

Abstract
Advances in free radical research show that reactive oxygen and nitrogen oxide species, for example superoxide, nitric oxide (NO) and peroxynitrite, play an important role in the pathogenesis of different viral infections, including dengue virus. The pathogenic mechanism of dengue haemorrhagic fever (DHF) is complicated and is not clearly understood. The hallmarks of the dengue disease, the antibody‐dependent enhancement, the shift from T‐helper type 1 (Th1) to Th2 cytokine response and the cytokine tsunami resulting in vascular leakage can now be explained much better with the knowledge gained about NO and peroxynitrite. This paper makes an effort to present a synthesis of the current opinions to explain the pathogenesis of DHF/shock syndrome with NO on centre stage.
Keywords: dengue virus, dengue haemorrhagic fever, pathogenesis, nitric oxide, peroxynitrite
Introduction
Nitric oxide (NO) is an important paracrine and autocrine signal used by different types of cells and produced by a variety of cells in the body, for example macrophages, vascular endothelial cells, Kupffer cells, adrenals and cerebellar tissues (Ignarro, 1991). NO has a wide range of functions in the body, from dilating blood vessels, aggregation of platelets, fighting infections and tumours, mediator of inflammation and macrophage cytotoxic activity to transmission of signals between nerve cells (1998, 1993, 1991). In optimal doses it has protective and regulatory action in cells, but higher concentrations have toxic effects. The enzyme NO synthase (NOS) contributes to signal transmission in different cell systems of body via synthesis of NO from l‐arginine in the presence of NADPH and dioxygen (O2). NOS binds flavin adenine dinucleotide, flavin mononucleotide, haeme, tetrahydrobiopterin and calmodulin. It activates cGMP, which inhibits calcium entry into the cell and decreases levels of intracellular calcium, and K+ channels, and stimulates a cGMP‐dependent protein kinase, etc. NOS has three isoforms (Table 1): the neuronal (nNOS or NOS1), inducible (iNOS or NOS2) and endothelial (eNOS or NOS3). Cytokines and other proinflammatory stimuli induce iNOS (1997a, 1997b). The activity of nNOS and eNOS is calcium‐ and calmodulin‐dependent, whereas that of iNOS is calcium‐independent. NO may regulate NOS expression and activity by negative feedback by the process of S‐nitrosylation.
Table 1.
Functions of NOS in DV infection
| Types of NOS | Location | Cell | Regulation | Dengue disease | |
|---|---|---|---|---|---|
| Lesions | * References | ||||
| Neuronal NOS (nNOS or NOS1) | Brain | Microglia | Ca2+/CAM | Blood–brain barrier damage | 1991, 2008 |
| Inducible NOS (iNOS or NOS2) | Immune system, cardiovascular system | Macrophage, dendritic cell | Ca2+ independent | Virus replication inhibited | 2005, 2006 |
| Endothelial NOS (eNOS or NOS3) | Endothelium | Endothelial cells | Ca2+/CAM | Increased capillary permeability | 2008, 1994, 1990 |
Only selected references have been cited.
Dengue viruses (DV) are transmitted by Aedes aegypti mosquitoes. They produce subclinical infection or a mild self‐limiting disease, the dengue fever (DF) and a serious life‐threatening dengue haemorrhagic fever (DHF) and dengue shock syndrome (DSS). Dengue is the most common arboviral disease of humans and is found in subtropical and tropical areas of the world located between 10°N and 10°S of the Equator. More than 2.5 billion persons are at risk of getting dengue infections. About 50–100 million cases of DF occur every year, with about 250 000–500 000 cases of DHF. The incidence of DHF has increased tremendously in India, Southeast Asia, the South Pacific and the American tropics in the past 25 years, with major epidemics occurring in many countries. The risk factors for developing DHF are cocirculation of all four DV strains in the same area, host factors such as immune status, age and genetic background and the virus genotype. Patients with DHF may develop capillary leakage, resulting in flow of serum proteins and fluid into the body cavities, for example the pleural and abdominal cavities. Patients have haemorrhages in different parts of the body. Investigations show thrombocytopoenia, neutropoenia and elevated liver enzymes, and sometimes disseminated intravascular coagulation. DHF may progress to a hypotensive state, the DSS, with cold clammy skin and unrecordable pulse and blood pressure. The course of shock is short, but life‐threatening. DHF is observed commonly in infants and children who are exposed to a second dengue infection. The pathogenesis of DHF is still not fully understood despite extensive studies and has been a subject of controversy from the time the syndrome was first recognized (reviewed by 2008, 2005, 2006a, 2006b, 2007).
The role of NO in different viral infections has been studied for many years and a number of interesting reviews have been published (2000, 2006). Chaturvedi and colleagues first reported the role of NO and peroxynitrite in DV infection in 1996 (1996a, 1996b, 1996), but its greater implications in dengue disease have been distinguished only recently. The hallmarks of the dengue disease, the antibody‐dependent enhancement (ADE; Halstead, 1970, 2007), the shift from T‐helper type 1 (Th1) to Th2 cytokine response (Chaturvedi et al., 1999) and the cytokine tsunami (2000, 2007) resulting in vascular leakage can now be explained much better with the knowledge gained about NO and peroxynitrite (2007, 2008a, 2008b). This paper makes an effort to present a synthesis of the current opinions to explain the pathogenesis of DHF/shock syndrome with NO on centre stage. Because of space constraints, all of the papers could not be cited.
NO
NO is a relatively stable free radical. It reacts with other free radicals (O2 −, superoxide anion; OH, hydroxyl ion) or metals like haeme iron (haemoglobin, myoglobin, cytochromes). NO is short‐lived and acts at the site of synthesis (Ignarro et al., 1993).
Mechanisms of the effects of NO
Even at high concentrations NO does not kill cells by itself. For acute toxicity, NO reacts with superoxide anion and forms the peroxynitrite (ONOO−), which has a wider range of chemical targets (Fig. 1). It can oxidize proteins, lipids, RNA and DNA. The toxic effects of NO are due to its ability to modulate mitochondrial respiration, DNA synthesis and energy metabolism. Peroxynitrite inhibits enzymes in the mitochondrial respiratory chain and the functions of manganese SOD (MnSOD), which results in increased formation of superoxide anion. ONOO− efficiently modifies and breaks DNA strands and inhibits DNA ligase, which increases DNA strand breaks, which activate DNA repair mechanisms, including the nuclear enzyme poly(ADP‐ribose) polymerase (PARP). Activation of PARP results in a drop in energy stores that impairs cellular metabolism and causes cell death. Reactions of NO differ between in vitro and in vivo systems. The main degradation product of NO in vitro is NO2 − (nitrite), whereas in vivo the main product is NO3 − (nitrate). The constitutive enzymes nNOS and eNOS induce low concentrations of NO, which modulates the activity of haeme‐containing proteins, for example guanylyl cyclase as well as proteins of the mitochondria such as cytochromes (reviewed by 1991, 1993, 2007, 2007). Induction of iNOS generates higher concentrations of NO, which can nitrosylate cysteine residues or produce tyrosine nitration in different proteins and also deamination of DNA. Nitrotyrosine is a marker for the generation of peroxynitrite (Hanafy et al., 2001).
Figure 1.
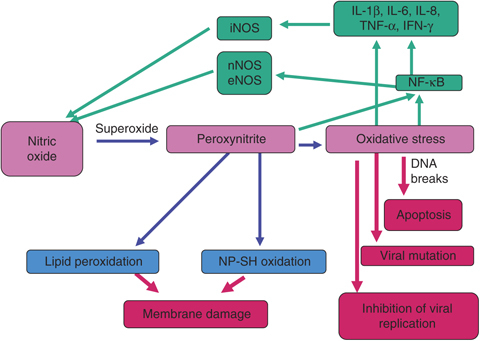
Mechanisms by which NO inhibits virus replication, induces virus mutation, damages cell membrane and breaks DNA, producing cell apoptosis.
Role of NO in the immune system
NO is known to have a strong immuno‐regulatory role (Bogdan, 2001). NO either activates or inhibits immune cell activation, proliferation, cytokine synthesis and cytokine signaling (1998, 1998, 1995). NO has been shown to upregulate proliferation and increases glucose uptake by T lymphocytes; on the other hand, it inhibits T‐cell activation. NO also modulates apoptosis; it promotes or prevents apoptosis and is involved in alteration of p53 levels (reviewed by Jiménez et al., 2001).
NO has variable effects on cytokine synthesis. Exogenous NO increases synthesis of tumour necrosis factor‐α (TNF‐α) in human peripheral blood mononuclear cells and lipopolysaccharide‐stimulated neutrophil preparations (Van Dervort et al., 1994). Endogenous NO is required for interleukin‐12 (IL‐12) production, whereas exogenous NO decreases IL‐12 production by macrophages (Huang et al., 1998). The production of IL‐6 and granulocyte colony‐stimulating factor mRNA is decreased in iNOS knockout mice or in normal animals treated with iNOS inhibitors (Hierholzer et al., 1998). NO is involved in cytokine signaling, as in iNOS‐deficient mice some cytokine signaling is lost. iNOS is required for IL‐12‐mediated T‐cell proliferation and activation and also for natural killer cell activation by interferon‐α/β (IFN‐α/β) (Diefenbach et al., 1999). Production of large amounts of NO by macrophages leads to inactivation of lymphocytes and induces a persistent immunosuppression in HIV infection (reviewed by Jiménez et al., 2001). NO plays a key role in the pathogenesis of inflammatory diseases. In the right situations and right concentrations it is anti‐inflammatory and has protective effects against various infections. On the other hand, in abnormal situations and higher concentrations NO is a proinflammatory mediator, inducing inflammation and pathological lesions. NO belongs to the labile radical entities, the reactive oxygen species (ROS), and reacts with oxygen and haeme‐iron‐containing groups, reducing nitrate compounds.
