

Abstract
The renin–angiotensin system (RAS) is a complex network that regulates blood pressure, electrolyte and fluid homeostasis, as well as the function of several organs. Angiotensin-converting enzyme 2 (ACE2) was identified as an enzyme that negatively regulates the RAS by converting Ang II, the main bioactive molecule of the RAS, to Ang 1–7. Thus, ACE2 counteracts the role of angiotensin-converting enzyme (ACE) which generates Ang II from Ang I. ACE and ACE2 have been implicated in several pathologies such as cardiovascular and renal disease or acute lung injury. In addition, ACE2 has functions independent of the RAS: ACE2 is the receptor for the SARS coronavirus and ACE2 is essential for expression of neutral amino acid transporters in the gut. In this context, ACE2 modulates innate immunity and influences the composition of the gut microbiota, which can explain diarrhea and intestinal inflammation observed in Hartnup disorder, Pellagra, or under conditions of severe malnutrition. Here we review and discuss the diverse functions of ACE2 and its relevance to human pathologies.
Keywords: Renin–angiotensin system, Virus receptor, Amino acid transport, Microbiota, Malnutrition
1. An overview of the renin–angiotensin system
The renin–angiotensin system (RAS) regulates several body functions on a systemic level or locally in various organs [1]. The systemic or circulatory RAS is involved in regulating blood pressure as well as electrolyte and liquid homeostasis, whereas local or tissue RAS regulates functions of numerous organs such as heart, kidney, or the lung. Within the RAS, regulation is achieved through a cascade of proteases that generate several bioactive peptides. The glycoprotein angiotensinogen is mainly produced and secreted by the liver and cleaved by renin, which is generated by the juxtaglomerular apparatus in the kidney, to result in the decapeptide angiotensin I (Ang I) [2]. Ang I can subsequently be cleaved to the octapeptide angiotensin II (Ang II) by angiotensin-converting enzyme (ACE). ACE is expressed in endothelial cells of the vasculature and also locally in a variety of tissues such as kidney, heart, lung, or the brain [3].
Ang II represents the main bioactive component within the RAS and signals through the G-protein-coupled receptors angiotensin II receptor type 1 (AT1) and angiotensin II receptor type 2 (AT2) [4]. Signaling through AT1 is a well established route to mediate vasoconstrictive effects, whereas AT2 receptor activation has opposing effects through activation of a vasodilatory cascade including effectors such as bradykinin, NO, or cGMP [5]. Ang II can further be processed by angiotensin-converting enzyme 2 (ACE2) to form the heptapeptide Ang 1–7, thereby removing the main activating peptide of the RAS from the system. Ang 1–7, in turn, binds to the Mas receptor [6] to counteract the activity of Ang II binding to AT1. Therefore, the ACE2/Ang 1–7/Mas axis is considered to be a negative regulator of the RAS, opposing the activity of the ACE/Ang II/AT1 axis [7] (Fig. 1 ).
Fig. 1.

Simplified diagram of the renin–angiotensin system. Angiotensinogen is secreted by the liver and gets converted to Ang I by renin, which is mainly produced in the kidneys. Ang I gets cleaved by ACE to result in Ang II. Ang II is a ligand for the AT1 receptor and promotes vasoconstriction and hypertension. Alternatively, Ang II can bind to the AT2 receptor to inhibit vasoconstriction. ACE2 processes Ang II to Ang 1–7 which binds to the Mas receptor to induce vasodilation.
Apart from signaling through the Mas receptor, Ang 1–7 can also act as an AT1 receptor antagonist, therefore further counteracting the ACE/Ang II/AT1 axis [8], [9]. In alternative pathways, Ang I can be cleaved by chymase to form Ang II or by other peptidases such as prolyl-endopeptidase (PEP), neutral endopeptidase (NEP), and thymet oligopeptidase (TOP), to directly generate Ang 1–7 [10]. However, in this review we will solely focus on the ACE/ACE2 peptidase system. ACE2 can potentially also act on other peptide systems, in particular on the apelin/APJ system [11]. Apelin is produced as a 77 amino acid pre-pro-hormone which is further processed to apelin-36 and apelin-13. Apelin signals through its receptor APJ and was shown to have vasodilatory effects [12]. ACE2 can cleave the carboxyterminal phenylalanine of apelin-36 and apelin-13 [11], thereby removing the vasodilator apelin, and might therefore counteract its own vasodilatory role within the RAS. Due to the fundamental role of Ang II in blood pressure control, several AT1 receptor antagonists and ACE inhibitors are used successfully in the clinic as a treatment for hypertension as well as for renal and cardiac diseases. Importantly, none of these inhibitors in clinical use have been reported to also block the enzymatic activity of ACE2 [13].
2. Angiotensin-converting enzyme 2 – ACE2
ACE2 was discovered as a homolog of ACE and mapped to the X chromosome in humans, rats, and mice [14], [15], [16]. ACE2 is expressed at high levels in kidney, heart, and testis [14], but is also found in many other tissues such as lung, small intestine, and liver [17], [18], [19]. The ace2 gene spans 18 exons and codes for an 805 amino acid type I transmembrane glycoprotein. ACE2 contains a short intracellular cytoplasmic tail and a longer extracellular domain that exhibits carboxymonopeptidase activity [1]. The active site of ACE2 contains the HEMGH motif, characteristic for zinc-metallopeptidases and shares approximately 42% sequence homology with the amino-terminal domain of ACE [15]. The carboxy-terminal domain of ACE2 is about 48% homologous with Collectrin (also known as Tmem27) [20]. Therefore, evolutionary, ACE2 is a chimeric protein consisting of the amino-terminal carboxypeptidase domain of ACE, whereas the carboxy-terminal part of ACE2 lacking enzymatic activity shares homology with Collectrin. Another homolog of ACE was discovered in 2007 and termed ACE3 [21]. However, due to the presence of several deletions and insertions in the genomic sequence, ACE3 seems to lack catalytic activity as a metalloprotease. So far no physiologic functions could be identified and it has been suggested that ace3 is a pseudogene [21], [22].
