

Abstract
Increased vascular permeability without morphological damage to the capillary endothelium is the cardinal feature of dengue haemorrhagic fever (DHF)/dengue shock syndrome (DSS). Extensive plasma leakage in various tissue spaces and serous cavities of the body, including the pleural, pericardial and peritoneal cavities in patients with DHF, may result in profound shock. Among various mechanisms that have been considered include immune complex disease, T‐cell‐mediated, antibodies cross‐reacting with vascular endothelium, enhancing antibodies, complement and its products, various soluble mediators including cytokines, selection of virulent strains and virus virulence, but the most favoured are enhancing antibodies and memory T cells in a secondary infection resulting in cytokine tsunami. Whatever the mechanism, it ultimately targets vascular endothelium (making it a battlefield) leading to severe dengue disease. Extensive recent work has been done in vitro on endothelial cell monolayer models to understand the pathophysiology of vascular endothelium during dengue virus (DV) infection that may be translated to help understand the pathogenesis of DHF/DSS. The present review provides a broad overview of the effects of DV infection and the associated host responses contributing towards alterations in vascular endothelial cell physiology and damage that may be responsible for the DHF/DSS.
Keywords: dengue virus, dengue haemorrhagic fever, vascular endothelium, vascular permeability, cytokines, pathogenesis
Introduction
Dengue is a mosquito‐borne Flavivirus of global public health importance; an estimated 2.5 billion people in more than 100 countries are at risk of acquiring dengue viral infection with more than 50 million new infections being projected annually (Halstead, 2007). The economic importance of dengue in the developing world is also a major concern (Clark et al., 2005). Dengue virus (DV) has four serotypes (1–4) and the virion has 10 proteins, the core (C) and membrane (M) proteins, the envelope (E) glycoprotein, and seven nonstructural (NS) proteins. Anti‐E antibodies neutralize DV infectivity in vitro and protect mice from DV challenge on passive transfer and show a variable degree of cross‐reactivity among the DV serotypes and mediate antibody‐dependent enhancement of infection at nonneutralizing levels. Antibodies against NS1 can trigger complement‐mediated lysis of DV‐infected cells in vitro and protect mice from DV challenge. At the same time, these antibodies may cross‐react with endothelial cells, leading to their activation and expression of cytokine, chemokine and adhesion molecules, resulting in cell damage. NS3 protein is the main antigen that stimulates DV‐reactive CD4+ and CD8+ T‐cell cells, which produce high levels of IFN‐γ as well as tumour necrosis factor (TNF)‐α, TNF‐β and chemokines including macrophage inhibitory protein‐1β upon interaction with DV‐infected antigen‐presenting cells, and are efficient at lysis of DV‐infected cells in vitro (reviewed by Chaturvedi et al., 2006b). The virus infection can result in a broad spectrum of clinical illness ranging from a mild disease, dengue fever (DF), to a serious life‐threatening dengue haemorrhagic fever/dengue shock syndrome (DHF/DSS) in a subset of patients. The characteristic features of DHF are increased capillary permeability without morphological damage to the capillary endothelium, thrombocytopenia, altered number and functions of leucocytes, altered haemostasis and liver damage. Sudden and extensive plasma leakage in tissue spaces and various serous cavities of the body including the pleura, pericardium and peritoneal cavities and associated haemorrhages, in patients with DHF grades III and IV may result in profound shock – DSS (Table 1) – that can be fatal if not clinically managed in time (Halstead, 2004, 2002a, 2007).
Table 1.
Clinical effects of endothelial cell dysfunction during DV infection
| Organ | Effects on endothelium * | Clinical effect |
|---|---|---|
| Lung pleura | Increased vascular permeability | Pleural effusion |
| Lung alveoli | Increased vascular permeability | Haemoptysis |
| Pericardium | Increased vascular permeability | Pericardial effusion |
| Abdomen | Increased vascular permeability | Ascitis |
| Brain | Damaged blood–brain barrier | Encephalopathy |
| Gut | Increased vascular permeability | Haemoptysis, Malaena |
| Urinary tract | Increased vascular permeability | Haematuria |
| Female reproductive tract | Increased vascular permeability | Menorrhagia, bleeding per vaginum |
The release of cytokines and other mediators released by activation of memory T cells acts on endothelium, resulting mainly in opening up of junctions between endothelial cells, thus increasing permeability.
The pathogenesis of DHF is not fully understood despite extensive work carried out in the last several decades, due mainly to absence of an appropriate animal model. Various mechanisms that have been suggested include enhancing antibodies in secondary dengue, memory T‐cell‐mediated pathogenesis, immune complex disease, complement and its products, anti‐NS1 antibodies that cross‐react with vascular endothelium, cytokine tsunami and other soluble mediators such as high concentrations of soluble IL‐2 receptor, soluble CD4, soluble CD8, TNF receptors, IL‐10 and macrophage migration inhibition factor, together with possible selection of virulent virus strains (South East Asian type DV‐2) and host genetic polymorphism (Table 2). Monocyte chemoattractant CC chemokine ligand 2 (CCL2) and monocyte chemoattractant protein‐1 (MCP‐1) are proteins that reduce tight junctions of vascular endothelium cells. High concentrations of these proteins have been reported in patients with DHF/DSS (Lee et al., 2006). Whatever the mechanism, it ultimately targets endothelium (using it as a battlefield) to produce pathological lesions and severe dengue disease (Table 1). Extensive work has been done in vitro to understand the pathophysiology of vascular endothelium in various conditions using an endothelial cell monolayer model. This has led to investigations on effects of DV infection in vitro on vascular endothelium. The purpose of this review is to provide a broad overview of the effects of DV infection and host responses contributing towards alterations in endothelial cell physiology.
Table 2.
Various mechanisms proposed for immunopathogenesis of DHF
| Group | Mechanism | Effects | References * |
|---|---|---|---|
| Antibody | Enhancement of infection | Increased cellular infection and viral load | Halstead (1989, 2007) |
| Immune complex | Complement activation | Theofilopoulos et al. (1976), Malasit (1987) | |
| Cross‐reactivity with endothelial cell and coagulation proteins | Bleeding | Chungue et al. (1994), Markoff et al. (1991), Falconar (1997) | |
| Cross‐reactivity with endothelial cell and NS1 protein | Apoptosis, inflammatory activation | Lin et al. (2005) | |
| T lymphocytes | Suppressor T cells | Prevention of DHF, increased pathology | Chaturvedi et al. (2007) |
| Memory T cell | Increased pathology | Mongkolsapaya et al. (2006) | |
| Shift from Th1 to Th2 | Increased pathology | Chaturvedi et al. (2000) | |
| Cytokine Tsunami | Increased capillary permeability | Chaturvedi et al. (1999, 2000, 2001) | |
| Bystander cell lysis | Liver injury | Gagnon et al. (1999) | |
| Macrophage and dendritic cells | Cytokine tsunami | Increased capillary permeability | Chaturvedi et al. (2006a) |
| Free radicals: NO, O2 –, etc. | Increased capillary permeability | Chaturvedi et al. (2006a) | |
| Metalloprotein | Increased capillary permeability | Luplertlop et al. (2006) | |
| Vascular endothelial cells (mainly in vitro work) | eNOS, NO | Increased capillary permeability | Yuan (2006) |
| Endothelin etc. | Increased capillary permeability | Jiang et al. (1999) | |
| IL‐8, IL‐6 | Increased capillary permeability | 1998, 2000 | |
| Decreased soluble vascular endothelial growth receptor 2 | Increased capillary permeability | Srikiatkhachorn et al. (2007) | |
| Virus virulence | Asian genotype | Increased transmissibility and cellular infection. | Leitmeyer et al. (1999), Rico‐Hesse (2003) |
| Selection of virulent strains in humans and mosquitoes. | Cologna et al. (2005) | ||
| American genotype | Do not produce DHF/DSS | Watts et al. (1999) | |
| Host genetics | Polymorphism of genes | Increased or decreased predisposition for DHF | Chaturvedi et al. (2006b) |
Mainly review papers, to save space.
Effects of DV infection on endothelial cells in vivo
The clinical presentations of patients with DHF/DSS summarized in Table 1 indicate that the severe dengue disease is due to extensive involvement of endothelial cells over the entire body. Studies on the pathogenesis of DHF/DSS are greatly restricted because no animal model is available. Although providing an inadequate model, mice have been used since the studies of dengue virologist Professor Susumu Hotta to answer some of the questions later validated in human patients. This section summarizes findings on dengue patients and DV‐infected mice (briefly), with an emphasis on direct or indirect effects on endothelium.
