

Abstract
Many viruses regulate a crucial point in the apoptotic pathway by expressing viral Bcl-2 homologues, which have become useful tools to investigate the mechanisms behind the control of the mitochondrial checkpoint of apoptosis. Concurrently, a number of viral inhibitors of innate immune signalling have been instrumental tools in the discovery of key host pathways. Here we discuss how viral inhibitors of the apoptotic and innate signalling pathways have further enhanced the understanding of both research fields and are beginning to shed light on how these two pathways converge.
Keywords: Apoptosis, Viral homologues, Inflammasome
1. Introduction
Viruses are intracellular parasites that require infected cells to survive for the appropriate length of time to propagate and thus ensure their successful transmission to the next host. For many viruses inhibiting host cell death, at least temporarily, is therefore paramount. For others, perhaps paradoxically, it is crucial that they induce cell death, for instance to facilitate viral exit or to eliminate cells that may hamper their propagation within the host. Viral genomes reflect their specific dependence on the induction or inhibition of cell death during their life cycle, with viral proteins targeting many key steps in cell death progression.
In response to the stress induced by viral infection, all metazoan cells have the intrinsic capacity to sense virus infection and restrict viral replication and spread. A staggering number of viral inhibitors of innate signalling have been discovered, and they have often been great tools in the discovery of key host pathways. Here we review the strategies that viruses employ to regulate a crucial point in the apoptotic pathway, the mitochondrial checkpoint, with an emphasis on the role of viral Bcl-2 homologues, and how these proteins remain useful in the investigation of the mechanisms of apoptosis. Furthermore, we discuss how viral inhibitors support the suggestion that the machinery that governs the detection of and response to viral infection shares features with cell death programs.
2. Inhibition of apoptosis: lessons learned from viral Bcl-2-like proteins
Apoptosis is a highly conserved process in multicellular organisms. Viruses employ strategies to regulate the mitochondrial checkpoint of apoptosis especially by altering the balance of pro-apoptotic and pro-survival proteins, either by producing pro-survival inhibitors to induce cellular death or by expressing viral Bcl-2 homologues to maintain cellular survival. Commitment to apoptosis can take place through two distinct but interconnected pathways: the intrinsic and extrinsic apoptotic pathways (see also Duprez et al. in this issue) [1].
The extrinsic apoptotic pathway is initiated by extracellular receptor-ligand interactions. Ligands such as FasL and TNFα induce oligomerisation of their respective receptors and the formation of death inducing signalling complexes (DISCs) in the cytoplasm, leading to association of fas associated death domain (FADD) and the cysteine protease caspase-8. Proteolytic activation of caspase-8 leads to dissociation of the DISC and activation of effector caspases (e.g. caspase-3,-6 and -7). Viruses from many different viral families inhibit the extrinsic pathway at different stages by mimicking TNF receptors and acting as decoys (such as CrmB from cowpox virus and MT-2 from Myxoma virus), by mimicking cellular inhibitors of caspase-8 activation (vFLIPs such as MC159 from Molluscum Contagiosum virus), and by inhibiting the proteolytic activity of caspase-8 (CrmA from cowpox virus) [2].
The intrinsic apoptotic pathway is activated by a variety of stress signals, such as DNA damage, hormone deprivation, unfolded protein response and pathogen infection. The intrinsic pathway is regulated by the Bcl-2 family of proteins, which control a checkpoint preventing the disruption of the mitochondrial outer membrane (MOM). MOM permeabilisation leads to the release of several apoptogenic proteins from the intermembrane space including cytochrome c. Upon release from mitochondria cytochrome c induces formation of the apoptosome in association with Apaf-1 and pro-caspase 9, leading to caspase-9 activation [1]. Direct inhibition of caspases by cellular Inhibitors of Apoptosis (IAPs) has also been found as a viral strategy although this seems to be limited to insect cells. IAPs were first characterised in the Baculovirus family, which express OpIAP and CpIAP [3]. Entomopoxviruses also express viral IAP's such as MsEPV and AmEPV [4].
The Bcl-2 family of proteins is subdivided on the basis of the structural conservation of the Bcl-2 Homology domains (BH) domains. For instance, the pro-apoptotic BH3-only proteins (including Bim, Bid, Bad, Bmf, Puma, Noxa) only share homology with the rest of the Bcl-2 family of proteins in the alpha-helical BH3 domain. These BH3-only proteins act as sensors of the intrinsic pathway by responding to diverse developmental and environmental death cues. The diversity of BH3-only proteins, together with their tissue specific distribution permits a variety of cytotoxic stress signals to initiate apoptosis [1].
In contrast, the pro-survival members of the Bcl-2 family (Bcl-2, Bcl-XL, Bcl-w, Mcl-1, A1 and perhaps Boo) contain a BH1, BH2 and BH3 domain, while some of these members also contain a BH4 domain at the N-terminus. The pro-survival proteins can interact with the BH3-only proteins in order to inhibit apoptosis. Interactions between BH3-only and pro-survival proteins are specific, implying that not all BH3-only proteins can induce apoptosis by themselves. The effectors of this pathway (Bak, Bax, and perhaps Bok) also contain a BH1, BH2 and BH3 domain but have a pro-apoptotic function. Following an apoptotic stimulus, BH3-only proteins inhibit the pro-survival proteins, allowing for Bak and Bax to kill the cell (Fig. 1 ) [1].
Fig. 1.
