role in viral budding [9]. Up to date, several classes of viral late domains have been described, namely P(T/S)AP, YXXL LXXLF and PPXY. They are present alone or in different combinations in the matrix proteins of negative-strand RNA viruses and in the Gag proteins of retroviruses. In several cases, viral late domains have been shown to be functionally interchangeable, can be positioned at various locations and can act in trans[10].
Recent studies show that the late domains serve as entry points into the vacuolar protein sorting (Vps) pathway and connect matrix proteins or Gag to cellular factors. The Vps machinery was first described in yeast and implicated in membrane protein trafficking from the Golgi and plasma membrane via the endosomal system to the lysosome for degradation (for review, see [11]). Mutation of any of the 17 different yeast Vps proteins leads to the formation of enlarged endosomal membrane compartments that cannot mature into multi vesicular bodies (MVB; class E compartment). Most of the class E Vps proteins exist as soluble proteins or small subcomplexes that are sequentially recruited to the site of MVB formation. Initial recognition of the cargo involves class E proteins Hrs (Vps27p), Stam, Eps15 (Ede1p) and clathrin. This leads to the recruitment of ESCRT-1 (endosome-associated complexes required for transport), composed of Vps23p, Vps28p and Vps37p to the endosomal membrane where it recognizes ubiquitinated cargo. ESCRT-I cargo recognition then induces the formation of ESCRT-II (Vps22p, Vps25p and Vps36p) and that in turn activates the assembly of ESCRT-III multi-protein complexes. Finally, another class E Vps protein, Vps4 an AAA-type ATPase, has been implicated in the disassembly of ESCRT-III a necessary step for MVB formation (reviewed by Katzmann et al. [11]). Although the mammalian system is more complex the framework is similar. Most of the components have now been described and their interactions have been mapped [12,13]. There are two human class E Vps proteins identified that recognize late domains, namely tsg101 (via P(T/S)AP), which is part of ESCRT-I and AIP-1/ALIX (via LXXLF and YXXL) which in turn interacts with Tsg101 and CHMP4 proteins (ESCRT-III), thus providing a link between ESCRT-I and ESCRT-III in retroviral budding [12,13]. It is now recognized that late domain interactions with cellular factors most likely recruit the whole Vps machinery to the site of budding.
The PPXY motif, the fourth identified late domain has been found in some retroviruses, in rhabdoviruses and filoviruses ([10] and references therein). The PPXY motif mediates interactions with proteins that contain WW domains, such as Nedd4 family ubiquitin ligases (E3 enzyme), which are part of an enzymatic cascade in which single ubiquitin moieties are transferred to lysine residues on the protein substrate.
Monoubiquitination of matrix proteins is consistent with recent findings that show that ubiquitin plays a role in retrovirus budding [14,15] and that Ebola virus VP40 can be ubiquitinated in vitro [16]. Ubiquitination may thus help to recruit the class E Vps components in a similar way as sorting of plasma membrane receptors requires monoubiquitination [11]. In addition, Nedd4, which can be also found in lipid rafts [17], has been linked genetically to ESCRT-III in yeast [18].
Finally, lipids and lipid modifying enzymes, such as endophilins that interact with AIP1/ALIX [19] might play an important role in viral budding [20]. Up to now not all enveloped viruses contain any of the known late domain sequences which could serve as entry points into the Vps machinery; however it is likely that they might yet use other short sequence elements which facilitate Vps recruitment. In addition this function may not reside within the matrix protein.
4. Structures of viral matrix proteins
To date there is still limited structural information on viral matrix proteins as only four different matrix protein structures have been solved, including VP40 from Ebola virus, M from VSV, a fragment of M1 from influenza virus and a number of retroviral matrix proteins.
4.1. Ebola virus matrix protein VP40
The Ebola virus matrix protein VP40 is an elongated, two-domain monomeric assembly, composed of two structurally related β-sandwich domains, which are connected by a flexible linker (Fig. 1(a)). Indeed the unique fold of both domains suggests that the two domains probably arose from a common ancestor by gene duplication [21]. Early work showed that this conformation of VP40 is metastable, which allows an easy transition into oligomeric ring-like structures in vitro [22,23]. The ring-structures are either octamers or hexamers [24] (Figs. 1(b) and (c)). In both cases, the N-terminal domain of VP40 constitutes the oligomerisation domain, which forms an anti-parallel dimer and is the building block for oligomerisation [23,25]. The C-terminal domains are flexibly attached to the rings and mediate membrane association in vitro and in vivo [5,22,23] (Figs. 1(b) and (c)).
1.