3. Physiologic functions of ACE2 within the RAS
The carboxy-peptidase domain of ACE2 efficiently cleaves the C-terminal phenylalanine of Ang II to produce the vasodilator Ang 1–7 [14], [15]. ACE2 also shows activity towards Ang I by cleaving the C-terminal leucine, which results in the supposedly biological inactive peptide Ang 1–9, which can further be processed by ACE to Ang 1–7. However, ACE2 shows a clear substrate preference for Ang II over Ang I [11]. Solving the crystal structure of ACE2 revealed a hinge-bending motion upon ligand binding, characteristic for several metallopeptidases. Moreover, compared to ACE a smaller and therefore less accommodating site within the active domain of ACE2 can explain the release of a single amino acid by ACE2 in contrast to ACE which cleaves off a dipeptide [23].
In the cardiovascular system, ACE and ACE2 are thought to be important players in blood pressure regulation through balancing the levels of the vasoconstrictor Ang II. Raised Ang II levels lead to increased blood pressure, therefore, inhibitors of ACE or antagonists of AT1 are highly effective treatment options for hypertension. Furthermore, it was reported that the ace2 gene maps to a quantitative trait locus on the X chromosome in three different rat models of hypertension and that hypertensive rats exhibit reduced ACE2 expression compared to wild type animals [16]. However, whereas targeted deletion of ACE in mice resulted in spontaneous hypotension [24], ACE2 knock-out mice showed no significant alterations in blood pressure at baseline [16], [25]. Upon chronic Ang II infusion, however as expected, ACE2 deficient mice do exhibit significantly increased blood pressure compared to control animals [25]. In addition, cardiac overexpression of ACE2 in rats was found to mediate resistance against the deleterious effects of hypertension and Ang II infusion [26], and vascular ACE2 overexpression resulted in decreased blood pressure in another study using rats [27]. Thus, ACE2 acts as a negative regulator of the RAS and has an antihypertensive role in blood pressure control.
ACE2 is expressed at substantial levels in cardiomyocytes in the heart. Gene deletion of ace2 resulted in impaired cardiac contractility in two different independent targeted mouse lines [16], [28]. The observed impaired heart function was associated with increased Ang II levels in heart and in the plasma. Indeed, double knock-out of ACE and ACE2 as well as treatment with AT1 antagonists rescued the cardiac malfunctions of ACE2 knock-out animals [16], [28], [29]. Of note, in a third ACE2 deficient mouse line aberrant cardiac contractility could not be observed [25], which turned out to be due to different mouse line backgrounds [30]. Importantly, in all mouse models generated, ACE2 protects from heart failure. In line with a beneficial effect of ACE2, treatment with Ang 1–7 has been shown to improve myocardial performance, cardiac remodeling, and survival in rodent heart failure models, including ischemia/reperfusion injury, myocardial infarction, or hypertension-induced cardiomyopathy [31], [32]. Of note, beside the beneficial effects of ACE2 in cardiovascular diseases, overexpression of ACE2 can have deleterious effects resulting in cardiac fibrosis and arrhythmia [33], [34]; whether this is physiologically relevant needs to be examined in future experiments.
In the lung, increased Ang II levels were shown to promote development of pulmonary hypertension [35] and pulmonary fibrosis [36] and ACE2 was reported to protect both from pulmonary hypertension and pulmonary fibrosis [37], [38], [39]. Moreover, ACE inhibitors and AT1 antagonists can attenuate the severity of experimentally induced pulmonary fibrosis [40], [41], corroborating the role of Ang II in this condition. Furthermore the RAS is also involved in acute lung injury and its severest form acute respiratory distress syndrome (ARDS) [42], [43]. ACE2 deficient animals develop histologically and functionally normal lungs, but upon experimentally induced acute lung injury they develop severe ARDS [44]. Importantly, ACE and ACE2 double knock-out, treatment with AT1 antagonists, or administration of recombinant ACE2 protein rescued the severe acute lung injury phenotype of ACE2 knock-out mice [44]. Therefore, therapy with recombinant ACE2 protein has the potential to be effective against acute lung injury as well as chronic lung diseases such as pulmonary hypertension or pulmonary fibrosis. Based on these data, recombinant soluble ACE2 has been developed as a possible future therapy to treat acute lung injury in humans and is currently being tested in phase 2 clinical trials.
In the kidney, ACE and ACE2 are expressed in the brush border of the proximal tubule epithelial cells and contribute to salt and fluid homeostasis as well as blood pressure control [45]. Besides systemic consequences of kidney fluid homeostasis, there is growing evidence that ACE2 protects from kidney disease by reducing local Ang II levels [46]. In this regard, ACE2 deficient mice develop late-onset nephrotic glomerulosclerosis [47] and display more severe diabetic kidney injury in experimental diabetic mice [48], [49]. These phenotypes emerged via Ang II because AT1 blockers or ACE inhibitors reversed the deleterious phenotypes [47], [48], [49]. Despite this data, the renoprotective role of ACE2 is still controversial because under certain experimental conditions ACE2 and Ang 1–7 were shown to promote renal injury [50]. Therefore, the precise role of ACE2 in the kidney awaits additional experiments and needs to be translated to human disease.
4. ACE2 is the SARS-coronavirus receptor
Interestingly, ACE2 also plays a major role in infections with severe acute respiratory syndrome corona-virus (SARS-CoV). This virus first emerged in 2003 and caused respiratory disease which could trigger ARDS and often ended fatal [51]. In the same year ACE2 was identified to be a potential receptor for the SARS-CoV in vitro [52], which was thereafter genetically confirmed in in vivo mouse studies [17], providing definitive evidence that ACE2 is the essential SARS receptor in vivo.
SARS infections are initiated by binding of spike protein trimers of the SARS-CoV to a hydrophobic pocket of the extracellular catalytic domain of ACE2 [53]. This interaction enables endocytosis, membrane fusion, and entry of the SARS-CoV into the host cell. Following virus entry, the ACE2 protein is downregulated which leads to local increase of Ang II levels, providing a molecular explanation for the frequent development of ARDS during SARS-CoV infections [17], [44], which was untypical for other coronaviruses. The significance of elevated Ang II in SARS pathogenesis was further substantiated by the fact that SARS-CoV infections of ACE2 knock-out mice do not induce lung injury, and recombinant ACE2 rescues from experimental lung injury upon SARS-CoV spike protein treatment and other triggers of lung injury [17]. Thus, the SARS-CoV virus might have shown us a novel treatment option for a previously untreatable disease, i.e. the use of ACE2 for the treatment of acute lung injury.