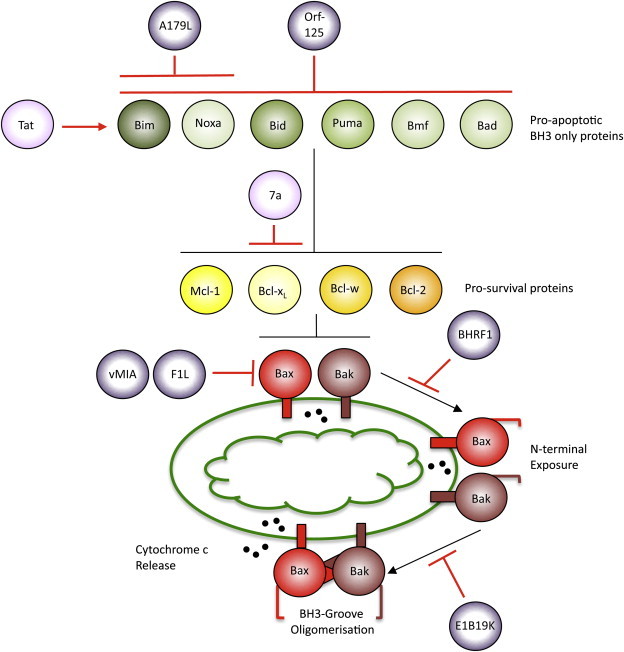
Viral proteins target specific elements of the mitochondrial checkpoint of apoptosis. According to a prevalent model, upon an apoptotic stimulus the BH3-only proteins (Bim, Noxa, Bid, Puma, Bmf, Bad) are induced or activated. They subsequently translocate to the mitochondria to inhibit the pro-survival Bcl-2 proteins (Bcl-2, Bcl-XL, Bcl-w, Mcl-1). Upon sequestration of pro-survival Bcl-2 proteins by BH3-only proteins, the pro-apoptotic Bcl-2 proteins Bak and Bax located in the outer mitochondrial membrane undergo structural rearrangements of their N-terminus, leading to their oligomerisation. The viral activators (pink) and inhibitors (purple) of apoptosis sequestering this pathway target specific subsets of Bcl-2 proteins, for details see text. Black lines indicate the cellular apoptotic pathway, red lines indicate viral inhibition, and red arrows indicate activation. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The structures of several cellular pro-survival members, as well as Bak and Bax have been solved. These proteins are characterised by a helical bundle structure in which a central hydrophobic helix is secluded from the cytosol by a set of 7-8 amphipathic helices. Furthermore, coalescence of the helices in the regions containing the BH1, BH2, and BH3 domains creates an elongated hydrophobic groove that behaves as an interaction domain with the various pro-apoptotic members of the Bcl-2 family of proteins [1].
Viruses have successfully mimicked elements of the intrinsic apoptotic pathway to either retain mitochondrial integrity or lead to MOM permeabilisation, depending on the viral life cycle, host, and the host immune response to the virus (for a detailed review of this field see; Galluzi et al. 2008). Techniques to induce MOM permeabilisation by interfering with pro-survival members of the intrinsic pathway are predominantly found within the RNA viruses. Vpr from Human immunodeficiency virus-1 (HIV-1) acts at mitochondria to induce swelling and MOM permeabilisation in lymphoid cells. Bcl-2 has been reported to inhibit Vpr induced MOM permeabilisation, however this process seems to be cell type dependent [5], [6]. Severe acute respiratory syndrome coronavirus (SARS-CoV) 7a protein has been reported to directly inhibit Bcl-XL pro-survival activity, along with its ability to bind all other pro-survival members (Fig. 1) [7]. Like other intrinsic forms of cellular stress, viral proteins may lead to BH3-only protein activation. Tat protein from HIV-1 is capable of inducing apoptosis by causing the disassociation of Bim from the cytoskeleton, where Bim is sequestered in healthy cells (Fig. 1) [8]. Interferon 1 production in response to vesicular stomatitits viral infection has been shown to induce expression of pro-apoptotic Noxa [9].
If viral infection induces apoptosis, this must depend on the recognition of viral molecules by cellular receptors. Recent evidence indicates that it is often viral nucleic acids that can be detected by these receptors (see also below) [10]. How such recognition is linked to the induction of apoptosis is not well understood but there is evidence that toll-like receptor 3 can at least in some situations induce apoptosis that appears to depend on caspase-8 activation, and the cytosolic receptors for viral RNA, RIG-I and MDA5, have recently been found to be capable of inducing not only a pro-inflammatory but also a pro-apoptotic response (Besch et al., J Clin Invest, in press) [11]. The role of this pro-apoptotic potential for viral propagation and host defence will need to be worked out in the future.