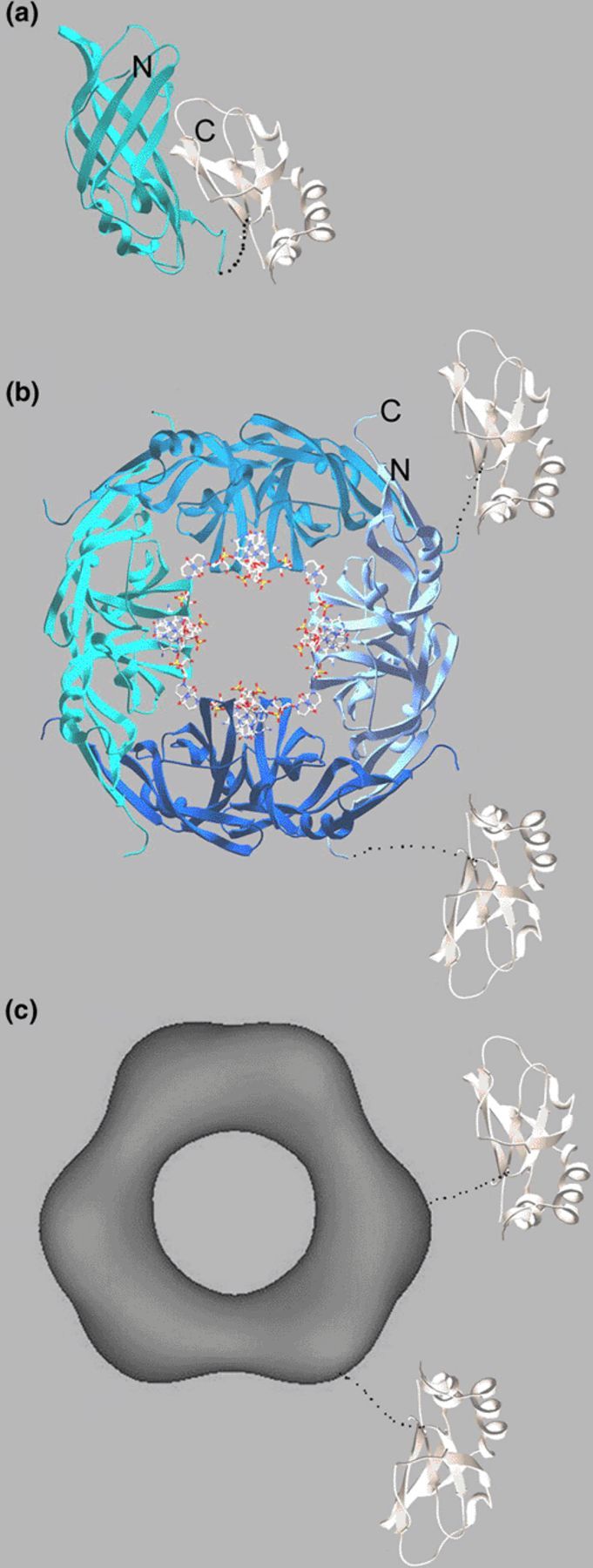
Ribbon diagrams and an electron microscopy reconstruction image of structures showing different conformations of the Ebola virus matrix protein VP40. (a) Monomeric VP40 is composed of two structurally related β sandwich domains. The N-terminal domain (N-terminal residue 44) mediates oligiomerisation and the C-terminal domain (C-terminal end residue 321) is responsible for membrane association. (b) The N-terminal domain of VP40 forms octamers by binding specific single-stranded RNA having the sequence 5-UGA-3′ at the dimer–dimer interface. The RNA is shown as all atom model. The N- (residue 69) and C-terminal (residue 190) ends are indicated. Note that the C-terminal ends connect to the C-terminal domain, which is missing in the structure. This is also indicated by the flexible attachment of two C-terminal domains (white). (c) Full-length VP40 can be also activated to form hexamers, which is mediated by the N-terminal domain. The electron microscopy reconstruction shows that two N-terminal domains assemble to create a ring-structure having threefold-symmetry. Like in the octamer structure the C-terminal domains are flexibly attached to the ring structure as indicated by two C-terminal domains and by the electron microscopy reconstructions [23].
The octamer was found to bind single stranded RNA at the dimer–dimer interface having the sequence 5′-UGA-3′ (Fig. 1(b)). RNA binding creates a new dimer–dimer interface and its binding stabilizes the protein–protein interaction generating the octamers [25]. Based on the crystal structure of the octamer, the complete N-terminal segment from residues 1 to 68 has to unfold in order to create the new ssRNA-binding interface. Interestingly recent studies suggest that the SDS resistant octamer is present in Ebola VLPs and in virus particles [26] in contrast to our previous finding which confirmed the presence of RNA containing octamers only in infected cells but not in virus particles [25].
