

Abstract
Aristolochic acid (AA) is a group of structurally related nitrophenanthrene carboxylic acids found in many plants that are widely used by many cultures as traditional herbal medicines. AA is a causative agent for Chinese herbs nephropathy, a term replaced later by AA nephropathy. Evidence indicates that AA is nephrotoxic, genotoxic, and carcinogenic in humans; and it also induces tumors in the forestomach, kidney, renal pelvis, urinary bladder, and lung of rats and mice. Therefore, plants containing AA have been classified as carcinogenic to humans (Group 1) by the International Agency for Research on Cancer. In our laboratories, we have conducted a series of genotoxicity and toxicogenomic studies in the rats exposed to AA of 0.1–10 mg/kg for 12 weeks. Our results demonstrated that AA treatments induced DNA adducts and mutations in the kidney, liver, and spleen of rats, as well as significant alteration of gene expression in both its target and nontarget tissues. AA treatments altered mutagenesis- or carcinogenesis-related microRNA expression in rat kidney and resulted in significant changes in protein expression profiling. We also applied benchmark dose (BMD) modeling to the 3-month AA-induced genotoxicity data. The obtained BMDL10 (the lower 95% confidence interval of the BMD10 that is a 10% increase over the background level) for AA-induced mutations in the kidney of rats was about 7 μg/kg body weight per day. This review constitutes an overview of our investigations on AA-induced genotoxicity and toxicogenomic changes including gene expression, microRNA expression, and proteomics; and presents updated information focused on AA-induced genotoxicity in rodents.
Keywords: Aristolochic acid, benchmark dose, genotoxicity, mutation, toxicogenomics
Introduction
Aristolochic acid (AA) refers to a group of structurally related nitrophenanthrene carboxylic acids (individual or mixture) found in plants of the Aristolochiaceae family, which is comprised of more than 450 species primarily in the Aristolochia and Asarum genera.[1–3] There are two major forms of AA, 8-methoxy-6-nitronaphtho[2,1-g][1,3] benzodioxole-5-carboxylic acid, and 6-nitronaphtho[2,1-g] [1,3]benzodioxole-5-carboxylic acid, according to the International Union of Pure and Applied Chemistry, which are commonly referred to as AAI (CAS# 313–67-7) and AAII (CAS# 475–80-9), respectively.[4] Since antiquity, herbal remedies containing AA have been widely used in African, Asian, European, and South American countries for medicinal purposes, such as for the treatments of snakebite, edema, and inflammatory diseases.[2,5] In traditional Chinese medicines, the Aristolochia species (such as Ma Dou Ling, Mu Tong, Guang Fang Ji, and Tian Xian Teng) were prescribed for hepatitis, headache, upper respiratory tract infection, heart diseases, and many other conditions.[6]
As early as 1825, the toxicity of certain Aristolochia species (e.g., Aristolochia serpentaria) in humans was reported, and in the 1950s, toxicity associated with Aristolochia clematitis in horses was observed.[7,8] In the 1980s, Mengs et al. reported that subchronical or chronical treatments of rats and mice with AA through gavage resulted in tumors in multiple tissues. When rats were gavaged with AA at the doses of 0.1, 1, and 10 mg/kg body weight (bw) for 3 months, the incidence of tumors in the forestomach, kidney, renal pelvis, and urinary bladder was about 25%, 85%, and 100% at these three doses, respectively.[9] Within 1 year after the mice were treated with 5 mg/kg AA for 3 weeks, carcinomas in the forestomach, glandular stomach, kidney, lung, and uteri were observed.[10] The toxicity and carcinogenicity of AA in humans first caught attention in the early 1990s. A group of Belgian women who consumed AA-containing herbal products including Aristolochia fangchi (Fang Ji) as part of a weight-loss regimen developed progressive interstitial renal fibrosis.[11,12] Urothelial cancer was identified in about half of these patients within a decade, and urothelial dysplasia was found in 19 of 21 patients without cancer.[13] Since then, similar cases of AA-associated renal fibrosis and upper urinary tract tumor formation were reported in other parts of the world.[14,15] Given its unique pathological and morphological features, the term “Chinese herbs nephropathy,” later replaced by a more accurate name “Aristolochic acid nephropathy” (AAN), was used to describe this “new” type of renal failure induced by AA.[16,17] It is worth noting that a high number of Balkan endemic nephropathy (BEN) cases, first described in the late 1950s, have been linked to AAN cases.[18,19] These findings suggest that the carcinogenic potential of AA was underestimated for a period of time.
Given evidence of AA’s toxicity and carcinogenicity in humans and in experimental animals, International Agency for Research on Cancer (IARC) classified products containing AA as Group 2A human carcinogen in 2002 (IARC 2002). Based on new data, a few years later, IARC reclassified AA as a Group 1 human carcinogen.[20] Therefore, AA-containing products have been banned by many regulatory agencies in most countries. In 2001, the United States Food and Drug Administration (U. S. FDA) issued a warning regarding the potential cancer risks of AA contained in some dietary supplements or botanical products,[21] and in 2011 and 2019, the FDA maintained an import alert that prohibited AA-containing dietary supplement products from entering the U. S. market.[22] Despite these efforts, traditional medicines containing AA are still marketed worldwide. For example, such products were/are available on the internet.[23] Using a liquid chromatography–mass spectrometry approach, 25 of 190 Chinese traditional herbal preparations from the Dutch market had AA contamination.[24] Similarly, AAI and AAII were present in 20% and 7% of 30 herbal products purchasable in the U. S. through the Internet.[25] Between 1997 and 2003, it was estimated that one-third of the population in Taiwan could potentially be exposed to AA through the prescription of traditional Chinese medicine, which may contribute to the high incidence of kidney failure and upper urinary tract tumor.[26,27] AAN still occurs occasionally in Korea, where AA has been prohibited since 2005, probably because of the availability of AA-containing drugs in oriental clinics.[28] Therefore, the human exposure to AA still exists widely and may be underestimated due to the complexity of the nomenclature of traditional Chinese medicines.
AA (either individual AA or mixtures of AA) was nominated by the National Institute of Environmental Health Sciences (located at Research Triangle Park, NC, USA) to the U. S. National Toxicology Program (NTP) for possible listing in the Report on Carcinogens, and in 2008, the NTP released its final report on AA.[1] During the last two decades, the toxic, genotoxic, and carcinogenic properties of AA have been studied in in vivo and in vitro experimental models,[29,30] and the data from human exposure and cancer cases, and experimental animals have been reviewed.[16,31–34] In our laboratories, we investigated DNA damage and mutations in the cII gene of target and nontarget organs of male Big Blue transgenic rats gavaged with AA (0.1–1 mg/kg bw) for 3 months.[35–37] We determined a significant dose-dependent induction of H-Ras mutant fraction of codon 61 CAA → CTA mutation in the kidney and liver.[38] We observed that more differentially expressed genes (DEGs) involved in cancer-related pathways occur in the kidney rather than in the liver of exposed rats using microarray analysis[39] and identified tissue-specific microRNA responses in AA-treated rats.[40] A review article focused on in vivo and in vitro genotoxicity of AA was published in 2007.[32] In the past decade, efforts have focused on assessing the potential mechanisms of AA-induced carcinogenesis, using in vivo models and toxicogenomic technologies. In the current review, we summarize these studies in rodents (mice and rats) and discuss the possible mechanisms underlying the genotoxicity and carcinogenicity of AA.
Metabolism of Aristolochic Acid In Vivo
Metabolic activation is often required for procarcinogens to become carcinogens in vivo. AAI and AAII, the major two components of AA, can be metabolized to their corresponding aristolactams under the anaerobic conditions by phase I metabolic enzymes.[41] While aristolactams are generally not genotoxic, aristolactam–nitrenium ions, the intermediate product generated during the metabolic procedure, can directly react with DNA and give rise to deoxyguanosine and deoxyadenosine adducts. Nitroreduction is the key step for the biotransformation of AA to reactive nitrenium ions. In the past several years, multiple enzymes have been studied using rodent models to examine their potential roles in nitroreduction reactions and the bioactivation of AA [Figure 1].
Figure 1:
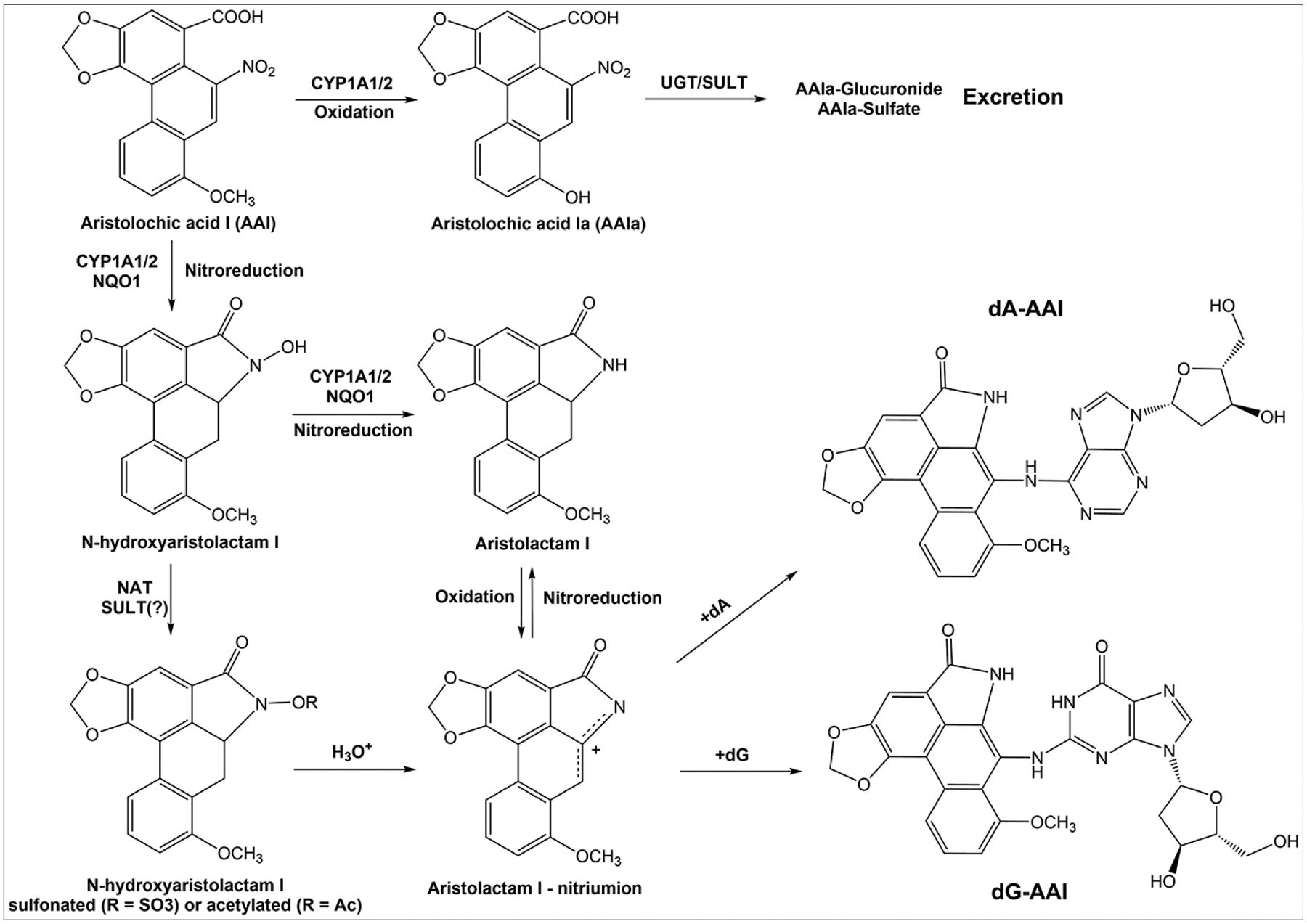
Bioactivation and detoxification of aristolochic acid I by metabolic enzymes. UGT: uridine 5’-diphospho-glucuronosyltransferase, SULT: sulfotransferase, NAT: N-acetyltransferase. The question mark (?) indicates that the role of sulfotransferase in the bioactivation of aristolochic acid is controversial
NAD(P)H:quinone oxidoreductase 1 (NQO1), a cytosolic reductase that is highly expressed in the kidney, has been implicated in mouse AAN.[42] The inhibition of NQO1 enzymatic activity by pretreatment with dicoumarol and phenindione (NQO inhibitors) attenuated AAI-induced nephrotoxicity in C57BL/6 mice. When NQO1 activity was suppressed, the formation of aristolactam I was significantly reduced along with an increase in levels of 8-OH-AAI (AAIa), a much less toxic metabolite of AAI. In mice, the protein level and enzymatic activity of NQO1 in both liver and kidney were significantly elevated by the administration of AAI at a dose of 50 mg/kg bw.[43] This result not only supports the role of NQO1 in AA bioactivation but also indicates that AAI may enhance its own genotoxicity through the induction of NQO1.