It should be noted that within the family of coronaviruses only SARS-CoV and human coronavirus NL63 were shown to utilize ACE2 for cell entry [54]. In 2012, a novel coronavirus was isolated from a patient in Saudi Arabia [55]. This virus was later termed Middle East respiratory syndrome Corona Virus (MERS-CoV), and currently has an alarming mortality rate of ∼60% [56]. Due to the high similarity of SARS-CoV and MERS-CoV, it was suggested that they both might enter the host via ACE2; however, recent findings have shown that MERS-CoV does not utilize ACE2 as a receptor [57], but enters the host cell via dipeptidyl peptidase 4 (DPP4 or CD26) [58].
5. ACE2 regulates amino acid transport in the intestine
Collectrin was discovered in 2001 as a protein expressed in the kidney with high sequence homology to the carboxy-terminal end of ACE2 [20]. Collectrin and ACE2 both are type 1 transmembrane proteins [59], but, in contrast to ACE2, Collectrin lacks catalytic activity. The collectrin gene is located on the X chromosome immediately upstream of the ace2 gene in all species examined and both genes share similar transcription factor binding sites [60]. In this regard, collectrin was shown to be under transcriptional control of hepatocyte nuclear factor α (HNF-1α) [59], [61], which is involved in pancreatic insulin secretion [62]. In vitro studies and in vivo overexpression experiments supported a role for Collectrin in insulin exocytosis through interaction with the SNARE complex [61]. However, Collectrin deficient mice did not exhibit impaired insulin secretion [63].
Surprisingly, analysis of Collectrin knock-out mice revealed that Collectrin is a regulator of neutral amino acid transporters expression on brush border membranes of renal proximal tubules [64]. In this study it was observed that urine of Collectrin deficient mice forms crystals when stored at 4 °C, which turned out to be mainly composed of tyrosine and phenylalanine [64]. Additional studies showed that Collectrin non-covalently associates with the Slc6 family of neutral amino acid transporters (B0AT1 (Slc6a19) and B0AT3 (Slc6a18)), the imino transporter SIT1 (Slc6a20) and the Slc1 glutamate and aspartate transporter EAAT3 (Slc1a1) [65]. Collectrin stabilizes amino acid transporter expression on the cell surface but does not affect mRNA expression of these transporters [64], [66]. These studies uncovered Collectrin as a key subunit of these amino acid transporters which controls polarized expression in the kidney required for renal reabsorption of amino acids.
B0AT1 is not only expressed in the kidney but also in the small intestine where Collectrin is absent. Interestingly, similar to the role of Collectrin in the kidney, we were able to show that the closest “relative” of Collectrin, i.e. ACE2, can bind and stabilize the neutral amino acid transporter B0AT1 in the small intestine [67]. Therefore, B0AT1 seems to rely on tissue specific interaction partners. In ACE2 deficient mice, B0AT1 is completely absent from the small intestine, but is expressed at normal levels in the kidney [67]. On the contrary, in Collectrin knock-out mice B0AT1 expression is lost from the kidney despite the presence of ACE2 [64]. B0AT1 and ACE2 colocalize on enterocytes of the small intestine, an interaction confirmed by co-immunoprecipitation experiments and overexpression in Xenopus laevis oocytes [64], [67], [68]. In the gut, ACE2 also interacts with SIT1 [65], [69], a transporter for proline, sarcosine, or betaine [70]. Interestingly, the enzymatic activity of ACE2 is not required for B0AT1 surface expression or transporter function [67]. B0AT1 is dispensable for expression of ACE2 or Collectrin [71].
Mutations in slc6a19 encoding B0AT1 have been identified to cause Hartnup disorder resulting in defective amino acid uptake in the kidney and small intestine [72], [73]. The main characteristic of Hartnup disorder is neutral aminoaciduria similar to the phenotype observed in Collectrin deficient mice [64]. Apart from aminoaciduria, patients with Hartnup disorder are often symptom free, but under stress conditions like malnutrition or infections develop pellagra-like symptoms such as light-sensitive rash, cerebellar ataxia, emotional instability, as well as diarrhea. More than twenty different mutations in slc6a19 have been identified so far in Hartnup disorder patients [65]. However no mutations in ace2 or collectrin have been reported in Hartnup disorder or aminoaciduria syndromes.
The aminoaciduria in Hartnup disorder is clearly caused by the impaired amino acid uptake in the kidney and small intestine, but how a defect in B0AT1 causes dermatitis or neurological phenotypes remains elusive. Our recent studies unraveled a novel connection between intestinal amino acid uptake and susceptibility to diarrhea and colitis [18], conditions also frequently observed in Hartnup disorder and Pellagra patients [74]. We assayed ACE2 deficient mice which cannot express B0AT1 in the small intestine and therefore exhibit dramatically reduced plasma levels of the essential amino acid tryptophan. Whereas the baseline intestinal architecture appears normal in ACE2 knock-out mice, treatment with chemical irritants revealed that ACE2 deficient mice are highly susceptible to experimentally induced colitis, while control mice only developed modest intestinal inflammation [18]. A direct involvement of the essential amino acid tryptophan was proven by administering a tryptophan free diet, which made wild-type mice highly susceptible to chemically induced colitis. Strikingly, in the reverse experiment the colitis phenotype could be rescued in ACE2 deficient mice by supplementing the diet with a glycine–tryptophan dipeptide which can be taken up by the proton-coupled peptide transporter PepT1 [75] and, therefore, circumvent the missing B0AT1 transporter system. Moreover, we could rescue the colitis phenotype of ACE2 mutant mice using nicotinamide, a downstream metabolite of tryptophan. Consistent with these results, nicotinamide has been used as a treatment for the vitamin B3 deficiency disease pellagra [74]. Collectrin deficient mice cannot efficiently reabsorb tryptophan from urine, but do not exhibit increased susceptibility to colitis. Therefore, only defective intestinal tryptophan uptake in ACE2 knock-out mice seems to mediate local intestinal inflammation and diarrhea [18].
