

Abstract
Purpose: To review information on the use of laboratory tests in screening, diagnosis, and monitoring of acute and chronic hepatic injury.
Data Sources and Study Selection: A MEDLINE search was performed for key words related to hepatic diseases, including acute hepatitis, chronic hepatitis, alcoholic hepatitis, cirrhosis, hepatocellular carcinoma, and etiologic causes. Abstracts were reviewed, and articles discussing use of laboratory tests selected for review. Additional articles were selected from the references.
Guideline Preparation and Review: Drafts of the guidelines were posted on the Internet, presented at the AACC Annual Meeting in 1999, and reviewed by experts. Areas requiring further amplification or literature review were identified for further analysis. Specific recommendations were made based on analysis of published data and evaluated for strength of evidence and clinical impact.
Recommendations: Although many specific recommendations are made in the guidelines, only some summary recommendations are listed here. In acute hepatic injury, prothrombin time and, to a lesser extent, total bilirubin are the best indicators of severity of disease. Although ALT is useful for detecting acute and chronic hepatic injury, it is not related to severity of acute hepatic injury and only weakly related to severity of chronic hepatic injury. Specific tests of viral markers should be the initial differential tests in both acute and chronic hepatic injury; when positive, they are also useful for monitoring recovery from hepatitis B and C.
This reports represents a continuation of the National Academy of Clinical Biochemistry Guidelines on Use of Laboratory Tests in the Diagnosis and Monitoring of Hepatic Injury. Part 1 (1) discusses performance characteristics for laboratory tests and also describes the methodology used to develop the guidelines. Table 2 in Part 1 outlines the codes used for characterizing the recommendations contained in both parts of the Guidelines.
Table 2.
Uncommon causes of acute hepatic injury.
| Disorder | Key feature | Laboratory tests | Associated findings | References |
|---|---|---|---|---|
| Wilson disease | Young individuals; low ALP, high bilirubin | Low ceruloplasmin in only 40%; abnormal gene on chromosome 13 | Urine, serum copper not reliable in acute Wilson disease; often associated with hemolysis and renal insufficiency | 95, 96, 210–212 |
| AIH | Young individuals; increased γ-globulins; low albumin; ascites often present | Positive ANA and/or ASMA | Other autoimmune disorders in some cases | 103, 213–215 |
| Hepatitis E | Travel to endemic area | Anti-HEV | Similar to hepatitis A | 216–219 |
| Other viruses | Clinical features of mononucleosis often present | Anti-CMV; anti-EBV | Increased ALP | 220 |
Hepatocyte injury is common worldwide. Despite a decreased incidence of acute viral hepatitis since introduction of vaccines for hepatitis A and B and testing of the blood supply for hepatitis C, almost 100 000 cases occur annually in the United States alone, most of them undiagnosed. An estimated 1.5% of the US population is chronically infected with hepatitis B or C virus. Individuals with chronic hepatitis are at increased risk for cirrhosis and hepatocellular carcinoma (HCC),1 the main causes of death from liver disease in Western countries. Chronic liver diseases are among the more common disorders seen by physicians. We present here guidelines for the use of laboratory tests in screening, diagnosis, and monitoring of acute and chronic hepatic injury.
Acute Hepatic Injury
Acute hepatic injury can be recognized by the presence of jaundice or nonspecific symptoms of acute illness accompanied by increases in the activities of aspartate aminotransferase (AST; EC 2.6.1.1) and/or alanine aminotransferase (ALT; EC 2.6.1.2). An estimated 80% of individuals with acute viral hepatitis are never diagnosed clinically, although some may be detected by increased aminotransferases in the face of nonspecific or absent clinical symptoms. AST and ALT activities are seldom >10 times the upper reference limit in liver diseases other than acute hepatic injury. Alkaline phosphatase (ALP; EC 3.1.3.1) is more than three times the upper reference limit in <10% of cases of acute hepatic injury (2)(3). With the decreasing incidence of acute viral hepatitis, other liver diseases are more commonly encountered as causes of increased AST or ALT activities; as many as 25% of those with AST >10 times the reference limit may have obstruction as a cause (4). Approximately 1–2% of patients with bile duct obstruction have transient increases in AST and/or ALT activities of >2000 U/L (5)(6); aminotransferase activities usually fall to within reference limits by 10 days even if obstruction persists (2)(4)(5). The distribution of direct bilirubin as a percentage of total bilirubin is similar in acute hepatic injury and obstructive jaundice (7). In patients with jaundice, only 16% of those with acute hepatic injury have direct bilirubin <50% of total bilirubin; values below this suggest another cause for jaundice, such as hemolysis (7). The best discriminant values for recognizing acute hepatic injury appear to be 200 U/L for AST (sensitivity, 91%; specificity, 95%) and 300 U/L for ALT (sensitivity, 96%; specificity, 94%) (3). AST is >10 times the upper reference limit in slightly over one-half of patients at the time of presentation (3). In uncomplicated alcoholic hepatitis, AST and ALT values are almost never >10 times the upper reference limit, the AST/ALT ratio is >2 in 80% of cases, and increased ALP is present in 20% of cases (8)(9)(10)(11)(12). Jaundice occurs in 60–70% of cases of alcoholic hepatitis (9)(10). The frequency of jaundice in patients with acute viral hepatitis differs both by age and etiologic agent. Jaundice is rare in children with viral hepatitis, and when it is present is less severe than in adults. In one study, only 1% of children with acute hepatitis had peak bilirubin >171 μmol/L (10 mg/dL), whereas 27% of adults did (13). In adults, jaundice develops in 70% of cases of acute hepatitis A (14), 33–50% of cases of acute hepatitis B (15)(16), and 20–33% of cases of acute hepatitis C (17)(18). In children, there is a direct correlation between age and peak serum bilirubin; an increase of 10 years in age is associated with an average increase of 85 μmol/L (5 mg/dL) in bilirubin. In adults, however, there is only a slight increase or no increase with increasing age (7).
Recommendation
Acute hepatic injury can be diagnosed by ALT >10 times the upper reference limits and ALP <3 times the appropriate upper reference limit (IIB).
markers of severity
Acute viral hepatitis A and B are usually self-limited illnesses, and most patients recover completely. In those with acute hepatitis C infection, ∼85% develop chronic hepatitis. Rarely, acute hepatic injury causes severe liver damage and acute liver failure. Testing should identify patients at highest risk for liver failure. Aminotransferase activities are related more to the cause of hepatic injury than to severity (Table 1 ). There is weak correlation between aminotransferase activities and bilirubin in viral hepatitis (3) and none in ischemic hepatic injury (19). Peak aminotransferase activities bear no relationship to prognosis and may fall with worsening of the patient’s condition (20). Prothrombin time (PT) is the most important predictor of prognosis; PT cutoff times >4 s beyond control or >20 s, or an international normalized ratio >6.5 identify patients at high risk of death (20)(21)(22)(23)(24). In alcoholic hepatitis, PT >5 s beyond control, bilirubin >428 μmol/L (>25 mg/dL), or albumin <25 g/L (2.5 g/dL) in a patient >55 years of age predicts 90% likelihood of death (10)(25). In ischemic or toxic hepatic injury, prolongation of PT is common early after injury; values peak by 24–36 h after injury and then rapidly return to normal. In acetaminophen injury, marked prolongation of PT does not by itself indicate likelihood of liver failure (26)(27), but a persistent increase in or increasing PT 4 days after acetaminophen ingestion does (28). Peak bilirubin is not a good indicator of prognosis in most forms of hepatic injury (20). Total bilirubin >257 μmol/L (15 mg/dL) or PT >3 s above the upper reference limit (29) in patients with viral hepatitis indicates severe liver injury and mandates close monitoring for encephalopathy.
Table 1.
Patterns of laboratory tests in types of acute hepatic injury.
| Disease | Peak ALT, × URL1 | AST:ALT ratio | Peak bilirubin, mg/dL | PT prolongation, s |
|---|---|---|---|---|
| Viral hepatitis | 10–40 | <1 | <15 | <3 |
| Alcoholic hepatitis | 2–8 | >2 | <15 | 1–3 |
| Toxic injury | >40 | >1 early | <5 | >5 (transient) |
| Ischemic injury | >40 | >1 early | <5 | >5 (transient) |
× URL, times the upper reference limit.
Recommendations
Total bilirubin >257 μmol/L (15 mg/dL) or PT >3 s above the upper reference limit in an individual with viral hepatitis, in the absence of other factors affecting results, indicates severe liver injury (IIB).
Direct bilirubin is needed to rule out other causes of increased total bilirubin, such as hemolysis, but it does not differentiate hepatic injury from obstructive jaundice (IIB).
With acetaminophen toxicity, a persistent increase in or increasing PT >4 days after ingestion indicates severe liver injury (IIB).
differential diagnosis
Initial laboratory evaluation of patients with acute hepatic injury should include a drug history and testing for antibodies to the hepatitis A, B, and C viruses (HAV, HBV, and HCV), using the Health Care Financing Administration-approved acute hepatitis panel [IgM against HAV (IgM anti-HAV) and the HBV core antigen (IgM anti-HBc), HBV surface antigen (HBsAg), and antibodies against HCV (anti-HCV); Fig. 1 ]. Most hepatic drug reactions occur within 3–4 months of initiating treatment. However, in some cases, hepatic injury may become manifest as late as 12 months after treatment is initiated, and in a few cases injury may become evident days to weeks after the responsible drug is stopped (30). Hence, it is important to ask about all drugs the patient may have received or has continued to receive during the past year or so.
Figure 1.
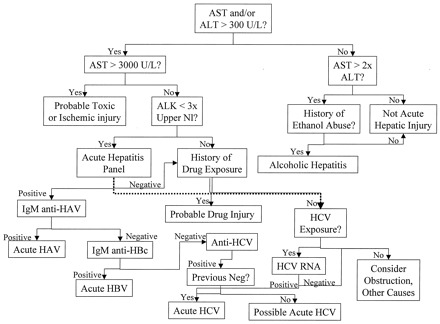
Evaluation of suspected acute hepatic injury.
Initial evaluation of patients with signs and symptoms such as jaundice, fever, and right upper quadrant abdominal pain should be by measurement of aminotransferases. Marked increases (>3000 U/L) of either enzyme are usually attributable to ischemic or toxic liver injury; if history is negative for either, then the diagnostic workup should continue as in persons with smaller increases. Viral serologies are the principal tests for evaluation of acute hepatic injury, although the falling incidence of viral diseases has made other causes proportionally more common. Because both prescription and nonprescription drugs can cause acute injury, a detailed drug history is critical, particularly in those with increased ALP. In those with coexistent increases in ALP, obstruction and other viruses such as EBV and CMV must be considered as well. ALK, alkaline phosphatase; Nl, normal.
IgM anti-HAV, the diagnostic test of choice for acute HAV infection, disappears by 4–6 months (31), whereas total HAV antibodies persist for life (32) and are found in a high percentage of the population (33). Because of its brief period of transmissibility, diagnosis of acute HAV infection should be made as soon as possible after presentation, ideally within 48 h, to allow immune globulin treatment of exposed individuals. IgM anti-HBc and HBsAg are the most reliable tests for acute HBV infection (29)(34); IgG (and thus total) anti-HBc persists for many years (35). Other HBV viral markers and antibodies are not of use in the diagnosis of acute HBV infection.
There currently is no test to definitively diagnose acute hepatitis C because anti-HCV and HCV RNA can be present in both acute and chronic HCV infections. Anti-HCV is detectable with second-generation enzyme immunoassays in only 57% of acute HCV cases at the time of initial increases in enzyme activities, whereas HCV RNA is positive in essentially all cases (36), although it is intermittently present in 15% (37)(38). By the time of clinical presentation, 80–90% of cases have detectable anti-HCV (38). Patterns that would support a diagnosis of acute hepatitis C are negative anti-HCV but positive HCV RNA, or (if HCV RNA was not tested) anti-HCV results that convert from negative to positive within a short period. Use of antibodies to hepatitis D virus (anti-HDV) to detect delta (HDV) infection should be limited to patients with positive HBsAg, particularly if accompanied by severe acute hepatitis, high risk factors (e.g., intravenous drug abuse or hemophilia), or a biphasic pattern of illness (39). If a patient with chronic hepatitis B becomes superinfected with HDV, a clinical picture resembling severe acute hepatic injury and hepatic failure may evolve (39).
Recommendations
Initial evaluation of acute hepatic injury should include a detailed drug history and viral markers (IgM anti-HAV, IgM anti-HBc, HBsAg, and anti-HCV; IIB).
Because of the need for postexposure prophylaxis, turnaround time of IgM anti-HAV should be <48 h (IIIC and IIIE).
If cost-effective (based on prevalence), laboratories may use total antibody to HAV and anti-HBc initially, performing IgM antibodies only if one or both is positive, if the turnaround time needs can be met (IIIE).
Diagnosis of acute HCV infection (in a patient with a clinical picture of acute hepatic injury) can be presumptively made by negative HAV and HBV markers, recent exposure, and either negative anti-HCV and positive HCV RNA or negative anti-HCV at initial presentation with development of positive anti-HCV within 1–3 months (IIIB).
Testing for HDV should be limited to patients with positive HBsAg, atypical clinical course, and high risk for HDV infection (IIB and IIE).
workup of patients without obvious cause for acute hepatic injury
Ischemic and toxic hepatic injury.
AST or ALT values >100-fold higher than normal are rare in viral hepatitis (2)(3) but common in both toxin ingestion, especially acetaminophen (27)(40)(41), and ischemic hepatic injury (19)(26)(42). In acetaminophen-induced hepatic injury, peak AST is >3000 U/L in 90% of cases (41), a value virtually never seen with acute viral hepatitis (3). Toxic or ischemic hepatic injury is the cause of >90% of cases of acute hepatic injury with AST activity >3000 U/L (43). In both ischemic and acetaminophen hepatic injury, AST and ALT activities typically peak early (often in the first 24 h after admission) with AST activity initially higher than that of ALT. After peaking, activities of both fall rapidly. AST may fall by 50% or more in the first 24 h (40)(41), and it declines more rapidly than ALT because of its shorter half-life (3); AST activity reaches near normal values an average of 7 days after injury (2). PT is >4 s above the reference limits in 90% of cases (26)(27) and falls rapidly after peak AST is reached (26). Bilirubin is <34 μmol/L (2 mg/dL) in 80% of cases of toxic or ischemic injury (26)(27)(42). Lactate dehydrogenase (EC 1.1.1.27) activity often is higher than that of AST at presentation in toxic or ischemic hepatic injury (26)(40)(41), whereas it is increased on initial determination in only 55% of cases of viral hepatitis, with average values being only slightly above the upper reference limit (3). Rarely, cocaine may cause hepatic injury, usually in patients with coexisting hypotension (44).