Phase I enzymes, particularly CYP1A1/2, can lead to both activation and detoxification of AAI in rodents.[44] Under aerobic conditions, AAI can be detoxified to AAIa by hepatic CYP1A1/2 through demethylation. AAIa appears to be minimally genotoxic. A previous in vivo study showed that the DNA adducts derived from AAIa represented only 1% of these derived from AAI in the renal cortex.[45] Pretreatment with β-naphthoflavone, a commonly used aryl hydrocarbon receptor agonist (CYP1A inducer), facilitated the clearance of AAI and thereby reduced the formation of AAI-DNA adducts.[46] This oxidative detoxification pathway can attenuate AA-associated renal failure in mice.[47] However, under anaerobic conditions, both AAI and AAII undergo reductive activation by CYP1A enzymes and form reactive nitrenium ions, which can bind directly to DNAs to form adducts. Therefore, oxygen concentration seems to be crucial in AA metabolism and has been suggested to contribute to AA-induced tissue-specific genotoxicity.[44] In addition, a recent study suggested that Phase II enzymes such as sulfotransferases and N-acetyltransferases appear to further increase the genotoxicity of AA by converting N-hydroxyaristolactam I to its sulfonated and acetylated conjugates, which then facilitate the formation of reactive nitrenium ions.[48] Nevertheless, another study did not find an increased formation of AAI- and AAII-DNA adducts in mice expressing human SULT1A1/1A2, suggesting that sulfotransferases may not be crucial in the bioactivation of AA in vivo.[49] Further studies are needed to confirm the roles of sulfotransferases and N-acetyltransferases in the metabolic activation of AA.
Aristolochic Acid-Induced DNA Damage
DNA adduct formation
The 7-(deoxyadenosin-N6-yl)-aristolactam I (dA-AAI) is the major DNA adduct found in multiple tissues of humans exposed to AA-containing products.[50,51] In fact, dA-AAI has been proposed to be a reliable biomarker of AA consumption since it can be detected in patients’ renal tissues after decades of exposures.[52] In rodents, AA-induced tumor formation only appears in the kidney, forestomach, and urinary tract. Therefore, an initial study was conducted to survey the tissue-wide levels of AA-derived DNA adducts. Male Wistar rats were treated with AAI and AAII at an oral dose of 10 mg/kg/day for 5 days, and the DNA adduct levels in different tissues were evaluated using the 32P-postlabeling method.[53] While the highest level of dA-AAI was showed in the forestomach (about 330 per 108 nucleotides) and a relatively low level in the kidney, dA-AAI was also found in other nontarget tissues including glandular stomach, liver, stomach, and urinary bladder epithelium. Although at lower levels, dA-AAII was detected in these tissues as well. Subsequently, two other studies from the same research group were carried out to examine the persistence of DNA adducts in several organs of rats that were administered a single oral dose of AAI.[54,55] These animals were sacrificed at various times after treatment (up to 36 weeks). The results showed both dA-AAI and 7-(deoxyguanosin-N2-yl)-aristolactam I (dG-AAI) were rapidly removed from the tissues (kidney, liver, lung, stomach, forestomach, and urinary bladder), with both DNA adducts reducing to about 50% in the kidney after 1 day of treatment. However, while dG-AAI was quickly diminished, about 25% of the dA-AAI remained in the tissues between 4 and 36 weeks.
In our laboratory, we compared the DNA adduct formation in the kidney, liver, and spleen of Big Blue transgenic rats treated with AA (containing 40% AAI and 56% AAII) at the doses of 0.1, 1, and 10 mg/kg for 3 months.[35,37] The selected doses and treatment schedule were matched to a previous carcinogenicity study in rats.[9] A subchronic repeated dose study can more closely approximate human exposure pattern than single-dose administration. Three major DNA adducts (dA-AAI, dG-AAI, and dA-AAII) were induced in the kidney, liver, and spleen in a dose-dependent manner, as measured by the 32P-postlabeling assay [Figure 2]. The kidney had the highest DNA adduct level among the three tissues (kidney vs. spleen, >20-fold; liver vs. spleen, 5–9-fold). The amount of dA-AAI in the liver and kidney was similar at all doses studied. Interestingly, the kidney (the target tissue) appeared to have more dG-AAI and dA-AAII adducts than the liver. At the carcinogenic dose of 10 mg/kg bw, dG-AAI and dA-AAII in the kidney were 2.3-and 3.6-fold higher than those in the liver, respectively.[35] In the spleen, dA-AAII seemed to be the major DNA adduct, and the level of dA-AAII was > 2-fold higher than dA-AAI and dG-AAI at all doses studied.[37] Whether the higher levels of dG-AAI and dA-AAII in the kidney eventually contribute to the tumor formation is still unclear. It should be noted that the tissues were harvested 1 day after the last dosing in these studies. As mentioned previously, dA-AAI is the major DNA adduct found in humans and its persistence has been proposed to be directly correlated with renal malignancy. To better interpret the role of dG-AAI and dA-AAII in renal cell carcinoma, future studies are required to examine their temporal occurrence across tissues during the subchronic exposure.
Figure 2:
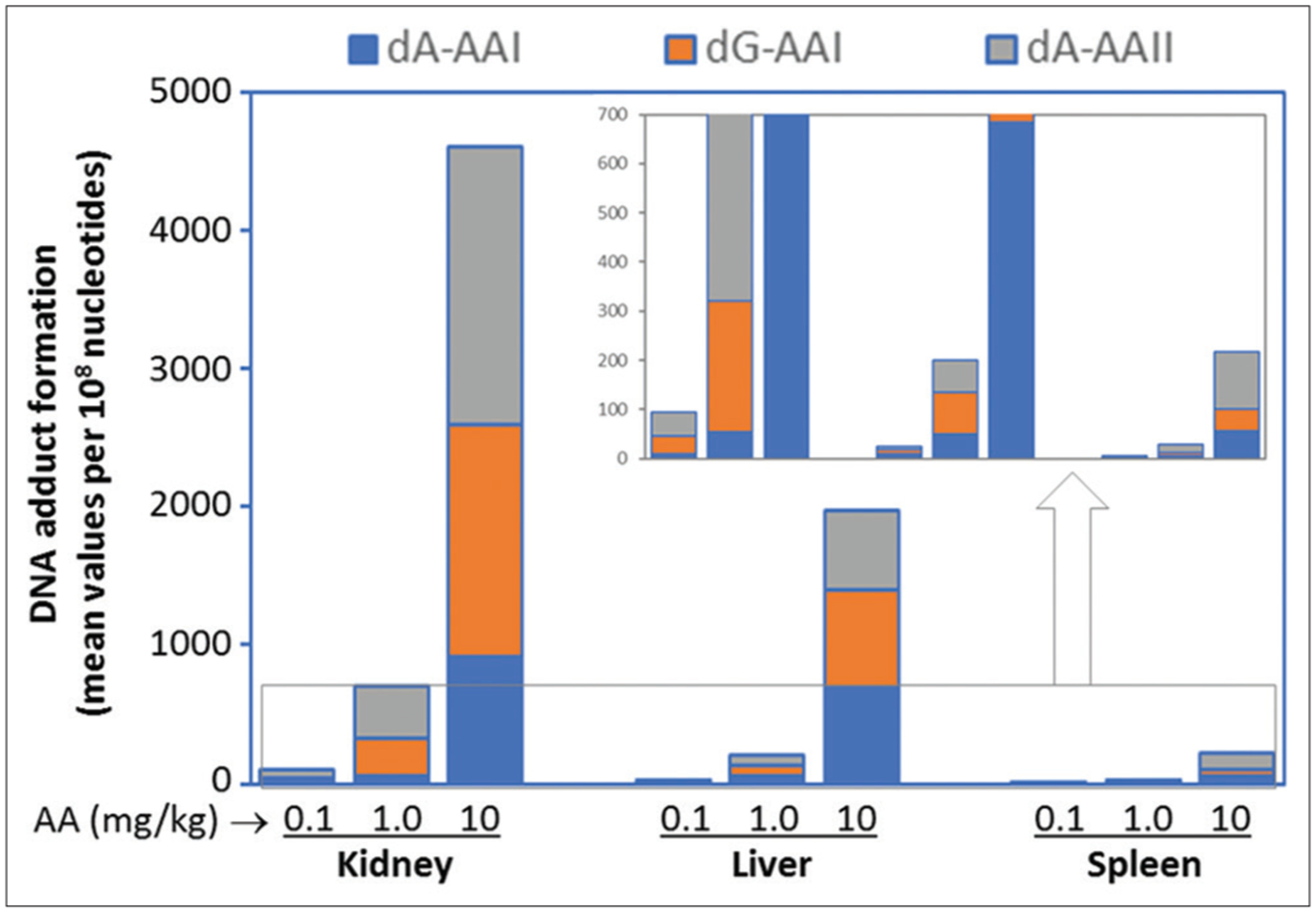
Total DNA adduct levels in the kidney, liver, and spleen of Big Blue rats treated with aristolochic acid at different doses (0.1, 1.0, and 10.0 mg/kg body weight) for 12 weeks. All DNA adducts (dA-AAI, dG-AAI, and dA-AAII) were detected by the nuclease P1 enrichment version of the 32P-postlabeling assay. The kidney and liver data are from Mei et al.[35] while the spleen data are from McDaniel et al.[37] All data represent the mean value of groups of 6 rats. The part of DNA adducts under 700/108 nucleotides has been enlarged in the inset graph
More recently, using the liquid chromatography with tandem mass spectrometry (LC-MS/MS) technique, dA-AAI and dG-AAI were identified in Sprague-Dawley rats given a single AAI dose of 10 or 30 mg/kg bw.[56] Similar to the previous studies using 32P-postlabeling, the highest amount of dA-AAI was found in the forestomach. In addition to the target tissues, dA-AAI was detected in the heart, small intestine, and large intestine 1 day after the dosing. A time-course kinetic analysis of the DNA adducts indicated that the average half-lives of dA-AAI were about 19.7 and 2.8 days in the target tissues (forestomach and kidney), which further explained the persistence of dA-AAI in animals. Using LC-MS/MS, the same research group also reported that the formation of AA-RNA adducts was significantly greater than AA-DNA adducts in both liver and kidney of rats.[57] The linkage between AA-RNA adducts and its carcinogenicity, however, remains unknown.
Chemical interactions may have further influence on the AA-DNA adduct formation. For example, ochratoxin A is a renal toxic food contaminant and that was proposed to be linked with the development of BEN.[58] Co-exposure to AA (about 33% AAI and 64% AAII) and ochratoxin A resulted in a significant increase of AA-DNA adduct formation in both liver and kidney in rats.[59] The suppression of detoxification pathways by ochratoxin A appears to account for the enhancement of AA’s genotoxicity.
Comet assay
The in vivo comet assay is often conducted to measure chemical-induced DNA damage and strand breaks in a target tissue. AA-induced DNA damage was examined using the comet assay in several in vivo studies. In isolated renal cells from male Sprague-Dawley rats treated with a single AA dose (20 or 40 mg/kg, containing 27% AAI and 65% AAII), the median Olive tail moment of DNA in the treated groups was significantly elevated 22–26 h after the treatments.[60] Sampling time appeared to play an important role in AA-induced DNA damage, as the increased Olive tail moment was not identified after 3–6 h of dosing. In another study, DNA double-strand breaks were detected by the alkaline comet assay in the bone marrow, liver, and kidney cells of F344 rats administered with 11, 22, and 30 mg/kg AA (1:1 mixture of AAI and AAII) for 4 consecutive days.[61] The magnitude of the AA-induced DNA damage, as measured by percentage of DNA in tail (% tail DNA), was similar across all tissues studied. No apparent dose-responsive pattern in the increased % tail DNA was observed, which could be partially due to a narrow dose range and sampling time window that were used in this study. Histopathological examination was performed to evaluate the potential cytotoxicity of AA in the kidney and liver. Given that only moderate nephrotoxicity was seen in AA-treated rats, it was unlikely that the observed DNA damages were caused by cytotoxicity, at least in the liver.