Strategies to maintain MOM integrity have predominantly evolved amongst DNA viruses by encoding functional homologues of Bcl-2 proteins, collectively termed vBcl-2s. vBcl-2s have been found in various members of the Poxviridae (F1, N1, M11L, A179L, ORFV125, M11L), Herpesviridae (BHRF1, BALF1, vMIA) Adenoviridae (E1B19K) and Birnaviridae (VP5) families of viruses [9]. Interestingly, while some vBcl-2s such as E1B19K are clear sequence homologues of cellular Bcl-2-like proteins, others share little primary sequence homology with their mammalian homologues. However, the crystal structure of some of these functional Bcl-2 homologues reveals conservation of the Bcl-2 family structural conformation. Examination of the latter class of vBcl-2 proteins now allows us to revisit the role of the conserved amino acids in the BH1, BH2, and BH3 domains of the pro-survival members as originally defined. The most highly conserved of these regions is the BH3 domain present in all members of the cellular Bcl-2 family of proteins. vBcl-2s allow us to question the strict requirement for the Bcl-2 pro-survival proteins to contain BH domains, including the core BH3 domain, to function. For example, Adenovirus E1B19K and African Swine Fever Virus (ASFV) A179L retain homology to all BH domains [12]. In contrast, FPV039 from Fowlpoxvirus only retains sequence homology with the BH1 and BH2 domains while Orf-125 of Parapoxvirus retains most sequence homology at the BH1 and BH3 [9], [13], [14]. Other vBcl-2's such as Vaccinia virus F1 and N1, Myxoma virus M11L and vMIA from Cytomegalovirus do not retain any significant homology to the BH domains found in mammalian Bcl-2's [9]. Therefore, vBcl-2s show that overall sequence homologies, and the sequence of the BH domains, are not strict requirements for the pro-survival function of Bcl-2 like proteins. Rather, they emphasize the requirement for their helical bundle structure.
vBcl-2s, like their mammalian counterparts, have been shown to vary in their anti-apoptotic potency. The variation in pro-survival potency of vBcl-2s and mammalian Bcl-2s has been debated and there is still no agreement [15], [16]. These different results are possibly due to differences in cell types used, protein expression levels or variations in apoptotic stimuli used to assess pro-survival function. While proteins such as Bcl-2, Bcl-XL and E1B19K are considered strong inhibitors of apoptosis, others such as N1, Bcl-w, and A1 are considered weak pro-survival members when over-expressed [17]. Two studies that compared the pro-survival activity of cellular pro-survivals and vBcl-2s have contradicted each other regarding the pro-survival activity of these proteins. Huang et al. describe the vBcl-2 E1B19K and the mammalian pro-survivals Bcl-2 and Bcl-XL as functionally equivalent by assessing different cell types and stimuli. In contrast, Cross et al. define the vBcl-2s E1B19K and BHRF1 as more efficient inhibitors of apoptosis than their mammalian homologues Bcl-2 and Bcl-XL [15], [16].
It is in part because of their different characteristics that vBcl-2s have proven useful tools in the dissection of the intrinsic apoptotic pathway. Distinct abilities to inhibit the different stages of the apoptotic pathway have been attributed to the various vBcl-2s. vMIA exclusively inhibits Bax at mitochondria by an interaction that does not depend on the sequestration of the Bax BH3 domain but that involves electrostatic interactions (Fig. 1) [18]. A recent report has suggested a similar alternative interaction to the typical sequestration of Bax BH3 domain for Bcl-XL [19]. Preferential inhibition of Bak has been reported for F1, although the interacting domains between these two proteins are still debated (Fig. 1) [20], [21]. Apoptotic inhibition by the sequestration of BH3-only proteins by vBcl-2s has been reported for A179L by binding both Bim and Noxa, and Orf-125 through binding of all tested BH3-only proteins (Fig. 1) [9], [12], [13].
The mechanisms behind the activation of Bak and Bax have been elucidated to an extent. Upon apoptotic signalling both Bak and Bax undergo conformational changes at their N-termini and oligomerise to permeabilise the MOM. Oligomerisation of Bax and Bak occurs via interactions involving the BH3 domain of the Bax or Bak monomers in a process that precedes, and directly allows for, the release of cytochrome c [22], [23]. E1B19K, BHRF1 and M11L have all been reported to bind both Bax and Bak, but despite the observation that they have the same binding partners, the stages at which these proteins bind Bak and Bax are distinct [16], [24]. M11L has been reported to bind both Bak and Bax by sequestering their BH3 domain [24]. BHRF1 inhibits Bak and Bax N-terminal exposure when both pro-apoptotic members are localised to mitochondria (Fig. 1) [16]. N-terminal exposure of Bak and Bax is a process that precedes the exposure of the BH3 domain, therefore suggesting an alternative form of binding between the vBcl-2s and the pro-apoptotic members [22], [23]. In contrast, E1B19K has been reported to inhibit the oligomerisation of Bak and Bax, a process that also involves Bak and Bax BH3 domain sequestration (Fig. 1) [16].
Together, the distinct mechanisms behind the function of the vBcl-2s show that interactions with the different pro-apoptotic members of this family are unique to the different proteins. Therefore, the targeted or combined use of vBcl-2s with their distinct properties will continue to add to our understanding of the modes of apoptotic inhibition involved in maintaining MOM integrity in a cellular and viral context.
3. Virus sensing and immune evasion
Clearly, activation of cellular suicide programmes represents an efficient host strategy to limit virus replication and spread. However, these programmes are merely one of the cellular responses that follow the detection of an invading virus: the innate immune system activates a variety of anti-viral responses, both in the infected cell and in professional immune cells [25]. This firmly places virus sensing at the centre of all anti-viral “outputs”, including initiation of cell death.
In order for the host cell to detect viruses, viral substrates have to be recognised as non-self to activate the innate immune system. It has become clear that viral recognition is achieved through the recognition of pathogen associated molecular patterns (PAMP). These specific pathogen protein and nucleic acid signature patterns are recognised at distinct sub-cellular locations by pattern recognition receptors (PRR) [25]. The term PRR generally tends to be reserved for those viral sensor proteins that signal to induce a transcriptional response. The most notable induced anti-viral gene products include the secreted type I interferons, which can induce apoptosis though the intrinsic pathway as mentioned above. Interferon-induced signalling pathways initiate an anti-viral transcriptional programme of IFN-stimulated genes (ISG) in infected cells and their neighbours, thus spreading an “anti-viral state” [26].