Other causes.
Rarely, Wilson disease and autoimmune hepatitis (AIH; discussed in more detail in Chronic Hepatic Injury) can present as acute hepatic injury (Table 2 ). Several viruses other than the classical agents [HAV, HBV, HCV, and hepatitis E virus (HEV)] have been associated with hepatitis, including herpesvirus, cytomegalovirus (CMV), enterovirus, coronavirus, reovirus (in neonates), adenovirus, parvovirus B6 (in pediatric populations), varicella-zoster virus, and Epstein-Barr virus (EBV). Syphilis, leptospirosis, and toxoplasmosis also may cause hepatic injury, as may other less common infectious agents. Rarely, other disorders, including lymphoma, Budd-Chiari syndrome, and venoocclusive disease, may present with a picture of acute hepatic injury. In general, hepatic injury associated with these etiologies either is unusual or is associated with a specific syndrome (chicken pox with varicella-zoster virus, mononucleosis with EBV, or CMV). Most patients with other infectious causes of hepatic injury have signs and symptoms that suggest a particular agent as the cause. Specific diagnosis of infection by other agents should be pursued when the etiology remains unknown after more common causes are excluded and when establishment of a specific diagnosis appears clinically indicated. Superinfection with other hepatitis viruses may occur in a patient with other forms of hepatic injury; for example, patients with chronic HCV or alcoholic hepatitis may become infected with either HAV or HBV and develop an acute hepatitis as a result of the superimposed infection. In chronic hepatitis, an acute increase in aminotransferases mimicking acute hepatic injury can occur with clearance of HBV e antigen (HBeAg) (45) or with emergence of quasispecies of HCV (46).
Recommendations
In patients with negative viral markers and initial AST >100 times the upper reference limit, toxic exposure or ischemia should be suspected (IIB).
In patients with negative viral markers and enzyme concentrations 8–100 times the upper reference limit, testing must exclude the possibility of Wilson disease and AIH (IIB).
Testing for antibody to hepatitis E is not recommended in the United States unless other viral serologies are negative and there is a history of recent travel to an endemic area (IIIE).
Tests for other infectious agents (EBV, CMV, syphilis, toxoplasmosis) may be used if no other causes are evident (IIB).
monitoring
Aminotransferases.
Aminotransferase activities tend to increase before and peak near onset of jaundice in viral hepatitis, falling gradually from that point onward (47). Activities tend to fall slowly in viral hepatitis and alcoholic hepatitis: AST and ALT decrease, on average, 11.7% and 10.5% per day, respectively, and remain increased 22 ± 16 and 27 ± 16 days, respectively (3). In hepatitis A, a secondary increase in enzymes occurs in 5–10% of cases before activities return to baseline and is associated with circulating HAV RNA and viral particles in stool, indicating a potential for transmission of infection (48)(49)(50). As discussed above, AST and ALT fall rapidly after reaching peak activities in ischemic and toxic hepatic injury. In all forms of hepatic injury, decreasing activities occur both with recovery and with massive necrosis, making enzyme activity a poor indicator of recovery (20). Once aminotransferases have shown a consistent pattern of decrease, they need not be checked again until the patient has clinically recovered. Return of aminotransferases to normal is not a reliable sign of recovery in hepatitis B or C. In patients with chronic HCV infection, 49% with normal ALT at the initial visit after seroconversion developed increased ALT on subsequent follow-up (51). In hepatitis B, AST and ALT may return to normal despite persistence of infection (52)(53).
Bilirubin.
Bilirubin peaks later than aminotransferases, often by a week or so, and then gradually decreases. Peak bilirubin >257–342 μmol/L (15–20 mg/dL) is unusual in viral hepatitis. Only 10–12% of patients with viral hepatitis have peak values >257 μmol/L (15 mg/dL), and only 4% have peak values >342 μmol/L (20 mg/dL); higher bilirubin is more common in HBV infection (3)(13). As total bilirubin declines, the proportion of δ-bilirubin increases, often reaching 70–80% of total bilirubin (54)(55). In adults with viral hepatitis, bilirubin remains increased 30.3 ± 19.7 days after peak concentrations are reached (3), but it clears more quickly in children (8); jaundice remains longer than 6 weeks in 34% of adult HBV cases but in only 15% of other forms of viral hepatitis (8). A prolonged increase in conjugated bilirubin occasionally occurs with viral hepatitis, particularly with HAV, but it does not signify a poor prognosis if synthetic function remains intact (56). Significantly increased bilirubin is uncommon in toxic and ischemic hepatic injury. Once serum bilirubin has begun to decrease, there is no reason to measure it again unless jaundice worsens clinically.
Coagulation tests.
Increased PT is a common finding in ischemic and toxic hepatic injury, often with results >15 s or 4 s above the reference limit before they rapidly return to normal. There are no data on the extent to which the increase affects prognosis in ischemic hepatic injury. An increase in PT to >15 s or >4 s above reference limits in viral or alcoholic hepatitis is a marker of more severe disease (8)(10)(57).
Recommendations
PT >4 s above reference limits, bilirubin >257 μmol/L (15 mg/dL), or development of encephalopathy identifies high-risk patients who require close monitoring and consideration of referral to a gastroenterologist or hepatologist (IIB).
In patients with acute hepatitis B, repeat HBsAg measurements should be performed within 6–12 months; if negative and tests for anti-HBV surface antigen antibody (anti-HBs) are positive, no additional follow-up is needed (IIE).
In patients with acute hepatitis C, ALT should be measured periodically over the next 1–2 years to assure continued normal results (IIB).
Chronic Hepatic Injury
Chronic hepatic injury is a relatively common disorder with minimal symptoms but long-term risk of significant morbidity and mortality. It is defined pathologically by ongoing hepatic necrosis and inflammation of the liver, often accompanied by fibrosis. Chronic hepatic injury may progress to cirrhosis (15–20% in the case of chronic HCV) and predisposes to HCC. Most commonly, it is the result of chronic viral infection. In the United States alone, there are an estimated 2.1–2.7 million people chronically infected with HCV (58). There are also ∼1–1.25 million chronic carriers of HBV in the United States. Although prevalence rates for HCV infection generally are between 0.5% and 5% in other parts of the world, prevalence rates for HBV vary markedly, and in many areas HBV is an endemic infection. The prevalence of endemic HBV in children is declining in many parts of the world because of the use of HBV vaccine. Clinical findings and laboratory investigation often are adequate to establish the most likely diagnosis, with a predictive value of 88% for alcoholic hepatitis and 81% for chronic viral hepatitis (before availability of HCV tests) compared with biopsy (59).
Recommendations
In the absence of liver biopsy showing chronic hepatitis, one of the following clinical definitions should be used to diagnose chronic hepatitis:
Persistence of increased ALT for >6 months after an episode of acute hepatitis; or
Increased ALT (without another explanation) on more than one occasion over a period of 6 months. A shorter time may be appropriate in patients with risk factors for chronic viral hepatitis, genetic causes of hepatic injury, or autoimmune liver injury, or in the presence of clinical signs or symptoms of liver disease (IIB).
Although the definition of chronic hepatic injury by increased ALT is widely accepted, 15–50% of patients with chronic hepatitis C have persistently normal ALT (51)(60)(61)(62). The likelihood of continuously normal ALT decreases with increasing number of measurements; even after three normal ALT values, 11% of patients with chronic HCV viremia subsequently developed persistently increased ALT (51). ALT often fluctuates between normal and abnormal, particularly in chronic hepatitis C; 60% of patients with multiple ALT measurements have at least occasional normal ALT values (D.R. Dufour, unpublished observations). The majority of patients with persistently normal ALT have histologic evidence of chronic hepatitis on biopsy but, in general, have milder inflammation, less fibrosis, and lower rates of progression to cirrhosis than do HCV patients with increased ALT (17)(61). Center for Disease Control and Prevention guidelines do not recommend treatment of patients with HCV and persistently normal ALT (63). Although long-term studies are needed, it appears that the clinical definition proposed will not miss a significant group of patients who require and benefit from treatment.
It is not always possible to distinguish acute from chronic hepatic injury. Most patients with chronic hepatitis C (the most common form of chronic hepatic injury) have ALT values one to four times the upper reference limit, and 90% have maximum ALT less than seven times the upper reference limits, values lower than typically seen in acute hepatitis. In ∼5% of cases, however, peak ALT may be >10 times the upper reference limit, often associated with jaundice, in a pattern similar to that seen in acute hepatic injury (D.R. Dufour, unpublished observations). In such cases, it often is necessary to do additional testing to rule out another cause of acute hepatic injury.
screening
General screening of the population for chronic hepatic injury is not cost-effective and should be limited to high-risk individuals (64). These include those with a family history of genetic diseases known to affect the liver, as discussed below, or risk factors for chronic viral infection (Table 3 ). ALT is consistently higher than AST with all causes of chronic hepatic injury except for alcohol; AST is normal in a substantial number of cases. ALT may be normal in patients with cirrhosis, whereas AST remains increased (65)(66)(67)(68). Total and direct bilirubin and ALP are normal in essentially all patients and are not useful in screening (59)(69)(70)(71)(72). If increased ALT is found on routine testing, this should be confirmed by repeat testing before further evaluation. A minority of individuals with only one increased ALT value are found to have liver disease (70)(73). Patients with slightly increased ALT (one to two times the upper reference limit) are more likely to have a transient increase not attributable to disease (59)(71)(73). Approximately 30% of patients with chronic hepatic injury attributable to HCV have peak ALT activities less than two times the upper reference limit (D.R. Dufour, unpublished observations). Because ALT is also found in skeletal muscle, it is advisable to consider history of exercise and, if positive, to consider measurement of creatine kinase (EC 2.7.3.2) to rule out skeletal muscle as the origin of ALT (73)(74).
Table 3.
Risk factors for chronic viral hepatitis.1
| Established risk factors |
| · Injection drug use |
| · Chronic hemodialysis |
| · Blood transfusion or transplantation prior to 1992 (HCV) |
| · Receipt of blood (including needlestick) from a donor subsequently testing positive for HCV |
| · Receipt of clotting factor concentrates produced before 1987 |
| · Asian ancestry (HBV) |
| · Unvaccinated healthcare workers (HBV) |
| · Birth to mother with chronic HBV or HCV |
| Possible risk factors |
| · Body piercing or tattooing |
| · Multiple sexual partners or sexually transmitted diseases |
| · Healthcare workers (HCV) |
| · Contacts of HCV positive persons |
From CDC (63).
In patients with risk factors for chronic HBV or HCV infection (Table 3 ), ALT will not identify all infected individuals; HBsAg and anti-HCV should be measured to screen for chronic infection. Chronic “carriers” of HBV typically have normal ALT (53), and 15–30% of patients with chronic HCV infection have persistently or intermittently normal ALT; however, the likelihood of only normal values decreases with frequency of testing (75). Because 15–25% of individuals with anti-HCV have no detectable viremia, persons with positive anti-HCV should have qualitative HCV RNA performed to identify those with persistent infection. HCV RNA may be transiently present in the early stages of infection. If a patient has persistently increased ALT and positive anti-HCV, but negative HCV RNA, the test should be repeated.
Recommendations
Screening for chronic hepatitis is recommended in asymptomatic high-risk individuals (IIB and IIE).
ALT is the most cost-effective screening test for metabolic or drug-induced liver injury; AST should also be measured with history of alcohol abuse (IIB and IIE).
Specific viral serologies (HBsAg and anti-HCV), as well as ALT, should be used in individuals at high risk for viral hepatitis (IB).
Confirmation of chronic HCV infection in an anti-HCV-positive individual should be made by HCV RNA tests; if negative and ALT is increased, HCV RNA should be repeated (IIB).
differential diagnosis
If the clinical history suggests alcohol abuse and/or AST activity is greater than ALT (especially if more than twofold higher than ALT activity), the most likely diagnosis is alcoholic hepatitis. Virtually no other form of chronic hepatic injury causes AST to be higher than ALT unless cirrhosis develops (69)(70)(71)(72). Although the majority of cases of chronic hepatic injury are caused by viruses, drugs, or ethanol, several other disorders may produce chronic hepatic injury. Additional tests are not needed if initial evaluation is consistent with hepatitis B or C or alcoholic hepatitis (71)(76). Prescription drugs may cause persistently increased ALT, most commonly with drugs such as sulfonamides, cholesterol-lowering agents, and isoniazid (30). In one study from an area with a low prevalence of viral hepatitis, a history of prescription drug use was common in those with chronic hepatic injury and no recognizable etiology despite extensive laboratory testing (77). In patients with increased ALT, negative viral markers, and a negative history for drug or alcohol ingestion, the workup should include less common causes of chronic hepatic injury (Table 4 ).
Table 4.
Other causes of chronic increases of ALT and/or AST.
| Cause | Key features | Screening test | Confirmatory test | References |
|---|---|---|---|---|
| NASH | Most common cause other than viral, alcoholic | None | Biopsy | 59, 68, 69, 78 |
| Hemochromatosis | Autosomal recessive trait; 1:200 among Northern European ancestry | Transferrin saturation 45% | HFE gene analysis for C282Y mutation | 83–88, 90, 93 |
| Wilson disease | Autosomal recessive trait; 1:30 000 individuals; hemolytic anemia, renal injury | Low ceruloplasmin in 65–95% of homozygotes, 20% of heterozygotes | Genetic analysis; low serum copper, high urine copper | 95–100 |
| AIH | Up to 18% of non-viral hepatitis, mainly in young women; increased γ-globulins | ANA and ASMA; false-positive anti-HCV common | Biopsy | 101–105 |
| PBC | Middle-aged women; usually mainly increase of ALP, often associated with Sjogren syndrome | AMA | Biopsy | 106–110 |
| Sclerosing cholangitis | Young to middle-aged men; usually mainly increase in ALP; often associated with inflammatory bowel disease | Anti-neutrophil cytoplasmic antibodies; ASMA, ANA may also be positive | Bile duct imaging | 111–114 |
| A1AT deficiency | Autosomal recessive trait; 1:1000 to 1:2000; controversial whether it causes chronic liver disease in adults | A1AT phenotyping | 115–123 |
Recommendations
Initial evaluation should include a detailed drug history along with measurement of HBsAg and anti-HCV. If anti-HCV is positive, chronic infection should be confirmed by qualitative HCV RNA measurement (IIB and IIE).
