

Abstract
The S1 and S2 subunits of the spike glycoprotein of the coronavirus which is responsible for the severe acute respiratory syndrome (SARS) have been modelled, even though the corresponding amino acid sequences were not suitable for tertiary structure predictions with conventional homology and/or threading procedures. An indirect search for a protein structure to be used as a template for 3D modelling has been performed on the basis of the genomic organisation similarity generally exhibited by coronaviruses. The crystal structure of Clostridium botulinum neurotoxin B appeared to be structurally adaptable to human and canine coronavirus spike protein sequences and it was successfully used to model the two subunits of SARS coronavirus spike glycoprotein. The overall shape and the surface hydrophobicity of the two subunits in the obtained models suggest the localisation of the most relevant regions for their activity.
Keywords: SARS, Protein structure, Structure prediction, Coronavirus, Molecular modelling
The recent outbreak of an atypical pneumonia, known as “severe acute respiratory syndrome” or SARS, has forced the scientific community to look for strategies to defeat the infective agent. In this respect, the identification of a coronavirus as responsible for the infection and its genomic characterisation [1], [2] represented the first milestone in this race to limit the world-wide diffusion of SARS infection.
The second milestone would be represented by the development of efficient vaccines and/or specific anti-viral drugs and, in the post-genomic era. Bioinformatics tools will play a primary role in giving rational bases for the development of anti-SARS molecular weapons.
So far, the search for drugs against SARS coronavirus, SARS_CoV, has been carried out on the basis of its putative receptor and, in this respect, the human aminopeptidase N, or CD13, has been proposed [3]. Thus, the inhibitors of the latter enzyme have been suggested in prophylaxis and therapeutics [4], even though some opposing data have been reported [5].
This type of approach to limit the viral infection should lead, hopefully soon, to an unambiguous assignment of the cell receptor, but alternative strategies should also be found to stop the diffusion of SARS_CoV.
In this respect, new specific antivirals, designed on the basis of structural knowledge of SARS_CoV proteins, cannot be synthesised yet, since, in spite of the already abundant genomic information, only one crystal structure [6] and one model [7] for the viral proteins are available so far. The principal reason for the apparent contradiction between the abundance of experimental data and lack of predicted 3D molecular models comes from the very poor sequence homology between SARS_CoV proteins and the ones available in the Protein Data Bank [8].
Here, we present the modelling of the S1 and S2 subunits of the spike glycoprotein of SARS_CoV, a highly antigenic viral envelope protein, involved in the host cell infection. The spike glycoprotein is translated as a large polypeptide that is subsequently cleaved by virus-encoded or host-encoded proteases to produce, like in other coronavirus, two functional subunits, S1 and S2 [9]. It has also been shown that S1 is the peripheral fragment and S2 is the membrane-spanning fragment [10]. Both chimeric S proteins appeared to cause cell fusion when expressed individually, suggesting that they were biologically fully active [9]. The spike is believed to be type I transmembrane protein, with N-terminal ectodomains and C-terminal hydrophobic anchor, and with an unusual cysteine-rich domain that bridges the putative junction of the anchor and the cytoplasmic tail [9]. The type I glycoprotein S of coronavirus, whose trimers constitute the typical viral spikes [10], is assembled into virions through non-covalent interactions with the M protein. In the SARS spike glycoprotein, the S2 domain can be easily identified by sequence alignment of other coronavirus proteins. As far as the remaining part of the S protein is concerned, the same procedure does not yield any information, suggesting that SARS_CoV has developed a new type of S1 domain which is not conserved among the other members of the S1 family.
As in the case of other viral membrane proteins, the S glycoprotein of SARS_CoV should play an important role in the interaction of the virus with its host cell receptor, having a primary role in eliciting antibodies in the host species [3]. Thus, it is apparent that knowledge of the 3D structure of SARS_CoV S protein, representing a critical toxic site and a candidate protective antigen, would be of great value in the search for a vaccine, in explaining existing data and in designing novel diagnostic kits and anti-viral drugs.