Oxidative DNA damage
Overproduction of reactive oxygen species (ROS) may lead to oxidative DNA damage that initiates and promotes chemical carcinogenesis.[62] The induction of oxidative DNA damage (as measured by the formation of 8-hydroxyguanosine) and depletion of glutathione by AA have been demonstrated using in vitro models.[63,64] In mice and rats, AA attenuated the overall antioxidant capacity in the kidney. A reduced level of glutathione accompanied by an impaired intrarenal antioxidant capacity was found in the kidney of mice treated with 10 mg/kg AA for 5 days.[65] Concordantly, the gene expression of ROS-generating enzymes such as Nox2 was upregulated in the plasma of male C57BL/6 mice exposed to a low-dose of AA.[66] The supplementation of l-arginine, a nitric oxide precursor with antioxidant effects, significantly decreased Nox2 expression and ROS production, and improved renal functions in mice treated with AA. This evidence clearly supports the conclusion that AA can induce oxidative stress, especially in renal tissues. However, no study has been systematically conducted to investigate whether AA can induce oxidative DNA damage in rodent models and whether oxidative stress can be a potential mechanism for AA’s carcinogenicity.
Micronucleus assay
The in vivo micronucleus test using bone marrow cells or peripheral blood cells is a critical tool to assess the genotoxicity of chemicals. Overall, the micronucleus response to AA exposure is not strong in rodents and appears to be impacted by treatment regimens. A previous study showed that a number of micronucleated polychromatic erythrocytes were significantly greater in both male and female mice treated with AA (77% AAI and 21% AAII) for 48 h than in the concurrent vehicle controls.[67] In that study, animals were dosed through a single intravenous injection, and the doses of AA ranged from 6 to 60 mg/kg. However, in another study using lambda/lacZ transgenic mice exposed to 15 mg/kg AA (56% AAI and 44% AAII) once a week for 4 weeks, the frequency of micronucleated reticulocytes (RETs) in the peripheral blood cells was not changed.[68] In rats, AA induced a minimum micronucleus response. The percentage of micronucleated RETs in peripheral blood increased slightly in F344 rats treated with 11 mg/kg AA (1:1 mixture of AAI and AAII) for 3 days as compared to controls but did not increase using lower doses.[61] On day 29 after the AA administration, no induction of micronucleated RETs was observed in any dose group. An immediate follow-up experiment attempting to examine the clastogenic effects of AA at higher doses (22 and 30 mg/kg/day) showed negative results in the micronucleus tests. Taken together, the current weight of evidence suggests that AA is a weak clastogen in vivo.
Aristolochic Acid-Induced Mutations in Gene Mutation Assays
cII transgene
Transgenic mutation assays have been used to detect gene mutations in multiple organs of transgenic animals (rats and mice). The Muta Mouse and Big Blue transgenic rodents are the most widely used screening systems for mutagens in vivo. The cII gene is a sensitive and efficient reporter of mutation in both assays.[69] Using the Muta Mouse, treatment with a single dose of 15 mg/kg AA once a week for 4 weeks increased the cII mutant frequency (MF) 15-, 9-, and 31-fold over the controls in the forestomach, kidney, and bladder, respectively.[68] The result from sequencing analysis revealed that AA caused a signature A:T to T:A transversion in the target tissues. On the other hand, cII MF was not significantly increased in the nontarget tissues, such as liver, spleen, and lung. Interestingly, the cII MF was increased about 9-fold in the colon, which was recognized recently as a possible AA target.[70]
We also investigated the mutagenicity of AA in Big Blue transgenic rats.[35–37] AA was administered to the rats through oral gavage at doses ranged from 0.1 to 10 mg/kg for 12 weeks. The Select-cII Mutation Detection System was used to identify the MF in the kidney, liver, and spleen. We found that AA increased the cII MF in a linear dose-responsive manner in all organs studied. The magnitude of MF ranged from 29 to 1319 × 10 − 6, 28–666 × 10 − 6, and 35–286 × 10 − 6 in the kidney, liver, and spleen, respectively [Figure 3]. As with the Muta Mouse model, A:T to T:A transversion was the predominant mutation type induced by AA while G: C to A:T transition appeared to be the major type of spontaneous mutations [Figure 4]. A:T to T:A transversion accounts for 54%, 50%, and 27% of all independent mutations in the liver, kidney, and spleen of rats exposed 10 mg/kg, respectively. In contrast, the A:T to T:A transversion in controls was below 5% in all the tissues studied. It is worth noting that in the spleen study, we sequenced all mutants from low-, middle-, and high-dose groups, and the percentages of A:T to T:A mutations increased dose dependently to 10%, 17%, and 27%, respectively.[37] These findings indicate that mutagenicity, particularly A:T to T:A transversion, is a key mode of action (MOA) for the tumor induction by AA.
Figure 3:
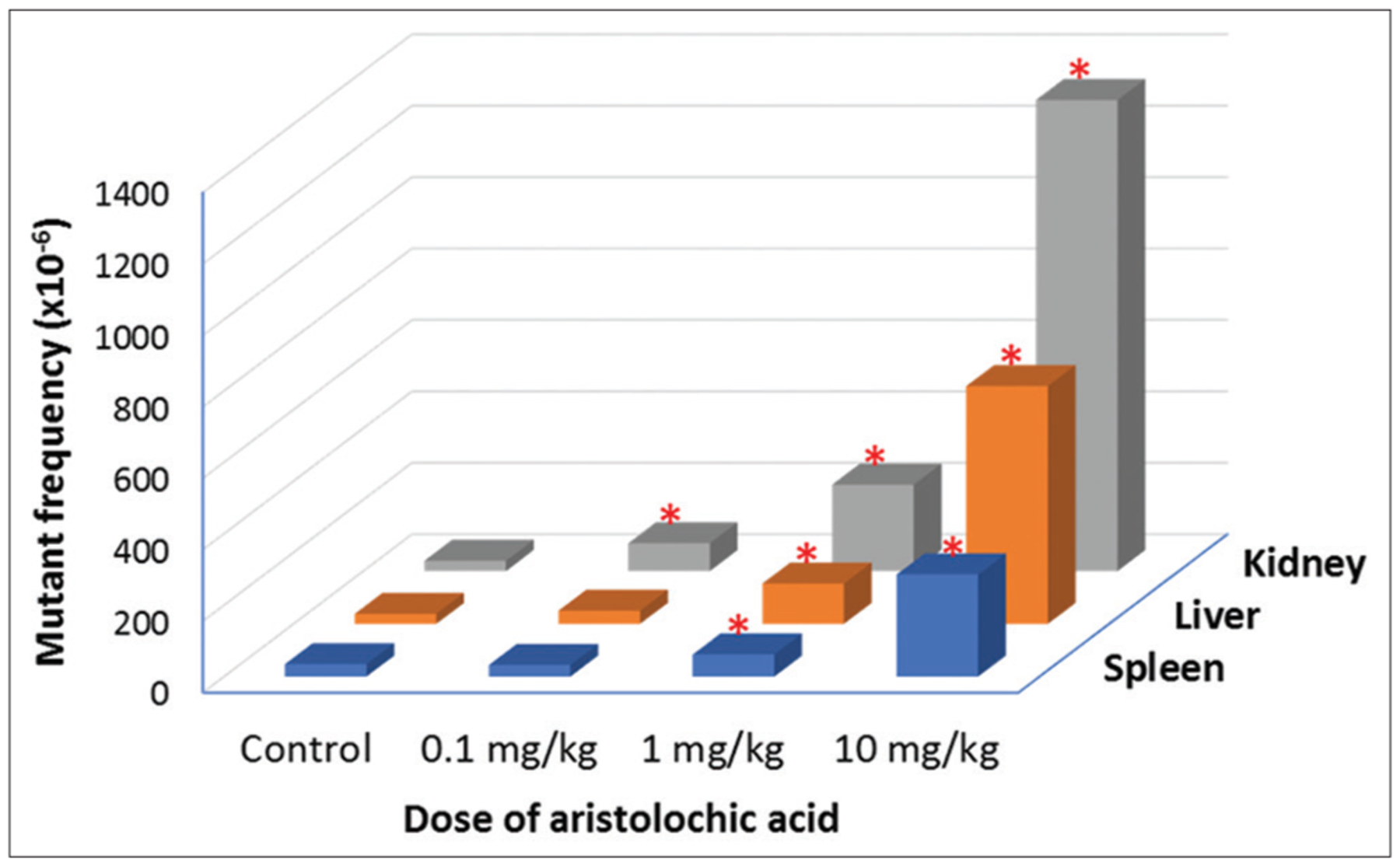
The cII mutant frequencies in the kidney, liver, and spleen of Big Blue rats treated with aristolochic acid at different doses (0.1, 1.0, and 10.0 mg/kg body weight) for 12 weeks. The data represent the mean value for groups of 6 rats. The kidney data are from Chen et al.,[36] while the liver and spleen data are from Mei et al.[35] and McDaniel et al.,[37] respectively. *Indicating a significant difference from the concurrent control group
Figure 4:
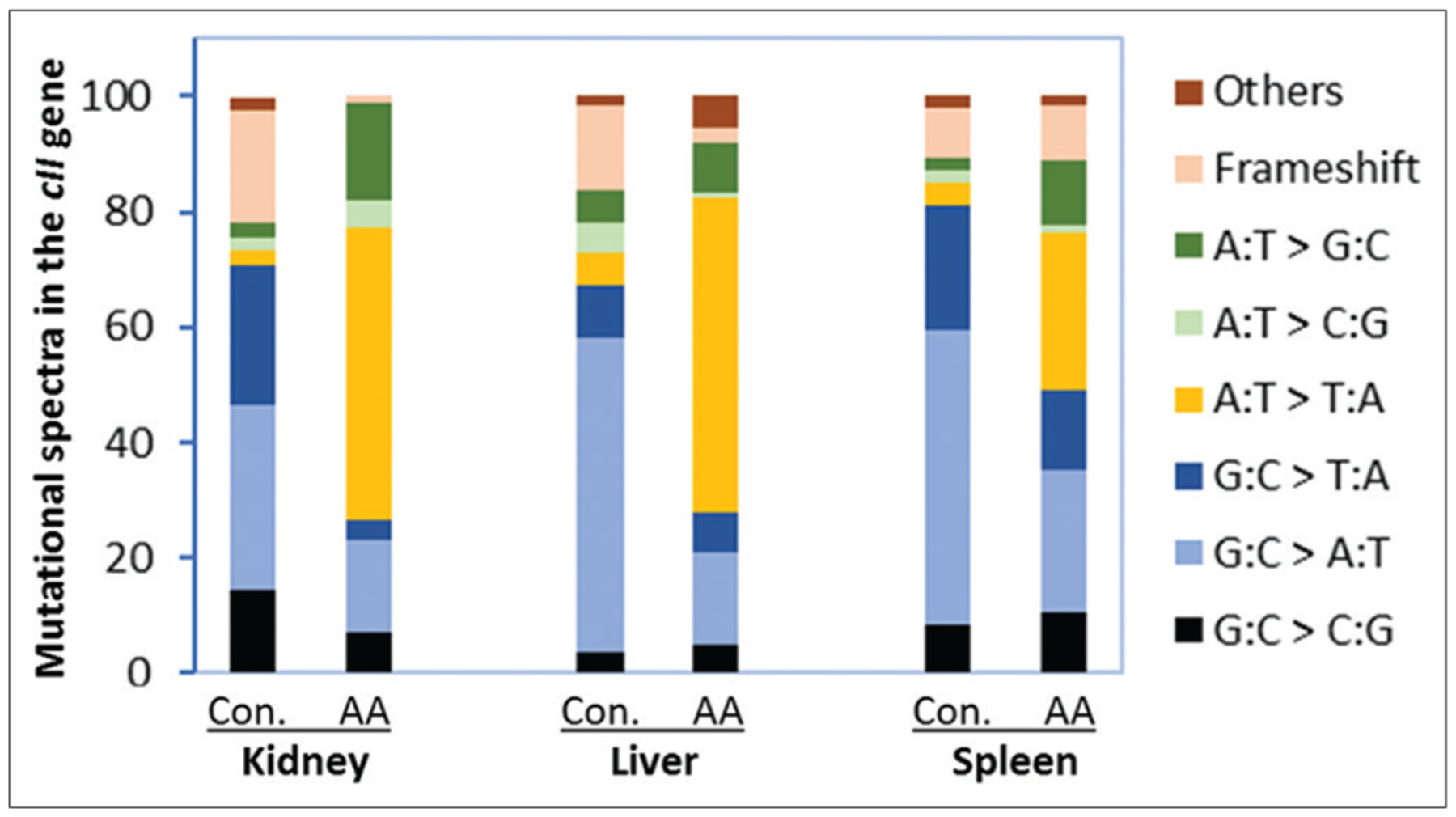
The summary of independent mutations in the cII gene of the kidney, liver, and spleen from control and 10 mg/kg aristolochic acid-treated rats. The kidney data are from Chen et al.,[36] while the liver and spleen data are from Mei et al.[35] and McDaniel et al.,[37] respectively. There are statistically significant differences between the mutation spectra in the control and AA-treated rats for each tissue. The group of “others” includes tandem base substitution and complex mutation
lacZ transgene
As a reporter gene for mutation, the lacZ transgene was incorporated into the genome of Muta Mouse within the lambda phage vector, and the lacZ MF can be determined by lacZ-positive selection of DNA samples recovered from tissues of interest. It is one of the transgenic rodent mutation assays that can detect tissue-specific mutations. Accordingly, the lacZ gene in Muta Mouse was used to evaluate the mutagenicity of AA in multiple tissues and organs. The mice were exposed to either vehicle or a weekly dose of 15 mg/kg AA for 4 weeks.[68] In AA target organs, including kidney, forestomach, and bladder, the lacZ MFs increased 10-, 33-, and 16-fold in the AA-treated groups over their concurrent controls. As expected, AA did not induce significantly higher MFs in the LacZ gene in the nontarget organs, except for colon. Notably, despite the close anatomical position, forestomach (1129 ± 262 × 10 −6) and granular stomach (141 ± 29 × 10 −6) had strikingly different lacZ MFs in AA-exposed mice, possibly because AA (as an acid) can quickly become solid and form deposits in the forestomach, leading to a higher local concentration. This observation further demonstrated that AA induces mutations in a tissue-specific manner.