Ding et al. (1988) screened the effect of 12 different cytokines on the production of nitrite by macrophages and found that the most effective was IFN‐γ. Further, NO production is inhibited by type 2 cytokines [IL‐4, IL‐10, IL‐13, transforming growth factor‐β (TGF‐β)] (MacMicking et al., 1997a). Therefore iNOS expression may depend on Th1–Th2 shift in the hosts. An enhanced Th1 response is observed in NO‐deficient mice during infections and antigenic stimulation, resulting in more production of interferon and less of IL‐4 (1998, 1998), indicating that NO selectively inhibits the expansion of Th1 cells by a negative feedback mechanism. Selective inhibition of IL‐12 synthesis by activated macrophages may be responsible for this to some extent (Huang et al., 1998). On the other hand, low concentrations of NO produced during the early stages of infection result in a strong Th1‐type response, which is effective in host defence against intracellular pathogens (Niedbala et al., 1999). A synergistic effect of IL‐18 and IL‐12 has been observed in the induction of Th1‐type cells (Robinson et al., 1997). The precise mechanism of the action of low concentrations of NO in the Th1 type is not fully known, but recently Niedbala et al. (2006) have suggested that the enhancing effect of low concentrations of NO is mediated by cGMP by exerting a direct and selective effect on Th1 cells and not via antigen‐presenting cells. Low concentrations of NO in endothelial cells (Umansky et al., 1998) and macrophages (Connelly et al., 2001) upregulates nuclear factor‐κB (NF‐κB) activity and the high doses downregulates it. cGMP may also act through the P‐Raf/MEK/ERK pathway. Thus, NO is also responsible for the induction of T‐cell subset response. Niedbala et al. (2007) have reported a new subset of CD4+CD25+ regulatory T cells (Tregs) derived from CD4+CD25− T cells induced by NO. The induction of Tregs (NO‐Tregs) is independent of cGMP but depends on p53, IL‐2 and OX40. NO‐Tregs produce IL‐4 and IL‐10, but not IL‐2, IFN‐γ or TGF‐β. The cells are GITR+, CD27+, T‐betlow, GATA3high and Foxp3. NO‐Tregs suppress the proliferation of CD4+CD25− T cells in vitro and function in an IL‐10‐dependent manner. NO‐Tregs are also induced in vivo in SCID mice adoptively transferred with CD4+CD25− T cells in the presence of lipopolysaccharide and IFN‐γ, and the induction is completely inhibited by N G‐monomethyl‐l‐arginine (NMMA), a pan NOS inhibitor (Niedbala et al., 2007).
Role of NO during viral infection
NO has multiple effects during viral infections; for example, it affects the virus or the host. NO can inhibit virus replication or can cause viral mutation and in the host it can cause cell damage and pathology. The induction of iNOS can occur during viral infections (Fig. 2) by two mechanisms, direct and indirect. It can occur directly by viral replication, for example respiratory syncytial virus, or by viral proteins, for example HIV‐1 glycoprotein, gp41 (Akaike & Maeda, 2000). The indirect mechanisms include induction by cytokines, the commonest being IFN‐γ and by other partly characterized cytokines (Table 2).
Figure 2.
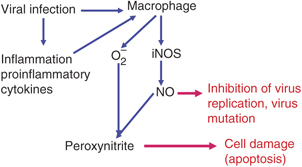
Mechanisms of the induction of NO during virus infections.
Table 2.
Induction of iNOS/NO by cytokines in viral infections
| Virus | Cytokine | Test system | References |
|---|---|---|---|
| Dengue | CF | Mouse spleen cells in vivo and in vitro | Misra et al. (1996a) |
| Macrophage cytotoxin | Mouse spleen cells cytotoxicity | 1996a, 1996b, 1996 | |
| Suppressor factor | Macrophage culture Signal transmission | Khare & Chaturvedi (1997) | |
| Ectromelia, vaccinia, and herpes simplex‐1 | IFN‐γ | Mouse macrophage | Karupiah et al. (1993) |
| Influenza A | IFN‐γ | Mouse lung | Akaike & Maeda (2000) |
| Sendai | IFN‐γ | Mouse lung | |
| Japanese encephalitis (JE) | Macrophage derived neutrophil chemotactic factor | Splenic macrophage of JE virus‐infected mice | Saxena et al. (2000) |
Effects of NO on virus replication
NO has a variable effect on the replication of viruses; it inhibits the replication of most of them, whereas it enhances some viruses, and has no effect on a few of the other viruses (Table 3). NO inhibits viral replication by reversible S‐nitrosylation of viral proteases (Zaragoza et al., 1997). Another antiviral mechanism of NO is via the formation of peroxynitrite, which blocks viral entry into the host cell (Padalko et al., 2004). NO has strong antiviral effects on hantavirus replication and peroxynitrite has strong antiviral effects on mature free virions, suggesting that different reactive nitrogen intermediates can have different effects on various parts of the replication cycle for the same virus (Klingström et al., 2006). Peroxynitrite enhances Sendai viral mutation in vitro and expands the quasispecies spectrum, facilitating the evolution of RNA viruses (Akaike et al., 2000). NO may accelerate viral mutation via formation of 8‐nitroguanosine, which may be a substantial contributor to erroneous RNA replication of the virus. NO‐generated 8‐nitroguanosine may cause viral mutation via two different mechanisms: directly, through incorporation into template RNAs for viral replication, and indirectly, by enhanced oxidative stress because of its potent redox‐active property (Yoshitake et al., 2004). NO has a unique biological effect on the genome of both pathogen and host via chemical modification of nucleic acids (2000, 2003). NO can act on viral proteins by different mechanisms. NO inhibits Coxsackie virus infection by nitration of the VP1 capsid protein (Padalko et al., 2004) or by S‐nitrosylation of the cysteine protease 3C (Saura et al., 1999). HIV‐1 is inhibited by S‐nitrosylation of the viral protease by NO (Persichini et al., 1998). Modification of cellular proteins can also inhibit viral infection, for example S‐nitrosylation of NF‐κB downregulates expression of the transactivator Zta, which is required for reactivation of Epstein–Barr virus (Mannick et al., 1994), and tyrosine nitration of microtubules attenuates respiratory syncytial virus infection (Huang et al., 2005).
Table 3.
Effect of NO on the replication of viruses grown in different cells *
| Virus | Test system | Mechanism of NO production | References |
|---|---|---|---|
| NO‐induced inhibition of virus replication | |||
| Influenza A, B | Kidney cells | Viral RNA synthesis inhibited | Rimmelzwaan et al. (1999) |
| Influenza PR8 | Murine macrophage | Induce iNOS RNA | Imanishi et al. (2005) |
| Hantavirus | A549 cells | IFNs induced | Stoltz et al. (2007) |
| Vero E6 cells | Cytokine induced | Klingström et al. (2006) | |
| iNOS(−/−) mice | NO not produced | ||
| Coronavirus (SARS CoV) | Vero E6 cells | Viral RNA synthesis inhibited | Akerström et al. (2005) |
| Hepatitis B | Liver of transgenic mice | IFN‐γ‐induced iNOS | Guidotti et al. (2000) |
| LCMV | Liver of transgenic mice | IFN‐γ‐induced iNOS | Guidotti et al. (2000) |
| Coxsackievirus B3 | Macrophage in vitro | IFN‐γ‐induced iNOS | Jarasch et al. (2005) |
| Sendai virus | Mice | NO and peroxynitrite | Akaike et al. (2000) |
| Rabies virus | Neuroblastoma cells | Transcription inhibition | Ubol et al. (2001) |
| Junin virus | Astrocyte culture | iNOS/NO generation | Pozner et al. (2008) |
| Japanese encephalitis virus (JEV) | Primarily JEV‐infected N18, human neuronal NT‐2, and BHK‐21 cells, as well as in persistently JEV‐infected C2‐2 cells. | Inhibit viral RNA synthesis | Lin et al. (1997) |
| DV | LLC‐MK2 monkey kidney cells | RdRp inhibition | Takhampunya et al. (2006) |
| Neuroblastoma cells | RNA production suppressed | Charnsilpa et al. (2005) | |
| NO‐induced increase of virus replication | |||
| HIV‐1 | T‐cell lines. Jurkat and MT‐2 | Activation of LTR‐mediated transcription | Jiménez et al. (2001) |
| Monocytes from PBMC of normal humans | Activation of NF‐κB by peroxynitrite | Aquaro et al. (2007) | |
| PBMC of HIV‐1 Patients | Decreased expression of iNOS RNA | Cairoli et al. (2008) | |
| Normal PBMC infected with HIV‐1 | Decreased expression of iNOS RNA | ||
| NO has no effect on virus replication | |||
| Vaccinia virus | Mice | iNOS‐deficient mice do not show increased viral replication | van den Broek et al. (2000) |
| Murine macrophage | Suppress iNOS through soluble viral protein | Bellows et al. (2003) | |
| Lymphocytic choriomeningitis virus | Mice | INOS/NO are redundant | Bartholdy et al. (1999) |
| Mouse hepatitis virus | Mice | iNOS‐deficient mice do not increase | Wu et al. (2000) |
| Tick‐borne encephalitis virus | Mice macrophage | Increased NO production has no effect | Kreil & Eibl (1996) |
| Hepatitis B virus | Hepatoma cells | Increased iNOS expression has no effect | Proto et al. (2008) |
All viruses could not be included.LCMV, lymphocytic choriomeningitis virus;LTR, long terminal repeat.
