

Abstract
The global outbreak in 2002–2003 of severe acute respiratory syndrome (SARS) posed a serious threat to public health and had a significant impact on socioeconomic stability. Although the global outbreak of SARS has been contained, there are serious concerns over its re-emergence and bioterrorism potential, and up to date, no specific treatment exists for this disease. Here we review the progress of studies on the pathogenesis of the disease, in particular, studies on the molecular level.
Keywords: Severe acute respiratory syndrome (SARS), SARS coronavirus (SARS-CoV), Angiotensin-converting enzyme 2 (ACE2)
1. Introduction
During the winter of 2002–2003, a new ‘plague’ emerged in Guangdong province, China, and quickly spread to other countries. Patients were characterized by fever, dry cough, dyspnea, headache, and hypoxemia; and death could be a result of progressive respiratory failure due to alveolar damage. The syndrome was designated ‘severe acute respiratory syndrome’ (SARS). The identification of the etiologic agent, a novel coronavirus, as SARS-associated coronavirus (SARS-CoV) was quickly made by international collaboration [1], [2]. By 31 July 2003, when the pandemic terminated, 8096 people in 26 countries had been diagnosed with probable SARS, 774 of whom died [http://www.who.int/csr/sars/country/table2004_04_21/en/index.html]. In the winter of 2003–2004, sporadic cases were reported, including four cases in Guangdong province, and laboratory acquired SARS from Singapore, Taiwan and Beijing [http://www.who.int/csr/don/archive/disease/severe_acute_respiratory_syndrome/en/index.html]. Although the death rate is low in comparison with fatalities during previous pandemics, the rapidity of spread due to air travel, the coverage in the media and the enormous economic and social impacts, the fear of renewed outbreaks as well as the potential misuse of the virus as a biological weapon all contribute to the far more pronounced impact of SARS-CoV. Since the identification of the etiological agent, rapid progress has been made towards understanding this newly emerged pathogen. Most previously published reviews focused on the epidemiology, clinical presentation and potential treatment of SARS-CoV infection; this review focuses on the molecular mechanisms of SARS pathogenesis.
2. General characteristics of SARS-CoV
2.1. SARS-CoV – a new coronavirus
The newly identified virus is a new member of the family Coronaviridae, genus Coronavirus. It is a family of large, enveloped, positive-sense single-stranded RNA viruses that replicate in the cytoplasm of animal host cells. The genome of SARS-CoV is about 29 kb with 13–15 open reading frames (ORFs). Downstream of ORF1a and 1b is ORFs that encode the four main structural proteins, namely, spike (S), envelope (E), membrane (M), and nucleocapsid (N) protein [3], [4]. The spike protein of SARS-CoV, which is involved in virus binding, fusion, and entry, is a typical class I viral fusion protein, similar to the transmembrane glycoproteins of many enveloped viruses [5]. The amino-terminal S1 and carboxyl-terminal S2 subunits of the SARS-CoV S protein can be identified through their homology with the S1 and S2 subunits of other coronaviruses.
Coronaviruses are usually subdivided into three phylogenetic groups. SARS-CoV is different from the known coronaviruses; depending on the method of sequence analysis used, SARS-CoV either constitutes a new phylogenetic group [1], [2], [3], [4] or a subgroup of group 2 [6].
2.2. Cellular receptors
Studies using pseudotyped lentiviruses carrying the S, M and E glycoproteins of SARS-CoV demonstrated that the spike protein is both necessary and sufficient for virus attachment to susceptible cells [5]. The cellular receptors were detected by taking advantage of this tag.
2.2.1. ACE2 as a functional cellular receptor
In an in vitro study, Li et al. demonstrated that the angiotensin-converting enzyme 2 (ACE2) is a functional cellular receptor of SARS-CoV, by using coimmunoprecipitation of the virus glycoprotein (S1) with lysates from cells that are susceptible to virus infection (Vero E6 cells) followed by mass spectrometry analysis [7]. Later, our group proved that ACE2 is crucial for SARS-CoV infection in vivo employing an ACE2 knockout mouse [8]. Later, the structure of SARS coronavirus spike receptor-binding domain (RBD) complexed with ACE2 was determined [9]. All these findings will influence the ongoing vaccine development and biological analysis.