Hprt gene
The Hprt gene, located on the X-chromosome and encoding hypoxanthine-guanine phosphoribosyltransferase (HPRT), has been widely used as a reporter gene for somatic mutations in vivo. More than three decades ago, the mutagenicity of AA on the Hprt locus was observed in rats using a granuloma pouch assay, in which AA (undefined composition of AAI and AAII) was directly injected to a subcutaneous air pouch.[71] Furthermore, a single oral dose of 45 or 90 mg/kg AA resulted in a significant induction in Hprt MF although only two rats were tested in the treated group. Using a more sophisticated technique, we reported that AA exposure increased MFs in the spleen T-cell Hprt gene.[61] A statistically significant increase was seen even in the low-dose group in which animals were treated with 2.75 mg/kg bw AA for 28 consecutive days. The Hprt MF in the high-dose group (11 mg/kg bw) was 55-fold higher than that in controls, confirming that AA is a potent mutagen in rats. The duration of exposure also plays an important role in determining AA’s mutagenicity. Given the same dose (11 mg/kg bw), the T-cell Hprt MF following a 28-day treatment was about 5-fold greater than that in a 3-day treatment.
Pig-a gene
The Pig-a assay, which uses the phosphatidylinositol glycan, Class A gene as a reporter of mutation, is a recommended in vivo tool by the International Workshop on Genotoxicity Testing (IWGT) for the evaluation of chemical-induced mutagenicity.[72] Two separate experiments were performed on male F344 rats to examine the mutagenicity of AA using the Pig-a assay.[61] In the first experiment, animals were treated with a daily dose of AA mixtures (0, 2.75, 5.5, and 11 mg/kg bw) for 28 days, while the second experiment used higher doses (0, 11, 22, and 30 mg/kg bw) for a shorter time (3 days). On the 4th day after the treatments, the relative RET fraction, indicating bone marrow toxicity, was reduced by AA in a dose-dependent manner in both experiments. The %RET frequencies recovered to the levels comparable to the controls by day 14 for all the dose groups. The Pig-a MFs, as measured by a number of CD59-deficient total red blood cells (RBCCD59−) and RET (RETCD59−), were evaluated on days 1, 15, and 29 in both experiments. The results showed the Pig-a MFs were significantly increased by AA in a dose- and time-dependent manner. The increases for RBCCD59− and RETCD59− MFs were up to 137- and 128-fold compared to the vehicle controls, respectively, supporting a mutagenic MOA for AA carcinogenesis.
Recently, another study examined the Pig-a MF in male Sprague-Dawley rats treated with a single dose of either 0, 15, 30, and 60 mg/kg bw AA (1:1 mixture of AAI and AAII) through oral gavage.[73] Peripheral blood was collected 7, 14, and 28 days after dosing. They found that AA treatment increased the Pig-a gene MF in a time-dependent manner. The elevated MF of RBCCD59− was identified at day 28 but not in earlier time points. The RETCD59− MF appeared to be a more sensitive marker, as statistically significant increases were found at days 7, 14, and 28 for the high-dose groups. It should be noted that the RETs were quickly regenerated after AA induced acute erythropoietic toxicity. Therefore, it is expected that the induction of MFs in the RETCD59− occurs earlier than in the RBCCD59− in peripheral blood.
Ras proto-oncogenes
Ras gene family members, including three isoforms, i.e. H-Ras, K-Ras, and N-Ras, are key genes regulating cell growth and division. Their association with various types of cancers has been studied extensively in the past decades.[74] Ras mutation often occurs at codons 12, 13, or 61. A single mutation in the Ras gene can activate the oncogene. For example, a mutation in the H-Ras leads to aberrant functions and is correlated with the development of urinary tract cancer.[75] The correlation between AA exposure and Ras mutation in vivo was initially identified in the 1990s. To investigate the mechanism of AAI-induced carcinogenicity, Schmeiser et al. treated male Wistar rats with 10 mg/kg bw AAI five times per week orally for 3 months.[76] A total of 35 tumors from various tissues were developed and analyzed. They found that AAI activated the Ras genes and A: T to T: A transversion at codon 61 was the predominant mutation type. Furthermore, this H-Ras mutation was identified in 100% of squamous cell carcinomas of the forestomach, 93% of forestomach tumors, and 100% of ear duct tumors. Concordantly, in mice, many A: T to T: A H-Ras transversions were observed in neoplastic tissue sections, while such a mutation was absent in adjacent normal tissues.[77] Given that dA-AAI is the major and persistent DNA adduct formed upon AAI exposure, this A: T to T: A transversion is not unexpected and consistent with a mutagenic MOA for AA-induced tumor induction.
DNA samples from Big Blue transgenic rats were used to investigate Ras mutation as a potential key event in the MOA for AA’s carcinogenesis. An unique allele-specific competitive blocker–polymerase chain reaction (ACB-PCR) technique was used to detect CAA to CTA (H-Ras) and GGT to GAT (K-Ras) mutations.[38] H-Ras mutant fraction at codon 61 increased in a dose-dependent manner in both liver and kidney of the AA-treated rats. Significant correlations between the H-Ras mutant fraction and the total burden of AA-DNA adducts and the cII gene MFs were identified [Figure 5], suggesting that H-Ras mutations play an important role in AA’s tumorigenesis. On the other hand, no correlation was observed between K-Ras mutation and AA-DNA adduct formation in either the kidney or the liver. It is argued that while at lower doses AA may amplify the spontaneous K-Ras mutation in target organs, the overt cytotoxicity at higher doses may trigger apoptosis in cells with preexisting K-Ras mutation, which can, in turn, influence the results of the mutation assays.
Figure 5:
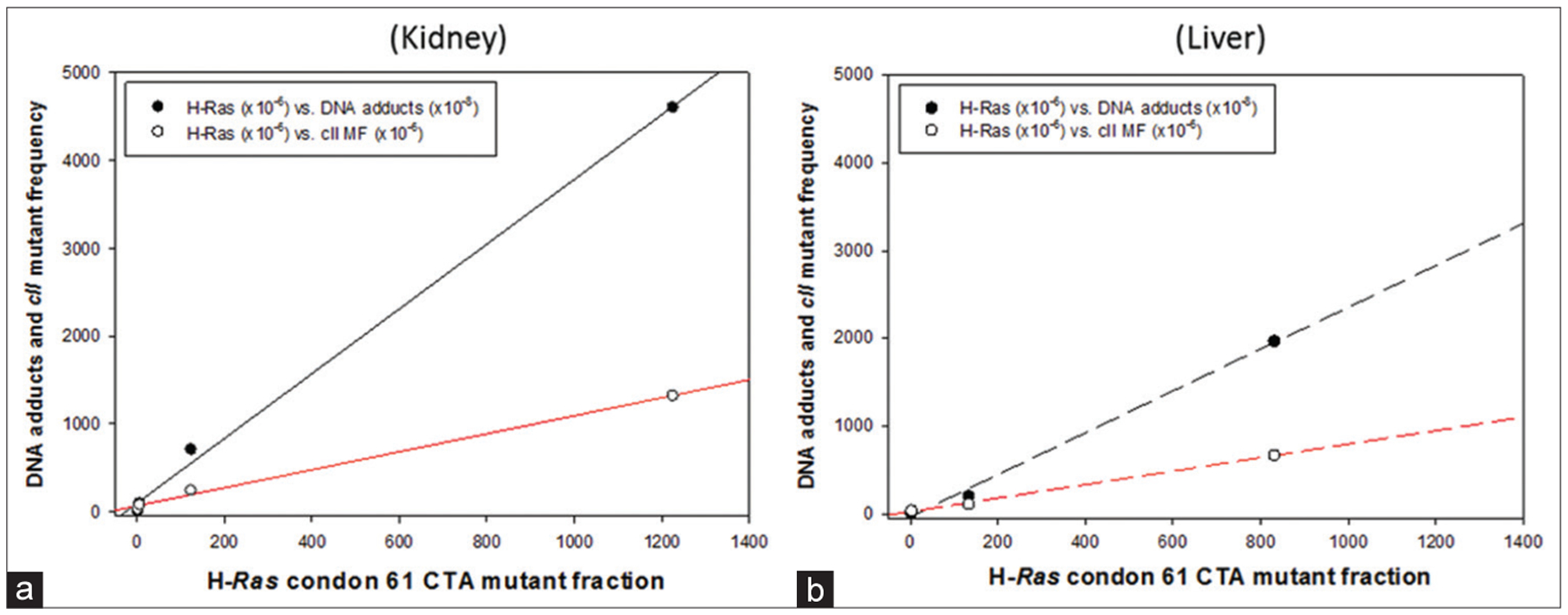
Correlations between H‑Ras codon 61 CTA mutant fraction and DNA adducts (black lines) or cII mutant frequencies (red lines) in the Kidney (a) and liver (b) from control and 10 mg/kg aristolochic acid‑treated rats. H‑Ras data are from Wang et al.,[38] while total DNA adduct levels and cII mutant frequencies are from Figures 2 and 3. Open dot: H-Ras mutant fraction vs. cII mutant frequency, Close dot: H-Ras mutant fraction vs. DNA adducts
p53 tumor suppressor gene
Multiple human studies demonstrated that AA induces mutations in the tumor suppressor gene p53 through the signature A: T to T: A transversion, which contributes to the overexpression of mutated p53 protein and carcinoma formation in the urinary tract.[78,79] A mouse model knocked-in with human p53 (Hupki) was used to evaluate the role of p53 in the carcinogenicity of AA.[80] Primary Hupki embryonic fibroblast cells were exposed to 100 μM AAI for 48 h and passaged for 8–10 weeks. Base substitution mutations were identified in 50% of the established embryonic fibroblast cell lines, with A to T transversion as the major mutation type. Particularly, this A to T mutation in codon 139 of the p53 DNA-binding domain matched with that identified in the urothelial tumor in a patient exposed to AA.[81] In addition, lacking/inhibiting p53 appears to attenuate AAN in mice. Tubular epithelial cell necrosis and apoptosis in the renal cortex is one of the major pathological features in chronic AAN. Tubular cell death was significantly decreased in p53 knockout mice, compared to wild-type controls treated with the same dose of AA.[82] Pretreatment of pifithrin-α, a p53 inhibitor, also ameliorated acute AAN in C57BL/6 mice. Phosphorylation of p53 increases the stability of the protein and thus enhances its activity. On activation, p53 can trigger cascades of downstream signaling pathways that can cause cell cycle arrest and induce apoptotic cell death. Indeed, p53 was found to be activated by phosphorylation in mice treated with 10 mg/kg AA for 3 days.[82] It has been well-documented that AA exposure results in an accumulation of DNA adducts in the kidney, initiating genotoxic stress. Previous evidence suggests that such genotoxic stress can potentially contribute to the posttranscriptional activation of p53.[83] Therefore, posttranscriptional modification of the tumor suppressor genes through genotoxicity may serve as another indirect mechanism for AA’s carcinogenic effects.