Effects of NO on host during viral infections
NO and peroxynitrite are double‐edged swords having both beneficial and harmful effects on the host during infections, resulting in pathological lesions (Table 4). Peroxynitrite is harmful to the host when present in high concentrations, oxidizing lipids and DNA, and oxidizing or nitrating proteins. However, peroxynitrite in low concentrations may be beneficial to the host during pathogen infection (Padalko et al., 2004). Nitrative stress‐mediated 8‐nitroguanosine formation during influenza or Sendai virus infection involves unique biochemical and pharmacological properties such as redox activity and mutagenic potential, which contributes to pathogenic processes during viral infection (Zaki et al., 2005). Central nervous system (CNS) inflammatory response to neurotropic virus infection is likely to be dependent upon the activity of peroxynitrite or its products on the blood–brain barrier (Hooper et al., 2001). Infection with virulent Coxsackie virus in mice increases expression of iNOS and produces inflammatory myocardial disease (Bevan et al., 2001). Adenovirus infection increases iNOS and peroxynitrite production in the lung, generating nitrotyrosine, and may contribute to inflammatory responses in the lung (Zsengellér et al., 2001). Hepatitis C virus (HCV) infection can stimulate the production of NO through activation of the iNOS gene by the viral core and NS3 proteins. NO causes DNA breaks and enhances DNA mutation. This sequence of events provides a mechanism for HCV pathogenesis and oncogenesis (Machida et al., 2004). On the other hand, enhanced oxidative stress may be involved in the pathogenesis of viral infections. HIV‐infected patients have a higher incidence of oxidative stress, endothelial dysfunction and cardiovascular disease than uninfected individuals (Table 4). Recent reports have demonstrated that viral proteins upregulate ROS, which may contribute to elevated cardiovascular risk in HIV‐1 patients (Kline et al., 2008). Using an HIV‐1 transgenic rat model it has been shown that HIV‐1 protein expression decreases NO and causes endothelial dysfunction. Diminished antioxidant capacity increases vascular superoxide levels, which reduce NO bioavailability and promote peroxynitrite generation. Restoring glutathione levels reverses HIV‐1 protein‐mediated effects on superoxide, NO and vasorelaxation (Kline et al., 2008). Experimental murine encephalitis induced by Junin virus increases expression of iNOS by unidentified cells, concomitant with the astrocyte reaction. The specific inhibition of iNOS is associated with greater mortality but lower astrocytosis (Table 4), suggesting that the protective role of NO is related to enhanced astrocyte activation, representing a beneficial cellular response to virus‐induced CNS damage (Gómez et al., 2003).
Table 4.
Viral infection in which pathogenesis/protection is through NO production *
| Virus | Test system | Site/lesion | Mechanisms of NO production | References |
|---|---|---|---|---|
| NO‐mediated pathology | ||||
| Rotavirus | Mice in vivo; ex vivo ileal loop | Ileum/diarrhoea | Upregulation of ileal iNOS mRNA by virus and by NS4 protein | Borghan et al. (2007) |
| Coxsackie B | Mice | Myocarditis | Increased iNOS/NO | Bevan et al. (2001) |
| Herpes simplex 1 | Mice | Liver/apoptosis | Increased production of NO, TNF‐α, IL‐6, IFN‐γ | Irie & Shiga (2005) |
| HIV | Cortical cell culture | Neurotoxicity | Activation of NOS by HIVgp120 and cytokines | Dawson et al. (1993) |
| PBMC monocyte | Lymphocyte inactivation | Large amount of NO production | Groeneveld et al. (1996) | |
| Cardiomyocyte culture, animals | Cardiomyopathy | NO induced cardiomyocyte apoptosis by TNF type 1 receptor activation | Monsuez et al. (2007) | |
| Microglia cells | Neurodegeneration | NO‐induced oxidative stress | Roy et al. (2008) | |
| Influenza A | Mice | Pneumonitis | iNOS presence | Karupiah et al. (1998) |
| Tick‐borne encephalitis virus | Mice | Encephalitis | Increased NO | Kreil & Eibl (1996) |
| Adenovirus | Mice | Lung inflammation | iNOS and peroxynitrite‐generated nitrotyrosine | Zsengellér et al. (2001) |
| Murine cytomegalovirus | Mice | Pneumonitis | Increased NO | Tanaka et al. (1997) |
| Hepatitis C | Hepatocyte culture | Hepatitis, oncogenesis | Increased iNOS/NO | Machida et al. (2004) |
| NO‐mediated protection of pathology | ||||
| Japanese encephalitis | Mice | Encephalitis protected | Production of macrophage‐derived neutrophil chemotactic factor increases iNOS | Saxena et al. (2001) |
| Junín virus | Mice, astrocyte culture | Brain damage protected | INOS inhibition increases brain damage | Gómez et al. (2003) |
Only a few examples have been cited.
DV
DV belong to the family Flaviviridae, genus Flavivirus, and have four genetically and antigenically related serotypes known as DV‐1 to DV‐4. Each serotype of DV contains abundant genetic variation and can be subdivided into subtypes or genotypes that may be responsible for the varying severity of clinical symptoms. DV replicates mainly in cells of monocyte/macrophage lineage (Chaturvedi et al., 2006a).
DV proteins and immune response
DV genome is a single‐stranded RNA of positive polarity that encodes a single polyprotein, which is translated into three structural proteins – C, prM and E – and seven nonstructural proteins, NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5, which are important for virus replication. Anti‐E antibodies are the main response against DV. The antibodies inhibit viral binding to cells, neutralize viral infectivity in vitro, protect mice from DV challenge on passive transfer and show a variable degree of cross‐reactivity among the DV serotypes. NS1 is a key glycoprotein involved in the production of infectious virus and the pathogenesis of dengue diseases. During the replication of DV, NS1 associates with the membrane on the cell surface and in the RNA replication complex (Noisakran et al., 2008). NS1 is expressed on the surface of the virus‐infected cells and is secreted into the circulation as a soluble multimer; it is an important target of antibodies against DV. Antibodies against NS1 can trigger complement‐mediated lysis of DV‐infected cells in vitro and protect mice from DV challenge. But the cross‐reaction of these antibodies with endothelial cells leads to expression of cytokine, chemokine and adhesion molecules, resulting in cell damage. NS3 is a multifunctional protein; it is the main antigen that stimulates DV‐reactive CD4+ and CD8+ T cell (reviewed by 2008, 2006b). NS3 has located at the active site of a trypsin‐like serine protease and, together with the NS2B cofactor, forms an active protease that is required for polyprotein processing. NS3 also has an RNA‐stimulated NTPase and RNA helicase that is important for viral RNA replication, as well as 5′‐RNA triphosphatase activity. NS5 is required for viral RNA replication and is an RNA‐dependent RNA polymerase (RdRp) (O'Reilly & Kao, 1998). In flavivirus‐infected cells, NS3 and NS5 exist in a complex and are thought to be components of the viral RNA replicase complex (1995, 2003). A role of NS3 and NS5 in virus replication and 5′‐capping has been suggested due to their enzymatic activities but the functions of the other nonstructural proteins are not clear (Benarroch et al., 2004).
Pathogenesis of DHF
The precise mechanism of DHF is not yet fully known. The important hypothesis put forward regarding the role of host factors are ADE of DV replication, shift of Th1‐ to Th2‐type cytokine response and other T‐cell responses resulting into cytokine tsunami. The viral factors include genotypic mutation, for example the Southeast Asian type of DV produces DHF in children, whereas the American genotype does not. Some of these mechanisms are discussed briefly. Non‐neutralizing anti‐DV antibodies from an earlier infection by a heterologous serotype of DV mediate ADE. They form complexes with the infecting DV leading to greater uptake by macrophages and consequent greater numbers of DV‐infected cells. The first suggestion of an association of increased risk for DHF with a secondary DV infection was made in 1970s (Halstead, 1970). A subpopulation of CD4+ T cells produce a unique cytokine, cytotoxic factor (CF) during DV infection of mice and man (hCF). CF induces H2‐A‐positive macrophages to produce another cytokine, macrophage cyototoxin (CF2), which amplifies its cytotoxic effects on target cells (Fig. 3). The hCF purified from the sera of DHF patients appear to be pathogenesis‐related proteins, capable of reproducing DHF‐like pathological lesions on inoculation in mice, such as increased capillary permeability, cerebral oedema and blood leukocyte changes (1998, 1991, 1997, 1997). The highest levels of hCF‐autoantibodies are seen in sera of patients with mild illness (DF) and the lowest in patients with DHF grade IV (Chaturvedi et al., 2001; reviewed by 2006a, 2006b).
Figure 3.
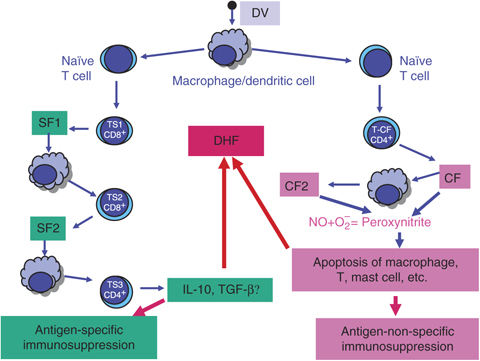
Role of NO in the proposed mechanisms of immunosuppression during DV infection.
A microorganism‐induced suppressor T‐cell (TS) cascade was delineated in DV‐infected mice in the late 1980s (Chaturvedi, 1984) and it has now been confirmed in a large number of viruses (Mills, 2004). The antigen‐specific TS cascade in DV‐infected mice consists of three generations of TS and their secretary soluble suppressor cytokines (SF), with in between macrophages transmitting the signals (Fig. 3). DV‐induced suppressor pathway suppresses antigen‐specific antibody production, including that of enhancing antibody. Thus increased replication of the virus mediated by the enhancing antibody and the immunopathology mediated by the immune complex is prevented. On the other hand, suppression of neutralizing antibody would delay elimination of DV from the body, causing pathological lesions. TS can also increase the severity by producing IL‐10/TGF‐β (1984, 2007). Both IL‐10 and TGF‐β suppress NO production (1992, 1993) and may increase the virus load and the severity of dengue disease. The outcome may depend upon the fine balance between them.