ACE2 is highly expressed in the small intestine but hardly detectable in the colon. How then does the impaired amino acid uptake in the small intestine explain the colitis phenotype? This contradiction was explained by a chain of events starting with the observation that reduced tryptophan levels lead to reduced mTOR pathway activity in the small intestine [18]. Aberrant mTOR activation resulted in impaired expression of antimicrobial peptides from small intestinal Paneth cells. The impaired expression of antimicrobial peptides in turn resulted in an altered composition of the intestinal microbiota (Fig. 2 ). That an alteration in the intestinal microbiome mediated the increased susceptibility to colitis was finally proven by microbiota transplantation experiments. These results also provide an explanation for the effectiveness of nicotinamide treatment of pellagra patients [18], [74]. It also provides an explanation for how protein malnutrition can lead to severe intestinal inflammation. Hundreds of millions of people worldwide are affected by malnutrition [76]. During famines acute diarrheal infection and intestinal inflammation are often the actual cause of death [77]. Furthermore, the observation that a reduction of mTOR activity leads to susceptibility of intestinal inflammation could provide an explanation why inhibition of mTOR with rapamycin was not successful in human clinical trials in inflammatory bowel disease [78].
Fig. 2.
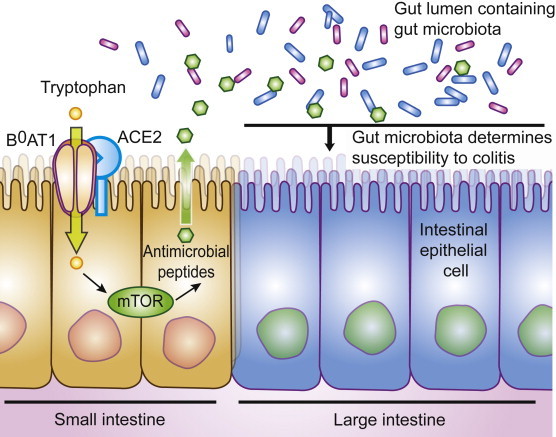
Role of ACE2 in the intestinal epithelium. ACE2 is necessary for the surface expression of the amino acid transporter B0AT1 in the epithelium of the small intestine. Tryptophan has to be obtained from the diet and uptake of tryptophan mainly depends on B0AT1. Tryptophan levels regulate via mTOR pathway activation the secretion of antimicrobial peptides which in turn influence the composition of the intestinal microbiota. In the absence of ACE2, tryptophan cannot get absorbed efficiently, which leads to aberrant secretion of antimicrobial peptides and consequently to an altered microbiota which confers susceptibility to inflammation of the large intestine.
Interestingly, following our initial experiments in mice, a connection between the microbiota and Kwashiorkor, a form of severe acute malnutrition, has recently been reported in humans [79]. The study was performed with twin pairs up to an age of three years in rural Malawi. Analysis of fecal microbiota revealed that gene diversity of the microbiota from children with Kwashiorkor did not develop with increasing age – in contrast to the microbiota of healthy children. To confirm a causal role of the microbiota, fecal content was transplanted to gnotobiotic mice. Only the microbiota from Kwashiorkor-affected children in combination with a Malawian diet, which lacks protein, lead to severe weight loss in recipient mice. Similarly, upon feeding a protein free diet or a tryptophan free diet to mice, we observed significant changes in the composition of gut microbiota resulting in colitis and weight loss [18]. Both studies show an intricate interaction between diets, the intestinal microbiota, and malnutrition [18], [79].
In another large study, Malawian children with uncomplicated severe acute malnutrition received ready-to-use therapeutic food with or without combined antibiotic treatment. Administration of antibiotics led to reduction of dehydrating diarrhea and also significantly lowered the mortality rates [80]. This data could implicate that antibiotic treatment protects better against fatal infections or it could point towards a critical role of the patients' microbiota in the progression of diarrhea and severe acute malnutrition. In this regard, antibiotic treatment rescued bloody diarrhea in a colitis model of ACE2 deficient mice [18].
6. Conclusions
With the discovery of ACE2, a homolog of the classic enzyme ACE, an important negative regulator of the RAS was identified. The enzymatic activity of ACE2 reduces Ang II levels and thereby counteracts the ACE/Ang II/AT1 axis. In the context of the RAS, ACE2 has vasodilator effects, regulates heart functions, exhibits a renoprotective role, counteracts fibrotic changes, and can protect from ARDS in the lung. Intriguingly, ACE2 has functions that are independent of the RAS and independent of the catalytic activity of ACE2. In this regard, ACE2 was identified as the receptor of the SARS coronavirus and recently it was discovered that ACE2 is essential for the expression of certain amino acid transporters in the small intestine. Via regulation of intestinal amino acid transport, ACE2 couples diet to the composition of the gut microbiome, also providing a molecular explanation how malnutrition in hundreds of millions of people could cause intestinal inflammation and diarrhea. How this function is linked to the regulation of the RAS needs to be further explored. Since the intestinal microbiota has been shown to influence host physiology and numerous conditions from obesity to autoimmune diseases [81], unraveling the complex network of interactions between host factors and their microbiota has the potential to uncover new therapeutic approaches.