Immunostaining techniques identified ACE2 on the surface of type 1 and type 2 alveolar epithelial cells, enterocytes of the small intestine and the brush border of the proximal tubular cells of the kidney [10]. These localizations explain the documented tissue tropism of SARS-CoV for the lung and gastrointestinal tract. However, it should be noted that colonic enterocytes as well as the liver tissue were largely negative for ACE2 protein expression, while SARS-CoV replication does occur therein. In contrast, whereas ACE2 is strongly expressed on the endothelial cells of arteries and veins of all tissues and the smooth muscle cells of the intestinal tract, there is no evidence of virus infection at any of these sites [10]. These observations suggest that another cellular factor is required for successful virus infection.
2.2.2. Lectins as potential receptors
Pseudotyped virus containing the spike protein has also been shown to bind to dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN) [5]. DC-SIGN is a type II transmembrane adhesion molecule found on dendritic cells. It consists of tandem repeats of a highly conserved 23-amino acid sequence and a C-type lectin domain that recognizes carbohydrate residues on a variety of pathogens. Unlike the ACE2 receptor on pneumocytes and enterocytes, DC-SIGN does not permit SARS-CoV infection of the dendritic cells but can transfer the virus to susceptible target cells through a synapse-like structure (trans infection), and this cell-mediated transfer can be blocked by the mouse anti-SARS-CoV spike protein antiserum [5].
Besides DC-SIGN, there is also a report showing that cells expressing a molecule which is 77% identical to DC-SIGN–L-SIGN (CD209L) can enhance SARS-CoV infection [11]. Another study proved that L-SIGN is a receptor for SARS-CoV with the expression cloning method [12]. This study also indicated that cells expressing L-SIGN became susceptible to SARS-CoV infection, but L-SIGN was a less efficient receptor when compared with ACE2 [12]. L-SIGN also is a type II transmembrane glycoprotein in the C-type lectin family. It has a structure similar to that of DC-SIGN, except that L-SIGN has considerable polymorphism in the tandem repeat domain. Since L-SIGN has much higher polymorphism, there must exist different combinations in the population. Chan et al. found that homozygous expression of polymorphic variants of L-SIGN plays a protective role in SARS-CoV infection, because cells homozygous for L-SIGN show higher binding capacity for SARS-CoV, higher proteasome-dependent viral degradation and a low capacity for trans infection [13].
All these findings indicate that lectins may play a role in the pathogen–host interaction. Whether there are other molecules involved in the SARS-CoV–host interaction remains open.
2.3. Entry into target cells
After the virus attaches to the host cells, the virus enters the cell either through pH-dependent receptor-mediated endocytosis or through direct membrane fusion. Currently, there is no consensus about this question in the SARS-CoV case. Some experiments demonstrated that SARS-CoV might gain cell entry via pH-dependent endocytosis, but there are also studies showing that the SARS coronavirus entered the cells through direct membrane fusion.
2.3.1. Cell entry by direct cell fusion
The N-terminal half of the S protein (S1) contains the receptor-binding domain, whereas the C-terminal half (S2) is the membrane-anchored membrane-fusion subunit, which contains two heptad repeat regions (HR1 and HR2). In the native state, spike proteins on the virus surface could be in oligomeric form and form the stems of the spikes on SARS-CoV; HR1 regions may be in random coil conformation covered by the S1 domain. After binding to ACE2 on the target cells, the S2 domain changes conformation by forming α-helices, extending and inserting its inert fusion peptide into the target cell membrane, and exposing HR1 and HR2 regions. Then the HR1 and HR2 regions form a six-helix oligomeric complex, with the HR2 trimer as a core. This fusion-active core structure brings the viral and target cell membranes into close proximity, resulting in fusion between the membranes and formation of fusion pores, which allows the virus genome to enter the target cell [14].
2.3.2. Cell entry by endocytosis
Apart from direct membrane fusion at the target cell surface, SARS-CoV might gain cell entry via pH-dependent endocytosis, which is also mediated by the S protein. The spike glycoproteins of most of the enveloped RNA viruses can mediate the attachment, fusion and entry of the virus. However, an activating trigger, which can result in a conformational change, is usually required to make the spike glycoproteins fulfill their functions. For example, binding to specific receptors/coreceptors is the activating trigger for MLV and HIV, while the influenza virus requires only an acidic milieu. However, this is different in the case of SARS-CoV, as some groups have found that a special endosomal protease, cathepsin L, was crucial in the activation of viral infectivity; inhibitors of cathepsin L can prevent SARS-CoV entry [15]. So SARS-CoV may first bind to ACE2 on the cell surface and be taken up into a vesicle (endocytosis). Later, cleavage of spike protein or ACE2 by cathepsin L facilitates fusion of the viral membrane and the vesicle membrane. These results are consistent with previous findings showing that the inhibitors of vacuolar acidification could block infection by S-bearing pseudotypes in a dose-dependent manner [5], as cathepsin L is a pH-dependent cysteine protease with its maximal activity in an acidic milieu and may lose its activity with increasing pH.