Recent advances have identified the Toll-like receptor family (TLR) and the Rig-like helicases (RLH) as PRRs. TLRs are classical single pass transmembrane proteins whose PAMP-binding domains contain leucine-rich repeats (LRR). Viral nucleic acid ligands, including long dsRNA, ssRNA or CpG DNA, are detected by a subset of TLRs (TLR3,7-9) that survey the endosomal compartment. Upon ligand activation, TLRs recruit adaptor proteins such as MyD88, TRIF or TRAM, which activate specific members of the TRAF adaptor and the IRAK kinase families. These signalling pathways ultimately lead to phosphorylation and nuclear translocation of the NF-κB and IRF3,7 transcription factors through respective activation of the IκB kinase (IKK) α,β,γ and IKKɛ/TBK1 complexes [25], [27].
It is vital for viruses to inhibit the signalling pathways downstream of TLRs (and RLHs, see below), and the viral evasion and subversion of these pathways continues to amaze. Inhibition occurs proximal to the TLR, as well as at every level towards transcription. For instance, Hepatitis C virus (HCV) NS3-4A blocks TLR3 mediated endosomal detection of dsRNA by cleaving the adaptor TRIF [28]. Vaccinia virus blocks TLR signalling through A46, an inhibitor of the adaptor proteins MyD88, MAL, TRIF and TRAM, while HCV NS5 inhibits MyD88. TRAF6, an adaptor downstream of MyD88 and IRAK, is inhibited by vaccinia A52, which in addition targets the kinase IRAK2 [27]. The IKK α,β,γ complex is also antagonized by viral proteins, such as vaccinia virus B14. HCV NS3, rabies virus phosphoprotein and hantavirus G1 protein all target the IKKɛ/TBK1 complex, while vaccinia K7 prevents the complex from activating IRFs by targeting the Dead-box protein 3 (DDX3) protein [27].
While TLRs predominantly recognise viral PAMPs within endosomes, cells also contain viral sensors in the cytosol [25]. The Rig-like helicase (RLH) family includes RIG-I, MDA5, and LGP-2. RIG-I and MDA5 both contain two caspase recruitment domains (CARDs), an ATPase and a helicase domain. Both use their helicase domains to recognise distinct viral nucleic acids. MDA5 is the main cytoplasmic receptor for longer viral dsRNA molecules. RIG-I on the other hand can be activated by short dsRNA fragments, as well as ssRNAs that have a 5' triphosphate [25]. Interestingly, these characteristics appear to explain why host RNAs are not recognised by either sensor protein: host RNA either have their 5' capped or contain monophosphates, and long dsRNA is only present in the context of viral infection. After binding of the RNA with their helicase domain, activated RIG-I and MDA5 use their CARD domains to bind the mitochondrion-located adaptor protein IPS-1 (also known as MAVS, VISA or Cardif). Recently, an additional adaptor protein localised to the ER and mitochondrial outer membrane, called MITA (or STING), was identified to recruit TBK1 and IRF3 to IPS-1 and thus enhance RIG-I specific signalling [29]. Via IPS-1 RLH signalling converges with the IKK α,β,γ and IKKɛ/TBK1 complexes leading to activation of the NF-κB and IRF3,7 transcription factors.
Unsurprisingly, the RIG-I and MDA5 pathways are heavily policed by viral inhibitors. Influenza NS1 binds the RIG-I/IPS1 complex to interfere with it’s signalling. MDA-5 is inhibited by the paramyxovirus V proteins, while poliovirus interferes with MDA5 function by inducing its cleavage. Furthermore, HCV NS3-4A abrogates IPS1 signalling by cleaving it from the mitochondrial membrane. In addition, hepatitis A virus 3ABC is targeted to the mitochondria where it degrades IPS1 [27].
It is worth noting that TLRs have also been shown to induce apoptosis in response to viral detection. TRIF, an adaptor for TLR3 and TLR4 signalling, has been shown to induce apoptosis via the extrinsic pathway in a FADD/caspase-8-dependent and mitochondrion-independent pathway [30]. This pathway could further be inhibited by the viral inhibitors of the extrinsic apoptotic pathway or novel inhibitors, but the mechanisms behind this remain beyond the scope of this review (For review see: Kaiser et al. 2005).
Another family of cytosolic PRRs are the so-called Nod-like receptors (NLR), sub-divided into three families on the basis of their N-terminal protein-protein interaction domains. The CARD domain NOD family, the pyrin domain (PYD) NALP family and the baculovirus inhibitory repeat (BIR) domain NAIP family [31]. These proteins are thought to maintain an auto-inhibitory interaction between a NOD domain and a C-terminal LRR domain to prevent spontaneous activation. Upon PAMP binding to their LRR domains, the NLR proteins oligomerize and expose their N-terminal domains to recruit effector proteins. The NOD family proteins recruit the kinase RICK via their CARD domains, leading to MAPK and NF-κB activation. The NALP family proteins recruit the adaptor ASC to their PYD domain, leading to the recruitment of pro-caspase-1 and it’s processing to caspase-1. Caspase-1 recruitment by ASC association has also been shown to be further induced by the interferon H1N-200 family member AIM2 (abscent in melanoma 2), due to its newly described NALP-like activity [32]. Caspase-1-activating complexes are called inflammasomes, and each inflammasome is designated according to the NLR within it [33]. The NLR system appears to in part work in concert with other pathogen detectors. For instance, PAMPs may engage the TLR- NF-κB pathway leading to pro-IL-1β production. Pro-IL-1β is then cleaved to IL-1β, a strong pro-inflammatory cytokine, by caspase-1 produced by inflammasome activation [34]. In addition, inflammasome activation can also contribute to pathogen-induced cell death in ways that are not well characterised (e.g. pyroptosis, see Duprez et al. in this issue) [35], [36].