Different types of T cells that become activated during DV infection and contribute to the development of DHF are summarized in Fig. 4. Serum levels of IFN‐γ and IL‐2 are the highest in DF and the lowest in severe DHF cases. On the other hand, serum levels of IL‐4, IL‐10 and IL‐6 are maximum in DHF patients and negligible in patients with DF. The levels of TNF‐α in the sera increase with the increase of severity of DHF (Chaturvedi et al., 1999). The most significant finding was a shift of the predominant Th1‐type response observed in 66% of the DF patients to Th2‐type response seen in 71% of DHF grade IV, indicating a role of Th2 cells in the pathogenesis of DHF (Chaturvedi et al., 1999). During acute infection in children, few dengue‐responsive CD8+ T cells are recovered that show an activated phenotype and undergo apoptosis. These DV‐specific T cells have a low affinity for the infecting virus and a higher affinity for previously encountered strains. Cross‐reactive DV‐specific T cells show high cytokine production, which may contribute to the vascular leakage. DV‐specific memory lymphocyte response has been detected even 20 years after a primary infection by DV. Induction of cross‐reactive memory T cells (mainly NS3‐specific) during a secondary infection leads to increased incidence of DHF and DSS. When viral peptides complexed with HLA molecules are presented to memory T cells, proliferation and the production of proinflammatory cytokines follow, which can directly affect vascular endothelial cells, resulting in plasma leakage (2003, 2006). Recently, Ubol et al. (2008b) have shown that during DF, genes in the interferon system and complement inhibitor play a role in lowering virus production and reducing tissue damage. In patients with DHF, the dysfunction of immune cells, complement, and cytokines increases viral load and tissue damage. Profound T‐cell activation and death may contribute to the systemic disturbances leading to DHF, and original antigenic sin in the T‐cell responses may suppress or delay viral elimination, leading to higher viral loads and increased immunopathology (reviewed by 2008, 2007, 2006, 2007, 2008). Further, the increased serum levels of IL‐8 (Raghupathy et al., 1998) and TGF‐β1 (Agarwal et al., 1999) are associated with increasing severity of DHF and death. Thus, the cascades of T‐cell activation resulting in a ‘tsunami’ of cytokines and other chemical mediators (‘cytokine tsunami’) released mainly from T cells, monocytes/macrophages and endothelial cells, ultimately cause an increase in vascular permeability and lead to DHF/DSS.
Figure 4.
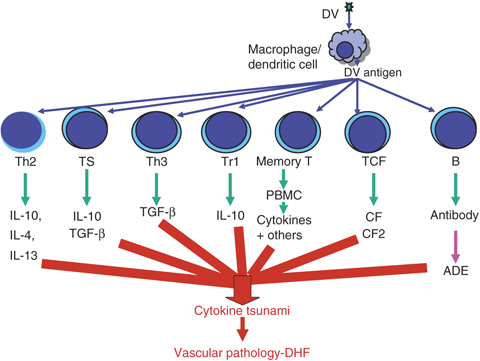
Role of various subsets of T cells in the pathogenesis of DHF through generation of cytokine tsunami. ADE (B cell) is included to emphasize its importance.
Role of NO in DV infection
During the 1990s a number of papers were published that showed some association between NO and viral infections, both in vivo and in vitro (reviewed by Reiss & Komatsu, 1998). But the role of NO and peroxynitrite in DV infection and the role in the pathogenesis of DHF was first suggested by Chaturvedi and colleagues (Misra et al., 1996a, b; Mukerjee et al., 1996). This generated a lot of interest in dengue virologists, resulting into interesting studies that are presented here briefly.
Studies in mice
Production of nitrite by the spleen cells of mice followed inoculation of DV or the DV‐induced cytokines CF and CF2. In DV‐infected mouse spleen, maximum NO2 production occurred at 8–11 days postinfection, which correlated with the peak appearance of CF in the spleen. Maximum NO2 − production occurred in the spleen cells 45 min after intravenous inoculation of mice with CF. The NO2 − was produced by macrophages (Misra et al., 1996a). In a similar study, CF2 was also shown to induce production of NO2 − in the macrophages of spleens of mice (Mukerjee et al., 1996). Further, production of superoxide anion (O2 −) and hydrogen peroxide (H2O2) by the spleen cells was shown following intracerebral inoculation of DV or intravenous inoculation of CF/CF2 in mice. It was concluded that the cell apoptosis was mediated by NO+O− (peroxynitrite) and not by H2O2 (1996b, 1998). NO also transmits the DV‐specific intracellular suppressor signal in macrophage (Khare & Chaturvedi, 1997).
Studies in patients
Rodriguez‐Ortega (1998) has discussed the clinical features of dengue that link NO with the pathology of the severe dengue disease. Valero et al. (2002) reported increased levels of NO in patients with DF, whereas in the patients with DHF, levels similar to those of healthy controls were found. In contrast, Trairatvorakul et al. (2005) reported that the levels of serum NO in DF patients were significantly lower than those of normal controls. Patients with DSS had higher NO levels than those with DHF I/II. Neves‐Souza et al. (2005) studied the expression of DV antigens and iNOS in human blood monocytes analysed by flow cytometry using cells either from patients with acute DF or after DV infection in vitro. Activation of DV‐infected monocytes based on induction of iNOS occurred both in vivo and in vitro, and the susceptibility of DV to NO production was noted. NO inhibits aggregation, recruitment and adhesion of platelets to the vascular endothelium. Mendes‐Ribeiro et al. (2008) have shown that an elevated rate of l‐arginine transport in DF patients is associated with enhanced NOS activity and elevated plasma fibrinogen levels, resulting in reduced platelet aggregation. Oxidative stress mediated changes in plasma proteins; for example, protein carbonylation and the ratio of protein carbonylation and protein‐bound sulphydryl group levels can be an early biomarker for prediction of severe dengue infection (Soundravally et al., 2008).
Studies in vitro
Production of nitrite by mouse spleen cell cultures was studied following inoculation of DV or CF/CF2. DV‐stimulated spleen cell culture supernatants showed a peak production of both CF and NO2 − at 72 h. Pretreatment of spleen cells with NMMA inhibited NO2 − production. NO2 − production was abrogated in a dose‐dependent manner by treatment of spleen cells with the Ca2+ channel blocking drug nifedipine. Thus, DV‐induced CF induces production of NO2 − in spleen cells, probably in a Ca2+‐dependent manner, and may be a mechanism of target cell killing (Misra et al., 1996a). Further, DV, CF or CF2 induce production of superoxide anion (O2 −) and hydrogen peroxide (H2O2) by the mouse spleen cell cultures. It was suggested that O2 − and nitrite are necessary for cell killing by CF/CF2 in a Ca2+‐dependent manner and the killing may possibly be by generation of peroxynitrite (1996b, 1998). In another study it was shown that NO transmits the DV‐specific suppressor signal intracellularly in macrophages (Khare & Chaturvedi, 1997). DV infection induces a biphasic activation of Kupffer cells in cultures: there is one peak of activation shortly after infection which involves the production of NO and IFN‐α, and a second peak after a few hours involving IL‐6 and TNF‐α synthesis. DV‐replicating Kupffer cells undergo apoptosis and are cleared by phagocytosis. The timing of the synthesis of these soluble mediators suggests the involvement of a very early step of infection in their activation (Marianneau et al., 1999). Valero et al. (2002) reported that in vitro incubation of human platelets with DV did not increase levels of NO. Chen & Wang (2002) reported that after DV infection, the in vitro‐differentiated monocyte/macrophages secrete multiple innate cytokines and chemokines, including TNF‐α, IFN‐α, IL‐1β, IL‐8, IL‐12, MIP‐1α and regulated upon activation, normal T cell expressed and secreted (RANTES) but not IL‐6, IL‐15 or NO. Human monocyte cultures infected with DV show increased numbers of apoptotic cells and increased production of TNF‐α. No increase in production of NO was observed. These results may be related to early primary viral infection in which virus could induce apoptosis in monocytes, but monocytes may contribute to host defence mechanisms against virus by viral phagocytosis, phagocytosis of infected apoptotic cells, and the release of proinflammatory cytokines (Espina et al., 2003).
Chareonsirisuthigul et al. (2007) reported that DV infection of THP‐1 cells via an ADE pathway suppresses NO radicals, by disrupting the transcription of the iNOS gene transcription factor, IRF‐1 and blocking the activation of STAT‐1. Further, it suppresses the transcription and translation of IL‐12, IFN‐γ and TNF‐α, whereas the expression and synthesis of the anti‐inflammatory cytokines IL‐6 and IL‐10 are enhanced. Thus, besides facilitating the entry process, ADE infection also modifies innate and adaptive intracellular antiviral mechanisms, resulting in unrestricted DV replication in THP‐1 cells (Chareonsirisuthigul et al., 2007). Brown et al. (2009) have shown that antibody‐enhanced DV infection of the FcR‐bearing mast cell/basophil KU812 cell line results in a massive induction of apoptosis.
Effect of NO on DV replication
Charnsilpa et al. (2005) investigated the effects of NO on DV production and RNA replication in mouse neuroblastoma cells in the presence of an exogenous NO donor, S‐nitroso‐N‐acetylpenicillamine (SNAP). NO inhibited viral replication via suppressed RNA synthesis, resulting in reduction of NS1 synthesis. Thus, NO may serve as a defence that diminishes viral load in patients. Further, the activity of recombinant DV NS5 in negative‐strand RNA synthesis is affected in the presence of 5 mM SNAP in in vitro RdRp assays, whereas the RNA helicase activity of DV NS3 is not inhibited (Takhampunya et al., 2006). Thus, the inhibitory effect of NO on DV infection is partly via inhibition of the RdRp activity, which then downregulates viral RNA synthesis. The clinical isolates of DV have been divided into two groups on the basis of their sensitivity to the inhibitory effect of NO radical on viral replication. Ubol et al. (2008a) have studied genotypic differences at the level of the amino acid sequence of the NS5 gene between NO‐sensitive viruses isolated from DF patients and NO‐resistant isolates from DHF patients. They found that these two groups of viruses contain different amino acid sequences at position 621–646 in the active site of NS5, suggesting that the response to the inhibitory effect of NO radical may, at least in part, be regulated by NS5. They have also studied the differences in global gene expression of NO‐producing host cells in response to infection by the two groups of isolates using cDNA array analysis. NO‐resistant viruses have a stronger influence on gene expression of the infected host cells in both the number and the type of genes.