Therefore, the spike protein mediated entry of SARS-CoV might be through direct membrane fusion or in a pH-dependent endocytosis fashion, and certain factors might influence this process.
3. General characteristics of SARS
3.1. Clinical features
The mean incubation period for this disease is estimated to be 6.4 days (95% CI 5.2–7.7). The mean time from onset of clinical symptoms to hospital admission varied from 3 to 5 days [16]. The most common symptoms included fever, chills, rigors, and myalgia. Cough and headache were also reported in more than 50% of the patients. Other common findings were lymphopenia, thrombocytopenia, and elevated lactate dehydrogenase and creatine kinase levels [17] watery diarrhea has also been reported [18].
Results from autopsied organs showed that the predominant damage occurred in lungs. The pathological characteristics of the lung include acute pulmonary exudative and hemorrhagic inflammation in lung tissue, greatly increased permeability of capillaries leading to a leakage of proteins such as cellulose, or even erythrocytes, into the alveolar wall and space, and accumulation of exudates and edema fluids in most of the alveoli spaces, and even hyaline membrane formation in some alveoli spaces [19].
Airspace opacity distributed peripherally in the lower lung zone is the most commonly encountered radiographic image in patients with SARS at presentation. Radiographic progression to multifocal or bilateral lung involvement followed by radiographic improvement occurs during treatment in about 70% of patients with SARS. No cavitation, lymphadenopathy, or pleural effusion was demonstrated [20].
3.2. Disease progression
Typically, SARS follows a three-phase clinical course [18]. Phase 1 is a viral replication phase that involves an initial presentation of high fever and myalgia of a few days' duration, which generally improve after a few days. The increasing viral load during this phase suggests that the symptoms are largely related to the effect of viral replication and cytolysis; however, this may also be due to antiviral and immunomodulatory therapies. In phase 2, which begins about 8 days after onset of fever, patients frequently had recurrence of fever, onset of diarrhea, and oxygen desaturation. The timing of IgG seroconversion, which starts on day 10, seems to correlate with falls in viral load, which occurs between days 10 and 15. Severe clinical worsening also occurs at this time. Therefore, the lung damage at this phase is most likely related to immunopathological damage as a result of host response, rather than uncontrolled viral replication itself [18].
Most cases improve after steroid treatment and enter a third phase of rehabilitation, while about 20% deteriorate with evidence of severe lung injury characterized by acute respiratory distress syndrome (ARDS) necessitating ventilation [18].
3.3. Prognostic factors
Epidemiological analysis of the patient population shows that some factors may influence the final outcome of the disease. Firstly, age: in patients older than 65 years, the mortality rate exceeds 50% [21], while SARS in children, especially those under 12 years, is generally associated with an uneventful course and a good outcome. Secondly, coexisting illnesses, especially diabetes mellitus and heart disease, are consistently found to be independent prognostic factors for poor outcome, which is defined as death, need for mechanical ventilation, or admission to an intensive care unit [18], [21]. Thirdly, in some studies, an increased lactate dehydrogenase level and elevated neutrophil count at the time of admission, as well as low CD4 and CD8 lymphocyte counts, were associated with a poor prognosis [22].
4. Molecular mechanisms of SARS pathogenesis
Although much has been learned about SARS in the three years since its discovery, aspects of the pathogenesis of the disease are still not fully understood. But it needs to be emphasized that since there is no specific drug or vaccine available, research on molecular mechanisms is crucial to identify potential treatment targets.
4.1. The function of the immune system in the progress of the disease
In phase 2 of the disease, the immune response plays an important role. The disease in children under the age of 12 and those with immunosuppressive conditions such as HIV infection or treatment with immunosuppressors is less severe, also indicating that immunopathology plays an important role in phase 2. The use of steroids for SARS in this phase seems to be beneficial, which is also evidence that the inflammation and immune response play a role in this phase.
4.1.1. Innate immune response to SARS-CoV
In general, during a viral infection, most cell types in the body respond by secreting high levels of type 1 interferons. This is not the case in the SARS-CoV infection. The immune-related genes that were overexpressed after the onset of SARS are usually associated with the innate immune response against bacterial infection and not against a viral infection. For example, the expression of lactoferrin is upregulated [23]. In addition, other innate immune defenses, such as the collectins, which can bind the glycosylated SARS-CoV S protein, may play an important role in host defense. This suggests that the response of SARS affected patients is mainly an innate inflammatory response rather than a specific immune response against a viral infection [23]. Besides the function of limiting virus spread, the immunological response against viral infection can also cause pathological damage to the host tissues. This is especially a concern in the case of pro-inflammatory cytokines. Nicholls and colleagues suggested that pro-inflammatory cytokines released by activated macrophages in alveoli could have a prominent role in the pathogenesis of SARS [24].