Materials and methods
A set of 14 different genomic sequences of SARS_CoV were collected using the NCBI [11] databases. The genomic regions codifying for the spike glycoprotein were identified from these sequences by using ORFfinder search [11]. The corresponding aminoacid sequences were aligned and, with the use of ClustalW v. 1.7 [12], 99–100% pairwise sequence identities were obtained. Then, a consensus sequence obtained from this alignment was used to find a subset of homologous S glycoprotein sequences of other coronaviruses using Psi-Blast v. 2.1 [11]. Some of these sequences were chosen for secondary structure prediction and fold recognition studies, using PsiPred v.2.1 [13] and GenTHREADER [14], respectively. With the fold recognition method the S glycoprotein of Human Coronavirus (SwissProt Accession No. SP_36334) [15] identifies the 1G9D pdb entry [16], while the S protein of Canine Coronavirus (SwissProt Accession No. SP_36300) [15] identifies the 1G5G pdb entry [16]. For each hit to a particular PDB entry, a SCOP v. 1.63 [17] classification of names and numbers was made. Model building of S1 and S2 subunits of SARS S glycoprotein was carried out with the ClustalW [12] alignment between the target sequence and that of the template structure. Models were subsequently optimised according to secondary structure predictions. Substitution of aminoacid residues and modelling of insertions and deletions in the target structure were performed using SwissPDBViewer software [18]. The 3D models were optimised by a 900 step minimisation run with AMBER [19] and finally validated with the PROCHECK v. 3.5.4 procedure [20]. Surface accessibility of the potential glycosylation site, mutation distribution, and localisation of CD13 binding regions were analysed by using MOLMOL software [21]. With the same software possible disulphide bridge formations were investigated by considering a threshold distance of 10 Å between Cα atoms of cysteine couples, after the prediction of the bonding/non-bonding state of all the cysteine present in S1 and S2 sequences [22]. The hydropathy profile of the protein was calculated according to Kyte and Doolittle plots [23] and hydrophilic/hydrophobic potentials were calculated with GRID [24], using a grid resolution of 1 Å and the standard “OH2” and “DRY” probes.
Results and discussion
The phylogenetic study for the structural proteins E, M, S, and the proteinase [1] indicates that the SARS coronavirus, even though it is most similar to group II of coronaviruses, which includes bovine and murine coronaviruses, should rather be considered as the first member of a new group IV. The sequence homology of the S glycoprotein of SARS_CoV with the ones of other coronaviruses, ranging from 20.39% to 27.63%, is rather low.
The genomic drift effects on the S glycoprotein have been analysed by aligning the available data. Thus, alignment of 14 different sequences obtained with ClustalW produced 99–100% identity. It is interesting to see that most of the amino acid mutations in SARS_CoV S glycoprotein sequences are predominantly located in the S1 subunit (1–680).
Since tertiary structure predictions by sequence homology and threading algorithms for fold recognition studies by using SARS_CoV S glycoprotein sequences could not yield reliable molecular models in the present report, the molecular models of the S1 and S2 subunits of SARS_CoV S glycoprotein have been obtained in an indirect way. In fact, a preliminary sequence alignment of various coronavirus S glycoproteins was performed and a fold recognition analysis was done on the aligned sequences. Among all the aligned S glycoprotein sequences, one of human coronavirus (SwissProt Accession Number SP36334) and one of canine coronavirus (SwissProt Accession Number SP36300) were chosen for fold recognition studies using GenTHREADER v2.1 [14]. The GenTHREADER results for human and canine CoV S proteins indicated two pdb entries, i.e., 1G9D of neurotoxin B of Clostridium botulinum and 1G5G of the Tetanus toxin, as possible templates for structure predictions. After sequence alignment of SARS S protein (SwissProt Accession Number P59594) with the corresponding ones from human and canine coronaviruses, model building of S1 and S2 subunits was obtained by using the latter two pdb files and shuffled PsiPred runs [13], with subsequent manual optimisation to enhance the overlapping between the predicted and observed secondary structure elements.
The structure of Tetanus toxin as a template allowed only a modelling of the carboxy terminus region by inserting also long gaps. On the contrary, as shown in Fig. 1 , by using the other candidate template, i.e., the C. botulinum toxin, a good level of similarity was achieved with 15% identity, 49% positives, and 5% gaps. Consequently, reliable 3D models of the S1 and S2 subunits were obtained, see Fig. 2 , as the small insertions, which still had to de done, did not cause major problems in the molecular modelling procedure. In addition, the secondary structure prediction [13] for both subunits and the template structure indicated that the distribution of α-helix, β-sheet, and random coil elements was comparable and located in the same regions.