Infection of endothelial cells with DV in patients with DHF and in mice
Electron microscopic studies of capillaries in skin biopsies from patients with DHF show marked distortion, but severely damaged vessels are not observed, and most of the endothelial junctions are intact, although swelling of stray endothelial cells is noted (Sahaphong et al., 1980). Histopathological and ultrastructural changes in skin eruptions of three DF patients showed swelling of the endothelial cells in small vessels of the papillary dermis, diapedesis of neutrophils, extravasation of erythrocytes, perivascular mononuclear cell infiltration, and degeneration of endothelial cells and neutrophils (Yu et al., 1989). Jessie et al. (2004) studied various tissues obtained from patients with DHF/DSS at postmortem, from ante‐mortem biopsies or from blood clots to demonstrate DV infection using fluorescent antibody (FA), immunohistochemistry (IHC) and in situ hybridization (ISH) and electron microscopic techniques. Viral antigens were demonstrated in Kupffer and sinusoidal endothelial cells of the liver; macrophages, multinucleated cells and reactive lymphoid cells in the spleen; macrophages and vascular endothelium in the lung; kidney tubules; and monocytes and lymphocytes in blood‐clot samples. Positive‐strand viral RNA was detected in the same IHC‐positive cells found in the spleen and blood‐clot samples. The strong, positive ISH signal in these cells indicated a high copy number of viral RNA, suggesting replication. No morphological damage to endothelial cells has been reported, although Limonta et al. (2007) have shown the presence of apoptotic cells in pulmonary, intestinal, cerebral and white blood cells in fatal cases of DHF/DSS from Cuba. In a mouse model, intradermal inoculation of high titres of DV predisposed endothelial cells to TNF‐α‐induced cell death, leading to endothelium damage and haemorrhage (Chen et al., 2007). Cardier et al. (2006) examined levels of soluble intercellular adhesion molecule and vascular cell adhesion molecule (sICAM‐1 and sVCAM‐1), and the presence of circulating endothelial cells (CECs), as evidence of vascular damage, in peripheral blood from DHF patients. They concluded that high levels of sICAM‐1 and sVCAM‐1, together with the presence of CECs, provided further evidence of endothelium damage and activation in DHF patients
Cytokine Tsunami in patients with DHF
Severe dengue disease, DHF/DSS, is the result of a ‘cytokine tsunami’ developing in the patients (Chaturvedi et al., 2007). The data summarized in Table 3 show that several cytokines are markedly increased in severe disease. The most significant finding reported for the first time from patients with DHF during a 1996 epidemic in India was the shift from the predominant helper T‐cell type 1 (Th1) response observed in cases of DF to the Th2‐type response in severe cases of DHF grade IV, a finding later confirmed in a number of studies (1999a, 2000). IL‐12 has a profound effect on the upregulation of Th1 cells while its absence shifts the balance towards Th2‐type cytokines. Elevated levels of IL‐12 are seen in patients with milder dengue illness (DF) and completely absent in patients with DHF grades III and IV (Pacsa et al., 2000). Thus, IL‐12 may play a role in preventing severe dengue disease by maintaining the Th1‐type response. Increased levels of IL‐8 in the sera and IL‐8 mRNA in peripheral‐blood mononuclear cell (PBMCs) are associated with increasing severity of DHF and death of patients. It has been suggested that the presence of high levels of IL‐8 may be a useful indicator of serious outcome for dengue illness (Raghupathy et al., 1998). Furthermore, the severity of disease and the duration of illness are correlated with levels of TGF‐β1; maximum levels of TGF‐β1 are detected in patients with DHF grade IV (Agarwal et al., 1999). Serum IL‐6 concentrations are higher in patients with DHF and DSS (1999a, 2000). Activated neutrophils release proteinases such as elastase, which may facilitate neutrophil‐mediated endothelial injury, and activate the complement, coagulation and fibrinolytic systems. As increased serum IL‐8 and elastase are found in patients with severe infections, they may have an important role in the pathogenesis of dengue infections (2000, 1998). The severity of plasma leakage in DV‐infected patients inversely correlates with plasma levels of soluble vascular endothelial growth factor A (VEGF‐A) receptors (VEGFR2). Furthermore, plasma viral load correlates with the degree of decline in plasma soluble VEGFR2 (Srikiatkhachorn et al., 2007). These results suggest that VEGF regulates vascular permeability and that binding to soluble VEGFR2 controls its activity. DV‐induced changes in surface and soluble VEGFR2 expression may be an important mechanism of plasma leakage in DHF.
Table 3.
Cytokine levels in patients with dengue *
| Cytokines | DF | DHF |
|---|---|---|
| IL‐1β | No change | No change |
| IL‐2 | Marked increase | Increase |
| IL‐4 | Decrease | Marked increase |
| IL‐6 | Increase | Marked increase |
| IL‐8 | Marked increase | Marked increase |
| IL‐10 | Decrease | Marked increase |
| IL‐12 | Marked increase | Decrease |
| IL‐13 | Decrease | Marked increase |
| IL‐18 | Increase | Marked increase |
| TNF‐α | Marked increase | Marked increase |
| IFN‐γ | Marked increase | Increase |
| Transforming growth factor‐β | Decrease | Marked increase |
| Cytotoxic factor | Increase | Marked increase |
*Modified from Chaturvedi et al. (2005).
Cytotoxic factor (CF) in patients with dengue infection
A unique cytokine, CF, is produced by CD4+ T cells during DV infection of mice and humans. The amino‐terminal sequence of CF has no homology with any known proteins. The majority of patients with dengue show the presence of CF in their sera, with peak amounts in the most severe cases of DHF grade IV (Chaturvedi et al., 1999b). CF selectively kills CD4+ T cells and H‐2A− macrophages (Mφ) and induces H‐2A+ Mφ to produce another cytokine, the Mφ cytotoxin (CF2) that amplifies the effect of CF (Fig. 1). CF was detected in about 70% of the stored sera from patient with dengue at AFRIMS, Bangkok (Mukerjee et al., 1997), and later at Lucknow, India, during the first extensive epidemic of DHF in India in 1996. In the majority of patients with dengue, sera was positive for CF, with peak levels in the most severe cases of DHF grade IV (Chaturvedi et al., 1999b). CF was purified from the pooled sera of the DHF patients. On intravenous inoculation into mice CF increased capillary permeability and damaged the blood–brain barrier, indicating the role of CF in pathogenicity (Mukerjee et al., 1997). CF and CF2 may be pathogenesis‐related proteins, capable of reproducing DHF‐like pathological lesions in mice (Fig. 1), such as increased capillary permeability, cerebral oedema and blood leukocyte changes (Chaturvedi et al., 1997). CF is produced in ex vivo cultures of CD4+ T cells obtained from peripheral blood of patients with severe dengue disease (Agarwal et al., 1998). The production of CF precedes the clinical illness in mice and humans. The DHF‐like pathological lesions produced by CF/CF2 can be prevented by pretreatment of mice with anti‐CF antibodies. Furthermore, active immunization of mice using CF as antigen protects them against subsequent challenge with CF. Challenge of such mice with a lethal intracerebral dose of DV results in death without the appearance of clinical symptoms of the disease (Chaturvedi et al., 1994). On the basis of data obtained from experimental mice and patients, we proposed that DV replicates in Mφ and induces CD4+ T cells to produce CF, which stimulates Mφ to produce CF2. CF/CF2 induce Mφ to produce free radicals, nitrite, reactive oxygen and peroxynitrite (2000, 2006a, 1998). The free radicals, besides killing the target cells via apoptosis, also directly upregulate production of proinflammatory cytokines IL‐1β, TNF‐α, IL‐8 and hydrogen peroxide in Mφ. The change in relative levels of IL‐12 and TGF‐β shifts a Th1‐dominant response to a Th2‐biased response, resulting in an exacerbation of dengue disease. Vascular permeability is increased due to the combined effect of a cytokine tsunami, release of histamine, free radicals, the products of the complement pathway, etc. Thus, the key player appears to be CF/CF2, but their activity is regulated by CF‐autoantibodies generated (Fig. 1) in patients with dengue disease (Chaturvedi et al., 2001).