A role for the ring structures in assembly and budding is also evident from the fact that only oligomeric VP40 interacted with WW3 from human Nedd4 in vitro via its N-terminal PPXY motif [27], an interaction which is important in vivo [28]. A number of studies also showed the importance of the PTAP motif present at the N-terminus for budding [29,30]. This motif binds to the UEV domain of Tsg101 independent of its oligomeric state. Thus, monomeric VP40 recruits Tsg101 to the site of budding [26,27], which in turn might recruit the complete Vps machinery for efficient budding as demonstrated in case of HIV-1 [12]. The late domain sequences are not present in the crystal structures of VP40 as they had been removed by proteolysis for efficient crystallisation purposes.
The structural studies of Ebola virus VP40 have now firmly established three conformations of Ebola virus VP40, although their role in assembly and budding or additional functions during the life cycle of Ebola virus is far from clear. It is, however, an interesting example of evolution that packs different functional aspects into one relatively small protein probably due to the limiting size of the viral genome.
4.2. Vesicular stomatitis virus M
The structure of the matrix protein form vesicular stomatitis virus consists of an N-terminal part composed of a five-stranded anti-parallel β-sheet packed against two β-helices and a small C-terminal part made up by a two-stranded β-sheet and an α-helix, which connects to the N-terminal region via a long linker (Fig. 2(a)) [31]. As with Ebola VP40, the vesiculovirus matrix protein displays a new fold. No defined oligomeric structures have been described for VSV M, which, however, polymerises in vitro [32].
2.
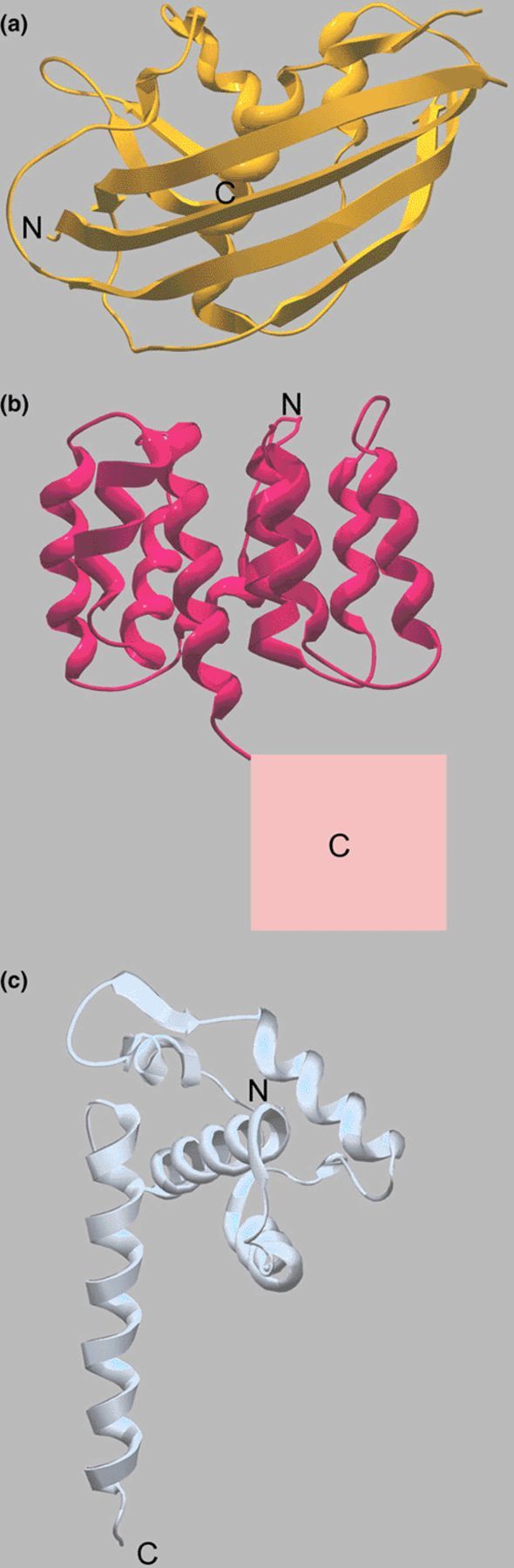
Ribbon diagrams of other known matrix protein structures. The N- and C-terminal ends are indicated. Note that the folds of Ebola virus VP40 (Fig. 1(a)) and the structures shown here are all different. (a) Crystal structure of the M protein from VSV. (b) Crystal structure of the influenza virus M1 N-terminal domain. The putative position of the C-terminal domain is indicated by the square. (c) Crystal structure of the matrix protein MA from HIV-1. The arrow indicates the suggested membrane-binding surface.