Role of NO in the pathogenesis of DHF
Alteration in NO production is associated with a variety of pathological effects, such as vasodilation, inflammation, thrombosis, immune response and neurotransmission. NO production may initiate or suppress apoptosis but the mechanism is not fully understood. Macrophage lineage cells replicate DV and are professional NO producers. NO produced by them downregulates replication of DV. DV‐infected mice spleen cells produce NO2 −. CF/CF2 induce production of NO2 − in the spleen cells in a Ca2+‐dependent manner, which may be a mechanism of target cell killing (Mukerjee et al., 1996). CF2 also induces production of superoxide anion (O2 −) and hydrogen peroxide (H2O2) by the spleen cells of mice in vitro and in vivo and the killing (Fig. 3) appears to be by generation of peroxynitrite (1996a, 1996b, 1998). Neves‐Souza et al. (2005) reported that activation of DV‐infected monocytes is based on induction of iNOS both in vivo and in vitro. A mechanism through which NO acts is via endothelin (ET‐1), which is produced by endothelial cells. Thrombin, cytokines and ROS, etc., stimulate the vascular endothelium to produce and release ET‐1. Release of ET‐1 is inhibited by prostacyclin and NO. Binding of ET‐1 to endothelial ETb receptors stimulates the formation of NO. In the absence of smooth muscle endothelin receptor stimulation, this NO causes vasodilation (reviewed by Iglarz & Clozel, 2007; Basu & Chaturvedi, 2008). DV infection of human umbilical cord endothelial cells inhibits the production of ET‐1 and prostacyclin 2, thus affecting their normal functions of secretion of vasoactive substances, resulting in increased vascular permeability and impairment of homoeostasis and blood coagulation (Jiang et al., 1999).
Anti‐NS1 antibodies act as autoantibodies that cross‐react with noninfected endothelial cells and trigger the intracellular signaling leading to the production of NO and to apoptosis. Endothelial cell damage may cause vascular leakage that contributes to the pathogenesis of dengue disease (Lin et al., 2002). Further, the mitochondria‐dependent pathway that is regulated by NO production causes endothelial cell apoptosis in this system (Lin et al., 2004). Yen et al. (2008) have reported macrophage infiltration into the vicinity of the endothelium, increased TNF‐α production and endothelial cell apoptosis in the tissues with increased expression of iNOS and nitrotyrosine in endothelium in the haemorrhagic tissues in DV‐infected mice. In vitro studies showed that primary mouse and human endothelial cells are productively infected by DV and produce reactive nitrogen and oxygen species (RNS and ROS) and apoptotic cell death, which is greatly enhanced by TNF‐α. N G‐nitro‐l‐arginine methyl ester and N‐acetyl cysteine reverse the effects of DV and TNF‐α on endothelial cells. Development and the severity of haemorrhage are markedly reduced in mice lacking iNOS or p47(phox) or in mice treated with oxidase inhibitor, indicating a possible use of antioxidant as a therapeutic agent for DHF (Yen et al., 2008). This has been confirmed in patients with DHF/DSS where antibodies against NS1 present in patient sera cross‐react with endothelial cells and induce apoptosis via a caspase‐dependent pathway and cause cell lysis in the presence of complement. DHF/DSS patient sera show higher percentages of cytotoxicity than DF patient sera (Lin et al., 2003). Antibodies directed against DV NS1 have been shown to cross‐react with human platelets and endothelial cells, leading to endothelial cell damage and inflammatory activation (Lin et al., 2006).
Conclusions and future perspectives
Multiple roles of NO in the pathogenesis of DV infections have been discussed as regards mutation of DV, nonspecific inflammatory responses and immunological host reactions modulated by NO that may cause DHF. A synthesis of the current understanding of the role of NO in the pathogenesis of DHF is presented in (Fig. 5). Replication of NO‐sensitive DV is inhibited by the generation of NO, resulting in lower virus load and consequently a milder dengue disease, DF (Fig. 5). The NO‐resistant DV are virulent, have a higher replication rate and have a stronger influence on host genetic response as compared with the NO‐susceptible DV. NO‐resistant DV induce greater expression of immune response‐related genes, for example genes involving cytokines/chemokines, activation of T cells, B cells, platelets and inflammatory cells. This may result in the interaction between platelets, vascular endothelial cells and inflammatory cells that induces vascular leakage. Further, NO‐resistant DV significantly upregulate IL‐6, IL‐7, IL‐8, RANTES and MCP‐3, which are correlated with increased DHF. Our findings in the sera of the patients with dengue and DHF show an inverse relationship between IL‐4 and IL‐10 to IFN‐γ (Chaturvedi et al., 1999). Higher levels of IL‐6 and IL‐8 that may be involved in vascular leakage and haemorrhage are found in DHF/DSS patients but not in DF patients (1999, 1998). Thus, NO has a central role in DV infection; inhibition of replication of DV by NO results in DF and failure to inhibit results in severe disease, DHF (Fig. 5).
Figure 5.
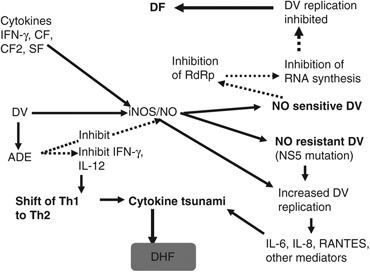
Roles of NO during DV infection that may determine whether it will lead to DF or DHF.
The ultimate role of NO on DV pathogenesis is still far from completely understood and a lot more work remains to be done. To determine whether the severity of DV infection is determined by mutation of NS5, in‐depth studies on NS5 genetic manipulation are needed. Are the phenotypes of DV determined by the differences in genotype of NS5? Effects of NO on DV NS5 have been investigated but what about other structural and nonstructural proteins of DV? In DV endemic areas all four serotypes are circulating in the population, resulting in concurrent infection with more than one DV or with other viruses. What is the role of NO in such infections? Infection of macrophages by DV–ADE complex suppresses innate immunity in infected cells, resulting in the higher production of virus per cell. Suppression of innate immunity is regulated by IL‐10, but is the master molecule NO? More studies are required to understand these aspects.
Statement
U.C.C. has retired and is now an INSA Honorary Scientist.
Acknowledgements
Professor Umesh C. Chaturvedi has retired from the position of the Head of the Department of Microbiology, K.G. Medical College (now CSM Medical University), Lucknow. The financial assistance to U.C.C. as INSA Honorary Scientist for the preparation of this manuscript by the Indian National Science Academy, New Delhi is gratefully acknowledged.
Editor: Willem van Leeuwen
References
- Agarwal R, Chaturvedi UC, Misra A, Mukerjee R, Kapoor S, Nagar R, Tandon R & Mathur A (1998) Production of cytotoxic factor by peripheral blood mononuclear cells (PBMC) in patients with dengue haemorrhagic fever. Clin Exp Immunol 112: 340–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal R, Elbishbishi EA, Chaturvedi UC, Nagar R & Mustafa AS (1999) Profile of transforming growth factor‐beta1 in patients with dengue haemorrhagic fever. Int J Exp Pathol 80: 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike T & Maeda H (2000) Nitric oxide and virus infection. Immunology 101: 300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike T, Fujii S, Kato A et al (2000) Viral mutation accelerated by nitric oxide production during infection in vivo . FASEB J 14: 1447–1454. [DOI] [PubMed] [Google Scholar]
- Akaike T, Okamoto S, Sawa T, Yoshitake J, Tamura F, Ichimori K, Miyazaki K, Sasamoto K & Maeda H (2003) 8‐Nitroguanosine formation in viral pneumonia and its implication for pathogenesis. P Natl Acad Sci USA 100: 685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerström S, Mousavi‐Jazi M, Klingström J, Leijon M, Lundkvist A & Mirazimi A (2005) Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome coronavirus. J Virol 79: 1966–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akuta T, Zaki MH, Yoshitake J, Okamoto T & Akaike T (2006) Nitrative stress through formation of 8‐nitroguanosine: insights into microbial pathogenesis. Nitric Oxide 14: 101–108. [DOI] [PubMed] [Google Scholar]
- Aquaro S, Muscoli C, Ranazzi A, Pollicita M, Granato T, Masuelli L, Modesti A, Perno CF & Mollace V (2007) The contribution of peroxynitrite generation in HIV replication in human primary macrophages. Retrovirology 4: 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholdy C, Nansen A, Christensen JE, Marker O & Thomsen AR (1999) Inducible nitric‐oxide synthase plays a minimal role in lymphocytic choriomeningitis virus‐induced, T cell‐mediated protective immunity and immunopathology. J Gen Virol 80: 2997–3005. [DOI] [PubMed] [Google Scholar]
- Basu A & Chaturvedi UC (2008) Vascular endothelium: the battle field of dengue viruses. FEMS Immunol Med Mic 53: 287–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows CF, Garry RF & Jaffe BM (2003) Vaccinia virus‐induced inhibition of nitric oxide production. J Surg Res 111: 127–135. [DOI] [PubMed] [Google Scholar]