Aristolochic Acid-Induced Toxicogenomic Changes
Gene expression profiles in aristolochic acid-treated rodents
Gene expression profiles generated through microarray or next-generation sequencing (NGS) technologies allow a better understanding of molecular mechanisms in chemical carcinogenesis at the transcriptomic level. To this end, our research group compared the gene expression profiles in the kidney (target tissue) and liver (nontarget tissue) of Big Blue transgenic rats exposed to 10 mg/kg AA (containing 40% AAI and 56% AAII) for 3 months[39] because rats treated with AA at this dose developed tumors in the kidney.[9] Principal component analysis (PCA) and hierarchical cluster analysis (HCA) clearly visualized the different gene expression profiles for the tissues and AA treatment. First, the liver samples (both the control and AA-treated samples) were well separated from all kidney samples, indicating a large difference between the two tissues. Second, kidney or liver samples from the rats exposed to AA were grouped together and clearly separated from their control group, suggesting that AA exposure can be identified based on gene expression profiles. Using the criteria of P < 0.01 and fold change >2, a total of 1174 and 838 differentially expressed genes (DEGs) were identified in the kidney and liver, respectively [Table 1]. In addition, AA-treated kidney had more DEGs with higher fold changes when compared to the liver, suggesting that AA treatments resulted in greater gene expression alterations in the kidney. Functional analysis of these DEGs indicated that many more DEGs in kidney were altered in cancer-related pathways, including cell growth and proliferation, defense response, immune response, apoptosis, and tumor morphology. We reported that tumor suppressor genes such as Inhba were increased as a potential defense mechanism against AA-associated genotoxicity in the kidney. In addition, a literature shows the activation of tumor suppressor gene p53 by AA in mice.[72] More recently, Arlt et al. examined the gene expression profiles of the kidney and liver in mice knocked in with human P53.[29] They identified a significant enrichment of nuclear factor-kappa B, p53, and cell cycle pathways in the kidney of AAI-treated animals. An increase in mRNA and protein levels of the cMyc oncogene was also observed. Collectively, gene expression profiles indicate that AA significantly altered multiple carcinogenic pathways in rodent kidney and the differential alterations between the target and nontarget tissues suggest tissue-specific mechanisms of AA-induced toxicity and carcinogenicity.
Table 1:
Fold changes of genotoxic endpoints and toxicogenomic changes after aristolochic acid treatments
| 10 mg/kg AA treatment (for 12 weeks) | Group | Fold change (AA vs. control) | Our study | |||
|---|---|---|---|---|---|---|
| Kidney | Liver | Spleen | Testis | |||
| DNA adducts* | dA-AAI | 99 | 75 | 53 | − | [35,37] |
| dG-AAI | 47 | 88 | 11 | − | ||
| dA-AAII | 41 | 73 | 47 | − | ||
| Total adducts | 49 | 79 | 47 | − | ||
| cII MF | All cII gene | 45.5 | 23.8 | 8.1 | 1.1 | [35–37,40] |
| A:T→T:A# | 25 | 11 | 6 | – | ||
| H-Ras mutant fraction | Median value | 412 | 280 | – | – | [38] |
| Mean value | 130 | 356 | – | – | ||
| Deferentially expressed genes (P<0.01) | 2–5 | 1050 | 788 | – | – | [39] |
| 5–10 | 95 | 34 | – | – | ||
| >10 | 29 | 16 | – | – | ||
| Sum | 1174 | 838 | ||||
| miRNA microarray assay (P<0.05) | 2–5 | 16 | 0 | – | – | [40] |
| 5–10 | 3 | 1 | – | – | ||
| >10 | 0 | 0 | – | – | ||
| Sum | 19 | 1 | ||||
| miRNA deep sequencing (P<0.05) | 2–5 | 25 | – | – | – | [87] |
| 5–10 | 12 | – | – | – | ||
| >10 | 14 | – | – | – | ||
| Sum | 51 | |||||
Fold changes for DNA adducts were calculated by the values in 10 mg/kg group divided by those in 0.1 mg/kg group due to no DNA adducts (“0”) in the control group,
Fold changes for A:T→T:A (as a signature mutation) were calculated based on the percentage in its mutation spectrum. −: Not determined, MF: Mutant frequency, miRNA: MicroRNA, AA: Aristolochic acid, dA-AA: 7-(deoxyadenosin-N6-yl)-aristolactam
In a later study, five different microarray platforms were compared using samples from AA-treated rat kidney and the results from the different platforms consistently showed carcinogenesis-related changes in the biological functions and pathways.[84] When NGS technologies became available, the gene expression profiles generated by NGS with those from microarray technologies using the same samples were compared.[85] A total of 1,416 DEGs were commonly identified by both methods, with 98.2% (1390) DEGs exhibiting changes in the same direction. In the biological process category, there were 62 gene ontology terms commonly enriched by two platforms, with more DEGs identified with NGS than microarray. These results suggest that the gene expression profiles can elucidate molecular mechanisms and provide an improved understanding of AA-induced toxicity.
MicroRNA expression profiles in aristolochic acid-treated rodents
MicroRNAs (miRNAs) are a class of small (~22 nucleotides in length) noncoding RNAs that regulate gene expression at the posttranscription level. Growing evidence indicates that miRNAs are extensively involved in chemical carcinogenesis and miRNA expression profiles are informative for identifying the genotoxicity and carcinogenicity of chemicals.[86] To determine if miRNAs could be used as tissue-specific biomarkers for AA-induced genotoxicity, miRNA expression profiles in the liver and kidney of rats treated with 10 mg/kg AA were examined using a miRNA microarray analysis (LC Sciences, Houston, TX, USA) that contains 359 rat miRNAs.[40]
A total of 247 miRNAs, expressed in at least one tissue sample, were used for the PCA and HCA analyses. The liver samples were clearly separated from the kidney samples, indicating that miRNA expression profiles in the two tissues are different, with the separation of the control and AA-treated kidney tissues and nonseparation between the control and AA-treated liver tissues. Of 247 detectable miRNAs, 10 mg/kg AA treatment resulted in 19 differentially expressed miRNAs in the kidney using criteria of P < 0.05 and fold change >2, whereas only one miRNA was changed in the liver [Table 1]. Among the 19 miRNAs differently expressed after AA treatment in the kidney, 11 of them were downregulated (all were <3-fold) and 8 were upregulated (five miRNAs, i.e. miR-21, miR-34a, miR-10b, miR-182, and miR-30e, were >3-fold). These 19 miRNAs altered in the kidney were all associated with carcinogenesis. For example, both miR-21 and miR-34a showed higher fold changes (>9) [Table 1] in AA-treated kidney. miR-34a was the only miRNA upregulated (5.2-fold) in AA-treated liver. Further studies indicated that miR-34a was upregulated dose dependently both in the kidney and liver, and the expression levels of miR-34a positively correlated with the DNA adduct levels and cII MF.[40] These data suggest that miRNA expression profiles can distinguish AA-induced genotoxic and carcinogenic insult in the target kidney from nontarget liver, and therefore, miRNA expression profile may be able to supplement genotoxicity endpoints.
Recently, deep-sequencing technologies (also called high-throughput sequencing or NGS) have become the powerful technology of choice. Our deep-sequencing data for global miRNA and mRNA expression showed that AA treatment resulted in 63 miRNAs and 6794 mRNAs altered significantly in rat kidney.[87] This miRNA deep-sequencing analysis identified 417 detectable miRNAs, which was higher than those identified through miRNA microarray assay (247 detectable miRNAs).[40] There were 13 miRNAs (including miR-34a) upregulated >10-fold and only one (i. e., miR-383) with >10-fold downregulation. Functional annotation indicated that the top diseases/functions related to these miRNAs were “cancer, organismal injury, and abnormalities,” suggesting that dysregulated miRNA expression plays an important role in AA-induced carcinogenesis in rat kidney.[87]
Proteomic expression profiles in aristolochic acid-treated rodents
Proteomics is the large-scale study of proteins, and a major effort, related to proteomics, has been biomarker development. Toxicoproteomics can be used to elucidate toxicological responses at the protein level, i.e., the analysis of protein expression, modifications, protein–protein/toxicant interactions, and protein activities.[88] A metabonomic study of AA-induced nephrotoxicity in Wistar rats found that certain metabolic pathways, such as homocysteine formation and folate cycle, were significantly accelerated.[89] Using trypsin-catalyzed 16O/18O labeling in conjunction with two-dimensional liquid chromatography separation and tandem mass spectrometry, proteomes of rat kidney were quantitatively analyzed in our proteomic study. Greater than 9,000 unique peptide sequences were identified, with 800 proteins that were significantly altered by AA treatment.[87] For most cancer-related targets, protein expression showed expression patterns similar to that observed from miRNA expression. For example, after a 12-week AA treatment, cancer-related targets that were associated with downregulated (12) and upregulated (54) miRNAs had their proteins expressed in the same direction (downregulation 75% [9/12] and upregulation 83% [45/54]). On the other hand, the correlation of expression between protein expression and mRNA was relatively weak, suggesting that more factors in addition to miRNAs are involved in posttranscriptional regulation.
Benchmark Dose Modeling of Aristolochic Acid-Induced Genotoxicity Data
Benchmark dose (BMD) modeling has been introduced to the genotoxicity field for quantitative analysis of in vitro and in vivo dose–response relationships.[90,91] The BMD is a dose used for estimating a predetermined change of adverse response over control (the benchmark response) and is preferred by the Working Group on Quantitative Approaches to Genetic Toxicology Risk Assessment of the IWGT over other point-of-departure (PoD) metrics, such as the no observed genotoxic effect level and the breakpoint dose.[92] The two-sided lower (BMDL) and upper (BMDU) bounds of BMD 95% confidence intervals can be generated and used for quantitative comparisons of test article-induced genotoxicity across multiple endpoints. The BMDLs are the preferred PoDs by toxicologists.[93] Currently, two software packages are generally acceptable for calculating the BMD values using mathematically modeled dose–response curves, i.e., BMD Software (BMDS) and PROAST, which were developed by the U. S. Environmental Protection Agency and the Netherlands’ National Institute for Public Health and the Environment, respectively.
We performed BMD analyses on two in vivo genotoxicity endpoints in three organs. Specifically, the continuous data model in BMDS (version 2.7, released in 2017; https://www.epa.gov/bmds/benchmark-dose-software-bmds-version-27-materials) was used to analyze cII MF dose–response curves for the kidney, liver, and spleen[35–37] and H-Ras mutations in the kidney and liver[38] of Big Blue transgenic rats gavaged with AA at 0.1, 1, and 10 mg/kg for 12 weeks. The BMDs producing a 10% (BMD10), 50% (BMD50), and 100% (2-fold; BMD100) increase over the background frequencies were calculated using the five models (i.e., exponential, Hill, linear, polynomial, and power) in BMDS used for modeling continuous data. The model that produced the lowest Akaike’s information criterion along with absolute values of scaled residuals ≤ ±2 was chosen as the best model for calculating the BMDs and their BMDLs and BMDUs.[94] Accordingly, the Hill model was used for analyzing the liver and kidney cII MF data, the linear model was selected for the spleen cII MFs and liver H-Ras mutation data, and the polynomial model was employed for the kidney H-Ras mutation data.