For structure determination, VSV M was purified from virus and solubilized by thermolysin cleavage, which removes residues 1–49 and 121–122/124, thus preventing polymerisation (Fig. 2(a)). Full length VSV M has a high tendency to self-associate into large multimers [33], which is sensitive to salt treatment in vitro [34]. Expression of M protein induces the formation and budding of vesicles, which is consistent with its membrane association and polymerisation activities. These particles, however, do not show the characteristic bullet-shaped structure of rhabdoviruses indicating that other viral components such as nucleocapsids are responsible for the distinct morphology of VSV [3].
The structure can be considered to represent the membrane-activated form that, however, no longer binds artificial membranes due to proteolytic removal of residues 121–122/124 [31]. It is conceivable that VSV M adopts a different conformation before activation, as cellular expression does not per se lead to polymerisation and membrane binding but produces mostly cytoplasmic M [33,35]. This putative other conformation(s) might then be also responsible for secondary functions of VSV M such as, inhibition of transcription and nucleocytoplasmic transport, nuclear localisation and cell rounding due to the induction of apoptosis [36–40]. VSV M has been also implicated in nucleocapsid condensation [41], which may be similar to the function of Ebola virus VP24 in nucleocapsid formation [42]. In addition, VSV M contains the late domain motifs PPPY and PSAP within the N-terminal 40 residues ([10] and references therein), which are missing in the crystal structure. The PPPY motif was shown to interact with mouse Nedd4 and proteasome inhibitors reduced virus titers implicating ubiquitin in virus budding [43]. The question remains whether any conformational diversity of VSV M might contribute to its different functions.
4.3. Influenza virus M1
The structure of the N-terminal domain of Influenza virus M1 protein has been solved at pH 4.0 [44] and at neutral pH [45]. Both structures are identical and consist of two four-helical bundle subdomains that pack against each other through a hydrophobic interface (Fig. 2(b)). The fold is again different from other known matrix protein structures. One side of the N-terminal domain of M1 is strictly positively charged while the opposite one exhibits an all-negative charge [45]. Proteolysis experiments suggest that the C-terminal domain is attached through a flexible linker. Established functions of M1 are membrane association and polymerisation of the N-terminal domain and RNP binding of the C-terminal domain [46]. The N-terminal domain also contains a stretch of basic residues, which have been implicated in nuclear localisation of M1 and export processes [47,48].
Overexpression of M1 in eukaryotic cells leads to the formation of intracellular tubular structures and the release of virus-like particles (VLPs), indicating that M1 contains all the information for assembly and budding [49]. VLP formation, however, is also enhanced by co-expression of HA which might augment M1 membrane association [49,50]. Although M1 has a high tendency to polymerise in vitro, its conformation upon cytoplasmic expression must be postulated to be monomeric as cytoplasmic and nuclear M1 pools have been reported in addition to membrane associated M1 [51,52]. The activation process and the structural changes that lead to specific M1 polymerisation at membranes in vivo have not yet been described.
M1 also exerts a number of additional functions, such as transport of RNP cores out of the nucleus [52]. No functional late domain sequences have yet been described for M1. As the complexity of the MVB machinery provides multiple possible entry points to contribute to its activation for budding processes, it is, however, unlikely that influenza uses another cellular machinery for budding.
4.4. HIV-1 matrix MA
The crystal structure of the matrix protein from HIV-1 and SIV folds into an arrangement of five α-helices and a three-stranded mixed β-sheet, a fold, which is unique to retroviral matrix proteins [53,54]. Helices 1–3 pack about a central helix (4) forming a compact globular domain that is capped by the β-sheet, while helix 5 extends away from the core (Fig. 2(c)). HIV-1 MA can form a trimer in solution and can polymerise into a crystalline lattice. The trimers have been suggested to form the building block for the mature MA shell. There, the individual trimers present a largely basic surface on one side that was proposed to interact with the inner membrane of the virus. Such an arrangement would pose the myristoylated N-terminal residue that is essential for membrane targeting close to the membrane and the C-terminal helix 5 would point towards the interior of the viral particle [53,54]. In addition to forming a protein shell underneath the viral membrane, MA is also part of the pre-integration reverse transcriptase complex that travels towards the nucleus along microtubules using the kinesin KIF-4 upon viral entry [55] and seems to be required for nuclear import [56].
Although the sequences of retrovirus matrix proteins differ significantly, the known structures of retroviral matrix proteins (SIV; bovine leukaemia virus, HTLV-II; Mason–Pfizer monkey virus; equine infectious anemia virus) exhibit the same common fold reflecting their evolutionary relationship [57].
Retroviral Gag proteins contain either one or two late domain sequences at different locations. HIV-1 Gag has two essential sequence motifs PTAP and LXXLF within the C-terminal fragment p6 while PPPY and PTAP motifs locate to the C-terminus of MA from HTLV-I [58].