4.1.2. The adaptive immune response to SARS-CoV
T cells are essential for adaptive immunity against viral infections in vivo. Antiviral CD4+ helper T cells help in the production of virus-specific antibodies by B cells, while CD8+ CTLs can kill virus-infected host cells. In the case of SARS-CoV infection, there is no conclusion as to the degree that T cells can influence the progress of the disease, since lymphopenia with a rapid decrease in both CD4 and CD8 T cells is common during the acute phase of SARS [22].
Neutralizing antibodies (IgG) have been detected in SARS patients, suggesting that humoral immunity plays an important role in the elimination of the virus. This is further evidenced by the activity of an anti-S1 human monoclonal antibody, 80R, in neutralizing SARS-CoV infection and inhibiting syncytia formation [25].
4.2. The role of apoptosis
Virus-induced apoptosis is a common phenomenon in viral infection, especially RNA virus infections, including some coronaviruses. Apoptosis can be used by the host cells to clear off the virus in the infected cells and also can assist virus dissemination by the release of viral particles, thereby facilitating its survival in vivo. Therefore, apoptosis can have two opposite roles on the pathogenicity of viral infection, enhancing or suppressing the viral infection. In the case of SARS, apoptosis was observed in patients' lung epithelial cells; thus, SARS-CoV induced apoptosis would certainly have a deleterious pathogenic role, leading to severe tissue damage [26]. This might explain the severe respiratory system damage in SARS patients.
Apoptosis may also play an important role in the hematological changes besides the direct injury in the lung. As mentioned above, hematological changes in patients with SARS are common and include lymphopenia, thrombocytopenia and occasionally leukopenia. The mechanism underlying this phenomenon remains unclear, but there are studies indicating that apoptosis as well as the immune response are involved [27].
Several studies have focused on the mechanism of SARS-CoV induced apoptosis, for example, proapoptotic components of the virus genes and apoptosis pathways. SARS-CoV infection in Vero E6 cells can lead to apoptosis. Further studies showed that a variety of signaling pathways were phosphorylated or dephosphorylated when the cells were infected with SARS-CoV. Specifically, p38 mitogen-activated protein kinase (MAPK) is thought to be involved in induction of apoptosis, since Mizutani et al. found that the p38 MAPK and its downstream targets, MAPKAPK-2 and HSP-27 were activated during viral replication [28]. A p38 inhibitor was able to partially prevent cytopathic effects induced by SARS-CoV infection. The signal transducer and activator of transcription (STAT)-3, which is usually constitutively phosphorylated at a tyrosine residue (705), is dephosphorylated by SARS-CoV-induced activation of p38 [28]. Although Akt, an inhibitor of apoptosis, was also partially activated, this weak activation cannot prevent SARS-CoV infection-induced apoptosis in Vero E6 cells. And recently, research on the 90 kDa ribosomal S6 kinases (p90RSKs), an important substrate of the ERK, showed that the specific serine residue (380) of p90RSKs, that has been reported to be involved in autophosphorylation by activation of the C-terminal kinase domain, was phosphorylated in confluent SARS-CoV infected cells, and this phosphorylation can be inhibited by an inhibitor of p38 MAPK [29]. All these results suggest that SARS-CoV induced apoptosis in Vero E6 cells is related to p38 MAPK.
Besides the whole virus, certain components of the virus can also induce apoptosis. It has been reported that SARS-CoV proteins 3a, 3b and 7a can induce apoptosis; the SARS coronavirus nucleocapsid protein can induce apoptosis in COS-1 cells in the absence of growth factors; the C-terminal domain of SARS-CoV spike protein is sufficient to induce apoptosis in Vero E6 cells. A recent study showed that the induction of apoptosis by the 7a protein also is related to its ability to activate p38 MAPK [30].
4.3. The role of RAS system
In adult SARS patients, respiratory distress is the principal cause of mortality. The histological change associated with ARDS is called diffuse alveolar damage (DAD), which is characterized by structureless non-cellular exudates filling the bronchioles. DAD is associated with a high mortality rate, and apart from supportive clinical care, there are few specific therapeutic options of proven benefit. Recent studies have shown that the renin-angiotensin system plays an important role in the SARS-CoV caused acute lung failure.