Figure 1.
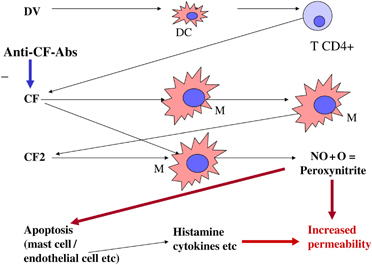
Schematic presentation of CF cascade leading to increased vascular permeability.
Role of T lymphocytes in patients with DHF
In an excellent review, Stephenson (2005) discussed the role of T cells in DHF. DHF occurs more frequently in patients with secondary infection with a heterologous DV serotype. This is due to increased uptake by macrophages of the DV‐enhancing antibody complex, resulting in increased replication. The presentation of viral peptides complexed with human leucocyte antigen (HLA) molecules to memory T cells leads to proliferation and the production of proinflammatory cytokines that can directly affect vascular endothelial cells, resulting in plasma leakage. The rapid induction of cross‐reactive memory T cells (mainly NS3‐specific) during a secondary infection would be consistent with the increased incidence of DHF and DSS. A DV‐specific memory lymphocyte response has been detected even 20 years following a primary DV infection (Sierra et al., 2002). The cascade of memory T‐cell activation and a ‘tsunami’ of inflammatory cytokines and other chemical mediators released mainly from T cells, monocytes/macrophages and endothelial cells ultimately cause an increase in vascular permeability. The interplay of different inflammatory cytokines induced during DV infection plays a role in either protection or increased disease severity (Chaturvedi et al., 2000). Suppressor T cells were demonstrated in DV‐infected mice that were considered to protect against development of DHF by suppressing enhancing antibody production (Chaturvedi et al., 1984) but could increase severity by producing IL‐10/TGF‐β (Chaturvedi et al., 2007). This remains to be investigated in human patients. A shift of the Th1 response to Th2 response in patients with DF results in a severe dengue disease, DHF/DSS (Chaturvedi et al., 1999a). Mongkolsapaya et al. (2003) have reported that during acute infection in children, few dengue‐responsive CD8+ T cells are recovered which show an activated phenotype and are undergoing apoptosis. These DV‐specific T cells have low affinity for the infecting virus and higher affinity for previously encountered strains. Cross‐reactive DV‐specific T cells show high cytokine production, which may contribute to vascular leakage (Mongkolsapaya et al., 2006). Mangada & Rothman (2005) have shown that TNF‐α‐/IFN‐γ‐positive CD4+ T‐cell ratios varies between peptides, but the ratio of the sum of responses was highest against heterologous serotypes. These results demonstrate epitope sequence‐specific differences in T‐cell effector function that may play a role in the immunopathogenesis of DHF. Profound T‐cell activation and death may contribute to the systemic disturbances leading to DHF, and original antigenic sin in the T‐cell responses may suppress or delay viral elimination, leading to higher viral loads and increased immunopathology. Appanna et al. (2007) investigated DV‐specific T‐cell responses to 32 peptide antigens from the structural and nonstructural regions from a DV isolate and found that predominantly NS3 422‐431 are important T‐cell targets. The findings also reveal high frequencies of T cells that recognize both serotype‐specific and cross‐reactive antigens in patients with DHF. Immunizing mice with different sequences of DV serotypes and measuring the frequency of peptide‐specific T cells after infection have supported this view. On adoptive transfer the memory T cells respond preferentially to the secondary infection, indicating that cross‐reactive T cells from a primary infection alter the immune response during a heterologous secondary infection (Beaumier et al., 2008).
Pathogenesis of DHF in infants
Age is an important variable in the outcome of secondary DV‐2 infections. DHF/DSS fatality and hospitalization rates are highest in young infants and the elderly. The risk that a child will die during a secondary DV‐2 infection is nearly 15‐fold higher than that in adults (Guzmán et al., 2002). Acute illness blood samples from infants with DHF/DSS show overproduction of both proinflammatory and anti‐inflammatory cytokines (Nguyen et al., 2004). Evidence suggests that enhancing and cross‐reactive neutralizing antibodies regulate dengue epidemics and disease severity. DHF/DSS in infants under 1 year of age have been reported from different countries. Curiously, all the infants had primary DV infection. Limited work has been done to explain this phenomenon in infants as DHF/DSS occurs mostly following secondary dengue infections. Classic DHF/DSS arises during initial dengue infections in infants with low circulating amounts of maternal dengue antibodies, an observation that precludes an exclusive causal role for secondary T‐cell responses (Halstead, 2007). PBMC apoptosis and plasma soluble levels of CD95, a mediator of apoptosis, were determined in sequential samples from children participating in a prospective study of DV infections by Myint et al. (2006). The level of PBMC apoptosis correlated with dengue disease severity. Apoptosis appears to be involved in modulation of the innate and adaptive immune responses to DV infection. Halstead et al. (2002b) have explained this phenomenon by comparison with a coronavirus, the feline infectious peritonitis virus (FIPV) infection in cats. Like the feline coronaviruses, DV also circulates as two biotypes: DV‐2 American genotype does not cause DHF/DSS, while DV‐2 SE Asian genotype does (Watts et al., 1999). Like feline coronaviruses, the severity of disease with the two DV biotypes may be regulated by cross‐reactive antibodies (Kochel et al., 2002). More work on viral–antibody interactions at the structural level is needed to clarify early pathogenesis events in DHF/DSS in infants. In an excellent review, Halstead (2007) discussed in detail the differences between DHF in adults and that in infants and the pathogenesis in the two groups of patients.
Effects of DV infection on endothelial cells in vitro
Endothelial cell monolayers have been used widely to study the pathophysiological changes that occur during DV infection to answer questions raised in patients with DHF/DSS. Most of the mechanistic information has come from in vitro experiments and remains to be translated to human patients. However, these in vitro experiments have generated useful knowledge about the mechanisms that may be occurring intracellularly. In this section, the relevant findings of such experiments performed on DV‐infected endothelial monolayers are described, with emphasis on the direct or indirect effects on endothelium.
Vascular endothelium
The vascular endothelial cells lining the inner lumen of blood vessels and capillaries constitute the primary anatomical blood–tissue barrier. The vascular endothelial cells are highly organized, physiologically specialized and form the major gatekeepers that control tissue nutrient flux, microcirculatory dynamics and permeability of macromolecules. Their role in immune functions and haemostasis is of equal importance. However, the integrity of the barrier and endothelial cell functions in the microcirculatory beds is not constant but is physiologically regulated by many factors such as electrolyte balance, metabolic status of the tissue, hormones, and neurotransmitters, and most importantly the pathophysiology of vascular disorders (Cines et al., 1998). Endothelial cells can dramatically alter their permeability in response to immune responses, especially to cytokines such TNF‐α, IFN‐γ, IL‐6, IL‐8 and other proinflammatory cytokines (Crone, 1986). Confluent monolayers of the human dermal microvascular endothelial cell line HMEC‐1 etc. are utilized as an experimental model to study the cellular responses induced by DV. Infected monolayers show increased permeability without evident cytopathic effects on the monolayer. The ontogeny of the vascular endothelial cells occurs from the blood islands of the primitive yolk sac (Risau & Flamme, 1995) and later as angioblasts from the cardiac crescent of the mesodermal plates. Further development, differentiation and tissue‐specific reorganization of these cells occur into organ‐specific vascular beds through complicated developmental pathways that include growth factors such as VEGF, factors like tie‐1, endothelins and vascular active peptides that interact through complex molecular signalling pathways (reviewed by Cines et al., 1998) leading to the final organization of the vascular tree. Recent studies have used transgenic mouse models and an in vitro model of embryonic stem cell differentiation, which have contributed to the current understanding of the cellular and molecular pathways regulating hemangioblast development and differentiation. Haematopoietic and endothelial cells develop from mesoderm via a transitional progenitor known as the hemangioblast. Flk‐1, a receptor tyrosine kinase, and Scl, a basic helix–loop–helix transcription factor, are two critical molecules functioning in this process. Recent studies have shown that Flk‐1‐expressing mesoderm contributes to the circulatory system, including haematopoietic, endothelial, smooth muscle, skeletal muscle and cardiac muscle cells. Furthermore, hemangioblast specification within Flk‐1‐expressing mesoderm is regulated by Scl expression (2005, 2007). The endothelial progenitor cells (EPCs) found in the bone marrow and peripheral blood are incorporated into injured vessels and become mature endothelial cells during re‐endothelialization and neovascularization processes. These cells express several surface markers, including CD34 and CD133 antigens, and may promote local angiogenesis by secreting angiogenic growth factors in a paracrine manner (Miller‐Kasprzak & Jagodziñski, 2007).