The BMD10 and its BMDL10 have historically been applied for quantitative risk assessment, and such a 10% increase may not be seen at some endpoints.[95] In agreement with the fact that AA induced the highest cII and H-Ras mutation responses in the kidney, the BMDL10s (the lower the more mutagenic) for both endpoints were the lowest in the kidney, followed by liver and spleen [Table 2 and Figure 6]. The BMDL10s for inducing cII mutations in kidney, liver, and spleen were 8, 41, and 286 μg/kg/day, respectively, i.e., a daily dose of 8 μg/kg AA will increase cII MF 10% above the background level in the kidney after 3 months of treatment. For the H-Ras mutations, the BMDL10 for kidney (7 μg/kg/day) was about 4-fold lower than that of the liver (28 μg/kg/day); this is to say that a daily dose of 7 μg/kg AA results in a 10% increase in H-Ras oncogene mutation over the control level in the kidney after 3-month treatment. The two BMDL10s calculated from the dose–response curves of cII MF and H-Ras mutation in the kidney were very close [Table 2]. Using the body surface area normalization method,[96] these BMDL10 of 7–8 μg/kg/day for both cII MF and H-Ras mutation in the rat kidney can be converted to a human equivalent dose of 1.1–1.3 μg/kg/day which equates to 66–78 μg/day for a 60 kg person. Therefore, a 60 kg person who consumes AA at 66–78 μg/day for 3 months may potentially have 10% more mutations that may have increased risk for cancer development. With modifying factors (uncertainty or safety factors), the permitted daily exposure defined as a pharmaceutically acceptable intake can also be calculated[97] and could be much lower than the BMDL10 for genotoxicity. It has been reported that the BMDL10 for AA-related end-stage renal disease is 420 μg cumulative AA exposure.[8] However, in a worst-case scenario, exposure to >130 mg of AA can be achieved in 1 week.[24] Considering the Belgian AAN cases, this 130 mg cumulative chronic consumption dose is associated with a higher risk of cancer; its conversion to a daily dose in rats (supposing 12-week treatment, 159 μg/kg/day) is about 2-fold higher than the BMDL100 for cII MF and H-Ras mutation in the kidney [Figure 6]. The more ingestion or inadvertent exposure to AA-containing herbal products, the higher the risk of human diseases, including cancer.
Table 2:
Comparison of the lower limits of benchmark doses (BMDLs) producing a 10%, 50%, and 100% increase over the background level (BMDL10–100) for the five dose responses
| BMDL (μg/kg) | cII MF | H-Ras mumtation | |||
|---|---|---|---|---|---|
| Kidney | Liver | Spleen | Kidney | Liver | |
| BMDL10* | 8 | 41 | 286 | 7 | 28 |
| BMDL50 | 45 | 225 | 1430 | 34 | 141 |
| BMDL100# | 98 | 500 | 2859 | 68 | 282 |
The BMDL10 has historically been used for the quantitative risk assessment,
The BMDL100 (i.e., a two-fold increase) has commonly used for the assessment of genotoxicity data. MF: Mutant frequency, BMDL: The lower limit of benchmark dose (BMD)
Figure 6:
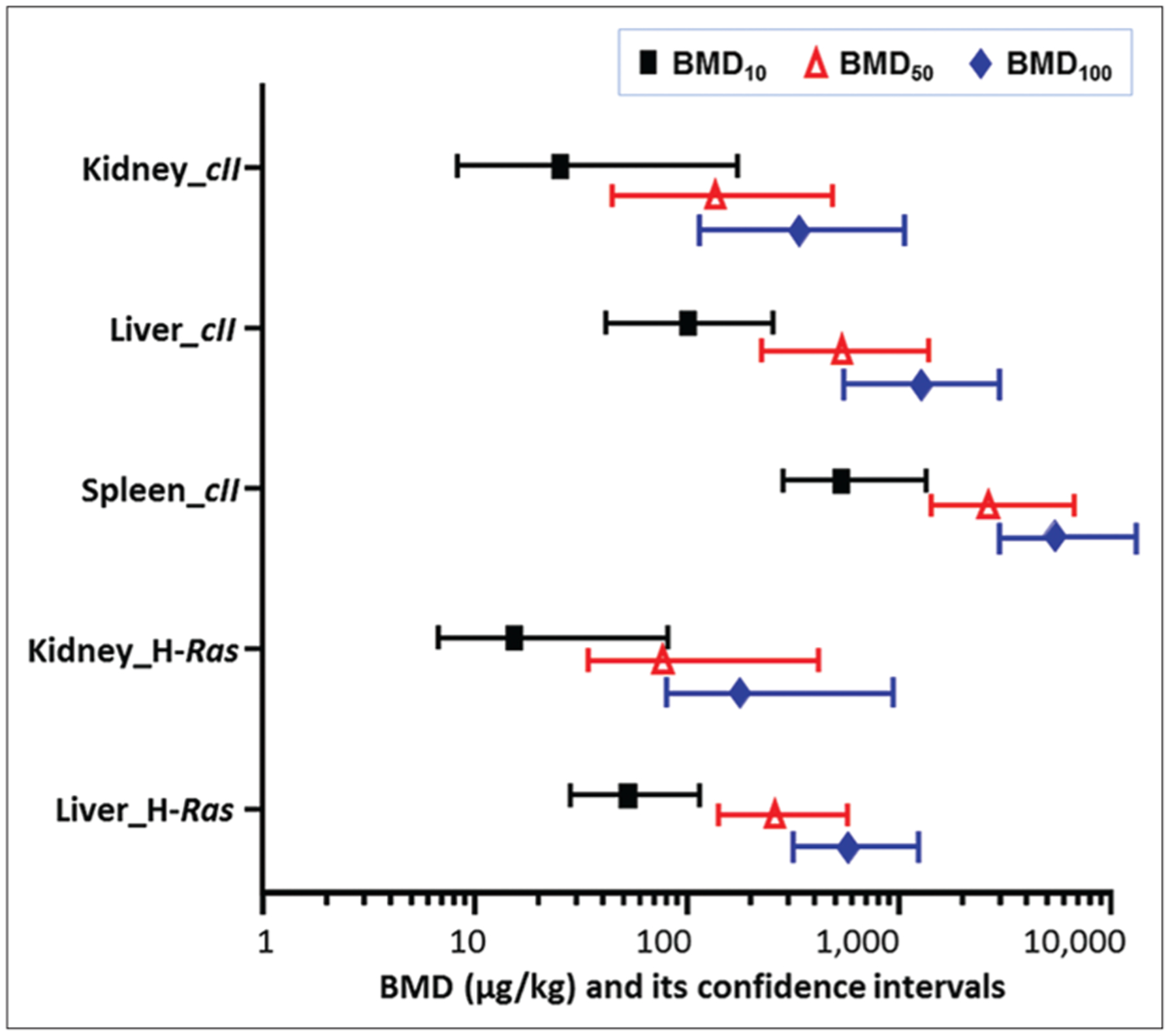
Comparison of benchmark dose values for the cII mutant frequencies and H-Ras mutant fractions induced by aristolochic acid. The benchmark dose values producing a 10%, 50%, and 100% increase over the background level were calculated using Benchmark dose software package. The bars indicate the calculated lower and upper 95% confidence interval (BMDL and BMDU) of each value. Black bar, BMD10; red bar, BMD50; and blue bar, BMD100
Concluding Remarks
Chemical carcinogenesis can be attributed to a genotoxic or nongenotoxic MOA. In vivo models, particularly rodents, have long been used to predict the carcinogenic potential of genotoxic compounds.[98] Since the genotoxic MOA was likely to be human relevant, the results from in vivo genotoxic tests (i.e., the in vivo comet, micronucleus, and reporter gene mutation assays) can be informative in cancer risk assessment. There was little research on AA-induced genotoxicity until the unfortunate incident in Belgium in the early 1990s, where a group of young women ingested a mixture of “slimming drugs” accidently containing A. fangchi. These AA exposures led to the formation of DNA adducts in multiple tissues in this group[50,51] and also induced the signature mutation (A: T to T: A) that contributed to the formation of urothelial cancer. Our interest in investigating AA-induced genotoxicity was inspired by these reports. During the last two decades, we have determined AA-induced DNA adducts, cII mutant frequencies, and H-Ras mutation fraction in the kidney, liver, and spleen of rats exposed to AA for 12 weeks [Figures 2, 5 and Table 1]. Since many new technologies have been developed and toxicogenomics has become an important subdiscipline in the field of toxicology,[99] we have also performed several toxicogenomic studies to better understand the biological processes driving AA-induced genotoxicity and carcinogenesis [Table 1]. In addition, we applied BMD modeling to find the lowest dose of AA that could induce mutations and discovered that the BMD lower confidence limit (BMDL10) for AA-induced mutations was about 7–8 μg/kg/day in rat kidneys [Table 2].
Recently, a molecular epidemiologic study indicated that AA exposure significantly contributed to upper urinary tract urothelial carcinoma in 151 patients in Taiwan.[27] In addition, the new results using whole-genome sequencing techniques have linked AA exposure to some cancers in Asia in additional tissues, such as bladder and liver.[100,101] Although the findings for hepatocellular carcinoma were based on a small sample size[102] and the A: T to T: A mutational signature was not observed after AA exposure alone,[103] these data are consistent with AA’s genotoxic MOA. The findings for liver cancers have renewed concerns about public health risk which is rapidly become a public debate in China.[104] In the early 2000s, the China Food and Drug Administration (CFDA) abolished the use of a few AA-containing medicinal materials, and in October of 2017 (just after publication of the AA liver cancer finding), the CFDA listed some Chinese medicines that contained AA on its official website for warning purposes.[104] It is worth noting that the market for traditional Chinese medicines is huge and Chinese herbs have also been exported as raw materials to over 175 countries, for example, Japan, South Korea, Germany, the Netherlands, and the USA.[105] Most herbal products are formulated in the USA, but the raw plant materials for herbal products are largely imported from China.[7]
Dietary exposure to AA and its derivatives is associated with the development of primary and secondary cancers in humans. Because AA-containing plants and products are not totally excluded in many countries and commonly used as dietary supplement products, AA-induced nephrotoxicity and carcinogenicity is now recognized as a worldwide health issue. While educating consumers and increasing global public awareness are very important for primary prevention, more investigation will help build the body of evidence and elucidate the molecular and cellular mechanisms of AA-induced toxicity, genotoxicity, and carcinogenicity. The generation, analysis, integration, and interpretation of the massively increased data obtained from in vitro, in vivo, and human studies are critical steps for better understanding AA-induced toxicity, providing new insights into the causes of human cancer, facilitating the development of biomarker screening for at-risk populations, and promoting public health.
Acknowledgments
XL was supported by an appointment to the Postgraduate Research Program at the National Center for Toxicological Research (NCTR) administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U. S. Department of Energy and the U. S. FDA. HW from Tianjin Center for New Drug Safety Assessment and Research (Tianjin, China) participated in the International Scientist Exchange Program at the NCTR. We thank Drs. Robert H. Heflich, Barbara L. Parsons, and Minjun Chen for their critical review of this manuscript. The information in this manuscript is not a formal dissemination of information by the U. S. FDA and does not represent the agency position or policy.
Footnotes
Conflicts of interest
There are no conflicts of interest.