5. Evolutionary sequence conservation
Sequence analysis between members of Filoviridae (Ebola and Marburg virus VP40), Paramyxoviridae (subfamily Paramyxovirinae: Sendai virus, SV5, measles virus; and subfamily pneumovirinae, human respiratory syncytial virus), Rhabdoviridae (VSV; rabies virus) and Bornaviridae (Borna disease virus) reveals no significant sequence identity (ranging from 2% to 7%). The same is true when comparing matrix proteins from the Mononegavirales with M1 from influenza virus and MA from retroviruses, which also underlines their observed structural diversity.
6. Conclusions
In summary, the known matrix protein structures indicate no structural homology between non-segmented negative strand RNA viruses such as Filoviridae (Ebola virus) and Rhabdoviridae (VSV) and segmented negative strand RNA viruses such as Orthomyxoviridae (influenza virus) as well as Retroviridae (HIV), although they share common functions. A minimal structural conservation is the presence of late domain sequences that mediate interaction with the class E Vps machinery for budding, although this still has to be shown in case of influenza virus M1. In addition, there is no striking common motif evident for membrane interaction, a common functional property of all viral matrix proteins.
This is in contrast to the functional and structural conservation of the fusion protein subunit of the glycoproteins derived from retroviruses (HIV-1; HTLV-I), filoviruses (Ebola), paramyxoviruses (SV5) and orthomyxoviruses (influenza virus) which show striking similarities [59]. They all fold into trimeric rod-like structures composed of a central triple-stranded coiled coil and an outer helical or non-helical layer. This arrangement places the fusion peptide and the transmembrane region at one end of the rod, which facilitates fusion of viral and cellular membranes during viral entry [60]. The conserved function and conformation of the fusion proteins may therefore be based on a common ancestral viral fusion protein [61].
The structural dissimilarity of matrix proteins might indicate two different scenarios: (i) Matrix proteins from related viruses have evolved from a common ancestor and have changed over time completely as they acquired new functions that helped to adapt to the hosts. Such dramatic changes could have been supported by their high mutation rate [62].
(ii) Secondly, matrix proteins have nothing in common with each other as they have been acquired independently during evolution. Although there is no doubt that matrix proteins are the major driving forces for assembly and budding there is some evidence that matrix protein-free viruses might have existed as matrix-less measles virus, VSV and rabies virus show infectivity, albeit severely impaired [63–65]. In addition, other enveloped viruses have solved their envelope acquirement differently, namely flaviviruses and alphaviruses use their glycoprotein to form a protein shell on the outside of the lipid bilayer envelope [66]. A third class of enveloped viruses, the corona virus uses an integral membrane protein that might function as a matrix protein [67]. In conclusion, different matrix protein conformations and/or their diverse oligomeric states or polymerisation features are most likely to contribute to the morphological differences between viral families.
Acknowledgements
We gratefully acknowledge critical comments on the manuscript by Dr. Stephan Becker and apologize to the many colleagues whose work we were unable to cite due to the publishers space limitations.
References
- [1]. Garoff H., Hewson R., Opstelten D.J. (1998) Virus maturation by budding. Microbiol Mol. Biol. Rev. 62, 1171–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]. Schmitt A.P., Lamb R.A. (2003) Escaping from the cell: assembly and budding of negative-strand RNA viruses. In: Reverse Genetics of Negative Stranded RNA Viruses (Kawaok Y. , Ed.) Curr. Top. Microbiol. Immunol. (in press) [DOI] [PubMed] [Google Scholar]
- [3]. Justice P.A., Sun W., Li Y., Ye Z., Grigera P.R., Wagner R.R. (1995) Membrane vesiculation function and exocytosis of wild-type and mutant matrix proteins of vesicular stomatitis virus. J. Virol. 69, 3156–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4]. Timmins J., Scianimanico S., Schoehn G., Weissenhorn W. (2001) Vesicular release of ebola virus matrix protein VP40. Virology 283, 1–6. [DOI] [PubMed] [Google Scholar]
- [5]. Jasenosky L.D., Neumann G., Lukashevich I., Kawaoka Y. (2001) Ebola virus VP40-induced particle formation and association with the lipid bilayer. J. Virol. 75, 5205–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6]. Krausslich H.G., Welker R. (1996) Intracellular transport of retroviral capsid components. Curr. Top. Microbiol. Immunol. 214, 25–63. [DOI] [PubMed] [Google Scholar]