In vitro alterations in endothelial cell biology by dengue viruses
Endothelial cells are permissive to DV infection in vitro. A significant amount of research has been carried out on pure endothelial cell cultures to study pathophysiology during DV infection in vitro. Infection of endothelial cells by DV has been studied but has resulted in conflicting findings; the role of endothelial cells in dengue disease pathogenesis remains incompletely understood. Vascular endothelial cell dysfunction may be caused by various stimuli, including infectious pathogens, cytotoxic reagents and pathophysiological mechanisms mediated by immune responses.
Replication of DV in endothelial cells from human umbilical cord vein (HUVEC) was demonstrated by virus titres and immunofluorescent antibody studies (Bunyaratvej et al., 1997). Replication of DV in HUVEC is inhibited by ribavirin, an antiviral synthetic guanosine analogue (Huang et al., 2000). Talavera et al. (2004) reported that 40 % of the HMEC‐1 cells expressed DV proteins. Several studies have shown susceptibility of endothelial cells to DV infection with induction of cytokine production, apoptosis of target cells and altered transcriptional factor activity (Avirutnan et al., 1998). The effects of DV infection on gene expression in HUVECs have been studied by Warke et al. (2003) using microarray and differential display analysis. They have showed evidence of upregulation of 269 genes that broadly included cellular defence pathway, immune and inflammatory response genes while a downregulated cluster of 169 genes mostly included cytoskeletal and membrane‐associated genes. Furthermore, recombinant TNF‐related apoptosis‐inducing ligand (rTRAIL) inhibited DV titres in DV‐infected dendritic cells (DCs) by an apoptosis‐independent mechanism (Warke et al. (2008). These data suggest that TRAIL plays an important role in the antiviral response to DV infection and may be a candidate for antiviral interventions against DV. The data from this study also showed significant activation of the myxovirus pathway (MxA), which has been characterized as a cellular antiviral response. In addition, other genes such as PLSCR1 associated with phosphotidylserine metabolism in cells are also affected, suggesting possible interference in basic signal‐transduction pathways by DV infection (Wiedmer et al., 2000). Kinglsey et al. (2006), using high density arrays, showed a more heterogeneous and differential gene expression in endothelial cell line ECV304 in response to DV infection that involved downregulation of apoptotic pathways. However, more consistent and possible physiologically relevant data come from recent studies that have shown significant alterations in the gene expression of haemostatically important molecules such as thrombomodulin, tissue plasminogen activator (tPA) and tissue factor inhibitor in DV‐infected endothelium (Jiang et al., 2007). Alterations in the β‐integrin expression after DV infection of HMEC‐1 might provide clues towards understanding the origin of the capillary barrier ‘breach’ that results in leakage given that integrands are crucial for cellular adherence (Zhang et al., 2007). DV infection induces the production of tPA but not of plasminogen activator inhibitor 1 in human endothelial cells, which is regulated by IL‐6 (Huang et al., 2003). Activation and damage of HMEC could be induced in vitro by inflammatory mediators present in the sera of dengue patients, as indicated by an increase in ICAM‐1 expression (Cardier et al., 2005). Recombinant DV envelope glycoprotein binds to three proteins on the surface of ECV304 cells. This virus–cell interaction may be associated with the receptor complex specific for DV infection of endothelial cells (Wei et al., 2003). Dewi et al. (2008) have analysed the interaction among PBMCs, DV and HUVEC in vitro by studying transendothelial electrical resistance (TEER) and transalbumin permeability. The results indicate that PBMCs increased the permeability of DV‐2‐infected HUVECs and that the increased permeability was concomitant with morphological change and a decrease in VE‐cadherin expression. The results suggest that functional impairment of the DV‐2‐infected HUVEC monolayer was caused by interaction with PBMCs.
Endothelial cell activation and production of IL‐8 during DV infection
Anderson et al. (1997) reported that HUVECs are activated when exposed to culture fluids (containing TNF‐α) from DV‐infected PBMCs. Activation is strongest for endothelial cell expression of VCAM‐1 and ICAM‐1. In contrast, activation of endothelial cell E‐selectin expression appears to be more transient. Endothelial cells inoculated directly with DV or with anti‐DV antibody combinations are poorly infectable. Among the proinflammatory cytokines and their mRNAs measured (IL‐6, IL‐1β, IL‐8, TNF‐α), in the culture supernatant of DV‐infected primary human monocytes, IL‐8 shows the greatest change. Similarly, two cell lines, 293T (a human epithelial cell line) and ECV304 (an endothelial cell line), respond to DV infection by increasing IL‐8 synthesis. IL‐8 produced by infected monocytes, endothelial or other epithelial cells is associated with the hyperacetylation of histones bound to the IL‐8 promoter in addition to the activation of transcription by NF‐κB (Bosch et al., 2002). Infection of human endothelial cells with DV induces the transcriptional upregulation and secretion of RANTES and IL‐8 and apoptosis of cells (Avirutnan et al., 1998). DV infection of HepG2 and primary DCs induces the production of IL‐8, RANTES, MIP‐1α and MIP‐1β, whereas only IL‐8 and RANTES are induced in HEK293 cells. Transfection of a plasmid expressing NS5 or a DV replicon induces IL‐8 gene expression and secretion while RANTES expression is not induced. Furthermore, IL‐8 induction by NS5 is principally through CAAT/enhancer binding protein, whereas DV infection also induces NF‐κB (Medin et al., 2005). Treatment with anti‐inflammatory drugs has variable effects on IL‐8 secretion in different cell types during DV infection (Medin & Rothman, 2006). The findings of Talavera et al. (2004) on confluent monolayers of the human dermal microvascular endothelial cell line HMEC‐1 suggest that IL‐8 production by DV‐infected monolayers contributes to the virus‐induced effect on the cytoskeleton and tight junctions and thereby modifies transendothelial permeability. In vitro studies show that mouse primary microvascular endothelial cells are susceptible to DV but that TNF‐α enhances DV‐induced apoptosis.
Effects of DV infection on tight junction complex of endothelium
The physiological basis of endothelial integrity lies in the presence of intercellular junctions known as tight junctions complexes (TJCs) that allow vectorial transport of solutes and other biological macromolecules (Blum et al., 1997). The TJCs also regulate the overall permeability of endothelial barrier functions. The cellular basis of this has been studied extensively and has been attributed to dynamic rearrangements of the cytoskeleton and its association with TJCs primarily through actin. Alterations in the sealing capacity of the junctions through modifications of the TJ can lead to enhanced permeability (Cereijido et al., 1989). Although the detailed molecular biology of the TJ is beyond the scope of this review, the most important molecules that make the TJ relevant to vascular pathophysiology include the zonula proteins (ZO 1‐3), occludins and claudins (Bazzoni & Dijana, 2004). All three protein families are integral to normal functioning of the TJC in maintaining endothelial permeability. They function at different levels from being purely structural scaffolds to interacting with complex signalling pathways within the cell, such as the ZO proteins that are members of membrane‐associated guanyl kinase family (MUGK). Increased production of IL‐8 occurs in DV‐infected primary human monocytes and in human epithelial and endothelial cell lines (Bosch et al., 2002). The findings of Talavera et al. (2004) suggest that IL‐8 production by infected monolayers contributes to the virus‐induced effect on the cytoskeleton and TJs and thereby modifies transendothelial permeability. Lee et al. (2006) reported that MCP‐1 expression is induced in DV2‐infected monocytes and lymphocytes, but not in liver or endothelial cells. Exposing monolayers of HUVECs to recombinant human MCP‐1 (rhMCP‐1) or to the culture supernatant of DV2‐infected human monocytes increased the vascular permeability of the cells. This was due to disrupted TJ protein ZO‐1 of HUVECs that was partially mediated by MCP‐1. Recently, Meertens et al. (2008) have reported that the TJ proteins claudins‐1, ‐6 and ‐9 function equally well as entry cofactors in endothelial cells for hepatitis C virus, another member of Flaviviridae.