Fig. 1.

Sequence alignment between SARS_CoV S protein and C. botulinum neurotoxin B (pdb ID 1G9D) used to construct the model. The regions exhibiting the same secondary structure, as predicted by PsiPred v.2.1, are also shown.
Fig. 2.
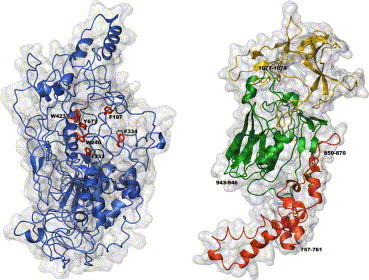
Surface and ribbon representations of the tertiary structure of the S1 and S2 subunits of SARS_CoV S glycoprotein. In the S1 representation (left) the residues forming the hydrophobic cluster are highlighted. In red, green, and yellow the I, II, and III domains of the S2 subunit are, respectively, shown (right), together with the putative CD13 binding regions. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this paper.)
In particular, the SARS S1 subunit has been modelled between residues 17 and 680, that is between the signal peptide and the S2 subunit, and the obtained structure has been deposited in the Protein Data Bank with the pdb ID 1Q4Z. In good analogy to the template fold of the corresponding domain, mainly anti-parallel β sheets with other segregated α and β regions were present in the 3D model.
As far as the S2 subunit is concerned, its outer moiety has been modelled between residues 727 and 1195, just before the predicted transmembrane segment, and identified between residues 1200 and 1223 [16]. In the obtained model, deposited in the Protein Data Bank with the pdb ID 1Q4Y, the three domains which are present in the template structure [17] may be clearly identified, see Fig. 2. Thus, domain I (residues 727–845) with a coiled coil fold contains a set of five anti-parallel helices, domain II (residues 846–1048) with a sandwich fold-like conacanavalin A is characterised by 12–14 strands in two sheets and, finally, domain III (residues 1049–1195) is six-stranded anti-parallel β barrel. A preliminary validation of this S2 model comes from the high surface exposure of the putative CD13 binding sites [3], respectively, located in a helix of the first domain and in two loops of the second and third domains.
A series of considerations should be taken into account to discuss the reliability of the predicted tertiary structures of the S1 and S2 subunits of SARS_CoV S glycoprotein. The first one implies that simple physicochemical rules are fulfilled by the modelled structures and, in this respect, the Ramachandran plots and the values of the corresponding G factors are excellent, as −0.3 and −0.2 were obtained, respectively, for the S1 and S2 subunits [20]. Furthermore, the fact that hydrophobic residues should be mostly buried in the molecular core with the hydrophilic ones located in surface exposed protein regions should be considered. This condition seems to be met by our models, as a good agreement between hydropathy profiles [24] and residue accessibility can be observed, see Fig. 3 .
Fig. 3.
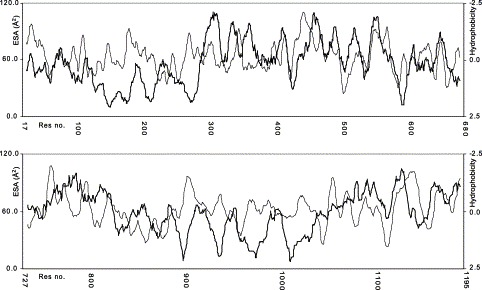
Hydropathy vs. residue exposed surface area (ESA) calculated for the models of S1 (top) and S2 (bottom) subunits of SARS_CoV S glycoprotein. In the graph hydrophobicity (light line) increases on descending the Y-axis, whereas residue accessibility (bold line) decreases.
In addition to the above-mentioned quality controls of our predicted structures, some of them were routinely performed upon structure deposition in the Protein Data Bank, while another set of criteria to validate the molecular models of the S1 and S2 subunits of SARS_CoV S protein comes from the observation of consistencies between the obtained structural features of the two models and some common experimentally derived findings, such as glycosylation site distribution and disulphide bridge formation.
As far as the N/O-glycosylation regions are concerned, these are expected to be surface exposed. In the S1 subunit, only three out of the total 14 predicted glycosylation sites [16] exhibit low surface accessibility and all of the five possible glycosylation sites result being exposed in the S2 subunit.