- [7]. Scheiffele P., Rietveld A., Wilk T., Simons K. (1999) Influenza viruses select ordered lipid domains during budding from the plasma membrane. J. Biol. Chem. 274, 2038–2044. [DOI] [PubMed] [Google Scholar]
- [8]. Gottlinger H.G., Dorfman T., Sodroski J.G., Haseltine W.A. (1991) Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc Natl. Acad. Sci. USA 88, 3195–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9]. Huang M., Orenstein J.M., Martin M.A., Freed E.O. (1995) p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 69, 6810–6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10]. Pornillos O., Garrus J.E., Sundquist W.I. (2002) Mechanisms of enveloped RNA virus budding. Trends Cell Biol. 12, 569–579. [DOI] [PubMed] [Google Scholar]
- [11]. Katzmann D.J., Odorizzi G., Emr S.D. (2002) Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell. Biol. 3, 893–905. [DOI] [PubMed] [Google Scholar]
- [12]. von Schwedler U.K., Stuchell M., Muller B., Ward D.M., Chung H.Y., Morita E., Wang H.E., Davis T., He G.P., Cimbora D.M., Scott A., Krausslich H.G., Kaplan J., Morham S.G., Sundquist W.I. (2003) The protein network of HIV budding. Cell 114, 701–713. [DOI] [PubMed] [Google Scholar]
- [13]. Strack B., Calistri A., Craig S., Popova E., Gottlinger H.G. (2003) AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114, 689–699. [DOI] [PubMed] [Google Scholar]
- [14]. Patnaik A., Chau V., Wills J.W. (2000) Ubiquitin is part of the retrovirus budding machinery. Proc. Natl. Acad. Sci. USA 97, 13069–13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]. Strack B., Calistri A., Accola M.A., Palu G., Gottlinger H.G. (2000) A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. USA 97, 13063–13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16]. Harty R.N., Brown M.E., Wang G., Huibregtse J., Hayes F.P. (2000) A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc. Natl. Acad. Sci. USA 97, 13871–13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17]. Lafont F., Simons K. (2001) Raft-partitioning of the ubiquitin ligases Cbl and Nedd4 upon IgE-triggered cell signaling. Proc. Natl. Acad. Sci. USA 98, 3180–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18]. Katzmann D.J., Sarkar S., Chu T., Audhya A., Emr S.D. (2003) Multivesicular body sorting: ubiquitin ligase Rsp5 is required for the modification and sorting of carboxypeptidase S. Mol. Biol. Cell 15 (2), 468–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19]. Chatellard-Causse C., Blot B., Cristina N., Torch S., Missotten M., Sadoul R. (2002) Alix (ALG-2-interacting protein X), a protein involved in apoptosis, binds to endophilins and induces cytoplasmic vacuolization. J. Biol. Chem. 277, 29108–29115. [DOI] [PubMed] [Google Scholar]
- [20]. Wang M.Q., Kim W., Gao G., Torrey T.A., Morse 3rd H.C., De Camilli P., Goff, S.P (2003) Endophilins interact with Moloney murine leukemia virus Gag and modulate virion production.J. Biol. 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21]. Dessen A., Volchkov V., Dolnik O., Klenk H.D., Weissenhorn W. (2000) Crystal structure of the matrix protein VP40 from Ebola virus. EMBO J. 19, 4228–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22]. Ruigrok R.W., Schoehn G., Dessen A., Forest E., Volchkov V., Dolnik O., Klenk H.D., Weissenhorn W. (2000) Structural characterization and membrane binding properties of the matrix protein VP40 of Ebola virus. J. Mol. Biol. 300, 103–112. [DOI] [PubMed] [Google Scholar]
- [23]. Scianimanico S., Schoehn G., Timmins J., Ruigrok R.H., Klenk H.D., Weissenhorn W. (2000) Membrane association induces a conformational change in the Ebola virus matrix protein. EMBO J. 19, 6732–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24]. Timmins J., Schoehn G., Kohlhaas C., Klenk H.-D., Ruigrok R.W.H., Weissenhorn W. (2003) Oligomerization and polimerization of the filovirus matrix protein VP40. Virology 312, 359–368. [DOI] [PubMed] [Google Scholar]
- [25]. Gomis-Ruth F.X., Dessen A., Timmins J., Bracher A., Kolesnikowa L., Becker S., Klenk H-D., Weissenhorn W. (2003) The matrix protein VP40 from Ebola virus octamerizes into pore-like structures with specific RNA binding properties. Structure (Cambridge) 11, 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26]. Panchal R.G., Ruthel G., Kenny T.A., Kallstrom G.H., Lane D., Badie S.S., Li L., Bavari S., Aman M.J. (2003) In vivo oligomerization and raft localization of Ebola virus protein VP40 during vesicular budding. Proc. Natl. Acad. Sci. USA 100, 15936–15941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27]. Timmins J., Schoehn G., Ricard-Blum S., Scianimanico S., Vernet T., Ruigrok R.W., Weissenhorn W. (2003) Ebola virus matrix protein VP40 interaction with human cellular factors Tsg101 and Nedd4. J. Mol. Biol. 326, 493–502. [DOI] [PubMed] [Google Scholar]