With persistently increased ALT and negative viral markers, the workup should include anti-nuclear antibodies (ANAs) and iron and iron-binding capacity (or unsaturated iron-binding capacity; IIIB).
In patients under age 40, ceruloplasmin should also be measured (IIIB).
In patients negative for these markers, α1-antitrypsin (A1AT) phenotype may be of use (IIIB).
If these tests are negative or inconclusive, diagnostic liver biopsy should be performed (IIIB).
workup of patients without obvious cause for chronic hepatic injury
Nonalcoholic steatohepatitis (NASH).
The occurrence of chronic liver disease histologically resembling alcoholic hepatitis in patients without alcohol abuse has been termed NASH. It is the most common cause of chronic hepatic injury other than viruses and alcohol and the most common cause of cryptogenic cirrhosis (59)(69)(70)(78). Although NASH occurs most commonly in middle-aged women with obesity and/or diabetes, it also occurs in men and in patients without these risk factors (78). Patients with NASH commonly have abnormal lipid profiles, although normal results do not rule out this disease. It differs from alcoholic hepatitis in that the ALT activity is higher than AST (except in patients with cirrhosis) (79)(80)(81). Weight loss may cause significant improvement in enzyme results; in one study, a 1% reduction in weight produced a 8.1% decrease in ALT activity (82).
Recommendation
Biopsy is necessary to establish the diagnosis of NASH (IIB).
Hemochromatosis.
An autosomal recessive trait, hemochromatosis is the most common inherited genetic defect in persons of northern European ancestry (∼1:200 to 1:300 in the United States) (83). The vast majority of cases are produced by one of two point mutations of the HFE gene on chromosome 6. The majority (60–90%) of affected individuals are homozygous for the C282Y (845A) mutation, whereas a minority has compound heterozygosity for this mutation and the H63D (187G) mutation (84)(85). Screening involves detection of increased transferrin saturation (saturation = serum iron × 100/total iron-binding capacity) (86) or low unsaturated iron-binding capacity (87). A transferrin saturation cutoff of ≥45% or unsaturated iron-binding capacity cutoff ≤28 μmol/L (155 μg/dL) has a sensitivity of 90–100% for homozygosity for the C282Y mutation; if fasting specimens are used, the specificity is 43% (88)(89). A recent consensus conference recommended that definitive diagnosis be made by genetic analysis (90). Although several recent publications have shown the feasibility of hemochromatosis screening using transferrin saturation, most organizations and researchers do not currently recommend screening because of unresolved issues regarding their ability to convince young adults to be tested, the specificity and reproducibility of screening tests, and questions about natural history of untreated disease (90)(91)(92). Screening has been advocated by the College of American Pathologists (93), and has been estimated to save $3.19 per blood donor screened (94).
Recommendations
Initial evaluation for hemochromatosis should be by fasting serum transferrin saturation or unsaturated iron-binding capacity (IIB).
Transferrin saturation ≥45% or unsaturated iron-binding capacity ≤28 μmol/L (155 μg/dL) should be followed by analysis for HFE gene mutations (IIB).
Screening of the population may be beneficial but is not currently recommended pending clarification of screening benefits (IIB and IIE).
Wilson disease.
An autosomal recessive disorder, Wilson disease occurs in ∼1 in 30 000 individuals in Europe and North America. It is caused by a mutation of a gene on chromosome 13 coding for an ATPase needed for copper transport (95). Wilson disease may present as liver disease, neurologic problems, or with psychiatric symptoms, almost always before age 40. Most patients who present with liver disease do not have neurologic manifestations (96). The most common diagnostic finding is low plasma ceruloplasmin. Low concentrations also occur with malnutrition, protein loss, and advanced liver disease, and falsely normal values can occur with pregnancy, estrogen administration, and acute inflammation (97). Most references report low ceruloplasmin in 95% of homozygotes and 20% of heterozygotes (97)(98)(99). One study found normal ceruloplasmin in 35% of patients with chronic liver disease attributable to Wilson disease (confirmed by genetic studies in 80%) but in only 15% of patients with Wilson disease without overt liver involvement (96). Other expected findings in Wilson disease include increased serum free copper, decreased total serum copper, increased urine copper excretion, and increased liver copper content. These tests may also provide misleading results in patients with Wilson disease (96)(100). Multiple tests frequently are needed to establish the diagnosis.
Recommendations
Testing for Wilson disease with ceruloplasmin is indicated in patients under age 40 with chronic hepatic injury or fatty liver and negative workup for viral hepatitis, drug-induced liver injury, and hemochromatosis (IIB).
Screening for Wilson disease in all patients with chronic hepatic injury is not indicated (IIB and IIE).
Genetic marker testing may be useful in equivocal cases, but testing must be able to detect multiple mutations in the Wilson disease gene (IIIB).
AIH.
AIH is responsible for up to 18% of chronic hepatitis not attributable to viruses or alcohol (101). Several variants of AIH have been described (102). Type 1, found primarily in young and middle-aged women, is the most common form; it is associated with high titers of ANA and/or anti-smooth muscle antibody (ASMA). Type 2, found primarily in children, is common in western Europe but rare in the United States; it is associated with antibodies to liver-kidney microsomal antigen, but rarely with positive ANA or ASMA. Many patients with type 2 also have HCV infection. Type 3, found primarily in young women, is associated with systemic autoimmune disease in many cases. Most affected individuals lack ANA, ASMA, or anti-liver-kidney microsomal antibodies, but are positive for antibodies for soluble liver antigen. Standardized diagnostic criteria and a scoring system have been defined by an international panel (103). The classic features of the most common type 1 include increased aminotransferases; minimal or no increases in ALP; polyclonal hypergammaglobulinemia (at least 1.5 times the upper reference limit); no evidence of viral infection, risk factors for viral infection, or exposure to drugs or alcohol; and positive ANA or ASMA (at least 1:80) (103). Approximately 40% of patients with chronic HCV infection have a positive ANA or ASMA, usually in low titers (102). False-positive anti-HCV results have been reported in 60% of patients with AIH when second-generation tests are used, and in 20% when third-generation assays are used (104); anti-HCV typically disappears with successful treatment (105). In equivocal cases, HCV RNA (or a recombinant immunoblot assay) can be used to establish the diagnosis (104).
Recommendations
AIH should be suspected in patients with chronic hepatic injury and increased immunoglobulins and absence of viral markers or risk factors for viral hepatitis (IIIB).
The diagnosis of type 1 AIH can be clinically supported by positivity for either ANA or ASMA in high titers (IIIB).
Primary biliary cirrhosis and primary sclerosing cholangitis.
Primary biliary cirrhosis (PBC) and primary sclerosing cholangitis (PSC) are autoimmune diseases that cause destruction of bile ducts. Although PBC and PSC characteristically cause increases in ALP and γ-glutamyltransferase (EC 2.3.2.2), patients with these diseases may have increased AST and ALT and be considered to have chronic hepatitis. PBC is associated with destruction of intrahepatic bile ducts; it is often associated with other autoimmune disorders, particularly Sjogren syndrome (up to 80% of cases) (106). An autoimmune marker, anti-mitochondrial antibody (AMA), is found in almost all patients with PBC; although other diseases may be associated with positive AMA, in PBC the antibody is directed against the pyruvate dehydrogenase complex (so-called M2 type of AMA), particularly to dihydrolipoamide acetyltransferase (E2) and E3-binding protein (107). Approximately 5–10% of patients have features of both PBC and AIH (108)(109). PBC often is detected in asymptomatic individuals by a finding of increased ALP. AST and ALT are increased in approximately one-half of cases, although values are more than two times the reference limit in 20% (110). PSC is associated with damage to both intra- and extrahepatic bile ducts; 70% of cases are associated with inflammatory bowel disease (Crohn disease or ulcerative colitis) (111). Perinuclear anti-neutrophil cytoplasmic antibodies are found in approximately two-thirds of all cases (112). In PSC, antibodies commonly are directed against bactericidal/permeability-increasing protein, cathepsin G, and/or lactoferrin. There appears to be no prognostic significance to the different antibody specificities, although patients with cirrhosis more commonly have antibody to multiple antigens and to antigens other than lactoferrin (113). ASMA and ANA are also present in up to 70% of cases (114).
Recommendations
PBC or PSC should be suspected in patients with chronic cholestasis (IIIB).
The diagnosis can be clinically supported by positivity for AMA (PBC) or anti-neutrophil cytoplasmic antibodies (PSC) in high titers (IIIB).
A1AT deficiency.
A1AT is the most important protease inhibitor; congenital deficiency occurs in ∼1 in 1000 to 1 in 2000 persons of European ancestry. The gene for A1AT is located on chromosome 14 (115); deficiency is usually attributable to a single amino acid substitution that alters carbohydrate binding and impairs release from hepatocytes (116). The most important deficiency involves homozygosity for the Z variant, termed Pi (for protease inhibitor) ZZ. Deficiency is associated with emphysema and neonatal hepatitis (117); chronic hepatic injury with cirrhosis and HCC (118) have also been reported. Almost all Pi ZZ neonates have evidence of liver injury at birth; this usually resolves by age 12 years (117). In adults, 50% of Pi Z-positive individuals (either homozygotes or heterozygotes) develop cirrhosis, and 31% develop HCC (116). There is also an excess of Pi Z heterozygotes among patients referred for liver transplants, particularly among patients with cryptogenic cirrhosis, where ∼25% of patients are Pi Z positive (119). There is evidence, however, that A1AT deficiency or heterozygosity for Pi Z phenotype may not directly cause liver disease, but increase susceptibility to liver damage by other agents, especially viruses. Two controlled studies found the same frequency of Pi Z (either homozygous or heterozygous) in patients with liver disease and controls (120)(121). In a study of 164 patients with Pi Z, 40% had chronic liver disease; 87% were also positive for HCV antibodies or HBV markers, and only 11% had no other liver disease risk factors (122). Because A1AT is an acute phase reactant, quantitative concentrations may be falsely normal with infection or inflammation, and falsely low concentrations may occur with malnutrition, protein-losing states, or end stage liver disease. In one study, quantitative concentrations were normal in 42% of heterozygous Pi Z patients with liver disease (123). Testing for A1AT deficiency should use phenotype analysis rather than quantitative plasma concentration (116).
Recommendations
Testing for A1AT deficiency may be of benefit in patients with chronic hepatic injury and no other apparent cause, although the role of A1AT deficiency in liver disease in adults is not clearly defined (IIB).
Testing is especially important in neonates with evidence of hepatic injury (IIB).
Testing for A1AT variants should be performed by determination of phenotype (IIB).
Screening patients with chronic hepatic injury for A1AT deficiency is not recommended (IIIB and IIIE).
other viruses
Two other viruses have been suggested as possibly involved in the pathogenesis of chronic hepatitis: hepatitis G (HGV) and TT virus (TTV). Both viruses can be transmitted by transfusion, and chronic viremia is present with both. To date, evidence has suggested that infection with these viruses is common, but there is no clear indication that they play a role in liver injury. HGV (and the related GB virus type C) are members of the flavivirus family, as is HCV. HGV was first isolated from patients following transfusion, although most showed no evidence of liver injury (124)(125). HGV can also frequently be found in chronic hepatitis (126), but it does not appear to be a common cause of cryptogenic chronic liver disease (127). This may be because HGV RNA is rarely found in the liver in chronically viremic patients (128). TTV was first identified in patients with posttransfusion hepatitis (129). TTV DNA is found in 1–7% of blood donors in the United States (130)(131). The presence of TTV DNA is no more common in persons with acute non-A-E hepatitis than in other causes of acute hepatitis or in control patients (131)(132).
Recommendation
Testing for HGV or TTV, in other than a research setting, is not recommended (IIIE).
monitoring
Although ALT is the most clinically used laboratory test for monitoring liver injury, there often are considerable fluctuations in enzyme activities over time (particularly in chronic HCV infection) (60)(61). It is important to measure ALT repeatedly in chronic HCV before concluding that ALT is normal (75); 43% of chronically infected individuals have ALT values fluctuating between normal and abnormal, and 16% of those with normal ALT on their first two visits and 11% of those with normal ALT on their first three visits subsequently develop increased ALT (51). In patients with chronic HBV infection without increased ALT (chronic carriers), ∼10% will develop increased ALT on follow-up (53); ALT should therefore be measured periodically even if initially normal.
With both chronic HBV and HCV, clearance of viral markers is the most reliable method for detecting resolution of infection. In untreated hepatitis B, a small percentage of patients spontaneously clear viral antigens; in long-term studies, loss of HBeAg occurred in one-third to one-half of patients (45)(133)(134). In those that lose HBeAg, 5–10% will subsequently clear HBsAg over 10 years of follow-up (53)(135). HBeAg should be rechecked periodically if initially positive. If HBeAg is negative and anti-HBV e antigen antibody (anti-HBe) is positive, this may indicate either the beginning of viral clearance from the body or integration of HBV DNA into host DNA and loss of ability to form replicating virus. HBsAg and anti-HBs should be measured periodically to look for viral clearance because HBsAg will remain positive in those with integration of HBV DNA. In treatment of HBV, the likelihood of viral clearance is related to baseline ALT activity; patients with increased ALT are more likely to respond than those with initially normal ALT activity (136)(137). Successful treatment is associated with loss of HBV DNA, HBsAg, and HBeAg.
Although there is evidence that quantitative HBeAg correlates well with HBV DNA (138)(139)(140), quantitative HBeAg assays are not commercially available. HBeAg may disappear even in patients who show no response to therapy (141). Moreover, there is an increasing frequency of “pre-core” mutants that cannot produce HBeAg, particularly in endemic areas in Asia and the Mediterranean region (142). Patients infected with such mutants have anti-HBe but continue to have circulating HBV DNA. In infection with normal strains, HBV DNA remains detectable longer than does HBsAg in recovery (143). When viral DNA integrates into the host genome, HBsAg is still produced, although HBeAg and HBV DNA commonly are negative in plasma (144)(145). With lamivudine treatment, however, production of viral nucleic acid through reverse transcriptase is inhibited (145), although viral DNA concentrations in the hepatocytes are not changed (137). For these reasons, use of HBV DNA, HBeAg, and HBsAg may all be useful in monitoring patients with chronic HBV because no single test provides unequivocal evidence of viral clearance.