4.3.1. The function of the renin-angiotensin system in acute lung injury
Acid aspiration or sepsis in wild-type mice, which mimics human acute lung injury, resulted in impairment of lung function as assessed by lung elasticity, blood oxygenation and pulmonary edema. The mice developed edema, alveolar wall thickening, bleeding, inflammatory cell infiltration and hyaline membrane formation. But in the ACE2 knockout mice, lung injury was much more severe than in the wild-type mice when assessed similarly. A rescue experiment using the recombinant human ACE2 protein showed a decreased degree of acute lung injury in both the knockout and wild-type mice. All these results demonstrated that loss of ACE2 is essential for lung injury [31].
Both ACE and ACE2 are key enzymes in the renin-angiotensin system. ACE cleaves the decapeptide angiotensinI (AngI) into octapeptide angiotensinII (AngII). ACE2 cleaves a single residue from AngI to generate Ang1–9, and a residue from AngII to generate Ang1–7. In this way, ACE2 counterbalances the function of ACE and negatively regulates the AngII level. Thus loss of ACE2 expression will lead to a high level of AngII, which will then, through receptor AT1a, have a causative role in acute lung failure ( Fig. 1).
Fig. 1.
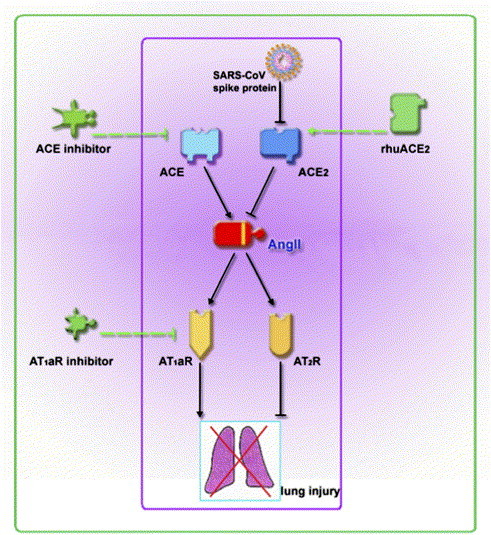
Schematic representation of the role of RAS in the SARS-CoV induced acute lung injury and the potential therapeutic drugs. ACE can cleave angiotensin I to produce angiotensin II, which can then either bind to AT1aR leading to lung injury, or bind to AT2R reducing the severity of lung injury. ACE2, on the contrary, can counteract ACE by converting the angiotensin II to a less damaging molecule. SARS-CoV infection or spike protein treatment can down-regulate the expression of ACE2, and thus aggravate lung injury. Based on these findings, ACE inhibitor (such as lisinopri, captopri), AT1aR inhibitor (such as losartan, valsartan) as well as recombinant human ACE2 (rhuACE2), are all potential drugs for this kind of acute lung injury.
4.3.2. The role of ACE2 in SARS-CoV caused lung injury
We speculated that the acute lung failure caused by SARS-CoV was mainly due to the function of ACE2. First we found that experimental infections of SARS-CoV in wild-type mice resulted in considerably reduced ACE2 expression in the lungs, while ACE expression was not changed. To further test our hypothesis, we established a defined model system using recombinant SARS-CoV spike protein. This model system allowed us to avoid possible secondary effects resulting from viral replication or infections in vivo and to directly test whether SARS-CoV spike protein might adversely affect acute lung injury through modulation of ACE2. With this system we found that binding of spike to endogenous ACE2 in Vero E6 cells also resulted in downregulation of ACE2 surface expression. Further studies showed that treatment with spike protein worsened the lung function in wild-type mice. Moreover, spike treatment of acid-challenged wild-type mice augmented the pathological changes in the lung parenchyma and increased lung edema, while in vivo spike protein administration did not affect the severity of lung failure in ACE2 knockout mice, indicating that the effect of spike protein on acute lung injury is ACE2 specific. Thus we hypothesize that infection with SARS-CoV can result in ACE2 downregulation through binding of SARS-CoV spike protein to ACE2. Given that ACE2 is a key negative regulatory factor for severity of lung edema and acute lung failure, SARS-CoV spike protein-mediated ACE2 downregulation then contributes to the severity of lung pathologies [8] (Fig. 1).
About one million people suffer from ARDS due to different disposing factors, and the death rate can reach 50%. As the recombinant ACE2 and angiotensin II receptor inhibitors are already in clinical use for control of blood pressure, this finding points to a possible therapy for the otherwise incurable disease (Fig. 1).