- [28]. Yasuda J., Nakao M., Kawaoka Y., Shida H. (2003) Nedd4 regulates egress of Ebola virus-like particles from host cells. J. Virol. 77, 9987–9992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29]. Martin-Serrano J., Zang T., Bieniasz P.D. (2001) HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 7, 1313–1319. [DOI] [PubMed] [Google Scholar]
- [30]. Licata J.M., Simpson-Holley M., Wright N.T., Han Z., Paragas J., Harty R.N. (2003) Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J. Virol. 77, 1812–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31]. Gaudier M., Gaudin Y., Knossow M. (2002) Crystal structure of vesicular stomatitis virus matrix protein. EMBO J. 21, 2886–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32]. Gaudin Y., Sturgis J., Doumith M., Barge A., Robert B., Ruigrok R.W. (1997) Conformational flexibility and polymerization of vesicular stomatitis virus matrix protein. J. Mol. Biol. 274, 816–825. [DOI] [PubMed] [Google Scholar]
- [33]. Jr. McCreedy B.J., McKinnon K.P., Lyles D.S. (1990) Solubility of vesicular stomatitis virus M protein in the cytosol of infected cells or isolated from virions. J. Virol. 64, 902–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]. Gaudin Y., Barge A., Ebel C., Ruigrok R.W. (1995) Aggregation of VSV M protein is reversible and mediated by nucleation sites: implications for viral assembly. Virology 206, 28–37. [DOI] [PubMed] [Google Scholar]
- [35]. Chong L.D., Rose J.K. (1993) Membrane association of functional vesicular stomatitis virus matrix protein in vivo. J. Virol. 67, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36]. Blondel D., Harmison G.G., Schubert M. (1990) Role of matrix protein in cytopathogenesis of vesicular stomatitis virus. J. Virol. 64, 1716–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37]. Petersen J.M., Her L.S., Varvel V., Lund E., Dahlberg J.E. (2000) The matrix protein of vesicular stomatitis virus inhibits nucleocytoplasmic transport when it is in the nucleus and associated with nuclear pore complexes. Mol. Cell. Biol. 20, 8590–8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38]. von Kobbe C., van Deursen J.M., Rodrigues J.P., Sitterlin D., Bachi A., Wu X., Wilm M., Carmo-Fonseca M., Izaurralde E. (2000) Vesicular stomatitis virus matrix protein inhibits host cell gene expression by targeting the nucleoporin Nup98. Mol. Cell 6, 1243–1252. [DOI] [PubMed] [Google Scholar]
- [39]. Glodowski D.R., Petersen J.M., Dahlberg J.E. (2002) Complex nuclear localization signals in the matrix protein of vesicular stomatitis virus. J. Biol. Chem. 277, 46864–46870. [DOI] [PubMed] [Google Scholar]
- [40]. Kopecky S.A., Lyles D.S. (2003) The cell-rounding activity of the vesicular stomatitis virus matrix protein is due to the induction of cell death. J. Virol. 77, 5524–5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41]. Newcomb W.W., Brown J.C. (1981) Role of the vesicular stomatitis virus matrix protein in maintaining the viral nucleocapsid in the condensed form found in native virions. J. Virol. 39, 295–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42]. Huang Y., Xu L., Sun Y., Nabel G.J. (2002) The assembly of Ebola virus nucleocapsid requires virion-associated proteins 35 and 24 and posttranslational modification of nucleoprotein. Mol. Cell 10, 307–316. [DOI] [PubMed] [Google Scholar]
- [43]. Harty R.N., Brown M.E., McGettigan J.P., Wang G., Jayakar H.R., Huibregtse J.M., Whitt M.A., Schnell M.J. (2001) Rhabdoviruses and the cellular ubiquitin–proteasome system: a budding interaction. J. Virol. 75, 10623–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44]. Sha B., Luo M. (1997) Structure of a bifunctional membrane-RNA binding protein, influenza virus matrix protein M1. Nat. Struct. Biol. 4, 239–244. [DOI] [PubMed] [Google Scholar]
- [45]. Arzt S., Baudin F., Barge A., Timmins P., Burmeister W., Ruigrok R.W.H. (2001) Combined results from solution studies on intact Influeenza virus M1 protein and from a new crystal form of its N-terminal domain show that M1 is an elongated monomer. Virology 279, 439–446. [DOI] [PubMed] [Google Scholar]
- [46]. Baudin F., Petit I., Weissenhorn W., Ruigrok R.W. (2001) In vitro dissection of the membrane and RNP binding activities of influenza virus M1 protein. Virology 281, 102–108. [DOI] [PubMed] [Google Scholar]