Although some herpesviruses may subvert IL-1β activity, only poxviruses encode known inhibitors of inflammasome signalling [27]. Vaccinia virus CrmA (B13R) is an inhibitor of caspase-1, although its role in vivo appears to be predominantly anti-apoptotic [37], Vaccinia also encodes the soluble IL1βR inhibitor B15, which largely prevents pyrogenic responses in vivo [28]. Another poxvirus, myxoma virus, encodes M13L, a PYD domain containing inhibitor of caspase-1 activation [38]. M13L is thought to compete with PYD-containing NLRs by binding ASC. Curiously, many NLR ligands are bacterial PAMPs, yet despite the fact that viral infection can induce IL-1β processing, few viral PAMPs that activate inflammasomes have thus far been discovered [27]. Mirroring this, few viral NLR or inflammasome inhibitors are known to date. Is this perhaps an indication of a fundamental difference in the way bacteria and viruses are sensed by inflammasomes? Even though it could establish a fundamental difference, it is more likely that we are still to uncover the diverse ways in which viruses modulate the inflammasome and NLR signalling.
4. Cell death and virus sensing: similarities revealed by viral inhibitors?
Subsequent to sensing of viral infection most cells will attempt to undergo cell death whilst also producing pro-inflammatory cytokines to spread the alarm amongst their neighbours. Recent advances indicate that some elements of the machineries governing cell death and the innate immune response overlap. In particular, viral inhibitors are providing insights into this overlap.
It is becoming increasingly evident that mitochondria are not merely regulating the metabolic status of the cell but are also a hub for signalling pathways that regulate cell death and innate immunity. The mitochondrial checkpoint for apoptosis is heavily targeted by vBcl2 anti-apoptotic proteins and, as discussed above, these viral proteins continue shedding light on the molecular nature of this checkpoint. In RLH signalling, the IPS1-associated complex recruits TBK1 and IRF3 via the adaptor MITA, which like IPS1 is localised to the outer mitochondrial membrane and the ER. In fact, the recent discovery that HCV cleavage of IPS1 off the mitochondria renders it non-functional proved that IPS1 functions at the mitochondria and represents a great example of viral proteins providing insights into host biology [27]. The central position of the mitochondria in both RLH and cell death signalling is intriguing: are mitochondria merely a convenient scaffold to attach signalling pathways onto or could it indicate that that these pathways are functionally interlinked?
It is thought that cell removal by apoptosis is the preferred cell death pathway as it prevents over-activation by the immune system. However, recent evidence suggests that, at least under certain conditions, anti-viral immunity could benefit from infected cells dying by other programmed, but non-apoptotic forms of cell death that lead to release of pro-inflammatory cell content into the extracellular milieu. [36]. NLR proteins have emerged as regulators of such infection-induced, pyrogenic forms of cell death, in part through inflammasome-mediated caspase-1 activation [35]. Caspase-1 processing produces mature IL-1β, but its substrates in the mediation of non-apoptotic cell death remain unclear. Interesting further similarities and cross-talk between inflammasome and cell death components have been described. For instance, the inflammasome shares characteristics with the apoptosome structure made up of Apaf-1 and caspase-9 (Fig. 2 ). Apaf-1 is maintained in an inactive state through the auto-inhibitory interactions of its C-terminal WD40 domains with the nucleotide-binding domain (NBD). Following cytochrome c release from the mitochondria the inhibition is relieved and Apaf-1 and pro-caspase-9 form the heptameric wheel-like apoptosome structure [35]. NLR proteins are structurally similar to Apaf-1, with C-terminal LRR domains instead of WD40 repeats, and are similarly activated by the release of an auto-inhibitory interaction [35].
Fig. 2.