Dengue viruses and changes in endothelium permeability
Dewi et al. (2004) studied the effect of proinflammatory cytokines and DV on the transendothelial electrical resistance and permeability alterations. They revealed susceptibility of endothelial cells to DV and also significant increases in permeability and electrical resistance of the cells after exposure to TNF‐α.
However, because the plasma leakage in DHF cases occurs at the time of defervesence, it becomes important to examine the profile of proinflammatory cytokines in a time‐course manner. Jose et al. (2005) showed differential effects of the sera from DHF cases on the activation of HMEC‐1, according to the day of disease evolution. Sera from the day of defervescence had the strongest activating potential in relation to later stages of disease and correlated with the risk for DHF. This study also showed the important but not essential role of TNF‐α in possibly affecting the permeability of endothelial cells in DHF that was further supported from a recent study using a mouse model of dengue (Chen et al., 2007). Although DV can infect different cell types, cells of the monocytic lineage such as Langerhans cells and interstitial DCs are thought to be the primary targets of DV. Immature monocyte‐derived DCs are also susceptible to DV with subsequent activation and release of vasoactive cytokines such as IL‐8 and TNF (Juffrie et al., 2000). An injury causes transendothelial migration of the DCs and expression of matrix metalloproteinases that can have deleterious effects of endothelial cell integrity (Asahi et al., 2001). In vitro, DV suppresses soluble VEGFR2 production by endothelial cells but upregulates surface VEGFR2 expression and promotes response to VEGF stimulation (Srikiatkhachorn et al., 2007). Luplertlop et al. (2006) showed that DV‐infected DCs overproduced soluble gelatinolytic matrix metalloproteinase MM9 and MMP2 with loss of the platelet endothelial adhesion molecule (PECAM) expression and increase in vascular permeability. These studies have initiated the need to examine in more detail the possible role of host factors in the origin of dengue‐associated vascular pathophysiology. Supernatants from DV‐infected monocyte‐derived macrophages contain factors that increase HUVEC permeability, but this is not accompanied by endothelial cell infection or directly correlated with release of DV or TNF‐α from monocyte‐derived macrophages (Carr et al., 2003). It was found that the increased permeability and disrupted TJs of HUVECs are affected through a mechanism partially dependent on MCP‐1, which is secreted by DV‐infected monocytes and lymphocytes (Lee et al., 2006). DV‐infected immature DCs overproduce soluble gelatinolytic MMP‐9 and to a lesser extent MMP‐2, which enhances endothelial permeability (Fig. 2). This permeability was associated with a loss of expression of PECAM‐1 and vascular endothelium–cadherin cell adhesion molecules and redistribution of F‐actin fibres. These in vitro observations have been confirmed in an in vivo vascular‐leakage mouse model (Luplertlop et al., 2006). Jiang et al. (2007) have shown that DV could promote the expression of thrombomodulin in cultured HUVEC, thus increasing anticoagulation, and enhance tPA activity, increasing risk of haemorrhage. Zhang et al. (2007) reported that DV infection induces upregulated expression of β3‐integrin, which is required for DV entry into human HMEC‐1. Permeability alteration of microvascular endothelium is a factor in the plasma leakage produced by DV infection, and β3‐integrin plays central roles in maintaining capillary integrity and regulating vascular permeability.
Figure 2.

Schematic representation of the possible interaction between DVs, cytokines and host factors leading to endothelial cell pathology.
Role of nitric oxide (NO) generated during DV infection
NO, generated from l‐arginine by NO synthase (NOS), is a multifunctional molecule that is involved in cytotoxic as well as cytoprotective processes. Its synthesis is highly regulated by the cell because an alteration in NO production is associated with a variety of pathological effects, such as vasodilatation, inflammation, thrombosis, immune response and neurotransmission. Generation of NO may result in either initiation or suppression of apoptosis. The involvement of NO in apoptosis induction has been reported in different cell systems, although the mechanisms are not fully understood. Rodriguez‐Ortega (1998) discussed the evidence that links NO with the pathology of the severe dengue disease. Macrophage lineage cells are professional NO producers. NO produced by them downregulates replication of DV. CF/CF2 induces production of NO2 – in the spleen cells in a Ca2+‐dependent manner, which may be a mechanism of target cell killing (Mukerjee et al., 1996). CF2 also induces production of superoxide anion (O2 –) and hydrogen peroxide (H2O2) by the spleen cells of mice in vitro and in vivo and the killing (Fig. 2) may possibly be by generation of peroxynitrite (1996a, 1996b, 1998). Neves‐Souza et al. (2005) reported that activation of DV‐infected monocytes is based on induction of iNOS both in vivo and in vitro.
Role of cross‐reacting anti‐DV NS1 antibody
Lin et al. (2002) have demonstrated that antibodies against DV NS1 generated in mice cross‐react with human and mouse vascular endothelium and platelets. After binding, mouse anti‐NS1 antibodies induce endothelial cell apoptosis in a caspase‐dependent manner. Inducible NOS expression shows a time‐ and dose‐dependent correlation with NO production. Endothelial cell apoptosis, characterized by exposure of phosphatidylserine on the cell surface and nuclear DNA fragmentation, is blocked by treatment with the NOS inhibitor N ω‐nitro‐l‐arginine methyl ester. Further studies have demonstrated that the expression of Bcl‐2 and Bcl‐xL decreased in both mRNA and protein levels, whereas p53 and Bax increased after anti‐NS1 treatment. Cytochrome c release is also observed. All of these effects could be inhibited by N ω‐nitro‐l‐arginine methyl ester. Taking these data together, anti‐NS1 antibodies act as autoantibodies that cross‐react with noninfected endothelial cells and trigger the intracellular signalling leading to the production of NO and to apoptosis (2002, 2004). This has been confirmed in patients with DHF/DSS where antibodies against NS1 present in patient sera cross‐react with endothelial cells and induce apoptosis via a caspase‐dependent pathway and cause cell lysis in the presence of complement. DHF/DSS patient sera show higher percentages of cytotoxicity than DF patient sera (Lin et al., 2003). Human platelets adhere to DV‐stimulated HUVEC and this interaction could contribute to the thrombocytopenia observed during infection (Krishnamurti et al., 2002). Complement activation was proposed to be responsible for DHF, but the cause of complement activation was not known. Avirutnan et al. (2006) have reported that complement activation mediated by NS1 leads to local and systemic generation of anaphylatoxins and SC5b‐9, which may contribute to the pathogenesis of the vascular leakage that occurs in patients with DHF/DSS. Antibodies directed against DV NS1 has been shown to cross‐react with human platelets and endothelial cells leading to endothelial cell damage and inflammatory activation (Lin et al., 2006).
Role of endothelin in endothelium permeability
Endothelial cells produce a number of factors that maintain their physiological activity. The findings summarized in Table 4 show that dysfunction of these factors during DV infections increases permeability. Endothelin (ET‐1) is produced by endothelial cells from a precursor, big‐ET‐1, by endothelin‐converting enzyme (ECE) found on the endothelial cell membrane. Thrombin, cytokines, reactive oxygen species, etc., stimulate the vascular endothelium to produce and release of ET‐1. Release of ET‐1 is inhibited by prostacyclin and nitric oxide. Binding of ET‐1 to endothelial ETb receptors stimulates the formation of NO. In the absence of smooth muscle endothelin receptor stimulation, this NO causes vasodilation (reviewed by Iglarz & Clozel, 2007). DV infection of HUVECs inhibits the production of endothelin 1 (ET‐1) and prostacyclin 2 (PGI2), thus affecting their normal functions of secretion of vasoactive substances, resulting in increased vascular permeability (Fig. 3) and impairment of homeostasis and blood coagulation (Jiang et al., 1999).
Table 4.
Effects of DV infection on vascular permeability through factors secreted by the endothelial cells
| Factors secreted by endothelial cells | Functions | Effects of DV infection |
|---|---|---|
| Prostacyclin | Vasodilation, inhibits platelet aggregation | Inhibited |
| Nitric oxide | Vasodilation, inhibits platelet adhesion and aggregation | Increased |
| Endothelin‐1 (ET‐1) | Vasoconstriction | Inhibited |
| Tissue plasminogen activator (tPA) | Regulates fibrinolysis | Increased |
| Thrombomodulin | Anticoagulant activity | Increased |
| von Willebrand Factor | Promotes platelet adhesion and activation of blood coagulation | Increased |
Figure 3.