- Benarroch D, Selisko B, Locatelli GA, Maga G, Romette J‐L & Canard B (2004) The RNA helicase, nucleotide 5′‐triphosphatase, and RNA 5′‐triphosphatase activities of Dengue virus protein NS3 are Mg2+‐dependent and require a functional Walker B motif in the helicase catalytic core. Virology 328: 208–218. [DOI] [PubMed] [Google Scholar]
- Bevan AL, Zhang H, Li Y & Archard LC (2001) Nitric oxide and Coxsackievirus B3 myocarditis: differential expression of inducible nitric oxide synthase in mouse heart after infection with virulent or attenuated virus. J Med Virol 64: 175–182. [DOI] [PubMed] [Google Scholar]
- Bogdan C (1998) The multiplex function of nitric oxide in (auto)immunity. J Exp Med 187: 1361–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan C (2001) Nitric oxide and the immune response. Nat Immunol 2: 907–916. [DOI] [PubMed] [Google Scholar]
- Borghan MA, Mori Y, El‐Mahmoudy AB, Ito N, Sugiyama M, Takewaki T & Minamoto N (2007) Induction of nitric oxide synthase by rotavirus enterotoxin NSP4: implication for rotavirus pathogenicity. J Gen Virol 88: 2064–2072. [DOI] [PubMed] [Google Scholar]
- Brown MG, Huang YY, Marshall JS, King CA, Hoskin DW & Anderson R (2009) Dramatic caspase‐dependent apoptosis in antibody‐enhanced dengue virus infection of human mast cells. J Leukocyte Biol. 85: 71–80. [DOI] [PubMed] [Google Scholar]
- Cairoli E, Scott‐Algara D, Pritsch O, Dighiero G & Cayota A (2008) HIV‐1 induced decrease of nitric oxide production and inducible nitric oxide synthase expression during in vivo and in vitro infection. Clin Immunol 127: 26–33. [DOI] [PubMed] [Google Scholar]
- Chareonsirisuthigul T, Kalayanarooj S & Ubol S (2007) Dengue virus (DENV) antibody‐dependent enhancement of infection upregulates the production of anti‐inflammatory cytokines, but suppresses anti‐DENV free radical and pro‐inflammatory cytokine production, in THP‐1 cells. J Gen Virol 88: 365–375. [DOI] [PubMed] [Google Scholar]
- Charnsilpa W, Takhampunya R, Endy TP, Mammen MP Jr, Libraty DH & Ubol S (2005) Nitric oxide radical suppresses replication of wild‐type dengue 2 viruses in vitro . J Med Virol 77: 89–95. [DOI] [PubMed] [Google Scholar]
- Chaturvedi UC (1984) Dengue virus‐induced suppressor pathway. Curr Sci 53: 971–976. [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi UC & Nagar R (2008) Dengue and dengue haemorrhagic fever: Indian perspective. J Biosci 33: 429–441. [DOI] [PubMed] [Google Scholar]
- Chaturvedi UC, Dhawan R, Khanna M & Mathur A (1991) Breakdown of the blood–brain barrier during dengue virus infection of mice. J GenVirol 72: 859–866. [DOI] [PubMed] [Google Scholar]
- Chaturvedi UC, Dhawan R & Mukerjee R (1997) Immunosuppression and cytotoxicity of dengue infection in the mouse model Dengue and Dengue Haemorrhagic Fever (Gubler DJ. & Kuno G, eds), pp. 291–312. CAB International Press, Wallingford, Oxon, UK. [Google Scholar]
- Chaturvedi UC, Raghupathy R, Pacsa AS et al (1999) Shift from a Th1‐type response to Th2‐type in dengue haemorrhagic fever. Curr Sci 76: 63–69. [Google Scholar]
- Chaturvedi UC, Agarwal R, Elbishbishi EA & Mustafa AS (2000) Cytokine cascade in dengue haemorrhagic fever: implications for pathogenesis. FEMS Immunol Med Mic 28: 183–188. [DOI] [PubMed] [Google Scholar]
- Chaturvedi UC, Elbishbishi EA, Agarwal R & Mustafa AS (2001) Cytotoxic factor‐autoantibodies: possible role in the pathogenesis of dengue haemorrhagic fever. FEMS Immunol Med Mic 30: 181–186. [DOI] [PubMed] [Google Scholar]
- Chaturvedi UC, Shrivastava R & Nagar R (2005) Dengue vaccines: problems and prospects. Indian J Med Res 121: 639–652. [PubMed] [Google Scholar]
- Chaturvedi UC, Nagar R & Shrivastava R (2006a) Macrophage and dengue virus: friend or foe? Indian J Med Res 124: 23–40. [PubMed] [Google Scholar]
- Chaturvedi UC, Nagar R & Shrivastava R (2006b) Dengue and dengue haemorrhagic fever: implications of host genetics. FEMS Immunol Med Mic 47: 155–166. [DOI] [PubMed] [Google Scholar]
- Chaturvedi UC, Shrivastava R, Tripathi RK & Nagar R (2007) Dengue virus‐specific suppressor T cells: current perspectives. FEMS Immunol Med Mic 50: 285–299. [DOI] [PubMed] [Google Scholar]
- Chen YC & Wang SY (2002) Activation of terminally differentiated human monocytes/macrophages by dengue virus: productive infection, hierarchical production of innate cytokines and chemokines, and the synergistic effect of lipopolysaccharide. J Virol 76: 9877–9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly L, Palacios‐Callender M, Ameixa C, Moncada S & Hobbs AJ (2001) Biphasic regulation of NF‐kappa B activity underlies the pro‐ and anti‐inflammatory actions of nitric oxide. J Immunol 166: 3873–3881. [DOI] [PubMed] [Google Scholar]
- Cunha FQ, Moncada S & Liew FY (1992) Interleukin‐10 (IL‐10) inhibits the induction of nitric oxide synthase by interferon‐γ in murine macrophages. Biochem Bioph Res Co 182: 1155–1159. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Uhl GR & Snyder SH (1993) Human immunodeficiency virus type Dawson 1 coat protein neurotoxicity mediated by nitric oxide in primary cortical cultures. P Natl Acad Sci USA 90: 3256–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan R, Chaturvedi UC, Khanna M & Mathur A (1994) Dengue virus‐induced cytokine damages the blood–brain barrier in mice. Proc Indian Natl Acad Sci 60: 45–52. [Google Scholar]
- Diefenbach A, Schindler H, Rollinghoff M, Yokoyama WM & Bogdan C (1999) Requirement for type 2 NO synthase for IL‐12 signaling in innate immunity. Science 284: 951–955. [DOI] [PubMed] [Google Scholar]
- Ding AH, Nathan CF & Stuehr DJ (1988) Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J Immunol 141: 2407–2412. [PubMed] [Google Scholar]
- Espina LM, Valero NJ, Hernández JM & Mosquera JA (2003) Increased apoptosis and expression of tumor necrosis factor‐alpha caused by infection of cultured human monocytes with dengue virus. Am J Trop Med Hyg 68: 48–53. [PubMed] [Google Scholar]
- Gómez RM, Yep A, Schattner M & Berría MI (2003) Junin virus‐induced astrocytosis is impaired by iNOS inhibition. J Med Virol 69: 145–149. [DOI] [PubMed] [Google Scholar]
- Green S & Rothman A (2006) Immunopathological mechanisms in dengue and dengue hemorrhagic fever. Curr Opin Infect Dis 19: 429–436. [DOI] [PubMed] [Google Scholar]
- Groeneveld PH, Kroon FP, Nibbering PH, Bruisten SM, Van Swieten P & Van Furth R (1996) Increased production of nitric oxide correlates with viral load and activation of mononuclear phagocytes in HIV‐infected patients. Scand J Infect Dis 28: 341–345. [DOI] [PubMed] [Google Scholar]
- Guidotti LG, McClary H, Loudis JM & Chisari FV (2000) Nitric oxide inhibits hepatitis B virus replication in the livers of transgenic mice. J Exp Med 191: 1247–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead SB (1970) Observations related to pathogenesis of dengue hemorrhagic fever. VI. Hypotheses and discussion. Yale J Biol Med 42: 350–362. [PMC free article] [PubMed] [Google Scholar]
- Halstead SB (2007) Dengue. Lancet 370: 1644–1652. [DOI] [PubMed] [Google Scholar]
- Hanafy KA, Krumenacker JS & Murad F (2001) NO, nitrotyrosine, and cyclic GMP in signal transduction. Med Sci Monitor 7: 801–819. [PubMed] [Google Scholar]
- Hatch S, Mathew A & Rothman A (2008) Dengue vaccine: opportunities and challenges. IDrugs 11: 42–45. [PubMed] [Google Scholar]
- Hierholzer C, Harbrecht B, Menezes JM et al (1998) Essential role of induced nitric oxide in the initiation of the inflammatory response after hemorrhagic shock. J Exp Med 187: 917–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper DC, Kean RB, Scott GS, Spitsin SV, Mikheeva T, Morimoto K, Bette M, Röhrenbeck AM, Dietzschold B & Weihe E (2001) The central nervous system inflammatory response to neurotropic virus infection is peroxynitrite dependent. J Immunol 167: 3470–3477. [DOI] [PubMed] [Google Scholar]
- Huang FP, Niedbala W, Wei XQ, Xu D, Feng GJ, Robinson JH, Lam C & Liew FY (1998) Nitric oxide regulates Th1 cell development through the inhibition of IL‐12 synthesis by macrophages. Eur J Immunol 28: 4062–4070. [DOI] [PubMed] [Google Scholar]
- Huang Y‐CT, Li Z, Brighton LE, Carson JL, Becker S & Soukup JM (2005) 3‐Nitrotyrosine attenuates respiratory syncytial virus infection in human bronchial epithelial cell line. Am J Physiol-Lung C 288: L988–L996. [DOI] [PubMed] [Google Scholar]
- Iglarz M & Clozel M (2007) Mechanisms of ET‐1‐induced endothelial dysfunction. J Cardiovasc Pharmacol 50: 621–628. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ (1991) Signal transduction mechanisms involving nitric oxide. Biochem Pharmacol 41: 485–490. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Fukuto JM, Griscavage JM et al (1993) Oxidation of nitric oxide in aqueous solution to nitrite but not nitrate: comparison with enzymatically formed nitric oxide from l-arginine. P Natl Acad Sci USA 90: 8130–8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanishi N, Andoh T, Sakai S, Satoh M, Katada Y, Ueda K, Terasawa K & Ochiai H (2005) Induction of inducible nitric oxide (NO) synthase mRNA and NO production in macrophages infected with influenza A/PR/8 virus and stimulated with its ether‐split product. Microbiol Immunol 49: 41–48. [DOI] [PubMed] [Google Scholar]