References
- 1.National Toxicology Program. Final report on carcinogens background document for aristolochic acids. Rep Carcinog Backgr Doc 2008;5976:1–246. [PubMed] [Google Scholar]
- 2.Luciano RL, Perazella MA. Aristolochic acid nephropathy: Epidemiology, clinical presentation, and treatment. Drug Saf 2015;38:55–64. [DOI] [PubMed] [Google Scholar]
- 3.Holden F, Amin V, Kuek D, Kopp JB, Hendry BM, Xu Q. Taming the fire of nephrotoxic botanicals. World J Tradit Chin Med 2019;5:151–63. [Google Scholar]
- 4.IARC. Plants containing aristolochic acid. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans 2012. Lyon, France: IARC 100A; 2012. Available from: https://monographs.iarc.fr/wp-content/uploads/2018/06/mono100A-23.pdf. [Last accessed on 2019 Jun 20]. [Google Scholar]
- 5.Heinrich M, Chan J, Wanke S, Neinhuis C, Simmonds MS. Local uses of Aristolochia species and content of nephrotoxic aristolochic acid 1 and 2 – A global assessment based on bibliographic sources. J Ethnopharmacol 2009;125:108–44. [DOI] [PubMed] [Google Scholar]
- 6.Lai MN, Lai JN, Chen PC, Hsieh SC, Hu FC, Wang JD. Risks of kidney failure associated with consumption of herbal products containing Mu Tong or Fangchi: A population-based case-control study. Am J Kidney Dis 2010;55:507–18. [DOI] [PubMed] [Google Scholar]
- 7.Stegelmeier BL, Brown AW, Welch KD. Safety concerns of herbal products and traditional Chinese herbal medicines: Dehydropyrrolizidine alkaloids and aristolochic acid. J Appl Toxicol 2015;35:1433–7. [DOI] [PubMed] [Google Scholar]
- 8.Wu F, Wang T. Risk assessment of upper tract urothelial carcinoma related to aristolochic acid. Cancer Epidemiol Biomarkers Prev 2013;22:812–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mengs U, Lang W, Poch JA. The carcinogenic action of aristolochic acid in rats. Arch Toxicol 1982;51:107–9. [Google Scholar]
- 10.Mengs U Tumour induction in mice following exposure to aristolochic acid. Arch Toxicol 1988;61:504–5. [DOI] [PubMed] [Google Scholar]
- 11.Vanherweghem JL, Depierreux M, Tielemans C, Abramowicz D, Dratwa M, Jadoul M, et al. Rapidly progressive interstitial renal fibrosis in young women: Association with slimming regimen including Chinese herbs. Lancet 1993;341:387–91. [DOI] [PubMed] [Google Scholar]
- 12.Cosyns JP, Jadoul M, Squifflet JP, De Plaen JF, Ferluga D, van Ypersele de Strihou C. Chinese herbs nephropathy: A clue to Balkan endemic nephropathy? Kidney Int 1994;45:1680–8. [DOI] [PubMed] [Google Scholar]
- 13.Nortier JL, Martinez MC, Schmeiser HH, Arlt VM, Bieler CA, Petein M, et al. Urothelial carcinoma associated with the use of a Chinese herb (Aristolochia fangchi) N Engl J Med 2000;342:1686–92. [DOI] [PubMed] [Google Scholar]
- 14.Yang CS, Lin CH, Chang SH, Hsu HC. Rapidly progressive fibrosing interstitial nephritis associated with Chinese herbal drugs. Am J Kidney Dis 2000;35:313–8. [DOI] [PubMed] [Google Scholar]
- 15.Lord GM, Cook T, Arlt VM, Schmeiser HH, Williams G, Pusey CD. Urothelial malignant disease and Chinese herbal nephropathy. Lancet 2001;358:1515–6. [DOI] [PubMed] [Google Scholar]
- 16.Debelle FD, Vanherweghem JL, Nortier JL. Aristolochic acid nephropathy: A worldwide problem. Kidney Int 2008;74:158–69. [DOI] [PubMed] [Google Scholar]
- 17.Gillerot G, Jadoul M, Arlt VM, van Ypersele De Strihou C, Schmeiser HH, But PP, et al. Aristolochic acid nephropathy in a Chinese patient: Time to abandon the term “Chinese herbs nephropathy”? Am J Kidney Dis 2001;38:E26. [DOI] [PubMed] [Google Scholar]
- 18.Arlt VM, Stiborová M, vom Brocke J, Simões ML, Lord GM, Nortier JL, et al. Aristolochic acid mutagenesis: Molecular clues to the aetiology of Balkan endemic nephropathy-associated urothelial cancer. Carcinogenesis 2007;28:2253–61. [DOI] [PubMed] [Google Scholar]
- 19.Grollman AP, Shibutani S, Moriya M, Miller F, Wu L, Moll U, et al. Aristolochic acid and the etiology of endemic (Balkan) nephropathy. Proc Natl Acad Sci U S A 2007;104:12129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grosse Y, Baan R, Straif K, Secretan B, El Ghissassi F, Bouvard V, et al. A review of human carcinogens – Part A: Pharmaceuticals. Lancet Oncol 2009;10:13–4. [DOI] [PubMed] [Google Scholar]
- 21.FDA. FDA Concerned About Botanical Products, Including Dietary Supplements, Containing Aristolochic Acid. FDA; 2001. Available from: https://www.fda.gov/food/recalls-outbreaks-emergencies/alertsadvisories-safety-information. [Last accessed on 2019 Jun 20].
- 22.FDA. Detention without Physical Examination of Bulk/Finished Dietary Supplements Products Containing Aristolochic Acid. US FDA; 2019. Available from: https://www.accessdata.fda.gov/cms_ia/importalert_141.html. [Last accessed on 2019 Jun 20].
- 23.Gold LS, Slone TH. Aristolochic acid, an herbal carcinogen, sold on the web after FDA alert. N Engl J Med 2003;349:1576–7. [DOI] [PubMed] [Google Scholar]
- 24.Martena MJ, van der Wielen JC, van de Laak LF, Konings EJ, de Groot HN, Rietjens IM. Enforcement of the ban on aristolochic acids in Chinese traditional herbal preparations on the dutch market. Anal Bioanal Chem 2007;389:263–75. [DOI] [PubMed] [Google Scholar]
- 25.Vaclavik L, Krynitsky AJ, Rader JI. Quantification of aristolochic acids I and II in herbal dietary supplements by ultra-high-performance liquid chromatography-multistage fragmentation mass spectrometry. Food Addit Contam Part A Chem Anal Control Expo Risk Assess 2014;31:784–91. [DOI] [PubMed] [Google Scholar]
- 26.Hsieh SC, Lin IH, Tseng WL, Lee CH, Wang JD. Prescription profile of potentially aristolochic acid containing Chinese herbal products: An analysis of national health insurance data in Taiwan between 1997 and 2003. Chin Med 2008;3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen CH, Dickman KG, Moriya M, Zavadil J, Sidorenko VS, Edwards KL, et al. Aristolochic acid-associated urothelial cancer in Taiwan. Proc Natl Acad Sci U S A 2012;109:8241–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ban TH, Min JW, Seo C, Kim DR, Lee YH, Chung BH, et al. Update of aristolochic acid nephropathy in Korea. Korean J Intern Med 2018;33:961–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arlt VM, Zuo J, Trenz K, Roufosse CA, Lord GM, Nortier JL, et al. Gene expression changes induced by the human carcinogen aristolochic acid I in renal and hepatic tissue of mice. Int J Cancer 2011;128:21–32. [DOI] [PubMed] [Google Scholar]
- 30.Zhang H, Cifone MA, Murli H, Erexson GL, Mecchi MS, Lawlor TE. Application of simplified in vitro screening tests to detect genotoxicity of aristolochic acid. Food Chem Toxicol 2004;42:2021–8. [DOI] [PubMed] [Google Scholar]
- 31.Arlt VM, Stiborova M, Schmeiser HH. Aristolochic acid as a probable human cancer hazard in herbal remedies: A review. Mutagenesis 2002;17:265–77. [DOI] [PubMed] [Google Scholar]
- 32.Chen T Genotoxicity of aristolochic acid: A review. J Food Drug Anal 2007;15:387–99. [Google Scholar]
- 33.Schmeiser HH, Stiborovà M, Arlt VM. Chemical and molecular basis of the carcinogenicity of Aristolochia plants. Curr Opin Drug Discov Devel 2009;12:141–8. [PubMed] [Google Scholar]
- 34.Stiborová M, Arlt VM, Schmeiser HH. DNA adducts formed by aristolochic acid are unique biomarkers of exposure and explain the initiation phase of upper urothelial cancer. Int J Mol Sci 2017;18 pii: E2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mei N, Arlt VM, Phillips DH, Heflich RH, Chen T. DNA adduct formation and mutation induction by aristolochic acid in rat kidney and liver. Mutat Res 2006;602:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen L, Mei N, Yao L, Chen T. Mutations induced by carcinogenic doses of aristolochic acid in kidney of big blue transgenic rats. Toxicol Lett 2006;165:250–6. [DOI] [PubMed] [Google Scholar]
- 37.McDaniel LP, Elander ER, Guo X, Chen T, Arlt VM, Mei N. Mutagenicity and DNA adduct formation by aristolochic acid in the spleen of big blue® rats. Environ Mol Mutagen 2012;53:358–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Meng F, Arlt VM, Mei N, Chen T, Parsons BL. Aristolochic acid-induced carcinogenesis examined by ACB-PCR quantification of H-ras and K-ras mutant fraction. Mutagenesis 2011;26:619–28. [DOI] [PubMed] [Google Scholar]
- 39.Chen T, Guo L, Zhang L, Shi L, Fang H, Sun Y, et al. Gene expression profiles distinguish the carcinogenic effects of aristolochic acid in target (kidney) and non-target (liver) tissues in rats. BMC Bioinformatics 2006;7 Suppl 2:S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meng F, Li Z, Yan J, Manjanatha M, Shelton S, Yarborough S, et al. Tissue-specific microRNA responses in rats treated with mutagenic and carcinogenic doses of aristolochic acid. Mutagenesis 2014;29:357–65. [DOI] [PubMed] [Google Scholar]
- 41.Schmeiser HH, Pool BL, Wiessler M. Identification and mutagenicity of metabolites of aristolochic acid formed by rat liver. Carcinogenesis 1986;7:59–63. [DOI] [PubMed] [Google Scholar]
- 42.Chen M, Gong L, Qi X, Xing G, Luan Y, Wu Y, et al. Inhibition of renal NQO1 activity by dicoumarol suppresses nitroreduction of aristolochic acid I and attenuates its nephrotoxicity. Toxicol Sci 2011;122:288–96. [DOI] [PubMed] [Google Scholar]
- 43.Levova K, Moserova M, Nebert DW, Phillips DH, Frei E, Schmeiser HH, et al. NAD(P)H:quinone oxidoreductase expression in Cyp1a-knockout and CYP1A-humanized mouse lines and its effect on bioactivation of the carcinogen aristolochic acid I. Toxicol Appl Pharmacol 2012;265:360–7. [DOI] [PubMed] [Google Scholar]
- 44.Arlt VM, Levová K, Bárta F, Shi Z, Evans JD, Frei E, et al. Role of P450 1A1 and P450 1A2 in bioactivation versus detoxication of the renal carcinogen aristolochic acid I: Studies in Cyp1a1−/−, Cyp1a2−/−, and Cyp1a1/1a2−/− mice. Chem Res Toxicol 2011;24:1710–9. [DOI] [PubMed] [Google Scholar]
- 45.Shibutani S, Bonala RR, Rosenquist T, Rieger R, Suzuki N, Johnson F, et al. Detoxification of aristolochic acid I by O-demethylation: Less nephrotoxicity and genotoxicity of aristolochic acid ia in rodents. Int J Cancer 2010;127:1021–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luan Y, Xing G, Ren J, Gu J. Role of hepatic cytochrome P450 enzymes in the detoxication of aristolochic acid I; effects on DNA adduct, mutation, and tumor formation. Genes Environ 2015;37:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao Y, Ge M, Xue X, Wang C, Wang H, Wu X, et al. Hepatic cytochrome P450s metabolize aristolochic acid and reduce its kidney toxicity. Kidney Int 2008;73:1231–9. [DOI] [PubMed] [Google Scholar]
- 48.Sidorenko VS, Attaluri S, Zaitseva I, Iden CR, Dickman KG, Johnson F, et al. Bioactivation of the human carcinogen aristolochic acid. Carcinogenesis 2014;35:1814–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arlt VM, Meinl W, Florian S, Nagy E, Barta F, Thomann M, et al. Impact of genetic modulation of SULT1A enzymes on DNA adduct formation by aristolochic acids and 3-nitrobenzanthrone. Arch Toxicol 2017;91:1957–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arlt VM, Alunni-Perret V, Quatrehomme G, Ohayon P, Albano L, Gaïd H, et al. Aristolochic acid (AA)-DNA adduct as marker of AA exposure and risk factor for AA nephropathy-associated cancer. Int J Cancer 2004;111:977–80. [DOI] [PubMed] [Google Scholar]
- 51.Schmeiser HH, Bieler CA, Wiessler M, van Ypersele de Strihou C, Cosyns JP. Detection of DNA adducts formed by aristolochic acid in renal tissue from patients with Chinese herbs nephropathy. Cancer Res 1996;56:2025–8. [PubMed] [Google Scholar]
- 52.Schmeiser HH, Nortier JL, Singh R, Gamboa da Costa G, Sennesael J, Cassuto-Viguier E, et al. Exceptionally long-term persistence of DNA adducts formed by carcinogenic aristolochic acid I in renal tissue from patients with aristolochic acid nephropathy. Int J Cancer 2014;135:502–7. [DOI] [PubMed] [Google Scholar]