Schematic representation of possible molecular events leading to altered permeability changes in endothelial cells.
Conclusions
Plasma leakage is a hallmark of DHF/DSS, characterized by a sudden and transient localized plasma leakage resulting in fluid accumulation in body cavities. Plasma leakage generally lasts no more than 48 h and is usually followed by rapid, complete recovery. Presence of DV has been detected in endothelial cells but it does not cause morphological damage, and the vascular leakage is mediated indirectly by various host factor cascades induced by the virus. Absence of tissue inflammation suggests a transient change in factors that regulate vascular permeability. In spite of a large number of mechanisms proposed from time to time, as discussed above, the precise mechanism of the pathogenesis is not clearly understood. As the ultimate target is the endothelial cells, a large body of data has accumulated from the in vitro studies on pathophysiology of endothelial cell monolayers during DV infection. As in mice, the endothelial cell monolayers are not the true model of the human disease, and only point to various possibilities that may operate in vivo. Efforts have been made to develop animal models but success has been limited. More intense studies are required to translate the knowledge obtained from animal models and in vitro experiments to human pathology.
Statement
U.C.C. has now retired.
Editor: Willem van Leeuwen
References
- Agarwal R, Chaturvedi UC, Misra A, Mukerjee R, Kapoor S, Nagar R, Tandon R & Mathur A (1998) Production of cytotoxic factor by peripheral blood mononuclear cells (PBMC) of the cases of dengue haemorrhagic fever. Clin Exp Immunol 112: 340–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal R, Elbishbishi EA, Chaturvedi UC, Nagar R & Mustafa AS (1999) Profile of transforming growth factor‐beta1 in patients with dengue haemorrhagic fever. Int J Exp Path 80: 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R, Wang S, Osiowy C & Issekutz AC (1997) Activation of endothelial cells via antibody‐enhanced dengue virus infection of peripheral blood monocytes. J Virol 71: 4226–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appanna R, Huat TL, See LL, Tan PL, Vadivelu J & Devi S (2007) Cross‐reactive T‐cell responses to the nonstructural regions of dengue viruses among dengue fever and dengue hemorrhagic fever patients in Malaysia. Clin Vaccine Immunol 14: 969–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME & Lo EH (2001) Effects of matrix metalloproteinase 9 gene knockout on the proteolysis of blood brain barrier and white matter components after cerebral ischaemia. J Neurosci 21: 7724–7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avirutnan P, Malasit P, Seliger B, Bhakdi S & Husmann M (1998) Dengue virus infection of human endothelial cells leads to chemokine production, complement activation, and apoptosis. J Immunol 161: 6338–6346. [PubMed] [Google Scholar]
- Avirutnan P, Punyadee N, Noisakran S et al (2006) Vascular leakage in severe dengue virus infections: a potential role for the nonstructural viral protein NS1 and complement. J Infect Dis 193: 1078–1088. [DOI] [PubMed] [Google Scholar]
- Bazzoni G & Dijana E (2004) Endothelial cell–cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 84: 860–891. [DOI] [PubMed] [Google Scholar]
- Beaumier CM, Mathew A, Bashyam HS & Rothman AL (2008) Cross‐reactive memory CD8(+) T cells alter the immune response to heterologous secondary Dengue virus infections in mice in a sequence‐specific manner. J Infect Dis 197: 608–617. [DOI] [PubMed] [Google Scholar]
- Blum MS, Toninelli E, Anderson JM, Balda MS, Zhou J, O'Donnell L, Pardi R & Bender JR (1997) Cytoskeletal rearrangements mediate human microvascular endothelial tight junction modulation by cytokines. Am J Physiol 273: H286–H294. [DOI] [PubMed] [Google Scholar]
- Bosch I, Xhaja K, Estevez L, Raines G, Melichar H, Warke RV, Fournier MV, Ennis FA & Rothman AL (2002) Increased production of interleukin‐8 in primary human monocytes and in human epithelial and endothelial cell lines after dengue virus challenge. J Virol 76: 5588–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunyaratvej A, Butthep P, Yoksan S & Bhamarapravati N (1997) Dengue viruses induce cell proliferation and morphological changes of endothelial cells. Southeast Asian J Trop Med Public Health 28 (suppl 3), 32–37. [PubMed] [Google Scholar]
- Cardier JE, Mariño E, Romano E, Taylor P, Liprandi F, Bosch N & Rothman AL (2005) Proinflammatory factors present in sera from patients with acute dengue infection induce activation and apoptosis of human microvascular endothelial cells: possible role of TNF-alpha in endothelial cell damage in dengue. Cytokine 30: 359–365. [DOI] [PubMed] [Google Scholar]
- Cardier JE, Rivas B, Romano E, Rothman AL, Perez‐Perez C, Ochoa M, Caceres AM, Cardier M, Guevara N & Giovannetti R (2006) Evidence of vascular damage in dengue disease: demonstration of high levels of soluble cell adhesion molecules and circulating endothelial cells. Endothelium 13: 335–340. [DOI] [PubMed] [Google Scholar]
- Carr JM, Hocking H, Bunting K, Wright PJ, Davidson A, Gamble J, Burrell CJ & Li P (2003) Supernatants from dengue virus type‐2 infected macrophages induce permeability changes in endothelial cell monolayers. J Med Virol 69: 521–528. [DOI] [PubMed] [Google Scholar]
- Cereijido M, Gonzalez‐mariscal L & Contreras G (1989) Tight‐junction: barrier between higher organisms and environment. News Physiol 4: 72–75. [Google Scholar]
- Chaturvedi UC (1984) Dengue virus‐induced suppressor pathway. Curr Sci 53: 971–976. [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi UC & Shrivastava R (2004) Dengue haemorrhagic fever: a global challenge. Indian J Med Microbiol 22: 5–6. [PubMed] [Google Scholar]
- Chaturvedi UC, Mukerjee R & Dhawan R (1994) Active immunization by a dengue virus‐induced cytokine. Clin Exp Immunol 96: 202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi UC, Dhawan R & Mukerjee R (1997) Immunosuppression and cytotoxicity of dengue infection in the mouse model Dengue and dengue haemorrhagic Fever (Gubler DJ. & Kuno G, eds), pp. 291–312. Oxon, UK CAB International Press, Wallingford. [Google Scholar]
- Chaturvedi UC, Raghupathy R, Pacsa AS et al (1999a) Shift from a Th1‐type response to Th2‐type in Dengue haemorrhagic fever. Curr Sci 76: 63–69. [Google Scholar]
- Chaturvedi UC, Agarwal R, Misra A, Mukerjee R, Kapoor S & Nagar R (1999b) Cytotoxic factor in dengue haemorrhagic fever. Med Principles Pract 8: 26–31. [Google Scholar]
- Chaturvedi UC, Agarwal R, Elbishbishi EA & Mustafa AS (2000) Cytokine cascade in Dengue haemorrhagic fever: implications for pathogenesis. FEMS Immunol Med Microbiol 28: 183–188. [DOI] [PubMed] [Google Scholar]
- Chaturvedi UC, Elbishbishi EA, Agarwal R & Mustafa AS (2001) Cytotoxic factor‐autoantibodies: possible role in the pathogenesis of dengue haemorrhagic fever. FEMS Immunol Med Microbiol 30: 181–186. [DOI] [PubMed] [Google Scholar]
- Chaturvedi UC, Shrivastava R & Nagar R (2005) Dengue vaccines: problems and prospects. Indian J Med Res 121: 639–652. [PubMed] [Google Scholar]
- Chaturvedi UC, Nagar R & Shrivastava R (2006a) Macrophage and dengue virus: friend or foe? Indian J Med Res 124: 23–40. [PubMed] [Google Scholar]