Most studies have shown that HCV RNA fluctuates over time but rarely varies by more than 1 log; in most cases, variation is <0.5 log (146)(147)(148). In patients tested repeatedly over several years, HCV RNA increases by an average of 0.25 log/year over time without treatment (149). In some series, however, up to a 3-log difference is seen in patients with increased ALT when HCV RNA is measured monthly (150); in approximately one-third of chronically infected patients, HCV RNA can fluctuate between a mean of 106 copies/mL and undetectable (151).
Currently, antiviral treatment is recommended for patients with chronic HCV infection who have increased ALT and more than mild inflammatory changes on biopsy. The most effective therapy currently available is combined ribavirin and interferon. Laboratory tests have been found helpful in predicting response to varying lengths of therapy and in detecting those who are not responding to treatment and in whom therapy should probably be discontinued. In those treated with combination therapy, both viral load and genotype have been found to identify patients who may respond to 24 rather than 48 weeks of therapy (152)(153).
In a combined analysis of these two studies, five factors were found useful in predicting response (Table 5 ). Persons with genotype 2 or 3, along with three or four other favorable risk factors, can be treated effectively with only 24 weeks of therapy; all other patients do better with 48 weeks of therapy (154). The best indicator of viral clearance is persistent absence of HCV RNA (determined by qualitative HCV RNA assays). The absence of HCV RNA 6 months after completion of treatment is associated with a <10% likelihood of recurrent HCV viremia (155). A decrease in viral load in the absence of clearance is not reliable evidence of treatment success; however, failure of HCV RNA to decline to <400 000 copies/mL by 12 weeks of therapy is associated with 100% likelihood of persistent HCV RNA at the end of treatment (154). An approach to monitoring treatment of patients with HCV by combination therapy is outlined in Fig. 2 . Some patients cannot take ribavirin; the only current treatment option for such patients is interferon monotherapy. With this form of treatment, failure of HCV RNA to fall to undetectable concentrations or failure of ALT to return to normal at 12 weeks after initiation of therapy is associated with a >95% likelihood of treatment failure, and is considered a reason to discontinue therapy (156).
Table 5.
Favorable and unfavorable risk factors in HCV treatment with interferon and ribavirin.
| Favorable factors |
| · Genotype 2 or 3 |
| · Viral load < median (3.5 × 106 copies/mL) |
| · Female gender |
| · Age <40 years |
| · No or only portal fibrosis |
| Unfavorable factors |
| · Genotype 1, 4, 5, 6 |
| · Viral load > median |
| · Male gender |
| · Age 40 or above |
| · Septal or more severe fibrosis |
From Poynard et al. (154).
Figure 2.

Management of therapy in chronic hepatitis C: laboratory testing depends on type of treatment given.
At present, combined ribavirin-interferon is recommended for all patients without contraindications. HCV RNA should be measured at 24 weeks of treatment; if positive, treatment should be discontinued. If negative, duration of treatment is based on number of favorable risk factors present (see Table 5 ). If four or more factors are favorable (and genotype is 2 or 3), treatment is stopped; in other cases, treatment is continued for 48 weeks. If −70 °C storage is available, specimens for HCV genotype and viral load can be obtained before treatment and frozen until after the 24-week testing is performed; if not, testing should be performed before therapy. In those patients who cannot tolerate ribavirin, monotherapy with interferon is used. ALT and qualitative HCV RNA should be checked after 12 weeks of therapy; if ALT remains increased or HCV RNA is detectable, treatment should be discontinued. There is no benefit to determination of quantitative HCV RNA or genotype when interferon monotherapy is used.
The optimal frequency of laboratory tests in patients with chronic hepatitis C has not been determined. The European Association for the Study of the Liver Consensus Conference on Hepatitis C recommends that complete blood counts and liver enzymes be performed every 6 months in untreated patients (157). The major complications of treatment with interferon are depression, thrombocytopenia, and hypothyroidism, whereas hemolytic anemia is the major complication of ribavirin therapy. The European Association for the Study of the Liver recommends complete blood counts weekly during the first 4 weeks of treatment and regular determinations after the first 4 weeks. They also recommend measurement of thyroid-stimulating hormone every 6 months during therapy.
Recommendations
In viral hepatitis, viral markers are the most reliable markers of resolution of hepatitis (IIB).
HCV RNA quantification and genotype are important determinants of duration of combination therapy. To reduce the expense of testing, if feasible, specimens should be obtained before treatment and stored at −70 °C pending results of treatment. If this is not possible, testing should be performed before treatment is begun (IIB and IIE).
In patients with HCV treated with interferon and ribavirin, qualitative HCV RNA should be measured after 24 weeks of treatment to determine potential responders. If genotype and quantitative HCV RNA were not performed but specimens were frozen for their analysis before treatment, those with negative HCV RNA and favorable risk factors should have those tests performed (IB and IE).
In patients with HCV treated with interferon monotherapy, qualitative HCV RNA and ALT should be measured after 12 weeks of treatment to determine nonresponders (IIB).
Following treatment in those with negative HCV RNA at 24 weeks, sensitive HCV RNA measurements (currently qualitative assays) should be performed 6 months after the end of treatment to document sustained virologic remission (IIB).
In untreated patients with HBV, HBeAg should be monitored periodically; once HBeAg is negative and anti-HBe is positive, HBsAg should be monitored periodically to determine viral clearance. With antiviral therapy, HBV DNA should also be used to document viral clearance (IIB).
In treated patients, a complete blood count with platelets should be measured every week for the first 4 weeks, then monthly thereafter. Thyroid-stimulating hormone should be measured every 3–6 months, or sooner if symptoms of thyroid dysfunction develop. Measurement of ALT should be performed at least monthly (IIIB).
ALT is the best marker of inflammatory activity available, but it is of limited utility in predicting degree of inflammation and of no use in estimating severity of fibrosis (IIB).
Cirrhosis
Chronic hepatitis is classified by histology based on activity of inflammation and degree of fibrosis; extent of fibrosis relates to likelihood of developing cirrhosis. Enzyme activities do not reflect severity of fibrosis, a major factor in prognosis, and there is at best a weak correlation between plasma ALT activity (158)(159)(160) or (in chronic HCV) HCV RNA concentrations (160)(161) and histological activity. At best, ALT explains only 30–50% of the variation in histologic activity, and there is considerable overlap in values in patients with mild, moderate, or severe activity (158)(159)(160), which relates to rate of progression of fibrosis (162).
Liver fibrosis is associated with deposition of several proteins in the liver. Among the proteins produced as part of fibrosis are collagen, laminin, elastin, and fibronectin; metalloproteinases and their inhibitors; and enzymes produced in collagen synthesis such as lysyl and prolyl hydroxylase. Various proteoglycans, such as hyaluronate, are also produced in the process of fibrosis. Numerous studies of plasma concentrations of proteoglycans, proteins of fibrosis, and their precursors (163)(164)(165)(166)(167)(168)(169)(170)(171)(172) have shown at best a weak correlation between marker concentrations and extent of fibrosis. Concentrations reflect degree of fibrogenesis at the time of sampling, and there is considerable overlap in values with varying degrees of fibrosis. A simple marker, calculated from routine monitoring tests, is the AST:ALT ratio. Several studies have shown that the ratio is typically <1 in patients with non-alcoholic hepatitis, but with progression to cirrhosis the ratio often increases to >1: the specificity of a ratio >1 for cirrhosis is 75–100%, with a sensitivity of 32–83% (65)(66)(67). In one study (67), the ratio increased with increasing fibrosis score. This appears to be attributable to a reduction of ALT production in damaged liver (173).
Other routine test results that predict likelihood of cirrhosis are thrombocytopenia and prolonged PT; an index using these two variables with the AST:ALT ratio has a sensitivity of 46% and a specificity of 98% for cirrhosis (65). Albumin is commonly measured in patients suspected of progressing to cirrhosis. Although it is not as sensitive as other markers, it is used as a marker of severity as part of the Child-Pugh classification of cirrhosis. α-Fetoprotein (AFP) is more likely to be increased as the degree of hepatic fibrosis increases (174), especially in cirrhosis; AFP >17.8 μg/L has a sensitivity of 35%, a specificity of 98.6%, and a positive predictive value of 97.7% for cirrhosis (175).
Recommendations
Biopsy is the only definitive marker of progression from chronic hepatitis to cirrhosis (IIB).
Laboratory markers of fibrosis should not be used except in research studies (IIIB and IIIE).
Markers of hepatic function that may indicate progression to cirrhosis (AST:ALT ratio, albumin, PT, platelet count) should be measured every 3–6 months in patients with chronic hepatitis (IIIB).
HCC
Primary liver cancer (HCC) is a serious late complication of chronic hepatic injury, particularly in cirrhosis caused by HBV, HCV, and hemochromatosis. Infrequently, HCC is seen in patients with chronic HCV and in asymptomatic HBV carriers without cirrhosis. It is the fifth most common malignancy worldwide and is particularly common in Eastern Asia and Africa (176). The incidence of HCC has increased by 70% in the United States over the past 20 years, particularly among younger patients (177), and it is increasing in other parts of the world as well (176). The risk of developing HCC in cirrhosis attributable to chronic HBV or HCV infection is 1.5% per year (178)(179). In a study of 448 cases of HCC, 75% occurred in patients with cirrhosis; however, in only 30% was cirrhosis recognized clinically before HCC was diagnosed (180). These data suggest that screening programs, if instituted, must include patients with chronic hepatic injury as well as patients with diagnosed cirrhosis. In one study, however, HCC developed only in 325 patients with severe chronic hepatitis or cirrhosis, and not in any of 800 patients with mild or moderate chronic hepatitis (181). Because patients with normal ALT generally have mild inflammation on biopsy (17)(61)(63), it is reasonable to exclude from screening those persons without cirrhosis and with normal ALT or less than severe hepatitis on biopsy. Other risk factors include male gender and age >55 years.
The prognosis of patients with HCC detected by development of symptoms is grim, with few patients surviving >6 months. Detection of small tumors offers the potential for curative resection and forms the rationale for considering screening. Current practice suggests measurement of AFP and ultrasound of the liver every 6 months (182). Unfortunately, AFP interpretation is complicated by intermittent increases in AFP concentrations in 12–13% of patients with chronic HBV or HCV (183), often (but not always) associated with transient increases in ALT (184). A Consensus Development workshop recommended screening chronic HBsAg carriers at least once, and preferably twice, yearly with AFP only, whereas patients with other risk factors (known cirrhosis, family history) should have both AFP and ultrasound (185). In chronic hepatic injury, a high risk of HCC is present in patients with hemochromatosis or with cirrhosis caused by HBV, HCV, and alcohol abuse. Other causes of chronic hepatic injury and cirrhosis have lower risks of HCC (186).
In Western countries, the predictive value of AFP is low, often in the range of 10–30%, with sensitivity of AFP between 40% and 80% (187)(188)(189)(190)(191)(192). In 147 patients with cirrhosis, none of the 30 patients with HCC had AFP >105 μg/L at the time of diagnosis and 60% had AFP <20 μg/L; however, the frequency of HCC in patients with AFP <50 μg/L was 17% compared with 42% in those with higher AFP (192). In another study of 260 patients with cirrhosis, HCC developed in 26% of patients with initial AFP <20 μg/L, but 46% in those with higher concentrations. Moreover, those with even transient increases above 100 μg/L had a significantly higher risk of HCC than those whose AFP was consistently <20 μg/L (193). A decision analysis on published reports of screening for HCC in Western patients with compensated cirrhosis concluded that, for patients with a likelihood of survival of 85% at 5 years, screening would likely add 3–9 months to average life expectancy at a cost of $26 000 to $55 000 per year of life gained, figures that compare favorably to those for colon cancer and breast cancer screening (194). In patients with a lower likelihood of survival, screening provided minimal or no gain in life expectancy and does not appear indicated. A systematic analysis of all published studies concluded that there are inadequate data to determine the benefit of screening for HCC among patients with chronic liver disease (195). If screening is used, a frequency of testing of every 6 months appears to be optimal based on observed doubling times of HCC, reported to average ∼3–5 months (196)(197)(198).
Des-γ-carboxy prothrombin has also been suggested as a screening test. Concentrations are increased occasionally in chronic liver disease, but there is less overlap with values seen in HCC than for AFP (199)(200). Occasional high concentrations are encountered in metastatic carcinoma to the liver, but they usually are minimally increased. Although des-γ-carboxy prothrombin appears less sensitive (50–70%) than AFP, it is more specific. There is poor correlation between AFP and des-γ-carboxy prothrombin, and some tumors are detected only by des-γ-carboxy prothrombin (199)(200)(201)(202)(203). Vitamin K deficiency can also cause substantial increases; repeating testing after administration of vitamin K improves specificity (199)(200)(204)(205). Recently, a more sensitive immunoassay has shown promise in detection of small HCC, with positivity in 27% of cases compared with 3% with older assays (206). Assays for des-γ-carboxy prothrombin are not widely available, in contrast to AFP assays. Other laboratory tests, including AFP variants (207) and lectin affinity chromatography of ALP (208), have been evaluated in too few patients to make definitive recommendations. A recent study identified high concentrations of abnormal forms of γ-glutamyltransferase in 78 of 91 patients with HCC but in only 2.5% of 116 patients with other liver diseases (209).
Recommendations
Screening for HCC is of questionable benefit in Western populations (IIB and IIE).
Screening should be confined to high-risk patients (those with severe chronic hepatitis or cirrhosis attributable to alcohol, HBV, HCV, or hemochromatosis) who are candidates for treatment of HCC, if detected (IIIB and IIIE).
If screening is used, measurement of AFP and ultrasound at intervals no more frequently than every 6 months are recommended (IIB).
There currently are few data to support the use of other tests (IIIB).
Acknowledgments
Development and publication of these guidelines were supported by grants from Abbot Diagnostics; Diasorin, Inc.; Bayer Diagnostics (formerly Chiron Diagnostics); Innogenetics, Inc.; and Ortho Clinical Diagnostics. The following individuals reviewed the guidelines at various stages of their development and offered helpful comments and modifications: Miriam Alter, Henry C. Bodenheimer, Thomas D. Boyer, Max A. Chernesky, Gary L. Davis, Jean C. Edmond, Stuart C. Gordon, Norman D. Grace, F. Blaine Hollinger, Donald M. Jensen, Lawrence A. Kaplan, Jacob Korula, Karen Lindsay, Brian J. McMahon, Jan M. Novak, Melissa Palmer, Eve A. Roberts, James R. Spivey, Thomas A. Shaw-Stiffel, and Myron Warshaw. Specific comments were provided by the following individuals during open discussion at the AACC Annual Meeting: Ed Ashwood, Bill Brock, Thomas Burgess, Jack Goldberg, Ajit Golwikar, Neal Greenberg, Michael Heinz, Richard Horowitz, Graham Johns, Ronald Lee, Steve Lobell, Greg Post, Phil Rosenthal, Norbert Tietz, Mark Walter, Earl Weissman, William Winter, and Jeffery Youn.