Besides ARDS that contributes significantly to SARS-CoV caused death, other SARS-CoV associated diseases, such as hypertension, glucose increase, and heart dysfunctions cannot be ignored [18], [21]. The mechanisms underlying the pathogenesis are still unclear and need to be understood.
5. Antiviral therapy and vaccine development
5.1. Antiviral therapy
Currently, there is no antiviral therapy of proven value for SARS. Clinically, treatment of SARS includes anti-SARS-CoV therapy and anti-inflammatory treatment to limit viral pneumonitis and subsequent pulmonary fibrosis. With insights into the field of SARS pathogenesis and SARS-CoV genome structure, novel potential therapeutic targets for antiviral therapy were evaluated. For example, inhibitors of each step of the virus life cycle, like binding inhibitors, fusion inhibitors, RNA transcription (replication) inhibitors as well as protease inhibitors, were designed and evaluated. Only a few potential anti-SARS agents have been tested in animal models, and their efficacy in humans is still unknown. Inconsistent results between different groups investigating the same compound may be related to testing methodology, in particular, differences between in vitro and in vivo antiviral mechanisms.
New technologies such as siRNA may be important. However, this new technology is still riddled with practical difficulties, like efficacious delivery systems and safety concerns, limiting its application to patient treatment.
Using sera from people convalescing from SARS to treat SARS patients was proved to be effective, implying that neutralizing antibody can serve as a therapeutic strategy. A retrospective study in a limited number of patients using human SARS convalescent plasma suggested that passive immunization had no obvious adverse effects [32]. However, the use of convalescent sera is not a practical therapy, especially when there is an outbreak worldwide.
There are still no effective antiviral therapies that can be used immediately on patients. The most promising method to prevent the disease is a protective vaccine.
5.2. Vaccine development
Immediately after the SARS outbreak, researchers began to investigate whether inactivated vaccine could be used to prevent SARS. In China, a collaborative group from the Chinese Academy of Medical Sciences, China's CDC, and the Sinovac Biotech Company passed the inactivated SARS-CoV strain PUMC001 vaccine clinical trial phase 1 from the SFDA in the summer of 2005. Although the inactivated SARS vaccine seems useful and reasonable in SARS prevention, the safety of the inactivated SARS-CoV vaccine is a serious concern.
Vaccines targeting structural genes, especially the recombinant spike protein, seem the safest strategy. A DNA vaccine encoding S protein can elicit neutralizing antibodies and induce T cell response, which can protect mice from SARS infection [33]. Varieties of vectors were used to express the spike protein and then used to immunize mice, African green monkeys, and hamsters. Neutralizing antibodies were detected which protected them from infection. The DNA SARS vaccine invented by scientists at the National Institute of Allergy and Infectious Diseases and produced by Vical Inc. of San Diego entered clinical trial phase 1 under US FDA guidelines at the end of 2004. Recently, a group proved that RBD could elicit much higher titers of neutralizing antibodies than did the full-length S protein [34], suggesting that RBD may be a potential vaccine candidate.
However, the discovery that spike RBD alone could worsen acute lung injury in mice indicated the potential safety issue of spike as protein vaccine. The harm of spike RBD to the lung was thought to be caused by its binding to SARS receptor ACE2 [8]. It is reported that S proteins from the 2002–2003 SARS outbreak and from the much less severe 2003–2004 outbreak have different binding affinities to the receptor ACE2. The latter has lower affinity to human ACE2, mainly due to an alteration of amino acid residues at sites 479 and 487 [35]. Therefore, in order to obtain safer recombinant spike protein vaccine, site-directed mutagenesis at these sites might be necessary to eliminate the binding affinity of spike to ACE2.
6. Concluding remarks
In summary, the global outbreak of SARS has led to the formation of a successful network of laboratories, and much has been learned about SARS in the three years since its discovery. However, some aspects of the molecular pathogenesis of the disease are still not fully understood. Further investigations of various aspects, including the life cycle of the virus, the molecular mechanisms of the disease, and the factors that can influence the progress of the disease, will result in a more thorough understanding of this new pathogen. Results from further research will certainly suggest more promising treatment strategies and may lead to prevention of this disease.
Acknowledgement
The authors thank Dr. Wenhui Li of Harvard Medical School for reading this manuscript and for his useful comments and suggestions.