References
- 1.Lambert D.W., Clarke N.E., Turner A.J. Not just angiotensinases: new roles for the angiotensin-converting enzymes. Cell Mol. Life Sci. 2010;67:89–98. doi: 10.1007/s00018-009-0152-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuba K., Imai Y., Penninger J.M. Multiple functions of angiotensin-converting enzyme 2 and its relevance in cardiovascular diseases. Circ. J. 2013;77:301–308. doi: 10.1253/circj.cj-12-1544. [DOI] [PubMed] [Google Scholar]
- 3.Kuba K., Imai Y., Ohto-Nakanishi T., Penninger J.M. Trilogy of ACE2: a peptidase in the renin–angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol. Ther. 2010;128:119–128. doi: 10.1016/j.pharmthera.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Imai Y., Kuba K., Ohto-Nakanishi T., Penninger J.M. Angiotensin-converting enzyme 2 (ACE2) in disease pathogenesis. Circ. J. 2010;74:405–410. doi: 10.1253/circj.cj-10-0045. [DOI] [PubMed] [Google Scholar]
- 5.Carey R.M., Padia S.H. Angiotensin AT2 receptors: control of renal sodium excretion and blood pressure. Trends Endocrinol. Metab. 2008;19:84–87. doi: 10.1016/j.tem.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Santos R.A., Simoes e Silva A.C., Maric C., Silva D.M., Machado R.P., de Buhr I., Heringer-Walther S., Pinheiro S.V., Lopes M.T., Bader M., Mendes E.P., Lemos V.S., Campagnole-Santos M.J., Schultheiss H.P., Speth R., Walther T. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. U S A. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santos R.A., Ferreira A.J., Verano-Braga T., Bader M. Angiotensin-converting enzyme 2, angiotensin-(1–7) and Mas: new players of the renin–angiotensin system. J. Endocrinol. 2013;216:R1–R17. doi: 10.1530/JOE-12-0341. [DOI] [PubMed] [Google Scholar]
- 8.Gironacci M.M., Coba M.P., Pena C. Angiotensin-(1-7) binds at the type 1 angiotensin II receptors in rat renal cortex. Regul. Pept. 1999;84:51–54. doi: 10.1016/s0167-0115(99)00067-1. [DOI] [PubMed] [Google Scholar]
- 9.Rowe B.P., Saylor D.L., Speth R.C., Absher D.R. Angiotensin-(1-7) binding at angiotensin II receptors in the rat brain. Regul. Pept. 1995;56:139–146. doi: 10.1016/0167-0115(95)00010-9. [DOI] [PubMed] [Google Scholar]
- 10.Passos-Silva D.G., Verano-Braga T., Santos R.A. Angiotensin-(1-7): beyond the cardio-renal actions. Clin. Sci. (Lond) 2013;124:443–456. doi: 10.1042/CS20120461. [DOI] [PubMed] [Google Scholar]
- 11.Vickers C., Hales P., Kaushik V., Dick L., Gavin J., Tang J., Godbout K., Parsons T., Baronas E., Hsieh F., Acton S., Patane M., Nichols A., Tummino P. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 12.Chandrasekaran B., Dar O., McDonagh T. The role of apelin in cardiovascular function and heart failure. Eur. J. Heart Fail. 2008;10:725–732. doi: 10.1016/j.ejheart.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Shi L., Mao C., Xu Z., Zhang L. Angiotensin-converting enzymes and drug discovery in cardiovascular diseases. Drug Discov. Today. 2010;15:332–341. doi: 10.1016/j.drudis.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tipnis S.R., Hooper N.M., Hyde R., Karran E., Christie G., Turner A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 15.Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N., Donovan M., Woolf B., Robison K., Jeyaseelan R., Breitbart R.E., Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000;87:E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 16.Crackower M.A., Sarao R., Oudit G.Y., Yagil C., Kozieradzki I., Scanga S.E., Oliveira-dos-Santos A.J., da Costa J., Zhang L., Pei Y., Scholey J., Ferrario C.M., Manoukian A.S., Chappell M.C., Backx P.H., Yagil Y., Penninger J.M. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 17.Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B., Huan Y., Yang P., Zhang Y., Deng W., Bao L., Zhang B., Liu G., Wang Z., Chappell M., Liu Y., Zheng D., Leibbrandt A., Wada T., Slutsky A.S., Liu D., Qin C., Jiang C., Penninger J.M. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hashimoto T., Perlot T., Rehman A., Trichereau J., Ishiguro H., Paolino M., Sigl V., Hanada T., Hanada R., Lipinski S., Wild B., Camargo S.M., Singer D., Richter A., Kuba K., Fukamizu A., Schreiber S., Clevers H., Verrey F., Rosenstiel P., Penninger J.M. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature. 2012;487:477–481. doi: 10.1038/nature11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paizis G., Tikellis C., Cooper M.E., Schembri J.M., Lew R.A., Smith A.I., Shaw T., Warner F.J., Zuilli A., Burrell L.M., Angus P.W. Chronic liver injury in rats and humans upregulates the novel enzyme angiotensin converting enzyme 2. Gut. 2005;54:1790–1796. doi: 10.1136/gut.2004.062398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H., Wada J., Hida K., Tsuchiyama Y., Hiragushi K., Shikata K., Wang H., Lin S., Kanwar Y.S., Makino H. Collectrin, a collecting duct-specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. J. Biol. Chem. 2001;276:17132–17139. doi: 10.1074/jbc.M006723200. [DOI] [PubMed] [Google Scholar]