Footnotes
An Approved Laboratory Medicine Practice Guideline of the National Academy of Clinical Biochemistry.
Presented in part at the American Association for Clinical Chemistry Annual Meeting, July 25–26, 1999, New Orleans, LA.
A monograph of these guidelines will be published by the National Academy of Clinical Biochemistry. Reprints are not available from the authors.
Nonstandard abbreviations: HCC, hepatocellular carcinoma; AST, aspartate aminotransferase (EC 2.6.1.1); ALT, alanine aminotransferase (EC 2.6.1.2); ALP, alkaline phosphatase (EC 3.1.3.1); PT, prothrombin time; HAV, hepatitis A virus; HBV, hepatitis B virus; HCV, hepatitis C virus; HBc, hepatitis B virus core antigen; HBsAg, hepatitis B virus surface antigen; HDV, hepatitis D virus; AIH, autoimmune hepatitis; HEV, hepatitis E virus; CMV, cytomegalovirus; EBV, Epstein-Barr virus; HBeAg, hepatitis B virus e antigen; ANA, anti-nuclear antibody; A1AT, α1-antitrypsin; NASH, nonalcoholic steatohepatitis; ASMA, anti-smooth muscle antibody; PBC, primary biliary cirrhosis; PSC, primary sclerosing cholangitis; AMA, anti-mitochondrial antibody; Pi, protease inhibitor; HGV, hepatitis G virus; TTV, TT virus; and AFP, α-fetoprotein.
References
- 1.Dufour DR, Lott JA, Nolte FS, Gretch DR, Koff RS, Seeff LB. Diagnosis and monitoring of hepatic injury. I. Performance characteristics of laboratory tests. Clin Chem 2000;46:2027-2049. [PubMed] [Google Scholar]
- 2.Ellis G, Goldberg DM, Spooner RJ, Ward AM. Serum enzyme tests in diseases of the liver and biliary tree. Am J Clin Pathol 1978;70:248-258. [DOI] [PubMed] [Google Scholar]
- 3.Rozen P, Korn RJ, Zimmerman HJ. Computer analysis of liver function tests and their interrelationship in 347 cases of viral hepatitis. Isr J Med Sci 1970;6:67-79. [PubMed] [Google Scholar]
- 4.Whitehead MW, Hawkes ND, Hainsworth I, Kingham JGC. A prospective study of the causes of notably raised aspartate aminotransferase of liver origin. Gut 1999;45:129-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anciaux ML, Pelletier AG, Attali P, Meduri B, Liguory C, Etienne JP. Prospective study of clinical and biochemical features of symptomatic choledocholithiasis. Dig Dis Sci 1986;31:449-453. [DOI] [PubMed] [Google Scholar]
- 6.Fortson WC, Tedesco FJ, Starnes EC, Shaw CT. Marked elevation of serum transaminase activity associated with extrahepatic biliary tract disease. J Clin Gastroenterol 1985;7:502-505. [DOI] [PubMed] [Google Scholar]
- 7.Borsch G, Baier J, Glocke M, Nathusius W, Gerhardt W. Graphical analysis of laboratory data in the differential diagnosis of cholestasis: a computer-assisted prospective study. J Clin Chem Clin Biochem 1988;26:509-519. [DOI] [PubMed] [Google Scholar]
- 8.Mendenhall CL. Alcoholic hepatitis. The VA Cooperative Study Group on Alcoholic Hepatitis. Clin Gastroenterol 1981;10:417-441. [PubMed] [Google Scholar]
- 9.Mihas AA, Doos WG, Spenney JG. Alcoholic hepatitis—a clinical and pathological study of 142 cases. J Chronic Dis 1978;31:461-472. [DOI] [PubMed] [Google Scholar]
- 10.Goldberg S, Mendenhall C, Anderson S, Garcia-Pont P, Kiernan T, Seeff L, et al. VA Cooperative Study on Alcoholic Hepatitis. IV. The significance of clinically mild alcoholic hepatitis—describing the population with minimal hyperbilirubinemia. Am J Gastroenterol 1986;81:1029-1034. [PubMed] [Google Scholar]
- 11.Nissenbaum M, Chedid A, Mendenhall C, Gartside P. Prognostic significance of cholestatic alcoholic hepatitis. The VA Cooperative Study Group No. 119. Dig Dis Sci 1990;35:891-896. [DOI] [PubMed] [Google Scholar]
- 12.Cohen JA, Kaplan MM. The SGOT/SGPT ratio—an indicator of alcoholic liver disease. Dig Dis Sci 1979;24:835-838. [DOI] [PubMed] [Google Scholar]
- 13.Stewart JS, Farrow LJ, Clifford RE, Lamb SG, Coghill NF, Lindon RL, et al. A three-year survey of viral hepatitis in West London. Q J Med 1978;47:365-384. [PubMed] [Google Scholar]
- 14.Lednar WM, Lemon SM, Kirkpatrick JW, Redfield RR, Fields ML, Kelley PW. Frequency of illness associated with epidemic hepatitis A virus infections in adults. Am J Epidemiol 1985;122:226-233. [DOI] [PubMed] [Google Scholar]
- 15.Gitlin N. Hepatitis B: diagnosis, prevention, and treatment. Clin Chem 1997;43:1500-1506. [PubMed] [Google Scholar]
- 16.McMahon BJ, Alward WL, Hall DB, Heyward WL, Bender TR, Francis DP, et al. Acute hepatitis B virus infection: relation of age to the clinical expression of disease and subsequent development of the carrier state. J Infect Dis 1985;151:599-603. [DOI] [PubMed] [Google Scholar]
- 17.Hoofnagel JH. Hepatitis C: the clinical spectrum of disease. Hepatology 1997;26(Suppl 1):15-20. [DOI] [PubMed] [Google Scholar]
- 18.Seeff LB, Wright EC, Zimmerman HJ, McCollum VA. Cooperative Study of Post-Transfusion Hepatitis, 1969–1974: incidence and characteristics of hepatitis and responsible risk factors. Am J Med Sci 1975;270:355-362. [DOI] [PubMed] [Google Scholar]
- 19.Gitlin N, Serio KM. Ischemic hepatitis: widening horizons. Am J Gastroenterol 1992;87:831-836. [PubMed] [Google Scholar]
- 20.Tygstrup N, Ranek L. Assessment of prognosis in fulminant hepatic failure. Semin Liver Dis 1986;6:129-137. [DOI] [PubMed] [Google Scholar]
- 21.Dymock IW, Tucker JS, Woolf IL, Poller L, Thomson JM. Coagulation studies as a prognostic index in acute liver failure. Br J Haematol 1975;29:385-395. [DOI] [PubMed] [Google Scholar]
- 22.Horak W, Waldram R, Murray-Lyon IM, Schuster E, Williams R. Kinetics of (14C) cholic acid in fulminant hepatic failure: a prognostic test. Gastroenterology 1976;71:809-813. [PubMed] [Google Scholar]
- 23.O’Grady JG, Alexander GJM, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989;97:439-445. [DOI] [PubMed] [Google Scholar]
- 24.Munoz SJ. Prothrombin time in fulminant hepatic failure. Gastroenterology 1991;100:1480-1481. [PubMed] [Google Scholar]
- 25.Kovacs MJ, Wong A, MacKinnon K, Weir K, Keeney M, Boyle E, et al. Assessment of the validity of the INR system for patients with liver impairment. Thromb Haemost 1994;71:727-730. [PubMed] [Google Scholar]
- 26.Dufour DR, Teot L. Laboratory identification of ischemic hepatitis (shock liver) [Abstract]. Clin Chem 1988;34:1287. [Google Scholar]
- 27.Singer AJ, Carracio TR, Mofenson HC. The temporal profile of increased transaminase levels in patients with acetaminophen-induced liver dysfunction. Ann Emerg Med 1995;26:49-53. [DOI] [PubMed] [Google Scholar]
- 28.Harrison PM, O’Grady JG, Keays RT, Alexander GJ, Williams R. Serial prothrombin time as prognostic indicator in paracetamol induced fulminant hepatic failure. Br Med J 1990;301:964-968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noskin GA. Prevention, diagnosis, and management of viral hepatitis: a guide for primary care physicians. Arch Fam Med 1995;4:923-934. [DOI] [PubMed] [Google Scholar]
- 30.Zimmerman HJ. Hepatotoxicity: the adverse effects of drugs and other chemicals on the liver, 2nd ed 1999:789pp Lippincott Williams & Wilkins Philadelphia. . [Google Scholar]
- 31.Lemon SM, Brown CD, Brooks DS, Simms TE, Bancroft WH. Specific immunoglobulin M response to hepatitis A virus determined by solid phase radioimmunoassay. Infect Immun 1980;28:927-936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skinhoj P, Mikkelsen F, Hollinger FB. Hepatitis A in Greenland: importance of specific antibody testing in epidemiologic surveillance. Am J Epidemiol 1977;105:140-147. [DOI] [PubMed] [Google Scholar]
- 33.Koff RS. Seroepidemiology of hepatitis A in the United States. J Infect Dis 1995;171(Suppl 1):19-23. [DOI] [PubMed] [Google Scholar]
- 34.Lemon SM, Gates NL, Simms TE, Bancroft WH. IgM antibody to hepatitis B core antigen as a diagnostic parameter of acute infection with hepatitis B virus. J Infect Dis 1981;143:803-809. [DOI] [PubMed] [Google Scholar]
- 35.Seeff LB, Beebe GW, Hoofnagle JH, Norman JE, Buskell-Bales Z, Waggoner JG, et al. A serologic follow-up of the 1942 epidemic of post-vaccination hepatitis in the United States Army. N Engl J Med 1987;316:965-970. [DOI] [PubMed] [Google Scholar]
- 36.Gretch DR, dela Rosa C, Carithers RL, Jr, Wilson RA, Williams B, Corey L. Assessment of hepatitis C viremia using molecular amplification technologies: correlations and clinical implications. Ann Intern Med 1995;123:321-329. [DOI] [PubMed] [Google Scholar]
- 37.Villano SA, Vlahov D, Nelson KE, Cohn S, Thomas DL. Persistence of viremia and the importance of long-term follow-up after acute hepatitis C infection. Hepatology 1999;29:908-914. [DOI] [PubMed] [Google Scholar]
- 38.Alter MJ, Margolis HS, Krawczynski K, Judson FN, Mares A, Alexander WJ, et al. The natural history of community acquired hepatitis C in the United States. N Engl J Med 1992;327:1899-1905. [DOI] [PubMed] [Google Scholar]
- 39.London WT, Evans AA. The epidemiology of hepatitis viruses B, C, and D. Clin Lab Med 1996;16:251-271. [PubMed] [Google Scholar]
- 40.Fry SW, Seeff LB. Hepatotoxicity of analgesics and anti-inflammatory agents. Gastroenterol Clin North Am 1995;24:875-905. [PubMed] [Google Scholar]
- 41.Zimmerman HJ, Maddrey WC. Acetaminophen (paracetamol) hepatotoxicity with regular intake of alcohol: analysis of instances of therapeutic misadventure. Hepatology 1995;22:767-773. [PubMed] [Google Scholar]
- 42.Fuchs S, Bogomolski-Yahalom V, Paltiel O, Ackerman Z. Ischemic hepatitis: clinical and laboratory observations of 34 patients. J Clin Gastroenterol 1998;26:183-186. [DOI] [PubMed] [Google Scholar]
- 43.Johnson RD, O’Connor ML, Kerr RM. Extreme serum elevations of aspartate aminotransferase. Am J Gastroenterol 1995;90:1244-1245. [PubMed] [Google Scholar]
- 44.Silva MO, Roth D, Reddy KR, Fernandez JA, Albores-Saavedra J, Schiff ER. Hepatic dysfunction accompanying acute cocaine intoxication. J Hepatol 1991;12:312-315. [DOI] [PubMed] [Google Scholar]
- 45.Liaw YF, Chu CM, Su IJ, Huang MJ, Lin DY, Chang-Chien CS. Clinical and histological events preceding hepatitis B e antigen seroconversion in chronic type B hepatitis. Gastroenterology 1983;84:216-219. [PubMed] [Google Scholar]
- 46.Yuki N, Hayashi N, Moribe T, Matsushita Y, Tabata T, Inoue T, et al. Relation of disease activity during chronic hepatitis C infection to complexity of hypervariable region 1 quasispecies. Hepatology 1997;25:439-444. [DOI] [PubMed] [Google Scholar]
- 47.Clermont RJ, Chalmers TC. The transaminase tests in liver disease. Medicine 1967;46:197-207. [DOI] [PubMed] [Google Scholar]
- 48.Tong MJ, el-Farra NS, Crew MI. Clinical manifestations of hepatitis A. Recent experience in a community teaching hospital. J Infect Dis 1995;171(Suppl 1):15-18. [DOI] [PubMed] [Google Scholar]
- 49.Sjogren MH, Tanno H, Foy O, Sileoni S, Cohen BD, Burke DS, et al. Hepatitis A virus in stool during clinical relapse. Ann Intern Med 1987;106:221-226. [DOI] [PubMed] [Google Scholar]
- 50.Glikson M, Galun E, Oren R, Tur-Kaspa R, Shouval D. Relapsing hepatitis A: review of 14 cases and literature survey. Medicine 1992;71:14-23. [DOI] [PubMed] [Google Scholar]
- 51.Inglesby TV, Rai R, Astemborski J, Gruskin L, Nelson KE, Vlahov D, Thomas DL. A prospective community-based evaluation of liver enzymes in individuals with hepatitis C after drug use. Hepatology 1999;29:590-596. [DOI] [PubMed] [Google Scholar]
- 52.Davis GL, Hoofnagle JH. Reactivation of chronic type B hepatitis presenting as acute viral hepatitis. Ann Intern Med 1985;102:762-765. [DOI] [PubMed] [Google Scholar]
- 53.de Franchis R, Meucci G, Vecchi M, Tatarella M, Colombo M, Del Ninno E, et al. The natural history of asymptomatic hepatitis B surface antigen carriers. Ann Intern Med 1993;118:191-194. [DOI] [PubMed] [Google Scholar]
- 54.Weiss JS, Gautam A, Lauff JJ, Sundberg MW, Jatlow P, Boyer JL, Seligson D. The clinical importance of a protein-bound fraction of serum bilirubin in patients with hyperbilirubinemia. N Engl J Med 1983;309:147-150. [DOI] [PubMed] [Google Scholar]