- [47]. Ye Z., Robinson D., Wagner R.R. (1995) Nucleus-targeting domain of the matrix protein (M1) of influenza virus. J. Virol. 69, 1964–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48]. Akarsu H., Burmeister W.P., Petosa C., Petit I., Muller C.W., Ruigrok R.W., Baudin F. (2003) Crystal structure of the M1 protein-binding domain of the influenza A virus nuclear export protein (NEP/NS2). EMBO J. 22, 4646–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49]. Gomez-Puertas P., Albo C., Perez-Pastrana E., Vivo A., Portela A. (2000) Influenza virus matrix protein is the major driving force in virus budding. J. Virol. 74, 11538–11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50]. Enami M., Enami K. (1996) Influenza virus hemagglutinin and neuraminidase glycoproteins stimulate the membrane association of the matrix protein. J. Virol. 70, 6653–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51]. Zhang J., Lamb R.A. (1996) Characterization of the membrane association of the influenza virus matrix protein in living cells. Virology 225, 255–266. [DOI] [PubMed] [Google Scholar]
- [52]. Martin K., Helenius A. (1991) Nuclear transport of influenza virus ribonucleoproteins: the viral matrix protein (M1) promotes export and inhibits import. Cell 67, 117–130. [DOI] [PubMed] [Google Scholar]
- [53]. Hill C.P., Worthylake D., Bancroft D.P., Christensen A.M., Sundquist W.I. (1996) Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl. Acad. Sci. USA 93, 3099–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54]. Rao Z., Belyaev A.S., Fry E., Roy P., Jones I.M., Stuart D.I. (1995) Crystal structure of SIV matrix antigen and implications for virus assembly. Nature 378, 743–747. [DOI] [PubMed] [Google Scholar]
- [55]. McDonald D., Vodicka M.A., Lucero G., Svitkina T.M., Borisy G.G., Emerman M., Hope T.J. (2002) Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 159, 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56]. Haffar O.K., Popov S., Dubrovsky L., Agostini I., Tang H., Pushkarsky T., Nadler S.G., Bukrinsky M. (2000) Two nuclear localization signals in the HIV-1 matrix protein regulate nuclear import of the HIV-1 pre-integration complex. J. Mol. Biol. 299, 359–368. [DOI] [PubMed] [Google Scholar]
- [57]. Conte M.R., Matthews S. (1998) Retroviral matrix proteins: a structural perspective. Virology 246, 191–198. [DOI] [PubMed] [Google Scholar]
- [58]. Pornillos O., Higginson D.S., Stray K.M., Fisher R.D., Garrus J.E., Payne M., He G.P., Wang H.E., Morham S.G., Sundquist W.I. (2003) HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J. Cell Biol. 162, 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59]. Skehel J.J., Wiley D.C. (2000) Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69, 531–569. [DOI] [PubMed] [Google Scholar]
- [60]. Weissenhorn W., Dessen A., Calder L.J., Harrison S.C., Skehel J.J., Wiley D.C. (1999) Structural basis for membrane fusion by enveloped viruses. Mol. Membr. Biol. 16, 3–9. [DOI] [PubMed] [Google Scholar]
- [61]. Rosenthal P.B., Zhang X., Formanowski F., Fitz W., Wong C.H., Meier-Ewert H., Skehel J.J., Wiley D.C. (1998) Structure of the haemagglutinin-esterase-fusion glycoprotein of influenza C virus. Nature 396, 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62]. Domingo E., Holland J.J. (1997) RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51, 151–178. [DOI] [PubMed] [Google Scholar]
- [63]. Lyles D.S., McKenzie M.O., Kaptur P.E., Grant K.W., Jerome W.G. (1996) Complementation of M gene mutants of vesicular stomatitis virus by plasmid-derived M protein converts spherical extracellular particles into native bullet shapes. Virology 217, 76–87. [DOI] [PubMed] [Google Scholar]
- [64]. Cathomen T., Mrkic B., Spehner D., Drillien R., Naef R., Pavlovic J., Aguzzi A., Billeter M.A., Cattaneo R. (1998) A matrix-less measles virus is infectious and elicits extensive cell fusion: consequences for propagation in the brain. EMBO J. 17, 3899–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65]. Mebatsion T., Weiland F., Conzelmann K.K. (1999) Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J. Virol. 73, 242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66]. Kuhn R.J., Strauss J.H. (2003) Enveloped viruses. Adv. Protein Chem. 64, 363–377. [DOI] [PubMed] [Google Scholar]
- [67]. Stadler K., Masignani V., Eickmann M., Becker S., Abrignani S., Klenk H-D., Rappuoli R. (2003) SARS: beginning to understand a new virus. Nat. Rev. Microbiol. 1, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]