- 21.Rella M., Elliot J.L., Revett T.J., Lanfear J., Phelan A., Jackson R.M., Turner A.J., Hooper N.M. Identification and characterisation of the angiotensin converting enzyme-3 (ACE3) gene: a novel mammalian homologue of ACE. BMC Genomics. 2007;8:194. doi: 10.1186/1471-2164-8-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inoue N., Kasahara T., Ikawa M., Okabe M. Identification and disruption of sperm-specific angiotensin converting enzyme-3 (ACE3) in mouse. PLoS One. 2010;5:e10301. doi: 10.1371/journal.pone.0010301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Towler P., Staker B., Prasad S.G., Menon S., Tang J., Parsons T., Ryan D., Fisher M., Williams D., Dales N.A., Patane M.A., Pantoliano M.W. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004;279:17996–18007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krege J.H., John S.W., Langenbach L.L., Hodgin J.B., Hagaman J.R., Bachman E.S., Jennette J.C., O'Brien D.A., Smithies O. Male-female differences in fertility and blood pressure in ACE-deficient mice. Nature. 1995;375:146–148. doi: 10.1038/375146a0. [DOI] [PubMed] [Google Scholar]
- 25.Gurley S.B., Allred A., Le T.H., Griffiths R., Mao L., Philip N., Haystead T.A., Donoghue M., Breitbart R.E., Acton S.L., Rockman H.A., Coffman T.M. Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J. Clin. Invest. 2006;116:2218–2225. doi: 10.1172/JCI16980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Der Sarkissian S., Grobe J.L., Yuan L., Narielwala D.R., Walter G.A., Katovich M.J., Raizada M.K. Cardiac overexpression of angiotensin converting enzyme 2 protects the heart from ischemia-induced pathophysiology. Hypertension. 2008;51:712–718. doi: 10.1161/HYPERTENSIONAHA.107.100693. [DOI] [PubMed] [Google Scholar]
- 27.Rentzsch B., Todiras M., Iliescu R., Popova E., Campos L.A., Oliveira M.L., Baltatu O.C., Santos R.A., Bader M. Transgenic angiotensin-converting enzyme 2 overexpression in vessels of SHRSP rats reduces blood pressure and improves endothelial function. Hypertension. 2008;52:967–973. doi: 10.1161/HYPERTENSIONAHA.108.114322. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto K., Ohishi M., Katsuya T., Ito N., Ikushima M., Kaibe M., Tatara Y., Shiota A., Sugano S., Takeda S., Rakugi H., Ogihara T. Deletion of angiotensin-converting enzyme 2 accelerates pressure overload-induced cardiac dysfunction by increasing local angiotensin II. Hypertension. 2006;47:718–726. doi: 10.1161/01.HYP.0000205833.89478.5b. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura K., Koibuchi N., Nishimatsu H., Higashikuni Y., Hirata Y., Kugiyama K., Nagai R., Sata M. Candesartan ameliorates cardiac dysfunction observed in angiotensin-converting enzyme 2-deficient mice. Hypertens. Res. 2008;31:1953–1961. doi: 10.1291/hypres.31.1953. [DOI] [PubMed] [Google Scholar]
- 30.Oudit G.Y., Kassiri Z., Patel M.P., Chappell M., Butany J., Backx P.H., Tsushima R.G., Scholey J.W., Khokha R., Penninger J.M. Angiotensin II-mediated oxidative stress and inflammation mediate the age-dependent cardiomyopathy in ACE2 null mice. Cardiovasc. Res. 2007;75:29–39. doi: 10.1016/j.cardiores.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 31.Ferreira A.J., Santos R.A., Almeida A.P. Angiotensin-(1-7): cardioprotective effect in myocardial ischemia/reperfusion. Hypertension. 2001;38:665–668. doi: 10.1161/01.hyp.38.3.665. [DOI] [PubMed] [Google Scholar]
- 32.Santos R.A., Ferreira A.J., Nadu A.P., Braga A.N., de Almeida A.P., Campagnole-Santos M.J., Baltatu O., Iliescu R., Reudelhuber T.L., Bader M. Expression of an angiotensin-(1-7)-producing fusion protein produces cardioprotective effects in rats. Physiol. Genomics. 2004;17:292–299. doi: 10.1152/physiolgenomics.00227.2003. [DOI] [PubMed] [Google Scholar]
- 33.Donoghue M., Wakimoto H., Maguire C.T., Acton S., Hales P., Stagliano N., Fairchild-Huntress V., Xu J., Lorenz J.N., Kadambi V., Berul C.I., Breitbart R.E. Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins. J. Mol. Cell Cardiol. 2003;35:1043–1053. doi: 10.1016/s0022-2828(03)00177-9. [DOI] [PubMed] [Google Scholar]
- 34.Masson R., Nicklin S.A., Craig M.A., McBride M., Gilday K., Gregorevic P., Allen J.M., Chamberlain J.S., Smith G., Graham D., Dominiczak A.F., Napoli C., Baker A.H. Onset of experimental severe cardiac fibrosis is mediated by overexpression of angiotensin-converting enzyme 2. Hypertension. 2009;53:694–700. doi: 10.1161/HYPERTENSIONAHA.108.122333. [DOI] [PubMed] [Google Scholar]
- 35.Morrell N.W., Morris K.G., Stenmark K.R. Role of angiotensin-converting enzyme and angiotensin II in development of hypoxic pulmonary hypertension. Am. J. Physiol. 1995;269:H1186–H1194. doi: 10.1152/ajpheart.1995.269.4.H1186. [DOI] [PubMed] [Google Scholar]
- 36.Marshall R.P., Gohlke P., Chambers R.C., Howell D.C., Bottoms S.E., Unger T., McAnulty R.J., Laurent G.J. Angiotensin II and the fibroproliferative response to acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2004;286:L156–L164. doi: 10.1152/ajplung.00313.2002. [DOI] [PubMed] [Google Scholar]
- 37.Ferreira A.J., Shenoy V., Yamazato Y., Sriramula S., Francis J., Yuan L., Castellano R.K., Ostrov D.A., Oh S.P., Katovich M.J., Raizada M.K. Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2009;179:1048–1054. doi: 10.1164/rccm.200811-1678OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamazato Y., Ferreira A.J., Hong K.H., Sriramula S., Francis J., Yamazato M., Yuan L., Bradford C.N., Shenoy V., Oh S.P., Katovich M.J., Raizada M.K. Prevention of pulmonary hypertension by angiotensin-converting enzyme 2 gene transfer. Hypertension. 2009;54:365–371. doi: 10.1161/HYPERTENSIONAHA.108.125468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X., Molina-Molina M., Abdul-Hafez A., Uhal V., Xaubet A., Uhal B.D. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2008;295:L178–L185. doi: 10.1152/ajplung.00009.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang R., Ibarra-Sunga O., Verlinski L., Pick R., Uhal B.D. Abrogation of bleomycin-induced epithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor. Am. J. Physiol. Lung Cell Mol. Physiol. 2000;279:L143–L151. doi: 10.1152/ajplung.2000.279.1.L143. [DOI] [PubMed] [Google Scholar]