- 55.Van Hootegem P, Fevery J, Blanckaert N. Serum bilirubins in hepatobiliary disease: comparison with other liver function tests and changes in the postobstructive period. Hepatology 1985;5:112-117. [DOI] [PubMed] [Google Scholar]
- 56.Gordon SC, Reddy KR, Schiff L, Schiff ER. Prolonged intrahepatic cholestasis secondary to acute hepatitis A. Ann Intern Med 1984;101:635-637. [DOI] [PubMed] [Google Scholar]
- 57.O’Grady JG, Alexander GJM, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989;97:4439-4445. [DOI] [PubMed] [Google Scholar]
- 58.Adolph L, Lorenz R. Enzyme diagnosis in diseases of the heart, liver, and pancreas 1982:9-27 Karger New York. . [Google Scholar]
- 59.Van Ness MM, Diehl AM. Is liver biopsy useful in the evaluation of patients with chronically elevated liver enzymes?. Ann Intern Med 1989;111:473-478. [DOI] [PubMed] [Google Scholar]
- 60.Alter HJ, Conry-Cantilena C, Melpolder J, Tan D, Van Raden M, Herion D, et al. Hepatitis C in asymptomatic blood donors. Hepatology 1997;26(Suppl 1):29-33. [DOI] [PubMed] [Google Scholar]
- 61.Mathurin P, Moussalli J, Cadranel J-F, Thibault V, Charlotte F, Dumouchel P, et al. Slow progression rate of fibrosis in hepatitis C patients with persistently normal alanine aminotransferase activity. Hepatology 1998;27:568-572. [DOI] [PubMed] [Google Scholar]
- 62.Kenny-Walsh E. Clinical outcomes after hepatitis C infection from contaminated anti-D immune globulin. The Irish Hepatology Research Group. N Engl J Med 1999;340:1228-1233. [DOI] [PubMed] [Google Scholar]
- 63. Centers for Disease Control and Prevention. Recommendations for the prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR 1998;47(No. RR-19):1-39. [PubMed] [Google Scholar]
- 64.Quinn PG, Johnston DE. Detection of chronic liver disease: costs and benefits. Gastroenterologist 1997;5:58-77. [PubMed] [Google Scholar]
- 65.Bonacini M, Hadi G, Govindarajan S, Lindsay KL. Utility of a discriminant score for diagnosing advanced fibrosis or cirrhosis in patients with chronic hepatitis C infection. Am J Gastroenterol 1997;92:1302-1304. [PubMed] [Google Scholar]
- 66.Williams AL, Hoofnagle JH. Ratio of serum aspartate to alanine aminotransferase in chronic hepatitis. Relationship to cirrhosis. Gastroenterology 1988;95:734-739. [DOI] [PubMed] [Google Scholar]
- 67.Sheth SG, Glamm SL, Gordon FD, Chopra S. AST/ALT ratio predicts cirrhosis in patients with chronic hepatitis C virus infection. Am J Gastroenterol 1998;93:44-48. [DOI] [PubMed] [Google Scholar]
- 68.Giannini E, Botta F, Fasoli A, Ceppa P, Risso D, Lantieri PB, et al. Progressive liver functional impairment is associated with an increase in AST/ALT ratio. Dig Dis Sci 1999;44:1249-1253. [DOI] [PubMed] [Google Scholar]
- 69.Sampliner RE, Deluk D, Harrow EJ. Blood donors with elevated alanine aminotransferase levels—persistence of elevation and liver histology [Abstract]. Gastroenterology 1983;84:1394. [Google Scholar]
- 70.Hultcrantz R, Glaumann H, Lindberg G, Nilsson LH. Liver investigation in 149 asymptomatic patients with moderately elevated activities of serum aminotransferases. Scand J Gastroenterol 1986;21:109-113. [DOI] [PubMed] [Google Scholar]
- 71.Friedman LS, Dienstag JL, Watkins E, Hinkle CA, Spiers JA, Rieder SV, Huggins CE. Evaluation of blood donors with elevated serum alanine aminotransferase levels. Ann Intern Med 1987;107:137-144. [DOI] [PubMed] [Google Scholar]
- 72.Hay JE, Czaja AJ, Rakela J, Ludwig J. The nature of unexplained chronic aminotransferase elevations of a mild to moderate degree in asymptomatic patients. Hepatology 1989;9:193-197. [DOI] [PubMed] [Google Scholar]
- 73.Kundrotas LW, Clement DJ. Serum alanine aminotransferase (ALT) elevation in asymptomatic US Air Force basic trainee blood donors. Dig Dis Sci 1993;38:2145-2150. [DOI] [PubMed] [Google Scholar]
- 74.Dufour DR. Effects of habitual exercise on routine laboratory tests [Abstract]. Clin Chem 1998;44:A136. [Google Scholar]
- 75.McMahon BJ, Christensen C, Gretch D, Williams JL, Sullivan DG, Bruden D, et al. Alanine aminotransferase (ALT) levels over time in anti-HCV-positive persons who are HCV RNA positive compared with persons who are HCV RNA negative but RIBA positive [Abstract]. Hepatology 1999;30:358A.10421641 [Google Scholar]
- 76.Cauza E, Maier-Dobersberger T, Polli C, Kaserer K, Kramer L, Ferenci P. Screening for Wilson’s disease in patients with liver diseases by serum ceruloplasmin. J Hepatol 1997;27:358-362. [DOI] [PubMed] [Google Scholar]
- 77.Mathiesen UL, Franzen LE, Fryden A, Foberg U, Bodemar G. The clinical significance of slightly to moderately increased liver transaminase values in asymptomatic patients. Scand J Gastroenterol 1999;34:85-91. [DOI] [PubMed] [Google Scholar]
- 78.Sheth SG, Gordon FD, Chopra S. Nonalcoholic steatohepatitis. Ann Intern Med 1997;126:137-145. [DOI] [PubMed] [Google Scholar]
- 79.Bacon BR, Farahvash MJ, Janney CJ, Neuschwander-Tetri BA. Nonalcoholic steatohepatitis: an expanded clinical entity. Gastroenterology 1994;107:1103-1109. [DOI] [PubMed] [Google Scholar]
- 80.Sorbi D, Boynton J, Lindor KD. The ratio of aspartate aminotransferase to alanine aminotransferase: potential value in differentiating nonalcoholic steatohepatitis from alcoholic liver disease. Am J Gastroenterol 1999;94:1018-1022. [DOI] [PubMed] [Google Scholar]
- 81.Pinto HC, Baptista A, Camilo ME, Valente A, Saragoca A, de Moura MC. Nonalcoholic steatohepatitis: clinicopathological comparison with alcoholic hepatitis in ambulatory and hospitalized patients. Dig Dis Sci 1996;41:172-179. [DOI] [PubMed] [Google Scholar]
- 82.Palmer M, Schaffner F. Effect of weight reduction on hepatic abnormalities in overweight patients. Gastroenterology 1990;99:1408-1413. [DOI] [PubMed] [Google Scholar]
- 83.McLaren CE, Gordeuk VR, Looker AC, Hasselblad V, Edwards CQ, Griffen LM, et al. Prevalence of heterozygotes for hemochromatosis in the white population of the United States. Blood 1995;86:2021-2027. [PubMed] [Google Scholar]
- 84.Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996;13:399-408. [DOI] [PubMed] [Google Scholar]
- 85.Crawford DH, Jazwinska EC, Cullen LM, Powell LW. Expression of HLA-linked hemochromatosis in subjects homozygous or heterozygous for the C282Y mutation. Gastroenterology 1998;114:1003-1008. [DOI] [PubMed] [Google Scholar]
- 86.Milman N, Albeck MJ. Distinction between homozygous and heterozygous subjects with hereditary haemochromatosis using iron status markers and receiver operating characteristic (ROC) analysis. Eur J Clin Chem Clin Biochem 1995;33:95-98. [DOI] [PubMed] [Google Scholar]
- 87.Witte DL. Mild liver enzyme abnormalities: eliminating hemochromatosis as cause. Clin Chem 1997;43:1535-1538. [PubMed] [Google Scholar]
- 88.Olynyk JK, Cullen DJ, Aquilia S, Rossi E, Summerville L, Powell LW. A population-based study of the clinical expression of the hemochromatosis gene. N Engl J Med 1999;341:718-724. [DOI] [PubMed] [Google Scholar]
- 89.Adams PC, Kertesz AE, McLaren CE, Barr R, Bamford A, Chakrabarti S. Population screening for hemochromatosis: a comparison of unbound iron binding capacity, transferrin saturation, and C282Y genotyping in 5,211 blood donors. Hepatology 2000;31:1160-1164. [DOI] [PubMed] [Google Scholar]
- 90.Bacon BR, Powell LW, Adams PC, Kresina TF, Hoofnagle JH. Molecular medicine and hemochromatosis: at the crossroads. Gastroenterology 1999;116:193-207. [DOI] [PubMed] [Google Scholar]
- 91.Burke W, Thomson E, Khoury MJ, McDonnell SM, Press N, Adams PC, et al. Hereditary hemochromatosis: gene discovery and its implications for population-based screening. JAMA 1998;280:172-178. [DOI] [PubMed] [Google Scholar]
- 92.Cogswell ME, McDonnell SM, Khoury MJ, Franks AL, Burke W, Brittenham G. Iron overload, public health, and genetics: evaluating the evidence for hemochromatosis screening. Ann Intern Med 1998;129:971-999. [DOI] [PubMed] [Google Scholar]
- 93.Witte DL, Crosby WH, Edwards CQ, Fairbanks VG, Mitros FA. Practice guideline development task force of the College of American Pathologists. Hereditary hemochromatosis. Clin Chim Acta 1996;245:139-200. [DOI] [PubMed] [Google Scholar]
- 94.Adams PC, Gregor JC, Kertesz AE, Valberg LS. Screening blood donors for hereditary hemochromatosis: decision analysis model based on a 30-year database. Gastroenterology 1995;109:177-188. [DOI] [PubMed] [Google Scholar]
- 95.Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATP-ase similar to the Menkes gene. Nat Genet 1993;5:327-337. [DOI] [PubMed] [Google Scholar]
- 96.Shilsky ML. Wilson disease: genetic basis of copper toxicity and natural history. Semin Liver Dis 1996;16:83-95. [DOI] [PubMed] [Google Scholar]
- 97.Gibbs K, Walshe JM. A study of the caeruloplasmic concentrations found in 75 patients with Wilson’s disease, their kindreds, and various control groups. Q J Med 1979;48:447-463. [PubMed] [Google Scholar]
- 98.Sass-Korstak A, Bearn AG. Hereditary disorders of copper metabolism. Stanbury JB Wyngaarden JB Fredrickson DS eds. The metabolic bases of inherited disease, 4th ed 1978:1098-1126 McGraw-Hill New York. . [Google Scholar]
- 99.Scheinberg JH, Sternlieb I. Wilson’s disease 1984:17-24 WB Saunders Philadelphia. . [Google Scholar]
- 100.Dufour J-F, Kaplan MM. Muddying the water: Wilson’s disease challenges will not soon disappear. Gastroenterology 1997;113:348-350. [PubMed] [Google Scholar]
- 101.Czaja AJ, Carpenter HA, Manns MP. Antibodies to soluble liver antigen, P450II5, and mitochondrial complexes in chronic hepatitis. Gastroenterology 1993;105:1522-1528. [DOI] [PubMed] [Google Scholar]
- 102.Czaja AJ. Autoimmune hepatitis: evolving concepts and treatment strategies. Dig Dis Sci 1995;40:435-456. [DOI] [PubMed] [Google Scholar]
- 103.Johnson PJ, McFarlane IG. Meeting report: International Autoimmune Hepatitis Group. Hepatology 1993;18:998-1008. [DOI] [PubMed] [Google Scholar]
- 104.Czaja AJ, Magrin S, Fabiano C, Fiorentino G, Diquattro O, Craxi A, Pagliaro L. Hepatitis C virus infection as a determinant of behavior in type I autoimmune hepatitis. Dig Dis Sci 1995;40:33-40. [DOI] [PubMed] [Google Scholar]
- 105.Czaja AJ, Taswell HF, Rakela J, Rabe D. Duration and specificity of antibodies to hepatitis C virus in chronic active hepatitis. Gastroenterology 1992;102:1675-1679. [DOI] [PubMed] [Google Scholar]
- 106.Neuberger J. Primary biliary cirrhosis. Lancet 1997;350:875-879. [DOI] [PubMed] [Google Scholar]
- 107.Yeaman SJ, Fussey SPM, Danner DJ, James OFW, Mutimer DJ, Bassendine MF. Primary biliary cirrhosis: identification of two major M2 mitochondrial autoantigens. Lancet 1988;i:1067-1070. [DOI] [PubMed] [Google Scholar]
- 108.Chazouilleres O, Wendum d, Serfaty L, Montembault S, Rosmordue O, Poupon R. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome: clinical features and response to therapy. Hepatology 1998;28:296-301. [DOI] [PubMed] [Google Scholar]
- 109.Czaja AJ. Frequency and nature of the variant syndromes of autoimmune liver disease. Hepatology 1998;28:360-365. [DOI] [PubMed] [Google Scholar]
- 110.Lohse AW, zum Buschenfelde KH, Franz B, Kanzler S, Gerken G, Dienes HP. Characterization of the overlap syndrome of primary biliary cirrhosis (PBC) and autoimmune hepatitis: evidence for it being a hepatitis form of PBC in genetically susceptible individuals. Hepatology 1999;29:1078-1084. [DOI] [PubMed] [Google Scholar]
- 111.Ponsioen CI, Tytgat GN. Primary sclerosing cholangitis: a clinical review. Am J Gastroenterol 1998;93:515-523. [DOI] [PubMed] [Google Scholar]
- 112.Mulder AH, Horst G, Haagsma EB, Limburg PC, Kleibeuker JH, Kallenberg CG. Prevalence and characterization of neutrophil cytoplasmic antibodies in autoimmune liver diseases. Hepatology 1993;17:411-417. [PubMed] [Google Scholar]
- 113.Chapman RW, Cottone M, Selby WS, Shepherd HA, Sherlock S, Jewell DP. Serum autoantibodies, ulcerative colitis, and primary sclerosing cholangitis. Gut 1986;27:86-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Roozendaal C, Van Milligen de Wit AW, Haagsma EB, Horst G, Schwarze C, Peter HH, et al. Antineutrophil cytoplasmic antibodies in primary sclerosing cholangitis: defined specificities may be associated with distinct clinical features. Am J Med 1998;105:393-399. [DOI] [PubMed] [Google Scholar]