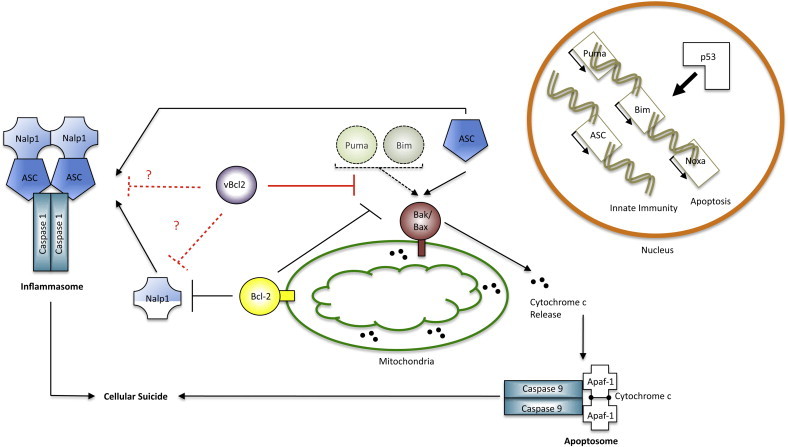
Cell death and innate immunity are intimately linked. ASC induction by p53 is capable of inducing Bax dependent apoptosis, hence leading to the formation of the apoptosome along with formation of the inflammasome. Similarities in the diagrammatic representation of the inflammasome and apoptosome are suggestive of the structural similarities between both molecular complexes. Indications of the BH3-only proteins (Bim, Puma, and Noxa) p53 mediated transcriptional control and possible interactions with Bax (depicted by the dashed arrow) highlight weaknesses in the ASC/Bax interaction pathway (see text). Bcl-2 plays a direct role in the inhibition of Bak/Bax activation and inflammasome formation by Nalp1. Findings regarding the innate and apoptotic inhibitory effect of vBcl-2s raise the question of whether these proteins may also be able to inhibit the inflammasome cascade. Black lines indicate the cellular apoptotic pathway, red lines indicate viral inhibition, and dashed lines indicate discrepancies or possibilities raised in the text. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Furthermore, direct interactions between cell death and inflammasome components have been reported. The adaptor ASC, an essential component of the inflammasome, facilitates p53-dependent cell death through activation of Bax, possibly at the mitochondria (Fig. 2) [39]. It is worth noting however, that the involvement of BH3-only proteins like Bim and Puma, which are also induced by p53, was not assessed in these studies (Fig. 2) [1]. Lastly, the pro-survival proteins Bcl-2 and Bcl-XL have recently been found to bind the NLR protein NALP1 and thus suppress the NALP1 inflammasome, leading to reduced caspase-1 activation and IL-1β production (Fig. 2) [40]. While it is not yet known which sub-cellular pool of these Bcl-2 proteins is involved in inflammasome interactions, it certainly raises the prospect of the NLR signalling pathway being regulated from the mitochondria (Fig. 2). Future work will undoubtedly elaborate on the role the Bcl-2 family plays in NLR signalling, especially since the ASC-Bax and Bcl-2-NALP1 interactions suggest competing roles of the Bcl-2 proteins in the regulation of the inflammasome. As discussed, many viruses contain sequence and structural Bcl-2 homologues. Given the obvious scarcity of known viral inhibitors of the inflammasome complex and NLR signalling in general, could some of these vBcl-2s represent such inhibitors?
Recent reports have revealed a further sequel to the story of vBcl-2 proteins. Myxoma virus M11L and vaccinia virus F1 and N1 all encode functional Bcl-2 homologues that directly target Bak (F1), Bax (N1) or both (M11L). As mentioned these poxvirus proteins share a helical bundle structure that includes a BH3-binding groove, but do not share sequence similarity with other vBcl-2s or their mammalian counterparts.
Graham et al. have solved the structures of the vaccinia virus TRAF6/IRAK2 inhibitor A52 and the IKKβ inhibitor B14. Intriguingly, these inhibitors of TLR and RLH signalling have the same structural helical bundle fold as the Bcl-2 family while neither have anti-apoptotic activity [41]. In line with this feature, B14 and A52 do not contain a hydrophobic groove that could accommodate BH3 peptides from pro-apoptotic Bcl-2 proteins. Both proteins belong to a family that includes vaccinia proteins A46, N1, C6, C16 and K7, some of which are proven innate signalling inhibitors, and it was proposed that these may all have a Bcl-2 structural fold [41]. Indeed, the structure of K7 was recently shown to have such a fold [42].
N1, a recognised vBcl-2, has an unusual, largely open hydrophobic groove when compared to other vBcl-2s and their mammalian counterparts. N1 has weak anti-apoptotic activity and is localised to the cytosol, where it is thought to bind Bax [41]. Possibly its weak apoptotic activity is explained by vaccinia-induced apoptosis being predominantly Bak-dependent, though this remains unexplored. Interestingly, N1 is also an inhibitor of TRAF6 and the IKKɛ/TBK1 complex, making it the only known vaccinia Bcl-2-like protein that inhibits both the intrinsic apoptotic pathway and some of the innate immunity pathways [43].
Based on the sequence divergence between these vaccinia Bcl-2-like proteins and their host counterparts Graham et al. propose that ancestral poxviruses have acquired a Bcl-2-like protein whose helical scaffold has been re-used to target multiple pathways [41]. In light of recent novel roles for both Bcl-2 proteins and the mitochondria in innate immunity signalling it remains however a fascinating possibility that vBcl-2s have shown us the way towards novel functions played by cellular Bcl-2 proteins.
5. Concluding remarks
Our understanding of the ways in which infection leads to cell suicide and induction of the innate immune system continues to undergo rapid advances. Viruses have evolved to counteract the effects of these anti-viral responses by inhibiting signalling cascades or mimicking intrinsic inhibitors of these pathways. The discovery of these innate immunity and apoptotic inhibitors have led to better understanding of viral biology, but have also produced considerable insight into the field of cellular biology. Viral modulators of cell death and innate signalling will continue to provide us with insights into the path from initial virus sensing to viral clearance.
In addition these viral proteins suggest that the two host programmes regulating the two so-far distinct pathways governing cell death and innate immunity are intimately linked. In a broader context, it is not surprising that these two pathways have evolved to intimately cooperate with each other. By integrating these pathways, metazoans have developed a finely tuned system allowing infected cells to warn the organism of infection and altruistically commit suicide to limit the parasitic requirements of viruses.