- Irie H & Shiga J (2005) Pathogenesis of herpes simplex hepatitis in macrophage‐depleted mice: possible involvement of tumor necrosis factor-alpha and inducible nitric oxide synthase in massive apoptosis. Anat Sci Int 80: 199–211. [DOI] [PubMed] [Google Scholar]
- Jarasch N, Martin U, Kamphausen E, Zell R, Wutzler P & Henke A (2005) Interferon‐gamma‐induced activation of nitric oxide‐mediated antiviral activity of macrophages caused by a recombinant coxsackievirus B3. Viral Immunol 18: 355–364. [DOI] [PubMed] [Google Scholar]
- Jiang L, Guo H & Fang D (1999) Effect of dengue virus infection on the production of ET‐1 and PGI2 by human vascular endothelial cells. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi 13: 239–242. [PubMed] [Google Scholar]
- Jiménez JL, González‐Nicolás J, Alvarez S, Fresno M & Angeles Muñoz‐Fernández MA (2001) Regulation of human immunodeficiency virus type 1 replication in human T lymphocytes by nitric oxide. J Virol 75: 4655–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor M, Zhang L, Ramachandra M, Kusukawa J, Ebner KE & Padmanabhan R (1995) Association between NS3 and NS5 proteins of dengue virus type 2 in the putative RNA replicase is linked to differential phosphorylation of NS5. J Biol Chem 270: 19100–19106. [DOI] [PubMed] [Google Scholar]
- Karupiah G, Xie Q, Buller RML, Nathan C, Duarte C & MacMicking JD (1993) Inhibition of viral replication by interferon‐γ‐induced nitric oxide synthase. Science 261: 1445–1448. [DOI] [PubMed] [Google Scholar]
- Karupiah G, Chen JH, Mahalingam S, Nathan CF & MacMicking JD (1998) Rapid interferon gamma‐dependent clearance of influenza A virus and protection from consolidating pneumonitis in nitric oxide synthase 2‐deficient mice. J Exp Med 188: 1541–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna M, Chaturvedi UC, Sharma MC, Pandey VC & Mathur A (1990) Increased capillary permeability mediated by a dengue virus‐induced lymphokine. Immunology 69: 449–454. [PMC free article] [PubMed] [Google Scholar]
- Khare M & Chaturvedi UC (1997) Transmission of dengue virus‐specific suppressor signal depends on the presence of calcium. Indian J Med Res 102: 1–8. [PubMed] [Google Scholar]
- Kline ER, Kleinhenz DJ, Liang B, Dikalov S, Guidot DM, Hart CM, Jones DP & Sutliff RL (2008) Vascular oxidative stress and nitric oxide depletion in HIV‐1 transgenic rats are reversed by glutathione restoration. Am J Physiol-Heart C 294: H2792–H2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingström J, Akerström S, Hardestam J, Stoltz M, Simon M, Falk KI, Mirazimi A, Rottenberg M & Lundkvist A (2006) Nitric oxide and peroxynitrite have different antiviral effects against hantavirus replication and free mature virions. Eur J Immunol 36: 2649–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreil TR & Eibl MM (1996) Nitric oxide and viral infection: no antiviral activity against a flavivirus in vitro, and evidence for contribution to pathogenesis in experimental infection in vivo. Virology 219: 304–306. [DOI] [PubMed] [Google Scholar]
- Lin CF, Lei HY, Shiau AL, Liu HS, Yeh TM, Chen SH, Liu CC, Chiu SC & Lin YS (2002) Endothelial cell apoptosis induced by antibodies against dengue virus nonstructural protein 1 via production of nitric oxide. J Immunol 169: 657–664. [DOI] [PubMed] [Google Scholar]
- Lin CF, Lei HY, Shiau AL, Liu CC, Liu HS, Yeh TM, Chen SH & Lin YS (2003) Antibodies from dengue patient sera cross‐react with endothelial cells and induce damage. J Med Virol 69: 82–90. [DOI] [PubMed] [Google Scholar]
- Lin CF, Wan SW, Cheng HJ, Lei HY & Lin YS (2006) Autoimmune pathogenesis in dengue virus infection. Viral Immunol 19: 127–132. [DOI] [PubMed] [Google Scholar]
- Lin YL, Huang YL, Ma SH, Yeh CT, Chiou SY, Chen LK & Liao CL (1997) Inhibition of Japanese encephalitis virus infection by nitric oxide: antiviral effect of nitric oxide on RNA virus replication. J Virol 71: 5227–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YS, Lin CF, Lei HY, Liu HS, Yeh TM, Chen SH & Liu CC (2004) Antibody‐mediated endothelial cell damage via nitric oxide. Curr Pharm Design 10: 213–221. [DOI] [PubMed] [Google Scholar]
- Machida K, Cheng KT, Sung VM, Lee KJ, Levine AM & Lai MM (2004) Hepatitis C virus infection activates the immunologic (type II) isoform of nitric oxide synthase and thereby enhances DNA damage and mutations of cellular genes. J Virol 78: 8835–8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean A, Wei XQ, Huang FP, Al Alem UA, Chan WL & Liew FY (1998) Mice lacking inducible nitric‐oxide synthase are more susceptible to herpes simplex virus infection despite enhanced Th1 cell responses. J Gen Virol 79: 825–830. [DOI] [PubMed] [Google Scholar]
- MacMicking J, Xie QW & Nathan C (1997a) Nitric oxide and macrophage function. Annu Rev Immunol 15: 323–350. [DOI] [PubMed] [Google Scholar]
- MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK & Nathan CF (1997b) Identification of nitric oxide synthase as a protective locus against tuberculosis. P Natl Acad Sci USA 94: 5243–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannick JB, Asano K, Izumi K, Kieff E & Stamler JS (1994) Nitric oxide produced by human B lymphocytes inhibits apoptosis and Epstein–Barr virus reactivation. Cell 79: 1137–1146. [DOI] [PubMed] [Google Scholar]
- Marianneau P, Steffan AM, Royer C, Drouet MT, Jaeck D, Kirn A & Deubel V (1999) Infection of primary cultures of human Kupffer cells by Dengue virus: no viral progeny synthesis, but cytokine production is evident. J Virol 73: 5201–5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes IB, Leung B, Wei XQ, Gemmell CC & Liew FY (1998) Septic arthritis following Staphylococcus aureus infection in mice lacking inducible nitric oxide synthase. J Immunol 160: 308–315. [PubMed] [Google Scholar]
- Mendes‐Ribeiro AC, Moss MB, Siqueira MA, Moraes TL, Ellory JC, Mann GE & Brunini TM (2008) Dengue fever reactivates the l‐arginine‐nitric oxide pathway: an explanation for reduced aggregation of human platelets. Clin Exp Pharmacol P 35: 1143–1146. [DOI] [PubMed] [Google Scholar]
- Mills KHG (2004) Regulatory T cells: friend or foe in immunity to infection? Nat Rev Immunol 4: 841–855. [DOI] [PubMed] [Google Scholar]
- Misra A, Mukerjee R & Chaturvedi UC (1996a) Production of nitrite by dengue virus‐induced cytotoxic factor. Clin Exp Immunol 104: 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra A, Mukerjee R & Chaturvedi UC (1996b) Release of reactive oxygen intermediates by dengue virus‐induced macrophage cytotoxin. Int J Exp Pathol 77: 237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra A, Mukerjee R & Chaturvedi UC (1998) Respiratory burst by dengue virus‐induced cytotoxic factor. Med Prin Pract 7: 251–260. [Google Scholar]
- Moncada S & Higgs A (1993) The l‐arginine–nitric oxide pathway. New Engl J Med 329: 2002–2012. [DOI] [PubMed] [Google Scholar]
- Moncada S, Palmer RM & Higgs EA (1991) Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev 43: 109–142. [PubMed] [Google Scholar]
- Mongkolsapaya J, Dejnirattisai W & Xu XN (2003) Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat Med 9: 921–927. [DOI] [PubMed] [Google Scholar]
- Mongkolsapaya J, Duangchinda T, Dejnirattisai W, Vasanawathana S, Avirutnan P & Jairungsri A (2006) T cell responses in dengue hemorrhagic fever: are cross-reactive T cells suboptimal? J Immunol 176: 3821–3829. [DOI] [PubMed] [Google Scholar]
- Monsuez JJ, Escaut L, Teicher E, Charniot JC & Vittecoq D (2007) Cytokines in HIV‐associated cardiomyopathy. Int J Cardiol 120: 150–157. [DOI] [PubMed] [Google Scholar]
- Mukerjee R, Misra A & Chaturvedi UC (1996) Dengue virus‐induced cytotoxin releases nitrite by spleen cells. Int J Exp Pathol 77: 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukerjee R, Chaturvedi UC, Vaughn DW & Kalayanarooj S (1997) Purification and pathogenicity of the cytotoxic factor from the cases of dengue haemorrhagic fever. Curr Sci 72: 494–501. [Google Scholar]
- Neves‐Souza PC, Azeredo EL, Zagne SM, Valls‐de‐Souza R, Reis SR, Cerqueira DI, Nogueira RM & Kubelka CF (2005) Inducible nitric oxide synthase (iNOS) expression in monocytes during acute Dengue Fever in patients and during in vitro infection. BMC Infect Dis 5: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedbala W, Wei XQ, Piedrafita D, Xu D & Liew FY (1999) Effects of nitric oxide on the induction and differentiation of Th1 cells. Eur J Immunol 29: 2498–2505. [DOI] [PubMed] [Google Scholar]