- 53.Pfau W, Schmeiser HH, Wiessler M 32P-postlabelling analysis of the DNA adducts formed by aristolochic acid I and II. Carcinogenesis 1990;11:1627–33. [DOI] [PubMed] [Google Scholar]
- 54.Fernando RC, Schmeiser HH, Scherf HR, Wiessler M. Formation and persistence of specific purine DNA adducts by 32P-postlabelling in target and non-target organs of rats treated with aristolochic acid I. IARC Sci Publ 1993;124:167–71. [PubMed] [Google Scholar]
- 55.Bieler CA, Stiborova M, Wiessler M, Cosyns JP, van Ypersele de Strihou C, Schmeiser HH. 32P-post-labelling analysis of DNA adducts formed by aristolochic acid in tissues from patients with Chinese herbs nephropathy. Carcinogenesis 1997;18:1063–7. [DOI] [PubMed] [Google Scholar]
- 56.Liu Y, Chan CK, Jin L, Wong SK, Chan W. Quantitation of DNA adducts in target and nontarget organs of aristolochic acid I-exposed rats: Correlating DNA adduct levels with organotropic activities. Chem Res Toxicol 2019;32:397–9. [DOI] [PubMed] [Google Scholar]
- 57.Leung EM, Chan W. Comparison of DNA and RNA adduct formation: Significantly higher levels of RNA than DNA modi epigenetic carcinogens or cytotoxic fications in the internal organs of aristolochic acid-dosed rats. Chem Res Toxicol 2015;28:248–55. [DOI] [PubMed] [Google Scholar]
- 58.Bui-Klimke TR, Wu F. Ochratoxin A and human health risk: A review of the evidence. Crit Rev Food Sci Nutr 2015;55:1860–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stiborová M, Bárta F, Levová K, Hodek P, Frei E, Arlt VM, et al. The influence of ochratoxin A on DNA adduct formation by the carcinogen aristolochic acid in rats. Arch Toxicol 2015;89:2141–58. [DOI] [PubMed] [Google Scholar]
- 60.Nesslany F, Zennouche N, Simar-Meintières S, Talahari I, Nkili-Mboui EN, Marzin D. In vivo comet assay on isolated kidney cells to distinguish genotoxic carcinogens from epigenetic carcinogens or cytotoxic compounds. Mutat Res 2007;630:28–41. [DOI] [PubMed] [Google Scholar]
- 61.Bhalli JA, Ding W, Shaddock JG, Pearce MG, Dobrovolsky VN, Heflich RH. Evaluating the weak in vivo micronucleus response of a genotoxic carcinogen, aristolochic acids. Mutat Res 2013;753:82–92. [DOI] [PubMed] [Google Scholar]
- 62.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annu Rev Pharmacol Toxicol 2004;44:239–67. [DOI] [PubMed] [Google Scholar]
- 63.Chen YY, Chung JG, Wu HC, Bau DT, Wu KY, Kao ST, et al. Aristolochic acid suppresses DNA repair and triggers oxidative DNA damage in human kidney proximal tubular cells. Oncol Rep 2010;24:141–53. [DOI] [PubMed] [Google Scholar]
- 64.Yu FY, Wu TS, Chen TW, Liu BH. Aristolochic acid I induced oxidative DNA damage associated with glutathione depletion and ERK1/2 activation in human cells. Toxicol In Vitro 2011;25:810–6. [DOI] [PubMed] [Google Scholar]
- 65.Li YC, Tsai SH, Chen SM, Chang YM, Huang TC, Huang YP, et al. Aristolochic acid-induced accumulation of methylglyoxal and nε-(carboxymethyl) lysine: An important and novel pathway in the pathogenic mechanism for aristolochic acid nephropathy. Biochem Biophys Res Commun 2012;423:832–7. [DOI] [PubMed] [Google Scholar]
- 66.Declèves AÉ, Jadot I, Colombaro V, Martin B, Voisin V, Nortier J, et al. Protective effect of nitric oxide in aristolochic acid-induced toxic acute kidney injury: An old friend with new assets. Exp Physiol 2016;101:193–206. [DOI] [PubMed] [Google Scholar]
- 67.Mengs U, Klein M. Genotoxic effects of aristolochic acid in the mouse micronucleus test. Planta Med 1988;54:502–3. [DOI] [PubMed] [Google Scholar]
- 68.Kohara A, Suzuki T, Honma M, Ohwada T, Hayashi M. Mutagenicity of aristolochic acid in the lambda/lacZ transgenic mouse (MutaMouse). Mutat Res 2002;515:63–72. [DOI] [PubMed] [Google Scholar]
- 69.Swiger RR, Cosentino L, Shima N, Bielas JH, Cruz-Munoz W, Heddle JA. The cII locus in the mutaMouse system. Environ Mol Mutagen 1999;34:201–7. [PubMed] [Google Scholar]
- 70.Colin P, Seisen T, Mathieu R, Shariat SF, Rouprêt M. Lynch syndrome and exposure to aristolochic acid in upper-tract urothelial carcinoma: Its clinical impact? Transl Androl Urol 2016;5:648–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maier P, Schawalder HP, Weibel B, Zbinden G. Aristolochic acid induces 6-thioguanine-resistant mutants in an extrahepatic tissue in rats after oral application. Mutat Res 1985;143:143–8. [DOI] [PubMed] [Google Scholar]
- 72.Gollapudi BB, Lynch AM, Heflich RH, Dertinger SD, Dobrovolsky VN, Froetschl R, et al. The in vivo pig-a assay: A report of the International Workshop on Genotoxicity Testing (IWGT) workgroup. Mutat Res Genet Toxicol Environ Mutagen 2015;783:23–35. [DOI] [PubMed] [Google Scholar]
- 73.Koyama N, Yonezawa Y, Nakamura M, Sanada H. Evaluation for a mutagenicity of aristolochic acid by pig-a and PIGRET assays in rats. Mutat Res 2016;811:80–5. [DOI] [PubMed] [Google Scholar]
- 74.Quinlan MP, Settleman J. Isoform-specific ras functions in development and cancer. Future Oncol 2009;5:105–16. [DOI] [PubMed] [Google Scholar]
- 75.Prior IA, Lewis PD, Mattos C. A comprehensive survey of ras mutations in cancer. Cancer Res 2012;72:2457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schmeiser HH, Janssen JW, Lyons J, Scherf HR, Pfau W, Buchmann A, et al. Aristolochic acid activates ras genes in rat tumors at deoxyadenosine residues. Cancer Res 1990;50:5464–9. [PubMed] [Google Scholar]
- 77.Schmeiser HH, Scherf HR, Wiessler M. Activating mutations at codon 61 of the c-ha-ras gene in thin-tissue sections of tumors induced by aristolochic acid in rats and mice. Cancer Lett 1991;59:139–43. [DOI] [PubMed] [Google Scholar]
- 78.Moriya M, Slade N, Brdar B, Medverec Z, Tomic K, Jelaković B, et al. TP53 mutational signature for aristolochic acid: An environmental carcinogen. Int J Cancer 2011;129:1532–6. [DOI] [PubMed] [Google Scholar]
- 79.Slade N, Moll UM, Brdar B, Zorić A, Jelaković B. P53 mutations as fingerprints for aristolochic acid: An environmental carcinogen in endemic (Balkan) nephropathy. Mutat Res 2009;663:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu Z, Hergenhahn M, Schmeiser HH, Wogan GN, Hong A, Hollstein M. Human tumor p53 mutations are selected for in mouse embryonic fibroblasts harboring a humanized p53 gene. Proc Natl Acad Sci U S A 2004;101:2963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Feldmeyer N, Schmeiser HH, Muehlbauer KR, Belharazem D, Knyazev Y, Nedelko T, et al. Further studies with a cell immortalization assay to investigate the mutation signature of aristolochic acid in human p53 sequences. Mutat Res 2006;608:163–8. [DOI] [PubMed] [Google Scholar]
- 82.Zhou L, Fu P, Huang XR, Liu F, Lai KN, Lan HY. Activation of p53 promotes renal injury in acute aristolochic acid nephropathy. J Am Soc Nephrol 2010;21:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem 2001;268:2764–72. [DOI] [PubMed] [Google Scholar]
- 84.Li Z, Su Z, Wen Z, Shi L, Chen T. Microarray platform consistency is revealed by biologically functional analysis of gene expression profiles. BMC Bioinformatics 2009;10 Suppl 11:S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Su Z, Li Z, Chen T, Li QZ, Fang H, Ding D, et al. Comparing next-generation sequencing and microarray technologies in a toxicological study of the effects of aristolochic acid on rat kidneys. Chem Res Toxicol 2011;24:1486–93. [DOI] [PubMed] [Google Scholar]
- 86.Chen T The role of MicroRNA in chemical carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 2010;28:89–124. [DOI] [PubMed] [Google Scholar]
- 87.Li Z, Qin T, Wang K, Hackenberg M, Yan J, Gao Y, et al. Integrated microRNA, mRNA, and protein expression profiling reveals microRNA regulatory networks in rat kidney treated with a carcinogenic dose of aristolochic acid. BMC Genomics 2015;16:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yu LR. Pharmacoproteomics and toxicoproteomics: The field of dreams. J Proteomics 2011;74:2549–53. [DOI] [PubMed] [Google Scholar]
- 89.Chen M, Su M, Zhao L, Jiang J, Liu P, Cheng J, et al. Metabonomic study of aristolochic acid-induced nephrotoxicity in rats. J Proteome Res 2006;5:995–1002. [DOI] [PubMed] [Google Scholar]
- 90.Wills JW, Johnson GE, Doak SH, Soeteman-Hernández LG, Slob W, White PA. Empirical analysis of BMD metrics in genetic toxicology part I: In vitro analyses to provide robust potency rankings and support MOA determinations. Mutagenesis 2016;31:255–63. [DOI] [PubMed] [Google Scholar]
- 91.Guo X, Mei N. Benchmark dose modeling of in vitro genotoxicity data: A reanalysis. Toxicol Res 2018;34:303–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.MacGregor JT, Frötschl R, White PA, Crump KS, Eastmond DA, Fukushima S, et al. IWGT report on quantitative approaches to genotoxicity risk assessment I. Methods and metrics for defining exposure-response relationships and points of departure (PoDs). Mutat Res Genet Toxicol Environ Mutagen 2015;783:55–65. [DOI] [PubMed] [Google Scholar]
- 93.Gollapudi BB, Johnson GE, Hernandez LG, Pottenger LH, Dearfield KL, Jeffrey AM, et al. Quantitative approaches for assessing dose-response relationships in genetic toxicology studies. Environ Mol Mutagen 2013;54:8–18. [DOI] [PubMed] [Google Scholar]
- 94.Davis JA, Gift JS, Zhao QJ. Introduction to benchmark dose methods and U.S. EPA’s benchmark dose software (BMDS) version 2.1.1. Toxicol Appl Pharmacol 2011;254:181–91. [DOI] [PubMed] [Google Scholar]
- 95.Guo X, Heflich RH, Dial SL, Richter PA, Moore MM, Mei N. Quantitative analysis of the relative mutagenicity of five chemical constituents of tobacco smoke in the mouse lymphoma assay. Mutagenesis 2016;31:287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J 2008;22:659–61. [DOI] [PubMed] [Google Scholar]
- 97.ICH. Impurities: Guideline for Residual Solvents Q3C (R6). ICH; 2016. Available from: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q3C/QC__R6___Step_4.pdf. [Last accessed on 2019 Jun 20]. [Google Scholar]
- 98.Kang SH, Kwon JY, Lee JK, Seo YR. Recent advances in in vivo genotoxicity testing: Prediction of carcinogenic potential using comet and micronucleus assay in animal models. J Cancer Prev 2013;18:277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ge Y, Wang D, Chiu J, Cristobal S, Sheehan D, Silvestre F, et al. Enviromental OMICS: Current status and future directions. OMICS 2013;3:75–87. [Google Scholar]
- 100.Ng AW, Poon SL, Huang MN, Lim JQ, Boot A, Yu W, et al. Aristolochic acids and their derivatives are widely implicated in liver cancers in Taiwan and throughout Asia. Sci Transl Med 2017;9 pii: eaan6446. [DOI] [PubMed] [Google Scholar]
- 101.Poon SL, Huang MN, Choo Y, McPherson JR, Yu W, Heng HL, et al. Mutation signatures implicate aristolochic acid in bladder cancer development. Genome Med 2015;7:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ji X, Feng G, Chen G, Shi T. Lack of correlation between aristolochic acid exposure and hepatocellular carcinoma. Sci China Life Sci 2018;61:727–8. [DOI] [PubMed] [Google Scholar]
- 103.Kucab JE, Zou X, Morganella S, Joel M, Nanda AS, Nagy E, et al. A compendium of mutational signatures of environmental agents. Cell 2019;177:821–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang HM, Zhao XH, Sun ZH, Li GC, Liu GC, Sun LR, et al. Recognition of the toxicity of aristolochic acid. J Clin Pharm Ther 2019;44:157–62. [DOI] [PubMed] [Google Scholar]
- 105.Teng L, Zu Q, Li G, Yu T, Job KM, Yang X, et al. Herbal medicines: Challenges in the modern world. Part 3. China and Japan. Expert Rev Clin Pharmacol 2016;9:1225–33. [DOI] [PubMed] [Google Scholar]