References
- 1.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 3.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 4.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 5.Yang Z.Y., Huang Y., Ganesh L., Leung K., Kong W.P., Schwartz O., Subbarao K., Nabel G.J. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 2004;78:5642–5650. doi: 10.1128/JVI.78.11.5642-5650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B., Huan Y., Yang P., Zhang Y., Deng W., Bao L., Zhang B., Liu G., Wang Z., Chappell M., Liu Y., Zheng D., Leibbrandt A., Wada T., Slutsky A.S., Liu D., Qin C., Jiang C., Penninger J.M. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 10.Hamming I., Timens W., Bulthuis M.L., Lely A.T., Navis G.J. H. van Goor, Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marzi A., Gramberg T., Simmons G., Moller P., Rennekamp A.J., Krumbiegel M., Geier M., Eisemann J., Turza N., Saunier B., Steinkasserer A., Becker S., Bates P., Hofmann H., Pohlmann S. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004;78:12090–12095. doi: 10.1128/JVI.78.21.12090-12095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeffers S.A., Tusell S.M., Gillim-Ross L., Hemmila E.M., Achenbach J.E., Babcock G.J., Thomas W.D., Jr., Thackray L.B., Young M.D., Mason R.J., Ambrosino D.M., Wentworth D.E., Demartini J.C., Holmes K.V. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. U.S.A. 2004;101:15748–15753. doi: 10.1073/pnas.0403812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan V.S., Chan K.Y., Chen Y., Poon L.L., Cheung A.N., Zheng B., Chan K.H., Mak W., Ngan H.Y., Xu X., Screaton G., Tarn P.K., Austyn J.M., Chan L.C., Yip S.P., Peiris M., Khoo U.S., Lin C.L. Homozygous L-SIGN (CLEC4M) plays a protective role in SARS coronavirus infection. Nat. Genet. 2006;38:38–46. doi: 10.1038/ng1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu S., Xiao G., Chen Y., He Y., Niu J., Escalante C.R., Xiong H., Farmar J., Debnath A.K., Tien P., Jiang S. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. 2004;363:938–947. doi: 10.1016/S0140-6736(04)15788-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang I.C., Bosch B.J., Li F., Li W., Lee K.H., Ghiran S., Vasilieva N., Dermody T.S., Harrison S.C., Dormitzer P.R., Farzan M., Rottier P.J., Choe H. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J. Biol. Chem. 2006;281:3198–3203. doi: 10.1074/jbc.M508381200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donnelly C.A., Ghani A.C., Leung G.M., Hedley A.J., Fraser C., Riley S., Abu-Raddad L.J., Ho L.M., Thach T.Q., Chau P., Chan K.P., Lam T.H., Tse L.Y., Tsang T., Liu S.H., Kong J.H., Lau E.M., Ferguson N.M., Anderson R.M. Epidemiological determinants of spread of causal agent of severe acute respiratory syndrome in Hong Kong. Lancet. 2003;361:1761–1766. doi: 10.1016/S0140-6736(03)13410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsang K.W., Ho P.L., Ooi G.C., Yee W.K., Wang T., Chan-Yeung M., Lam W.K., Seto W.H., Yam L.Y., Cheung T.M., Wong P.C., Lam B., Ip M.S., Chan J., Yuen K.Y., Lai K.N. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003;348:1977–1985. doi: 10.1056/NEJMoa030666. [DOI] [PubMed] [Google Scholar]
- 18.Peiris J.S., Chu C.M., Cheng V.C., Chan K.S., Hung I.F., Poon L.L., Law K.I., Tang B.S., Hon T.Y., Chan C.S., Chan K.H., Ng J.S., Zheng B.J., Ng W.L., Lai R.W., Guan Y., Yuen K.Y. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lang Z., Zhang L., Zhang S., Meng X., Li J., Song C., Sun L., Zhou Y. Pathological study on severe acute respiratory syndrome. Chin. Med. J. (Engl.) 2003;116:976–980. [PubMed] [Google Scholar]
- 20.Wong K.T., Antonio G.E., Hui D.S., Lee N., Yuen E.H., Wu A., Leung C.B., Rainer T.H., Cameron P., Chung S.S., Sung J.J., Ahuja A.T. Severe acute respiratory syndrome: radiographic appearances and pattern of progression in 138 patients. Radiology. 2003;228:401–406. doi: 10.1148/radiol.2282030593. [DOI] [PubMed] [Google Scholar]