- 41.Otsuka M., Takahashi H., Shiratori M., Chiba H., Abe S. Reduction of bleomycin induced lung fibrosis by candesartan cilexetil, an angiotensin II type 1 receptor antagonist. Thorax. 2004;59:31–38. doi: 10.1136/thx.2003.000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marshall R.P., Webb S., Bellingan G.J., Montgomery H.E., Chaudhari B., McAnulty R.J., Humphries S.E., Hill M.R., Laurent G.J. Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2002;166:646–650. doi: 10.1164/rccm.2108086. [DOI] [PubMed] [Google Scholar]
- 43.Raiden S., Nahmod K., Nahmod V., Semeniuk G., Pereira Y., Alvarez C., Giordano M., Geffner J.R. Nonpeptide antagonists of AT1 receptor for angiotensin II delay the onset of acute respiratory distress syndrome. J. Pharmacol. Exp. Ther. 2002;303:45–51. doi: 10.1124/jpet.102.037382. [DOI] [PubMed] [Google Scholar]
- 44.Imai Y., Kuba K., Rao S., Huan Y., Guo F., Guan B., Yang P., Sarao R., Wada T., Leong-Poi H., Crackower M.A., Fukamizu A., Hui C.C., Hein L., Uhlig S., Slutsky A.S., Jiang C., Penninger J.M. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Warner F.J., Lew R.A., Smith A.I., Lambert D.W., Hooper N.M., Turner A.J. Angiotensin-converting enzyme 2 (ACE2), but not ACE, is preferentially localized to the apical surface of polarized kidney cells. J. Biol. Chem. 2005;280:39353–39362. doi: 10.1074/jbc.M508914200. [DOI] [PubMed] [Google Scholar]
- 46.Batlle D., Wysocki J., Soler M.J., Ranganath K. Angiotensin-converting enzyme 2: enhancing the degradation of angiotensin II as a potential therapy for diabetic nephropathy. Kidney Int. 2012;81:520–528. doi: 10.1038/ki.2011.381. [DOI] [PubMed] [Google Scholar]
- 47.Oudit G.Y., Herzenberg A.M., Kassiri Z., Wong D., Reich H., Khokha R., Crackower M.A., Backx P.H., Penninger J.M., Scholey J.W. Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin II-dependent glomerulosclerosis. Am. J. Pathol. 2006;168:1808–1820. doi: 10.2353/ajpath.2006.051091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tikellis C., Bialkowski K., Pete J., Sheehy K., Su Q., Johnston C., Cooper M.E., Thomas M.C. ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes. 2008;57:1018–1025. doi: 10.2337/db07-1212. [DOI] [PubMed] [Google Scholar]
- 49.Wong D.W., Oudit G.Y., Reich H., Kassiri Z., Zhou J., Liu Q.C., Backx P.H., Penninger J.M., Herzenberg A.M., Scholey J.W. Loss of angiotensin-converting enzyme-2 (Ace2) accelerates diabetic kidney injury. Am. J. Pathol. 2007;171:438–451. doi: 10.2353/ajpath.2007.060977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zimmerman D., Burns K.D. Angiotensin-(1-7) in kidney disease: a review of the controversies. Clin. Sci. (Lond.) 2012;123:333–346. doi: 10.1042/CS20120111. [DOI] [PubMed] [Google Scholar]
- 51.Peiris J.S., Yuen K.Y., Osterhaus A.D., Stohr K. The severe acute respiratory syndrome. N. Engl. J. Med. 2003;349:2431–2441. doi: 10.1056/NEJMra032498. [DOI] [PubMed] [Google Scholar]
- 52.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 54.Hofmann H., Pyrc K., van der Hoek L., Geier M., Berkhout B., Pohlmann S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. U S A. 2005;102:7988–7993. doi: 10.1073/pnas.0409465102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 56.de Groot R.J., Baker S.C., Baric R.S., Brown C.S., Drosten C., Enjuanes L., Fouchier R.A., Galiano M., Gorbalenya A.E., Memish Z., Perlman S., Poon L.L., Snijder E.J., Stephens G.M., Woo P.C., Zaki A.M., Zambon M., Ziebuhr J. Middle East respiratory syndrome coronavirus (MERS-CoV); announcement of the coronavirus study group. J. Virol. 2013;87:7790–7792. doi: 10.1128/JVI.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muller M.A., Raj V.S., Muth D., Meyer B., Kallies S., Smits S.L., Wollny R., Bestebroer T.M., Specht S., Suliman T., Zimmermann K., Binger T., Eckerle I., Tschapka M., Zaki A.M., Osterhaus A.D., Fouchier R.A., Haagmans B.L., Drosten C. Human coronavirus EMC does not require the SARS-coronavirus receptor and maintains broad replicative capability in mammalian cell lines. mBio. 2012;3 doi: 10.1128/mBio.00515-12. e00515-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raj V.S., Mou H., Smits S.L., Dekkers D.H., Muller M.A., Dijkman R., Muth D., Demmers J.A., Zaki A., Fouchier R.A., Thiel V., Drosten C., Rottier P.J., Osterhaus A.D., Bosch B.J., Haagmans B.L. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495:251–254. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Akpinar P., Kuwajima S., Krutzfeldt J., Stoffel M. Tmem27: a cleaved and shed plasma membrane protein that stimulates pancreatic beta cell proliferation. Cell Metab. 2005;2:385–397. doi: 10.1016/j.cmet.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Y., Wada J. Collectrin, a homologue of ACE2, its transcriptional control and functional perspectives. Biochem. Biophys. Res. Commun. 2007;363:1–5. doi: 10.1016/j.bbrc.2007.08.136. [DOI] [PubMed] [Google Scholar]
- 61.Fukui K., Yang Q., Cao Y., Takahashi N., Hatakeyama H., Wang H., Wada J., Zhang Y., Marselli L., Nammo T., Yoneda K., Onishi M., Higashiyama S., Matsuzawa Y., Gonzalez F.J., Weir G.C., Kasai H., Shimomura I., Miyagawa J., Wollheim C.B., Yamagata K. The HNF-1 target collectrin controls insulin exocytosis by SNARE complex formation. Cell Metab. 2005;2:373–384. doi: 10.1016/j.cmet.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 62.Pontoglio M. Hepatocyte nuclear factor 1, a transcription factor at the crossroads of glucose homeostasis. J. Am. Soc. Nephrol. 2000;11(Suppl. 16):S140–S143. [PubMed] [Google Scholar]