- 115.Carrell RW, Jeppsson JO, Laurell CB, Brennan SO, Owen MC, Vaughan L, Boswell DR. Structure and variation of human α1-antitrypsin. Nature 1982;298:329-334. [DOI] [PubMed] [Google Scholar]
- 116.Propst T, Propst A, Dietze O, Judmaier G, Braunsteiner H, Vogel W. α1-Antitrypsin deficiency and liver disease. Dig Dis 1994;12:139-149. [DOI] [PubMed] [Google Scholar]
- 117.Sveger T. The natural history of liver disease in α1-antitrypsin deficient children. Acta Paediatr Scand 1988;77:847-851. [DOI] [PubMed] [Google Scholar]
- 118.Carlson J, Eriksson S. Chronic ‘cryptogenic’ liver disease and malignant hepatoma in intermediate α1-antitrypsin deficiency identified by a Pi Z-specific monoclonal antibody. Scand J Gastroenterol 1985;20:835-842. [DOI] [PubMed] [Google Scholar]
- 119.Graziadei IW, Joseph JJ, Wiesner RH, Therneau TM, Batts KP, Porayko MK. Increased risk of chronic liver failure in adults with heterozygous α1-antitrypsin deficiency. Hepatology 1998;28:1058-1063. [DOI] [PubMed] [Google Scholar]
- 120.Fisher RL, Taylor L, Sherlock S. α1-Antitrypsin deficiency in liver disease: the extent of the problem. Gastroenterology 1976;71:646-651. [PubMed] [Google Scholar]
- 121.Morin T, Martin JP, Feldmann G, Rueff B, Benhamou JP, Ropartz C. Heterozygous α1-antitrypsin deficiency and cirrhosis in adults, a fortuitous association. Lancet 1975;1:250-251. [DOI] [PubMed] [Google Scholar]
- 122.Propst T, Propst A, Dietze O, Judmaier G, Braunsteiner H, Vogel W. High prevalence of viral infection in adults with homozygous and heterozygous α1-antitrypsin deficiency and chronic liver disease. Ann Intern Med 1992;117:641-645. [DOI] [PubMed] [Google Scholar]
- 123.Hodges JR, Millward-Sadler GH, Baarbatis C, Wright R. Heterozygous MZ α1-antitrypsin deficiency in adults with chronic active hepatitis and cryptogenic cirrhosis. N Engl J Med 1981;304:557-560. [DOI] [PubMed] [Google Scholar]
- 124.Linnen J, Wages J, Jr, Zhang-Keck ZY, Fry KE, Krawczynski KZ, Alter H, et al. Molecular clone in disease association of hepatitis G virus: a transfusion transmissible agent. Science 1996;271:505-508. [DOI] [PubMed] [Google Scholar]
- 125.Alter HJ, Nakatsuji Y, Melpolder J, Wages J, Wesley R, Shih JW, et al. The incidence of transfusion-associated hepatitis G virus infection and its relation to liver disease. N Engl J Med 1997;336:747-754. [DOI] [PubMed] [Google Scholar]
- 126.Brandhagen DJ, Gross JB, Jr, Poterucha JJ, Charlton MR, Detmer J, Kolberg J, et al. The clinical significance of simultaneous infection with hepatitis G virus in patients with chronic hepatitis C. Am J Gastroenterol 1999;94:1000-1005. [DOI] [PubMed] [Google Scholar]
- 127.Hollingsworth RC, Minton EJ, Fraser-Moodie C, Metivier E, Rizzi PM, Irving WL, et al. Hepatitis G infection: role in cryptogenic liver disease and primary liver cancer in the UK. Trent Hepatitis C Virus Study Group. J Viral Hepat 1998;4:165-169. [DOI] [PubMed] [Google Scholar]
- 128.Laskus T, Radkowski M, Wang LF, Vargas H, Rakela J. Lack of evidence for hepatitis G virus replication in the livers of patients coinfected with hepatitis C and G viruses. J Virol 1997;71:7804-7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nishizawa AT, Okamoto H, Konishi K, Yoshizawa H, Miyakawa Y, Mayumi M. A novel DNA virus (TTV) associated with elevated transaminase levels in posttransfusion hepatitis of unknown etiology. Biochem Biophys Res Commun 1997;241:92-97. [DOI] [PubMed] [Google Scholar]
- 130.Charlton M, Adjei P, Poterucha J, Zein N, Moore B, Therneau T, et al. TT-Virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998;28:839-842. [DOI] [PubMed] [Google Scholar]
- 131.Matsumoto A, Yeo AET, Shih JWK, Tanaka E, Kiyosawa K, Alter HJ. Transfusion-associated TT virus infection and its relationship to liver disease. Hepatology 1999;30:283-288. [DOI] [PubMed] [Google Scholar]
- 132.Kanda T, Yokosuka O, Ikeuchi T, Seta T, Kawai S, Imazeki F, Saisho H. The role of TT virus infection in acute hepatitis. Hepatology 1999;29:1905-1908. [DOI] [PubMed] [Google Scholar]
- 133.Hoofnagle JH, Dusheiko GM, Seeff LB, Jones EA, Waggoner JG, Bales ZB. Seroconversion from hepatitis B e antigen to antibody in chronic type B hepatitis. Ann Intern Med 1981;94:744-748. [DOI] [PubMed] [Google Scholar]
- 134.Brown SD, Barbara AJ, Lambert T, Wilson DV. Spontaneous loss of HBeAg and development of anti-HBe during long-term follow-up of blood donors found to be HBsAg positive. Br J Biomed Sci 1995;52:106-109. [PubMed] [Google Scholar]
- 135.Ruiz-Moreno M, Otero M, Millan A, Castillo I, Carerizo M, Jimenez FJ, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999;29:572-575. [DOI] [PubMed] [Google Scholar]
- 136.Lok AS, Ghany MG, Watson G, Ayola B. Predictive value of aminotransferase and hepatitis B virus DNA levels on response to interferon therapy for chronic hepatitis B. J Viral Hepat 1998;5:171-178. [DOI] [PubMed] [Google Scholar]
- 137.Lai CL, Chien RN, Leung NW, Chang TT, Guan R, Tai DI, et al. A one-year trial of lamivudine for chronic hepatitis B. N Engl J Med 1998;339:61-68. [DOI] [PubMed] [Google Scholar]
- 138.Bernard F, Raymond G, Willems B, Villeneuve JP. Quantitative assessment of serum hepatitis B e antigen, IgM hepatitis B core antibody and HBV DNA in monitoring the response to treatment in patients with chronic hepatitis B. J Viral Hepat 1997;4:265-272. [DOI] [PubMed] [Google Scholar]
- 139.Hayashi PH, Beames MP, Kuhns MC, Hoofnagle JH, Di Bisceglie AM. Use of quantitative assays for hepatitis B e antigen and IgM antibody to hepatitis B core antigen to monitor therapy in chronic hepatitis B. Am J Gastroenterol 1996;91:2323-2328. [PubMed] [Google Scholar]
- 140.Heijtink RA, Kruining J, Honkoop P, Kuhns MC, Hop WC, Osterhaus AD, Schalm SW. Serum HBeAg quantitation during antiviral therapy for chronic hepatitis B. J Med Virol 1997;53:282-287. [PubMed] [Google Scholar]
- 141.Dienstag JL, Perillo RP, Schiff ER, Bartholomew M, Vicary C, Rubin M. A preliminary trial of lamivudine for chronic hepatitis B infection. N Engl J Med 1995;333:1704-1705. [DOI] [PubMed] [Google Scholar]
- 142.Shobokshi OA, Serebour FE, Skakni L. Hepatitis B surface gene mutants and their emerging role in the efficacy of HBV vaccination programs. Ann Saudi Med 1999;19:87-92. [DOI] [PubMed] [Google Scholar]
- 143.Loriot MA, Marcellin P, Walker F, Boyer N, Degott C, Randrianatoavina I, et al. Persistence of hepatitis B virus DNA in serum and liver from patients with chronic hepatitis B after loss of HBsAg. J Hepatol 1997;27:251-258. [DOI] [PubMed] [Google Scholar]
- 144.Eleftheriou N, Thomas HC, Heathcote J, Sherlock S. Incidence and clinical significance of e antigen and antibody in active and chronic liver disease. Lancet 1975;2:1171-1173. [DOI] [PubMed] [Google Scholar]
- 145.Omata M. Treatment of chronic hepatitis B infection. N Engl J Med 1998;339:114-115. [DOI] [PubMed] [Google Scholar]
- 146.Nguyen TT, Sedghi-Vaziri A, Wilkes LB, Mondala T, Pockros PJ, Lindsay KL, McHutchison JG. Fluctuations in viral load (HCV RNA) are relatively insignificant in untreated patients with chronic HCV infection. J Viral Hepat 1996;3:75-78. [DOI] [PubMed] [Google Scholar]
- 147.Gordon SC, Dailey PJ, Silverman AL, Khan BA, Kodali VP, Wilber JC. Sequential serum hepatitis C viral RNA levels longitudinally assessed by branched DNA signal amplification. Hepatology 1998;28:1702-1706. [DOI] [PubMed] [Google Scholar]
- 148.Halfon P, Bourliere M, Halimi G, Khiri H, Bertezene P, Portal I, et al. Assessment of spontaneous fluctuations of viral load in untreated patients with chronic hepatitis C by two standardized quantitation methods: branched DNA and Amplicor Monitor. J Clin Microbiol 1998;36:2073-2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fanning L. Natural fluctuations of hepatitis C viral load in a homogeneous patient population: a prospective study. Hepatology 2000;31:225-229. [DOI] [PubMed] [Google Scholar]
- 150.Pontisso P, Bellati G, Brunetto M, Chemello L, Colloredo G, Di Stefano R, et al. Hepatitis C RNA virus profiles in chronically infected individuals: do they relate to disease activity?. Hepatology 1999;29:585-589. [DOI] [PubMed] [Google Scholar]
- 151.Beld M, Penning M, McMorrow M, Gorgels J, van den Hoek A, Goudsmit J. Different hepatitis C virus (HCV) RNA load profiles following seroconversion among injecting drug users without correlation with HCV genotype and serum alanine aminotransferase levels. J Clin Microbiol 1998;36:872-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.McHutchison JG, Gordon SC, Schiff ER, Shiffman ML, Lee WM, Rustgi VK, et al. Interferon α-2b or in combination with ribavirin as initial treatment for chronic hepatitis C. N Engl J Med 1998;339:1485-1492. [DOI] [PubMed] [Google Scholar]
- 153.Poynard T, Marcellin P, Lee SS, Niederau C, Minuk GS, Ideo G, et al. Randomised trial of interferon α2b plus ribavirin for 48 weeks or for 24 weeks versus interferon α2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. International Hepatitis Interventional Therapy Group. Lancet 1998;352:1426-1432. [DOI] [PubMed] [Google Scholar]
- 154.Poynard T, McHutchison J, Goodman Z, Ling M-H, Albrecht J. Is an “A la Carte” combination interferon α-2b plus ribavirin regimen possible for first line treatment in patients with chronic hepatitis C? The ALGOVIRC Project Group. Hepatology 2000;31:211-218. [DOI] [PubMed] [Google Scholar]
- 155.Camma C, Giunta M, Pinzello G, Morabito A, Verderio P, Pagliaro L. Chronic hepatitis C and interferon α: conventional and cumulative meta-analyses of randomized clinical trials. Am J Gastroenterol 1999;94:581-595. [DOI] [PubMed] [Google Scholar]
- 156.Management of hepatitis C. NIH Consens Statement 1997;15:1–41.. [PubMed] [Google Scholar]
- 157.Anonymous. EASL International Consensus Conference on Hepatitis C. Paris, 26–28, February 1999, Consensus statement. European Association for the Study of the Liver. J Hepatol 1999;30:956–61.. [PubMed] [Google Scholar]
- 158.Haber MM, West AB, Haber AD, Reuben A. Relationship of aminotransferases to liver histological status in chronic hepatitis C. Am J Gastroenterol 1995;90:1250-1257. [PubMed] [Google Scholar]
- 159.Healey CJ, Chapman RWG, Fleming KA. Liver histology in hepatitis C infection: a comparison between patients with persistently normal or abnormal transaminases. Gut 1995;37:274-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.McCormick SE, Goodman ZD, Maydonovitch CL, Sjogren MH. Evaluation of liver histology, ALT elevation, and HCV RNA titer in patients with chronic hepatitis C. Am J Gastroenterol 1996;91:1516-1522. [PubMed] [Google Scholar]
- 161.Perrillo RP. The role of liver biopsy in hepatitis C. Hepatology 1997;26(Suppl 1):57-61. [DOI] [PubMed] [Google Scholar]
- 162.Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. Lancet 1997;349:825-832. [DOI] [PubMed] [Google Scholar]
- 163.Oberti F, Valsesia E, Pilette C, Rousselet MC, Bedossa P, Aube C, et al. Noninvasive diagnosis of hepatic fibrosis or cirrhosis. Gastroenterology 1997;113:1609-1616. [DOI] [PubMed] [Google Scholar]
- 164.Kropf J, Gressner AM, Negwer A. Efficacy of serum laminin measurement for diagnosis of fibrotic liver diseases. Clin Chem 1988;34:2026-2030. [PubMed] [Google Scholar]
- 165.Tsutsumi M, Takase S, Urashima S, Ueshima Y, Kawahara H, Takada A. Serum markers for hepatic fibrosis in alcoholic liver disease: which is the best marker, type III procollagen, type IV collagen, laminin, tissue inhibitor of metalloproteinase, or prolyl hydroxylase?. Alcohol Clin Exp Res 1996;20:1512-1517. [DOI] [PubMed] [Google Scholar]
- 166.Yamada S, Suou T, Kawasaki H, Yoshikawa N. Clinical significance of serum 7S collagen in various liver diseases. Clin Biochem 1992;25:467-470. [DOI] [PubMed] [Google Scholar]