References
- 1.Youle R.J., Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 2.Chen Y.B., Seo S.Y., Kirsch D.G., Sheu T.T., Cheng W.C., Hardwick J.M. Alternate functions of viral regulators of cell death. Cell Death Differ. 2006;13:1318–1324. doi: 10.1038/sj.cdd.4401964. [DOI] [PubMed] [Google Scholar]
- 3.Verhagen A.M., Coulson E.J., Vaux D.L. Inhibitor of apoptosis proteins and their relatives: IAPs and other BIRPs. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-7-reviews3009. REVIEWS3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor J.M., Barry M. Near death experiences: poxvirus regulation of apoptotic death. Virology. 2006;344:139–150. doi: 10.1016/j.virol.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 5.Muthumani K., Hwang D.S., Desai B.M., Zhang D., Dayes N., Green D.R., Weiner D.B. HIV-1 Vpr induces apoptosis through caspase 9 in T cells and peripheral blood mononuclear cells. J. Biol. Chem. 2002;277:37820–37831. doi: 10.1074/jbc.M205313200. [DOI] [PubMed] [Google Scholar]
- 6.Jacotot E., Ravagnan L., Loeffler M., Ferri K.F., Vieira H.L., Zamzami N., Costantini P., Druillennec S., Hoebeke J., Briand J.P., Irinopoulou T., Daugas E., Susin S.A., Cointe D., Xie Z.H., Reed J.C., Roques B.P., Kroemer G. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med. 2000;191:33–46. doi: 10.1084/jem.191.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan Y.X., Tan T.H., Lee M.J., Tham P.Y., Gunalan V., Druce J., Birch C., Catton M., Fu N.Y., Yu V.C., Tan Y.J. Induction of apoptosis by the severe acute respiratory syndrome coronavirus 7a protein is dependent on its interaction with the Bcl-XL protein. J. Virol. 2007;81:6346–6355. doi: 10.1128/JVI.00090-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giacca M. HIV-1 Tat, apoptosis and the mitochondria: a tubulin link? Retrovirology. 2005;2:7. doi: 10.1186/1742-4690-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galluzzi L., Brenner C., Morselli E., Touat Z., Kroemer G. Viral control of mitochondrial apoptosis. PLoS Pathog. 2008;4 doi: 10.1371/journal.ppat.1000018. e1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishii K.J., Koyama S., Nakagawa A., Coban C., Akira S. Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe. 2008;3:352–363. doi: 10.1016/j.chom.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Ruckdeschel K., Pfaffinger G., Haase R., Sing A., Weighardt H., Hacker G., Holzmann B., Heesemann J. Signaling of apoptosis through TLRs critically involves toll/IL-1 receptor domain-containing adapter inducing IFN-beta, but not MyD88, in bacteria-infected murine macrophages. J. Immunol. 2004;173:3320–3328. doi: 10.4049/jimmunol.173.5.3320. [DOI] [PubMed] [Google Scholar]
- 12.Galindo I., Hernaez B., Diaz-Gil G., Escribano J.M., Alonso C. A179L, a viral Bcl-2 homologue, targets the core Bcl-2 apoptotic machinery and its upstream BH3 activators with selective binding restrictions for Bid and Noxa. Virology. 2008;375:561–572. doi: 10.1016/j.virol.2008.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Westphal D., Ledgerwood E.C., Hibma M.H., Fleming S.B., Whelan E.M., Mercer A.A. A novel Bcl-2-like inhibitor of apoptosis is encoded by the parapoxvirus ORF virus. J. Virol. 2007;81:7178–7188. doi: 10.1128/JVI.00404-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banadyga L., Gerig J., Stewart T., Barry M. Fowlpox virus encodes a Bcl-2 homologue that protects cells from apoptotic death through interaction with the proapoptotic protein Bak. J. Virol. 2007;81:11032–11045. doi: 10.1128/JVI.00734-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang D.C., Cory S., Strasser A. Bcl-2, Bcl-XL and adenovirus protein E1B19kD are functionally equivalent in their ability to inhibit cell death. Oncogene. 1997;14:405–414. doi: 10.1038/sj.onc.1200848. [DOI] [PubMed] [Google Scholar]
- 16.Cross J.R., Postigo A., Blight K., Downward J. Viral pro-survival proteins block separate stages in Bax activation but changes in mitochondrial ultrastructure still occur. Cell Death Differ. 2008;15:997–1008. doi: 10.1038/cdd.2008.14. [DOI] [PubMed] [Google Scholar]
- 17.Cooray S., Bahar M.W., Abrescia N.G., McVey C.E., Bartlett N.W., Chen R.A., Stuart D.I., Grimes J.M., Smith G.L. Functional and structural studies of the Vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J. Gen. Virol. 2007;88:1656–1666. doi: 10.1099/vir.0.82772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pauleau A.L., Larochette N., Giordanetto F., Scholz S.R., Poncet D., Zamzami N., Goldmacher V.S., Kroemer G. Structure-function analysis of the interaction between Bax and the cytomegalovirus-encoded protein vMIA. Oncogene. 2007;26:7067–7080. doi: 10.1038/sj.onc.1210511. [DOI] [PubMed] [Google Scholar]
- 19.Fletcher J.I., Meusburger S., Hawkins C.J., Riglar D.T., Lee E.F., Fairlie W.D., Huang D.C., Adams J.M. Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc. Natl. Acad. Sci. U S A. 2008;105:18081–18087. doi: 10.1073/pnas.0808691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Postigo A., Cross J.R., Downward J., Way M. Interaction of F1L with the BH3 domain of Bak is responsible for inhibiting Vaccinia-induced apoptosis. Cell Death Differ. 2006;13:1651–1662. doi: 10.1038/sj.cdd.4401853. [DOI] [PubMed] [Google Scholar]