- Niedbala W, Cai B & Liew FY (2006) Role of nitric oxide in the regulation of T cell functions. Ann Rheum Dis 65 (suppl 3): iii37–iii40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedbala W, Cai B, Liu H, Pitman N, Chang L & Liew FY (2007) Nitric oxide induces CD4+CD25+ Foxp3 regulatory T cells from CD4+CD25 T cells via p53, IL‐2, and OX40. P Natl Acad Sci USA 104: 15478–15483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noisakran S, Dechtawewat T, Avirutnan P, Kinoshita T, Siripanyaphinyo U, Puttikhunt C, Kasinrerk W, Malasit P & Sittisombut N (2008) Association of dengue virus NS1 protein with lipid rafts. J Gen Virol 89: 2492–2500. [DOI] [PubMed] [Google Scholar]
- O'Reilly EK & Kao CC (1998) Analysis of RNA‐dependent RNA polymerase structure and function as guided by known polymerase structures and computer predictions of secondary structure. Virology 252: 287–303. [DOI] [PubMed] [Google Scholar]
- Padalko E, Ohnishi T, Matsushita K, Sun H, Fox‐Talbot K, Bao C, Baldwin WM III & Lowenstein CJ (2004) Peroxynitrite inhibition of Coxsackievirus infection by prevention of viral RNA entry. P Natl Acad Sci USA 101: 11731–11736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persichini T, Colasanti M, Lauro GM & Ascenzi P (1998) Cysteine nitrosylation inactivates the HIV‐1 protease. Biochem Bioph Res Co 250: 575–576. [DOI] [PubMed] [Google Scholar]
- Pozner RG, Collado S, De Giusti CJ, Ure AE, Biedma ME, Romanowski V, Schattner M & Gómez RM (2008) Astrocyte response to Junín virus infection. Neurosci Lett 445: 31–35. [DOI] [PubMed] [Google Scholar]
- Proto S, Taylor JA, Chokshi S, Navaratnam N & Naoumov NV (2008) APOBEC and iNOS are not the main intracellular effectors of IFN‐gamma‐mediated inactivation of Hepatitis B virus replication. Antivir Res 78: 260–267. [DOI] [PubMed] [Google Scholar]
- Raghupathy R, Chaturvedi UC, Al‐Sayer H, Elbishbishi EA, Agarwal R, Nagar R, Nusrat H, Mustafa AS, Azizieh F & Khan MAY (1998) Elevated levels of IL‐8 in dengue hemorrhagic fever. J Med Virol 56: 280–285. [DOI] [PubMed] [Google Scholar]
- Reiss CS & Komatsu T (1998) Does nitric oxide play a critical role in viral infections? J Virol 72: 4547–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimmelzwaan GF, Baars MM, De Lijster P, Fouchier RA & Osterhaus AD (1999) Inhibition of influenza virus replication by nitric oxide. J Virol 73: 8880–8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Shibuya K, Mui A et al (1997) IGIF does not drive Th1 development but synergizes with IL‐12 for interferon‐gamma production and activates IRAK and NFkappaB. Immunity 7: 571–581. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Ortega M (1998) Nitric oxide in dengue pathology. Acta Cient Venez 49 (suppl 1): 8–12. [PubMed] [Google Scholar]
- Roy A, Jana A, Yatish K, Freidt MB, Fung YK, Martinson JA & Pahan K (2008) Reactive oxygen species up‐regulate CD11b in microglia via nitric oxide: implications for neurodegenerative diseases. Free Radical Bio Med 45: 686–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura M, Zaragoza C, McMillan A, Quick RA, Hohenadl C, Lowenstein JM & Lowenstein CJ (1999) An antiviral mechanism of nitric oxide: inhibition of a viral protease. Immunity 10: 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena SK, Singh A & Mathur A (2000) Antiviral effect of nitric oxide during Japanese encephalitis virus infection. Int J Exp Pathol 81: 165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena SK, Mathur A & Srivastava RC (2001) Induction of nitric oxide synthase during Japanese encephalitis virus infection: evidence of protective role. Arch Biochem Biophys 391: 1–7. [DOI] [PubMed] [Google Scholar]
- Sharma JN, Al‐Omran A & Parvathy SS (2007) Role of nitric oxide in inflammatory diseases. Inflammopharmacology 15: 252–259. [DOI] [PubMed] [Google Scholar]
- Soundravally R, Sankar P, Hoti SL, Selvaraj N, Bobby Z & Sridhar MG (2008) Oxidative stress induced changes in plasma protein can be a predictor of imminent severe dengue infection. Acta Trop 106: 156–161. [DOI] [PubMed] [Google Scholar]
- Stoltz M, Ahlm C, Lundkvist A & Klingström J (2007) Lambda interferon (IFN‐lambda) in serum is decreased in hantavirus‐infected patients, and in vitro‐established infection is insensitive to treatment with all IFNs and inhibits IFN‐gamma‐induced nitric oxide production. J Virol 81: 8685–8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takhampunya R, Padmanabhan R & Ubol S (2006) Antiviral action of nitric oxide on dengue virus type 2 replication. J Gen Virol 87: 3003–3011. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Nakazawa H, Okada K, Umezawa K, Fukuyama N & Koga Y (1997) Nitric oxide mediates murine cytomegalovirus‐associated pneumonitis in lungs that are free of the virus. J Clin Invest 100: 1822–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trairatvorakul P, Chongsrisawat V, Ngamvasinont D, Asawarachun D, Nantasook J & Poovorawan Y (2005) Serum nitric oxide in children with dengue infection. Asian Pac J Allergy 23: 115–119. [PubMed] [Google Scholar]
- Tripathi P, Tripathi P, Kashyap L & Singh V (2007) The role of nitric oxide in inflammatory reactions. FEMS Immunol Med Mic 51: 443–452. [DOI] [PubMed] [Google Scholar]
- Ubol S, Hiriote W, Anuntagool N & Utaisincharoen P (2001) A radical form of nitric oxide suppresses RNA synthesis of rabies virus. Virus Res 81: 125–132. [DOI] [PubMed] [Google Scholar]
- Ubol S, Chareonsirisuthigul T, Kasisith J & Klungthong C (2008a) Clinical isolates of dengue virus with distinctive susceptibility to nitric oxide radical induce differential gene responses in THP‐1 cells. Virology 376: 290–296. [DOI] [PubMed] [Google Scholar]
- Ubol S, Masrinoul P, Chaijaruwanich J, Kalayanarooj S, Charoensirisuthikul T & Kasisith J (2008b) Differences in global gene expression in peripheral blood mononuclear cells indicate a significant role of the innate responses in progression of dengue fever but not dengue hemorrhagic fever. J Infect Dis 197: 1459–1467. [DOI] [PubMed] [Google Scholar]
- Umansky V, Hehner SP, Dumont A, Hofmann TG, Schirrmacher V, Droge W & Schmitz ML (1998) Co‐stimulatory effect of nitric oxide on endothelial NF‐kappaB implies a physiological self‐amplifying mechanism. Eur J Immunol 28: 2276–2282. [DOI] [PubMed] [Google Scholar]
- Valero N, Espina LM, Añez G, Torres E & Mosquera JA (2002) Short report: increased level of serum nitric oxide in patients with dengue. Am J Trop Med Hyg 66: 762–764. [DOI] [PubMed] [Google Scholar]
- Van Den Broek M, Bachmann MF, Höhler G, Barner M, Escher R, Zinkernagel R & Kopf M (2000) IL‐4 and IL‐10 antagonize IL‐12‐mediated protection against acute vaccinia virus infection with a limited role of IFN‐γ and nitric oxide synthetase 2. J Immunol 164: 371–378. [DOI] [PubMed] [Google Scholar]
- Van Dervort AL, Yan L, Madara PJ, Cobb JP, Wesley RA, Corriveau CC, Tropea MM & Danner RL (1994) Nitric oxide regulates endotoxin‐induced TNF‐α production by human neutrophils. J Immunol 152: 4102–4109. [PubMed] [Google Scholar]
- Vodovotz Y, Bogdan C, Paik J, Xie Q & Nathan C (1993) Mechanisms of suppression of macrophage nitric oxide release by transforming growth factor β. J Exp Med 178: 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, Xu D, Muller W, Moncada S & Liew FY (1995) Altered immune responses in mice lacking inducible nitric oxide synthase. Nature 375: 408–411. [DOI] [PubMed] [Google Scholar]
- Westaway EG, Mackenzie JM & Khromykh AA (2003) Kunjin RNA replication and applications of Kunjin replicons. Adv Virus Res 59: 99–140. [DOI] [PubMed] [Google Scholar]
- Winter S, Konter J, Scheler S, Lehmann J & Fahr A (2008) Permeability changes in response to NONOate and NONOate prodrug derived nitric oxide in a blood‐brain barrier model formed by primary porcine endothelial cells. Nitric Oxide 18: 229–239. [DOI] [PubMed] [Google Scholar]
- Wu GF, Pewe L & Perlman S (2000) Coronavirus‐induced demyelination occurs in the absence of inducible nitric oxide synthase. J Virol 74: 7683–7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen YT, Chen HC, Lin YD, Shieh CC & Wu‐Hsieh BA (2008) Enhancement by tumor necrosis factor alpha of dengue virus‐induced endothelial cell production of reactive nitrogen and oxygen species is key to hemorrhage development. J Virol 82: 12312–12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitake J, Akaike T, Akuta T, Tamura F, Ogura T, Esumi H & Maeda H (2004) Nitric oxide as an endogenous mutagen for Sendai virus without antiviral activity. J Virol 78: 8709–8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki MH, Akuta T & Akaike T (2005) Nitric oxide‐induced nitrative stress involved in microbial pathogenesis. J Pharmacol Sci 98: 117–129. [DOI] [PubMed] [Google Scholar]
- Zaragoza C, Ocampo CJ, Saura M, McMillan A & Lowenstein CJ (1997) Nitric oxide inhibition of coxsackievirus replication in vitro . J Clin Invest 100: 1760–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zsengellér ZK, Ross GF, Trapnell BC, Szabó C & Whitsett JA (2001) Adenovirus infection increases iNOS and peroxynitrite production in the lung. Am J Physiol-Lung C 280: L503–L511. [DOI] [PubMed] [Google Scholar]