- 63.Malakauskas S.M., Kourany W.M., Zhang X.Y., Lu D., Stevens R.D., Koves T.R., Hohmeier H.E., Muoio D.M., Newgard C.B., Le T.H. Increased insulin sensitivity in mice lacking collectrin, a downstream target of HNF-1alpha. Mol. Endocrinol. 2009;23:881–892. doi: 10.1210/me.2008-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Danilczyk U., Sarao R., Remy C., Benabbas C., Stange G., Richter A., Arya S., Pospisilik J.A., Singer D., Camargo S.M., Makrides V., Ramadan T., Verrey F., Wagner C.A., Penninger J.M. Essential role for collectrin in renal amino acid transport. Nature. 2006;444:1088–1091. doi: 10.1038/nature05475. [DOI] [PubMed] [Google Scholar]
- 65.Singer D., Camargo S.M. Collectrin and ACE2 in renal and intestinal amino acid transport. Channels (Austin) 2011;5:410–423. doi: 10.4161/chan.5.5.16470. [DOI] [PubMed] [Google Scholar]
- 66.Malakauskas S.M., Quan H., Fields T.A., McCall S.J., Yu M.J., Kourany W.M., Frey C.W., Le T.H. Aminoaciduria and altered renal expression of luminal amino acid transporters in mice lacking novel gene collectrin. Am. J. Physiol. Ren. Physiol. 2007;292:F533–F544. doi: 10.1152/ajprenal.00325.2006. [DOI] [PubMed] [Google Scholar]
- 67.Camargo S.M., Singer D., Makrides V., Huggel K., Pos K.M., Wagner C.A., Kuba K., Danilczyk U., Skovby F., Kleta R., Penninger J.M., Verrey F. Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. Gastroenterology. 2009;136:872–882. doi: 10.1053/j.gastro.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kowalczuk S., Broer A., Tietze N., Vanslambrouck J.M., Rasko J.E., Broer S. A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J. 2008;22:2880–2887. doi: 10.1096/fj.08-107300. [DOI] [PubMed] [Google Scholar]
- 69.Singer D., Camargo S.M., Ramadan T., Schafer M., Mariotta L., Herzog B., Huggel K., Wolfer D., Werner S., Penninger J.M., Verrey F. Defective intestinal amino acid absorption in Ace2 null mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012;303:G686–G695. doi: 10.1152/ajpgi.00140.2012. [DOI] [PubMed] [Google Scholar]
- 70.Broer S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol. Rev. 2008;88:249–286. doi: 10.1152/physrev.00018.2006. [DOI] [PubMed] [Google Scholar]
- 71.Broer A., Juelich T., Vanslambrouck J.M., Tietze N., Solomon P.S., Holst J., Bailey C.G., Rasko J.E., Broer S. Impaired nutrient signaling and body weight control in a Na+ neutral amino acid cotransporter (Slc6a19)-deficient mouse. J. Biol. Chem. 2011;286:26638–26651. doi: 10.1074/jbc.M111.241323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kleta R., Romeo E., Ristic Z., Ohura T., Stuart C., Arcos-Burgos M., Dave M.H., Wagner C.A., Camargo S.R., Inoue S., Matsuura N., Helip-Wooley A., Bockenhauer D., Warth R., Bernardini I., Visser G., Eggermann T., Lee P., Chairoungdua A., Jutabha P., Babu E., Nilwarangkoon S., Anzai N., Kanai Y., Verrey F., Gahl W.A., Koizumi A. Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nat. Genet. 2004;36:999–1002. doi: 10.1038/ng1405. [DOI] [PubMed] [Google Scholar]
- 73.Seow H.F., Broer S., Broer A., Bailey C.G., Potter S.J., Cavanaugh J.A., Rasko J.E. Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. Nat. Genet. 2004;36:1003–1007. doi: 10.1038/ng1406. [DOI] [PubMed] [Google Scholar]
- 74.Segal I., Ou Tim L., Demetriou A., Paterson A., Hale M., Lerios M. Rectal manifestations of pellagra. Int. J. Colorectal Dis. 1986;1:238–243. doi: 10.1007/BF01648345. [DOI] [PubMed] [Google Scholar]
- 75.Meredith D. Review. The mammalian proton-coupled peptide cotransporter PepT1: sitting on the transporter-channel fence? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009;364:203–207. doi: 10.1098/rstb.2008.0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Khan Y., Bhutta Z.A. Nutritional deficiencies in the developing world: current status and opportunities for intervention. Pediatr. Clin. North Am. 2010;57:1409–1441. doi: 10.1016/j.pcl.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 77.Weisstaub G., Araya M. Acute malnutrition in Latin America: the challenge of ending avoidable deaths. J. Pediatr. Gastroenterol. Nutr. 2008;47(Suppl. 1):S10–S14. doi: 10.1097/MPG.0b013e3181818e78. [DOI] [PubMed] [Google Scholar]
- 78.Reinisch W., Panes J., Lemann M., Schreiber S., Feagan B., Schmidt S., Sturniolo G.C., Mikhailova T., Alexeeva O., Sanna L., Haas T., Korom S., Mayer H. A multicenter, randomized, double-blind trial of everolimus versus azathioprine and placebo to maintain steroid-induced remission in patients with moderate-to-severe active Crohn's disease. Am. J. Gastroenterol. 2008;103:2284–2292. doi: 10.1111/j.1572-0241.2008.02024.x. [DOI] [PubMed] [Google Scholar]
- 79.Smith M.I., Yatsunenko T., Manary M.J., Trehan I., Mkakosya R., Cheng J., Kau A.L., Rich S.S., Concannon P., Mychaleckyj J.C., Liu J., Houpt E., Li J.V., Holmes E., Nicholson J., Knights D., Ursell L.K., Knight R., Gordon J.I. Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science. 2013;339:548–554. doi: 10.1126/science.1229000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Trehan I., Goldbach H.S., LaGrone L.N., Meuli G.J., Wang R.J., Maleta K.M., Manary M.J. Antibiotics as part of the management of severe acute malnutrition. N. Engl. J. Med. 2013;368:425–435. doi: 10.1056/NEJMoa1202851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sommer F., Backhed F. The gut microbiota–masters of host development and physiology. Nat. Rev. Microbiol. 2013;11:227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]