- 167.Guechot J, Laudat A, Loria A, Serfaty L, Poupon R, Giboudeau J. Diagnostic accuracy of hyaluronan and type III procollagen amino-terminal peptide serum assays as markers of liver fibrosis in chronic viral hepatitis C evaluated by ROC curve analysis. Clin Chem 1996;42:558-563. [PubMed] [Google Scholar]
- 168.Korner T, Kropf J, Gressner AM. Serum laminin and hyaluronan in liver cirrhosis: markers with high prognostic value. J Hepatol 1996;25:684-688. [DOI] [PubMed] [Google Scholar]
- 169.Fabris C, Falleti E, Federico E, Toniutto P, Pirisi M. A comparison of four serum markers of fibrosis in the diagnosis of cirrhosis. Ann Clin Biochem 1997;34:151-155. [DOI] [PubMed] [Google Scholar]
- 170.Sopena B, Martinez-Vazquez C, Fernandez-Rodriguez CM, de la Fuente J, Rivera A, Rodriguez M, et al. Serum angiotensin converting enzyme and C4 protein of complement as a combined diagnostic index in alcoholic liver disease. Liver 1996;16:305-308. [DOI] [PubMed] [Google Scholar]
- 171.Falleti E, Pirisi M, Fabris C, Bortolotti N, Soardo G, Gonano F, Bartoli E. Circulating standard CDP9D isoform in patients with liver disease: relationship with other soluble adhesion molecules and evaluation of diagnostic usefulness. Clin Biochem 1997;30:69-73. [DOI] [PubMed] [Google Scholar]
- 172.Trinchet JC. Clinical use of serum markers of fibrosis in chronic hepatitis. J Hepatol 1995;22(Suppl 2):89-95. [PubMed] [Google Scholar]
- 173.Schmidt E, Schmidt FW. Progress in the enzyme diagnosis of liver disease: reality or illusion. Clin Biochem 1990;23:375-382. [DOI] [PubMed] [Google Scholar]
- 174.Goldstein NS, Blue DE, Hankin R, Hunter S, Bayati N, Silverman AL, Gordon SC. Serum α-fetoprotein levels in patients with chronic hepatitis C—relationship with serum alanine aminotransferase values, histologic activity index, and hepatocyte MIB-1 scores. Am J Clin Pathol 1999;111:811-816. [DOI] [PubMed] [Google Scholar]
- 175.Bayati N, Silverman AL, Gordon SC. Serum α-fetoprotein levels and liver histology in patients with chronic hepatitis C. Am J Gastroenterol 1998;93:2452-2456. [DOI] [PubMed] [Google Scholar]
- 176.Bosch FX, Ribes J, Borras J. The epidemiology of primary liver cancer. Semin Liver Dis 1999;19:271-285. [DOI] [PubMed] [Google Scholar]
- 177.El-Serag HB, Mason AC. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med 1999;340:745-750. [DOI] [PubMed] [Google Scholar]
- 178.Fattovich G, Giustina G, Schalm SW, Hadziyannis S, Sanchez-Tapias J, Almasio P, et al. Occurrence of hepatocellular carcinoma and decompensation in western European patients with cirrhosis type B. Hepatology 1995;21:77-82. [DOI] [PubMed] [Google Scholar]
- 179.Fattovich G, Giustina G, Degos F, Tremolada F, Diodati G, Almasio P, et al. Morbidity and mortality in compensated cirrhosis type C: a retrospective follow-up study of 384 patients. Gastroenterology 1997;112:463-472. [DOI] [PubMed] [Google Scholar]
- 180.Zaman SN, Johnson PJ, Williams R. Silent cirrhosis in patients with hepatocellular carcinoma. Implications for screening in high-incidence and low-incidence areas. Cancer 1990;65:1607-1610. [DOI] [PubMed] [Google Scholar]
- 181.Izzo F, Cremona F, Ruffolo F, Palaia R, Parisi V, Curley SA. Outcome of 67 patients with hepatocellular carcinoma detected during screening or 1125 patients with chronic hepatitis. Ann Surg 1998;227:513-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chalasani N, Said A, Ness R, Hoen H, Lumeng L. Screening for hepatocellular carcinoma in patients with cirrhosis in the United States: results of a national survey. Am J Gastroenterol 1999;94:2224-2229. [DOI] [PubMed] [Google Scholar]
- 183.Tong MJ, Blatt LM, Kao VW. Screening and surveillance of chronic viral hepatitis patients for hepatocellular carcinoma: a seven year study using serum α-fetoprotein and abdominal ultrasound [Abstract]. Hepatology 1999;30:209A.10385658 [Google Scholar]
- 184.Di Bisceglie AM, Hoofnagle JH. Elevations in serum α-fetoprotein in patients with chronic hepatitis B. Cancer 1989;64:2117-2120. [DOI] [PubMed] [Google Scholar]
- 185.McMahon BJ, London T. Workshop on screening for hepatocellular carcinoma. J Natl Cancer Inst 1991;83:916-919. [DOI] [PubMed] [Google Scholar]
- 186.Riegler JL. Preneoplastic conditions of the liver. Semin Gastrointest Dis 1996;7:74-87. [PubMed] [Google Scholar]
- 187.Pateron D, Ganne N, Trinchet JC, Aurousseau MH, Mal F, Meicler C, et al. Prospective study of screening for hepatocellular carcinoma in Caucasian patients with cirrhosis. J Hepatol 1995;22:708-709. [DOI] [PubMed] [Google Scholar]
- 188.Sherman M, Peltekian KM, Lee C. Screening for hepatocellular carcinoma in chronic carriers of hepatitis B virus: incidence and prevalence of hepatocellular carcinoma in a North American population. Hepatology 1995;22:432-438. [PubMed] [Google Scholar]
- 189.Villeneuve JP, Desrochers M, Infante-Rivard C, Willems B, Raymond G, Bourcier M, et al. A long-term follow-up study of asymptomatic hepatitis B surface antigen-positive carriers in Montreal. Gastroenterology 1994;108:1000-1005. [DOI] [PubMed] [Google Scholar]
- 190.Colombo M, de Franchis R, Del Ninno E, Sangiovanni A, De Fazio C, Tommasini M, et al. Hepatocellular carcinoma in Italian patients with cirrhosis. N Engl J Med 1991;325:675-680. [DOI] [PubMed] [Google Scholar]
- 191.Mazzaferro V, Regalia E, Doci R, Andreola S, Pulvirenti A, Bozzetti F, et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med 1996;334:693-699. [DOI] [PubMed] [Google Scholar]
- 192.Cottone M, Turri M, Caltagirone M, Parisi P, Orlando A, Fiorentino G, et al. Screening for hepatocellular carcinoma in patients with Child’s A cirrhosis: an 8 years prospective study by ultrasound and α fetoprotein. J Hepatol 1994;21:1029-1034. [DOI] [PubMed] [Google Scholar]
- 193.Oka H, Tamori A, Kuroki T, Kobayashi K, Yamamoto S. Prospective study of α-fetoprotein in cirrhotic patients monitored for development of hepatocellular carcinoma. Hepatology 1994;19:61-66. [PubMed] [Google Scholar]
- 194.Sarasin FP, Giostra E, Hadengue A. Cost-effectiveness of screening for detection of small hepatocellular carcinoma in western patients with Child-Pugh class A cirrhosis. Am J Med 1996;101:422-434. [DOI] [PubMed] [Google Scholar]
- 195.Collier J, Sherman M. Screening for hepatocellular carcinoma. Hepatology 1998;27:273-278. [DOI] [PubMed] [Google Scholar]
- 196.Sheu JC, Sung JL, Chen DS, Yang PM, Lai MY, Lee CS, et al. Growth rate of asymptomatic hepatocellular carcinoma and its clinical significance. Gastroenterology 1985;89:259-266. [DOI] [PubMed] [Google Scholar]
- 197.Cottone M, Virdone R, Fusco G, Orlando A, Turri M, Caltagirone M, et al. Asymptomatic hepatocellular carcinoma in Child’s A cirrhosis. Gastroenterology 1989;96:1566-1571. [DOI] [PubMed] [Google Scholar]
- 198.Barbara L, Benzi G, Gaiani S, Fusconi F, Zironi G, Siringo S, et al. Natural history of small untreated hepatocellular carcinoma in cirrhosis: a multivariate analysis of prognostic factors of tumor growth rate and patient survival. Hepatology 1992;16:132-137. [DOI] [PubMed] [Google Scholar]
- 199.Fujiyama S, Morishita T, Hashiguchi O, Sato T. Plasma abnormal prothrombin (des-γ-carboxy prothrombin) as a marker of hepatocellular carcinoma. Cancer 1988;61:1621-1628. [DOI] [PubMed] [Google Scholar]
- 200.Liebman HA, Furie BC, Tong MJ, Blanchard RA, Lo KJ, Lee SD, et al. Des-γ-carboxy (abnormal) prothrombin as a serum marker of primary hepatocellular carcinoma. N Engl J Med 1984;310:1427-1431. [DOI] [PubMed] [Google Scholar]
- 201.Aoyagi Y, Oguro M, Yanagi M, Mita Y, Suda T, Suzuki Y, et al. Clinical significance of simultaneous determinations of α-fetoprotein and des-γ-carboxy prothrombin in monitoring recurrence in patients with hepatocellular carcinoma. Cancer 1996;77:1782-1796. [DOI] [PubMed] [Google Scholar]
- 202.Izuno K, Fujiyama S, Yamasaki K, Sato M, Sato T. Early detection of hepatocellular carcinoma associated with cirrhosis by combined assay of des-γ-carboxy prothrombin and α-fetoprotein: a prospective study. Hepatogastroenterology 1995;42:387-393. [PubMed] [Google Scholar]
- 203.Mita Y, Aoyagi Y, Yanagi M, Suda T, Suzuki Y, Asakura H. The usefulness of determining des-γ-carboxy prothrombin by sensitive enzyme immunoassay in the early diagnosis of patients with hepatocellular carcinoma. Cancer 1998;82:1643-1648. [DOI] [PubMed] [Google Scholar]
- 204.Lefrere JJ, Gozin D, Soulier JP, Bettan L, Mavier P, Dhumeaux D, et al. Specificity of increased des-γ-carboxyprothrombin in hepatocellular carcinoma after vitamin K1 injection. J Hepatol 1987;5:27-29. [DOI] [PubMed] [Google Scholar]
- 205.Sakon M, Monden M, Gotoh M, Kobayashi K, Kanai T, Umeshita K, et al. The effects of vitamin K on the generation of des-γ-carboxy prothrombin in patients with hepatocellular carcinoma. Am J Gastroenterol 1991;86:339-345. [PubMed] [Google Scholar]
- 206.Nomura F, Ishijima M, Kuwa K, Tanaka N, Nakai T, Ohnishi K. Serum des-γ-carboxy prothrombin levels determined by a new generation of sensitive immunoassays in patients with small-sized hepatocellular carcinoma. Am J Gastroenterol 1999;94:650-654. [DOI] [PubMed] [Google Scholar]
- 207.Shiraki K, Takase K, Tameda Y, Hamada M, Kosaka Y, Nakano T. A clinical study of lectin-reactive α-fetoprotein as an early indicator of hepatocellular carcinoma in the follow-up of cirrhotic patients. Hepatology 1995;22:802-807. [PubMed] [Google Scholar]
- 208.Lu Y, Lu Q, Chen HL. Diagnosis of primary liver cancer using lectin affinity chromatography of serum alkaline phosphatase. J Exp Clin Cancer Res 1997;16:75-80. [PubMed] [Google Scholar]
- 209.Yao DF, Huang ZW, Chen SZ, Huang JF, Lu JX, Xiao MB, Meng XY. Diagnosis of hepatocellular carcinoma by quantitative detection of hepatoma-specified bands of serum γ-glutamyltransferase. Am J Clin Pathol 1998;110:743-749. [DOI] [PubMed] [Google Scholar]
- 210.Steindl P, Ferenci P, Dienes HP, Grimm G, Pabinger I, Madl C, et al. Wilson’s disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology 1997;113:212-218. [DOI] [PubMed] [Google Scholar]
- 211.Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991;100:1129-1134. [DOI] [PubMed] [Google Scholar]
- 212.Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet 1995;9:210-217. [DOI] [PubMed] [Google Scholar]
- 213.Crapper RM, Bhathal PS, Mackay IR, Frazer IH. “Acute” autoimmune hepatitis. Digestion 1986;34:216-225. [DOI] [PubMed] [Google Scholar]
- 214.Davis GL, Czaja AJ, Baggenstoss AH, Taswell HF. Prognostic and therapeutic implications of extreme aminotransferase elevation in chronic hepatitis. Mayo Clin Proc 1982;57:303-309. [PubMed] [Google Scholar]
- 215.Czaja AJ. Diagnosis and therapy of autoimmune liver disease. Med Clin North Am 1996;80:973-994. [DOI] [PubMed] [Google Scholar]
- 216.Mast EE, Alter MJ, Holland PV, Purcell RH. Evaluation of assays for antibody to hepatitis E virus by a serum panel. Hepatitis E Virus Antibody Serum Panel Evaluation Group. Hepatology 1998;27:857-861. [DOI] [PubMed] [Google Scholar]
- 217.Thomas DL, Yarbough PO, Vlahov D, Tsarev SA, Nelson KE, Saah AJ. Seroreactivity to hepatitis E virus in areas where the disease is not endemic. J Clin Microbiol 1997;35:1244-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Clayson ET, Myint KS, Snitbhan R, Vaughn DW, Innis BL, Chan L, et al. Viremia, fecal shedding, and IgM and IgG responses in patients with hepatitis E. J Infect Dis 1995;172:927-933. [DOI] [PubMed] [Google Scholar]
- 219.Chobe LP, Chadha MS, Banerjee K, Arankalle VA. Detection of HEV RNA in faeces, by RT-PCR during the epidemics of hepatitis E in India (1976–1995). J Viral Hepat 1997;4:129-133. [DOI] [PubMed] [Google Scholar]
- 220.Horwitz CA, Burke MD, Grimes P, Tombers J. Hepatic function in mononucleosis induced by Epstein-Barr virus and cytomegalovirus. Clin Chem 1980;26:243-246. [PubMed] [Google Scholar]