- Chaturvedi UC, Nagar R & Shrivastava R (2006b) Dengue and dengue haemorrhagic fever: implications of host genetics. FEMS Immunol Med Microbiol 47: 155–166. [DOI] [PubMed] [Google Scholar]
- Chaturvedi UC, Shrivastava R, Tripathi RK & Nagar R (2007) Dengue virus‐specific suppressor T cells: current perspectives. FEMS Immunol Med Microbiol 50: 285–299. [DOI] [PubMed] [Google Scholar]
- Chen HC, Hofman FM, Kung JT, Lin YD & Wu‐Hsieh BA (2007) Both virus and tumor necrosis factor alpha are critical for endothelium damage in a mouse model of dengue virus‐induced hemorrhage. J Virol 81: 5518–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chungue E, Poli L, Roche C, Gestas P, Glaziou P & Markoff LJ (1994) Correlation between detection of plasminogen cross‐reactive antibodies and hemorrhage in dengue virus infection. J Infect Dis 170: 1304–1307. [DOI] [PubMed] [Google Scholar]
- Cines DB, Pollak ES, Buck CA et al (1998) Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 91: 3527–3561. [PubMed] [Google Scholar]
- Clark DV, Mammen JRMP, Nisalak A, Pithimethee V & Endy TP (2005) Economic impact of dengue fever/dengue hemorrhagic fever in Thailand at the family and population levels. Am J Trop Med Hyg 72: 786–791. [PubMed] [Google Scholar]
- Cologna R, Armstrong PM & Rico‐Hesse R (2005) Selection for virulent dengue viruses occurs in humans and mosquitoes. J Virol 79: 853–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crone C (1986) Modulation of solute permeability in microvascular endothelium. Fed Proc 45: 77–83. [PubMed] [Google Scholar]
- Dewi B, Takasaki T & Kurane I (2004) In vitro assessment of human endothelial cell permeability: effects of inflammatory cytokines and dengue virus infection. J Virol Methods 121: 171–180. [DOI] [PubMed] [Google Scholar]
- Dewi BE, Takasaki T & Kurane I (2008) Peripheral blood mononuclear cells increase the permeability of dengue virus‐infected endothelial cells in association with downregulation of vascular endothelial cadherin. J Gen Virol 89: 642–652. [DOI] [PubMed] [Google Scholar]
- Gagnon SJ, Ennis FA & Rothman AL (1999) Bystander target cell lysis and cytokine production by dengue virus‐specific human CD4+ cytotoxic T lymphocyte clones. J Virol 73: 3623–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán MG, Kouri G, Bravo J, Valdes L, Vazquez S & Halstead SB (2002) Effect of age on outcome of secondary dengue 2 infections. Int J Infect Dis 6: 118–124. [DOI] [PubMed] [Google Scholar]
- Halstead SB (1989) Antibody, macrophages, dengue virus infection, shock, and hemorrhage: a pathogenetic cascade. Rev Infect Dis 11: S830–S839. [DOI] [PubMed] [Google Scholar]
- Halstead SB (2002a) Dengue. Curr Opin Infect Dis 15: 471–476. [DOI] [PubMed] [Google Scholar]
- Halstead SB (2007) Dengue. Lancet 370: 1644–1652. [DOI] [PubMed] [Google Scholar]
- Halstead SB, Lan NT, Myint TT et al (2002b) Dengue hemorrhagic fever in infants: research opportunities ignored. Emerg Infect Dis 8: 1474–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Lei HY, Liu HS, Lin YS, Liu CC & Yeh TM (2000) Dengue virus infects human endothelial cells and induces IL‐6 and IL‐8 production. Am J Trop Med Hyg 63: 71–75. [DOI] [PubMed] [Google Scholar]
- Huang YH, Lei HY, Liu HS, Lin YS, Chen SH, Liu CC & Yeh TM (2003) Tissue plasminogen activator induced by dengue virus infection of human endothelial cells. J Med Virol 70: 610–616. [DOI] [PubMed] [Google Scholar]
- Iglarz M & Clozel M (2007) Mechanisms of ET‐1‐induced endothelial dysfunction. J Cardiovasc Pharmacol 50: 621–628. [DOI] [PubMed] [Google Scholar]
- Jessie K, Fong MY, Devi S, Lam SK, Thong K & Wong KT (2004) Localization of Dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J Infect Dis 189: 1411–1418. [DOI] [PubMed] [Google Scholar]
- Jiang L, Guo H & Fang D (1999) Effect of dengue virus infection on the production of ET‐1 and PGI2 by human vascular endothelial cells. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi 13: 239–242. [PubMed] [Google Scholar]
- Jiang Z, Tang X, Xiao R, Jiang L & Chen X (2007) Dengue virus regulates the expression of hemostasis‐related molecules in human vein endothelial cells. J Infect 55: 23–28. [DOI] [PubMed] [Google Scholar]
- Jose EC, Marino E, Romano E et al (2005) Proinflammatory factors present in sera from patients with acute dengue infection induced activation and apoptosis of human microvascular endothelial cells: possible role of TNF alpha in endothelial cell damage in dengue. Cytokine 30: 359–365. [DOI] [PubMed] [Google Scholar]
- Juffrie M, Van Der Meer GM, Hack CE, Haasnoot K, Sutaryo VAJ & Thijs LG (2000) Inflammatory mediators in dengue virus infection in children: interleukin-8 and its relationship to neutrophil degranulation. Infect Immun 68: 702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinglsey J, Liew L & Chow VTK (2006) Microarray and real time PCR analysis of a novel set of differentially expressed human geneses in ECV304 endothelial‐like cells infected with dengue virus type 2. J Virol Methods 131: 47–57. [DOI] [PubMed] [Google Scholar]
- Kochel TJ, Watts DM, Halstead SB et al (2002) Neutralization of American genotype dengue 2 viral infection by dengue l antibodies may have prevented dengue hemorrhagic fever in Iquitos, Peru. Lancet 360: 310–312. [DOI] [PubMed] [Google Scholar]
- Krishnamurti C, Peat RA, Cutting MA & Rothwell SW (2002) Platelet adhesion to dengue‐2 virus‐infected endothelial cells. Am J Trop Med Hyg 66: 435–441. [DOI] [PubMed] [Google Scholar]
- Lee YR, Liu MT, Lei HY, Liu CC, Wu JM, Tung YC, Lin YS, Yeh TM, Chen SH & Liu HS (2006) MCP‐1, a highly expressed chemokine in dengue haemorrhagic fever/dengue shock syndrome patients, may cause permeability change, possibly through reduced tight junctions of vascular endothelium cells. J Gen Virol 87: 3623–3630. [DOI] [PubMed] [Google Scholar]
- Leitmeyer KC, Vaughn DW, Watts DM, Salas R, Villalobos I, De Chacon RC & Rico‐Hesse R (1999) Dengue virus structural differences that correlate with pathogenesis. J Virol 73: 4738–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limonta D, Capó V, Torres G, Pérez AB & Guzmán MG (2007) Apoptosis in tissues from fatal dengue shock syndrome. J Clin Virol 40: 50–54. [DOI] [PubMed] [Google Scholar]
- Lin CF, Lei HY, Shiau AL, Liu HS, Yeh TM, Chen SH, Liu CC, Chiu SC & Lin YS (2002) Endothelial cell apoptosis induced by antibodies against Dengue virus nonstructural protein 1 via production of nitric oxide. J Immunol 169: 657–664. [DOI] [PubMed] [Google Scholar]
- Lin CF, Lei HY, Shiau AL, Liu CC, Liu HS, Yeh TM, Chen SH & Lin YS (2003) Antibodies from dengue patient sera cross‐react with endothelial cells and induce damage. J Med Virol 69: 82–90. [DOI] [PubMed] [Google Scholar]
- Lin CF, Chiu SC, Hsiao YL et al (2005) Expression of cytokine, chemokine, and adhesion molecules during endothelial cell activation induced by antibodies against dengue virus nonstructural protein 1. J Immunol 174: 395–403. [DOI] [PubMed] [Google Scholar]
- Lin CF, Wan SW, Cheng HJ, Lei HY & Lin YS (2006) Autoimmune pathogenesis in dengue virus infection. Viral Immunol 19: 127–132. [DOI] [PubMed] [Google Scholar]
- Lin YS, Lin CF, Lei HY, Liu HS, Yeh TM, Chen SH & Liu CC (2004) Antibody‐mediated endothelial cell damage via nitric oxide. Curr Pharm Des 10: 213–221. [DOI] [PubMed] [Google Scholar]
- Lugus JJ, Park C & Choi K (2005) Developmental relationship between hematopoietic and endothelial cells. Immunol Res 32: 57–74. [DOI] [PubMed] [Google Scholar]