- 21.Booth C.M., Matukas L.M., Tomlinson G.A., Rachlis A.R., Rose D.B., Dwosh H.A., Walmsley S.L., Mazzulli T., Avendano M., Derkach P., Ephtimios I.E., Kitai I., Mederski B.D., Shadowitz S.B., Gold W.L., Hawryluck L.A., Rea E., Chenkin J.S., Cescon D.W., Poutanen S.M., Detsky A.S. Clinical features and short-term outcomes of 144 patients with SARS in the greater Toronto area. JAMA. 2003;289:2801–2809. doi: 10.1001/jama.289.21.JOC30885. [DOI] [PubMed] [Google Scholar]
- 22.Wong R.S., Wu A., To K.F., Lee N., Lam C.W., Wong C.K., Chan P.K., Ng M.H., Yu L.M., Hui D.S., Tam J.S., Cheng G., Sung J.J. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. BMJ. 2003;326:1358–1362. doi: 10.1136/bmj.326.7403.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reghunathan R., Jayapal M., Hsu L.Y., Chng H.H., Tai D., Leung B.P., Melendez A.J. Expression profile of immune response genes in patients with severe acute respiratory syndrome. BMC Immunol. 2005;6:2. doi: 10.1186/1471-2172-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicholls J.M., Poon L.L., Lee K.C., Ng W.F., Lai S.T., Leung C.Y., Chu C.M., Hui P.K., Mak K.L., Lim W., Yan K.W., Chan K.H., Tsang N.C., Guan Y., Yuen K.Y., Peiris J.S. Lung pathology of fatal severe acute respiratory syndrome. Lancet. 2003;361:1773–1778. doi: 10.1016/S0140-6736(03)13413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sui J., Li W., Roberts A., Matthews L.J., Murakami A., Vogel L., Wong S.K., Subbarao K., Farzan M., Marasco W.A. Evaluation of human monoclonal antibody 80R for immunoprophylaxis of severe acute respiratory syndrome by an animal study, epitope mapping, and analysis of spike variants. J. Virol. 2005;79:5900–5906. doi: 10.1128/JVI.79.10.5900-5906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan H., Xiao G., Zhang J., Hu Y., Yuan F., Cole D.K., Zheng C., Gao G.F. SARS coronavirus induces apoptosis in Vero E6 cells. J. Med. Virol. 2004;73:323–331. doi: 10.1002/jmv.20094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang M., Li C.K., Li K., Hon K.L., Ng M.H., Chan P.K., Fok T.F. Hematological findings in SARS patients and possible mechanisms. Int. J. Mol. Med. 2004;14:311–315. [review] [PubMed] [Google Scholar]
- 28.Mizutani T., Fukushi S., Murakami M., Hirano T., Saijo M., Kurane I., Morikawa S. Tyrosine dephosphorylation of STAT3 in SARS coronavirus-infected Vero E6 cells. FEBS Lett. 2004;577:187–192. doi: 10.1016/j.febslet.2004.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizutani T., Fukushi S., Saijo M., Kurane I., Morikawa S. Regulation of p90RSK phosphorylation by SARS-CoV infection in Vero E6 cells. FEBS Lett. 2006;580:1417–1424. doi: 10.1016/j.febslet.2006.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kopecky-Bromberg S.A., Martinez-Sobrido L., Palese P. 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J. Virol. 2006;80:785–793. doi: 10.1128/JVI.80.2.785-793.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imai Y., Kuba K., Rao S., Huan Y., Guo F., Guan B., Yang P., Sarao R., Wada T., Leong-Poi H., Crackower M.A., Fukamizu A., Hui C.C., Hein L., Uhlig S., Slutsky A.S., Jiang C., Penninger J.M. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soo Y.O., Cheng Y., Wong R., Hui D.S., Lee C.K., Tsang K.K., Ng M.H., Chan P., Cheng G., Sung J.J. Retrospective comparison of convalescent plasma with continuing high-dose methylprednisolone reatment in SARS patients. Clin. Microbiol. Infect. 2004;10:676–678. doi: 10.1111/j.1469-0691.2004.00956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Z.Y., Kong W.P., Huang Y., Roberts A., Murphy B.R., Subbarao K., Nabel G.J. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature. 2004;428:561–564. doi: 10.1038/nature02463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang D.M., Wang G.L., Lu J.H. Severe acute respiratory syndrome: vaccine on the way. Chin. Med. J. (Engl.) 2005;118:1468–1476. [PubMed] [Google Scholar]
- 35.Li W., Zhang C., Sui J., Kuhn J.H., Moore M.J., Luo S., Wong S.K., Huang I.C., Xu K., Vasilieva N., Murakami A., He Y., Marasco W.A., Guan Y., Choe H., Farzan M. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24:1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]