- 21.Kvansakul M., Yang H., Fairlie W.D., Czabotar P.E., Fischer S.F., Perugini M.A., Huang D.C., Colman P.M. Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ. 2008;15:1564–1571. doi: 10.1038/cdd.2008.83. [DOI] [PubMed] [Google Scholar]
- 22.Dewson G., Kratina T., Sim H.W., Puthalakath H., Adams J.M., Colman P.M., Kluck R.M. To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol. Cell. 2008;30:369–380. doi: 10.1016/j.molcel.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 23.George N.M., Evans J.J., Luo X. A three-helix homo-oligomerization domain containing BH3 and BH1 is responsible for the apoptotic activity of Bax. Genes Dev. 2007;21:1937–1948. doi: 10.1101/gad.1553607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kvansakul M., van Delft M.F., Lee E.F., Gulbis J.M., Fairlie W.D., Huang D.C., Colman P.M. A structural viral mimic of prosurvival Bcl-2: a pivotal role for sequestering proapoptotic Bax and Bak. Mol. Cell. 2007;25:933–942. doi: 10.1016/j.molcel.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 25.Pichlmair A., Reis e Sousa C. Innate recognition of viruses. Immunity. 2007;27:370–383. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 26.Haller O., Kochs G., Weber F. The interferon response circuit: induction and suppression by pathogenic viruses. Virology. 2006;344:119–130. doi: 10.1016/j.virol.2005.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bowie A.G., Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roy C.R., Mocarski E.S. Pathogen subversion of cell-intrinsic innate immunity. Nat. Immunol. 2007;8:1179–1187. doi: 10.1038/ni1528. [DOI] [PubMed] [Google Scholar]
- 29.Zhong B., Yang Y., Li S., Wang Y.Y., Li Y., Diao F., Lei C., He X., Zhang L., Tien P., Shu H.B. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 30.Kaiser W.J., Offermann M.K. Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 2005;174:4942–4952. doi: 10.4049/jimmunol.174.8.4942. [DOI] [PubMed] [Google Scholar]
- 31.Meylan E., Tschopp J., Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 32.Fernandes-Alnemri T., Yu J.W., Datta P., Wu J., Alnemri E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanneganti T.D., Lamkanfi M., Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27:549–559. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 34.Creagh E.M., O'Neill L.A. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends. Immunol. 2006;27:352–357. doi: 10.1016/j.it.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Ting J.P., Willingham S.B., Bergstralh D.T. NLRs at the intersection of cell death and immunity. Nat. Rev. Immunol. 2008;8:372–379. doi: 10.1038/nri2296. [DOI] [PubMed] [Google Scholar]
- 36.Bergsbaken T., Fink S.L., Cookson B.T. Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haga I.R., Bowie A.G. Evasion of innate immunity by vaccinia virus. Parasitology. 2005;130(Suppl):S11–S25. doi: 10.1017/S0031182005008127. [DOI] [PubMed] [Google Scholar]
- 38.Johnston J.B., Barrett J.W., Nazarian S.H., Goodwin M., Ricciuto D., Wang G., McFadden G. A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity. 2005;23:587–598. doi: 10.1016/j.immuni.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 39.Ohtsuka T., Ryu H., Minamishima Y.A., Macip S., Sagara J., Nakayama K.I., Aaronson S.A., Lee S.W. ASC is a Bax adaptor and regulates the p53-Bax mitochondrial apoptosis pathway. Nat. Cell Biol. 2004;6:121–128. doi: 10.1038/ncb1087. [DOI] [PubMed] [Google Scholar]
- 40.Bruey J.M., Bruey-Sedano N., Luciano F., Zhai D., Balpai R., Xu C., Kress C.L., Bailly-Maitre B., Li X., Osterman A., Matsuzawa S., Terskikh A.V., Faustin B., Reed J.C. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell. 2007;129:45–56. doi: 10.1016/j.cell.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 41.Graham S.C., Bahar M.W., Cooray S., Chen R.A., Whalen D.M., Abrescia N.G., Alderton D., Owens R.J., Stuart D.I., Smith G.L., Grimes J.M. Vaccinia virus proteins A52 and B14 Share a Bcl-2-like fold but have evolved to inhibit NF-kappaB rather than apoptosis. PLoS Pathog. 2008;4 doi: 10.1371/journal.ppat.1000128. e1000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalverda A.P., Thompson G.S., Vogel A., Schroder M., Bowie A.G., Khan A.R., Homans S.W. Poxvirus K7 protein adopts a Bcl-2 fold: biochemical mapping of its interactions with human DEAD box RNA helicase DDX3. J. Mol. Biol. 2009;385:843–853. doi: 10.1016/j.jmb.2008.09.048. [DOI] [PubMed] [Google Scholar]
- 43.DiPerna G., Stack J., Bowie A.G., Boyd A., Kotwal G., Zhang Z., Arvikar S., Latz E., Fitzgerald K.A., Marshall W.L. Poxvirus protein N1L targets the I-kappaB kinase complex, inhibits signaling to NF-kappaB by the tumor necrosis factor superfamily of receptors, and inhibits NF-kappaB and IRF3 signaling by toll-like receptors. J. Biol. Chem. 2004;279:36570–36578. doi: 10.1074/jbc.M400567200. [DOI] [PubMed] [Google Scholar]