- Luplertlop N, Missé D, Bray D, Deleuze V, Gonzalez JP, Leardkamolkarn V, Yssel H & Veas F (2006) Dengue‐virus‐infected dendritic cells trigger vascular leakage through metalloproteinase overproduction. EMBO Rep 7: 1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangada MM & Rothman AL (2005) Altered cytokine responses of dengue‐specific CD4+ T cells to heterologous serotypes. J Immunol 175: 2676–2683. [DOI] [PubMed] [Google Scholar]
- Medin CL & Rothman AL (2006) Cell type‐specific mechanisms of interleukin‐8 induction by dengue virus and differential response to drug treatment. J Infect Dis 193: 1070–1077. [DOI] [PubMed] [Google Scholar]
- Medin CL, Fitzgerald KA & Rothman AL (2005) Dengue virus nonstructural protein NS5 induces interleukin‐8 transcription and secretion. J Virol 79: 11053–11061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meertens L, Bertaux C, Cukierman L, Cormier E, Lavillette D, Cosset FL & Dragic T (2008) The tight junction proteins claudin‐1, ‐6 and ‐9 are entry cofactors for the Hepatitis C virus. J Virol 82: 3555–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller‐Kasprzak E & Jagodziński PP (2007) Endothelial progenitor cells as a new agent contributing to vascular repair. Arch Immunol Ther Exp (Warsz) 55: 247–259. [DOI] [PubMed] [Google Scholar]
- Misra A, Mukerjee R & Chaturvedi UC (1996a) Production of nitrite by dengue virus‐induced cytotoxic factor. Clin Exp Immunol 104: 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra A, Mukerjee R & Chaturvedi UC (1996b) Release of reactive oxygen intermediates by dengue virus‐induced macrophage cytotoxin. Int J Exp Pathol 77: 237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra A, Mukerjee R & Chaturvedi UC (1998) Respiratory burst by dengue virus‐induced cytotoxic factor. Med Principles Pract 7: 251–260. [Google Scholar]
- Mongkolsapaya J, Dejnirattisai W & Xu XN (2003) Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat Med 9: 921–927. [DOI] [PubMed] [Google Scholar]
- Mongkolsapaya J, Duangchinda T, Dejnirattisai W, Vasanawathana S, Avirutnan P & Jairungsri A (2006) T cell responses in dengue hemorrhagic fever: are cross-reactive T cells suboptimal? J Immunol 176: 3821–3829. [DOI] [PubMed] [Google Scholar]
- Mukerjee R, Misra A & Chaturvedi UC (1996) Dengue virus‐induced cytotoxin releases nitrite by spleen cells. Int J Exp Pathol 77: 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukerjee R, Chaturvedi UC, Vaughn DW & Kalayanarooj S (1997) Purification and pathogenicity of the cytotoxic factor from the cases of dengue haemorrhagic fever. Curr Sci 72: 494–501. [Google Scholar]
- Myint KS, Endy TP, Mongkolsirichaikul D et al (2006) Apoptosis is an important modulator of cellular immune responses during systemic viral infections. J Infect Dis 194: 600–607. [DOI] [PubMed] [Google Scholar]
- Neves‐Souza PC, Azeredo EL, Zagne SM, Valls‐de‐Souza R, Reis SR, Cerqueira DI, Nogueira RM & Kubelka CF (2005) Inducible nitric oxide synthase (iNOS) expression in monocytes during acute Dengue fever in patients and during in vitro infection. BMC Infect Dis 5: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TH, Lei HY, Nguyen TL, Lin YS, Huang KJ, Le BL & Lin CF (2004) Dengue hemorrhagic fever in infants: a study of clinical and cytokine profiles. J Infect Dis 189: 221–232. [DOI] [PubMed] [Google Scholar]
- Pacsa AS, Agarwal R, Elbishbishi EA, Chaturvedi UC, Nagar R & Mustafa AS (2000) Interleukin‐12 in patients with dengue haemorrhagic fever. FEMS Immunol Med Microbiol 28: 151–155. [DOI] [PubMed] [Google Scholar]
- Raghupathy R, Chaturvedi UC, Al‐Sayer H, Elbishbishi EA, Agarwal R, Nagar R, Nusrat H, Mustafa AS, Azizieh F & Khan MAY (1998) Elevated levels of IL‐8 in dengue hemorrhagic fever. J Med Virol 56: 280–285. [DOI] [PubMed] [Google Scholar]
- Risau W & Flamme I (1995) Vasculogenesis. Annu Rev Cell Dev Biol 11: 73–91. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Ortega M (1998) Nitric oxide in dengue pathology. Acta Cient Venez 49 (suppl 1), 8–12. [PubMed] [Google Scholar]
- Sahaphong S, Riengrojpitak S, Bhamarapravati N & Chirachariyavej T (1980) Electron microscopic study of the vascular endothelial cell in dengue hemorrhagic fever. Southeast Asian J Trop Med Public Health 11: 194–204. [PubMed] [Google Scholar]
- Sierra B, Garcia G, Perez AB, Morier L, Rodriguez R, Alvarez M & Guzman MG (2002) Long‐term memory cellular immune response to dengue virus after a natural primary infection. Int J Infect Dis 6: 125–128. [DOI] [PubMed] [Google Scholar]
- Srikiatkhachorn A, Ajariyakhajorn C, Endy TP, Kalayanarooj S, Libraty DH, Green S, Ennis FA & Rothman AL (2007) Virus‐induced decline in soluble vascular endothelial growth receptor 2 is associated with plasma leakage in dengue hemorrhagic fever. J Virol 81: 1592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson JR (2005) Understanding dengue pathogenesis: implications for vaccine design. Bull World Health Organ 83: 308–314. [PMC free article] [PubMed] [Google Scholar]
- Talavera D, Castillo AM, Dominguez MC, Gutierrez AE & Meza I (2004) IL8 release, tight junction and cytoskeleton dynamic reorganization conducive to permeability increase are induced by dengue virus infection of microvascular endothelial monolayers. J Gen Virol 85: 1801–1813. [DOI] [PubMed] [Google Scholar]
- Theofilopoulos AN, Wilson CB & Dixon FJ (1976) The Raji cell radioimmune assay for detecting immune complexes in human sera. J Clin Invest 57: 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warke RV, Xhaja K, Martin KJ et al (2003) Dengue virus induces novel changes in gene expression of human umbilical vein endothelial cells. J Virol 77: 11822–11832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warke RV, Martin KJ, Giaya K, Shaw SK, Rothman AL & Bosch I (2008) TRAIL is a novel anti‐viral protein against dengue virus. J Virol 82: 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts DM, Porter KR, Putvatana P et al (1999) Failure of secondary infection with American genotype dengue 2 to cause dengue haemorrhagic fever. Lancet 354: 1431–1434. [DOI] [PubMed] [Google Scholar]
- Wei HY, Jiang LF, Fang DY & Guo HY (2003) Dengue virus type 2 infects human endothelial cells through binding of the viral envelope glycoprotein to cell surface polypeptides. J Gen Virol 84: 3095–3098. [DOI] [PubMed] [Google Scholar]
- Wiedmer T, Zhou Q, Kwoh DY & Sims PJ (2000) Identification of three new members of the phospholipid scramblase gene family. Biochim Biophysics Acta 1467: 244–253. [DOI] [PubMed] [Google Scholar]
- Yao HY, Liu B & Mao N (2007) Relationship between hematopoietic development and endothelial cells (hemangioblast or hemogenic endothelium) – review. Zhongguo Shi Yan Xue Ye Xue Za Zhi 15: 198–201. [PubMed] [Google Scholar]
- Yu HS, Wang MT, Tai CL, Yang SA & Chien CH (1989) Skin eruption and histopathological changes in dengue fever. Gaoxiong Yi Xue Ke Xue Za Zhi 5: 17–23. [PubMed] [Google Scholar]
- Yuan SY (2006) New insights into eNOS signaling in microvascular permeability. Am J Physiol Heart Circ Physiol 291: H1029–H1031. [DOI] [PubMed] [Google Scholar]
- Zhang JL, Wang JL, Gao N, Chen ZT, Tian YP & An J (2007) Up‐regulated expression of beta3 integrin induced by dengue virus serotype 2 infection associated with virus entry into human dermal microvascular endothelial cells. Biochem Biophys Res Commun 356: 763–768. [DOI] [PubMed] [Google Scholar]