The disulphide bridge formation is a very critical point to assess the reliability of a molecular model and the fact that cysteine residues have to be within a suitable bonding distance to form disulphide bridges must be considered. In the S1 subunit there are 20 cysteines, while in the S2 subunit are eight in the external portion and nine in the internal portion [11] of the C terminus. The latter cysteines seem not to have a bridging role, as they are predicted in a non-bonding state from neural network predictions [22] and, in any case, do not belong to the presently modelled part of the molecule.
It is worth noting that all of the possible cystine bridges, 10 and four disulphide bridges, respectively, in the S1 and S2 subunits, can be formed in the two proposed models, since always the corresponding cysteine Cα atoms are found at a maximum distance of 10 Å. In particular, for the S1 subunit disulphide bridges are predicted between C19–C128, C133–C467, C159–C288, C278–C474, C323–C657, C348–C511, C366–C378, C419–C603, C524–C576, and C635–648 and in the model of the S2 subunit disulphide bridges between C731–C833, C742–C822, C1014–C1025, and C1064–C1108 can be obtained.
The two molecular model models were analysed in terms of surface hydrophobic potential to identify possible binding sites. According to the calculations performed for the S1 subunit a highly hydrophobic pocket is outlined. In this molecular moiety six hydrophobic residues, i.e., Phe 187, Phe 253, Phe 334, Trp 340, Trp 423, and Tyr 677, contribute to an extensive hydrophobic cluster formation, where long-range interactions can contribute to the S1 structure stability. This feature, not present in the used template structure, should also be of fundamental relevance in the S1 folding process [25] and could represent a suitable target for anti-viral drugs.
In the case of the S2 subunit, the hydrophobic potential analysis identifies two putative binding sites in the Phe850–Phe870 and in the Phe1077–Phe1079 regions, both located in the putative binding sites of CD13 [3].
As the analysis of the mutation distribution among SARS_CoV proteins could be important to predict its antigenic drift, this has been done by aligning the 14 S protein sequences so far present in the NCBI database for this virus [11] and 10 mutation sites were identified. Seven of these mutations are in the S1 subunit; the ones in position 77 and 244 are both localised in α helices, while the others, 239, 311, 344, 501, and 577, are in loop regions. It is interesting to note that, apart from 244 and 501 residue positions, all the other ones are located in well-exposed regions of the protein surface. High surface exposure is exhibited also by the sites of the remaining three mutations, i.e., 778, 794, and 1148 located in domains I and II of the S2 subunit.
As a final remark, it should be underlined that the consistent series of structural features, exhibited by the proposed models for the S1 and S2 subunits of the SARS_CoV spike protein, supports their reliability as a possible rational starting point for anti-viral drug design.
Acknowledgements
Thanks are due to the University of Siena for financial support and to Mrs. Graziella Pietrini for technical assistance.
References
- 1.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 2.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 3.Yu X.J., Luo C., Lin J.C., Hao P., He Y.Y., Guo Z.M., Qin L., Su J., Liu B.S., Huang Y., Nan P., Li C.S., Xiong B., Luo X.M., Zhao G.P., Pei G., Chen K.X., Shen X., Shen J.H., Zou J.P., He W.Z., Shi T.L., Zhong Y., Jiang H.L., Li Y.X. Putative hAPN receptor binding sites in SARS–CoV spike protein. Acta Pharmacol. Sin. 2003;24:481–488. [PubMed] [Google Scholar]
- 4.Kontoyiannis D.P., Pasqualini R., Arap W. Aminopeptidase N inhibitors and SARS. Lancet. 2003;361:1558. doi: 10.1016/S0140-6736(03)13186-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams R.K., Yeager C.L., Holmes K.V. Potential for receptor-based antiviral drugs against SARS. Lancet. 2003;362:77. doi: 10.1016/S0140-6736(03)13818-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 7.Shen X., Xue J.H., Yu C.Y., Luo H.B., Qin L., Yu X.J., Chen J., Chen L.L., Xiong B., Yue L.D., Cai J.H., Shen J.H., Luo X.M., Chen K.X., Shi T.L., Li Y.X., Hu G.X., Jiang H.L. Small envelope protein E of SARS: cloning, expression, purification, CD determination, and bioinformatics analysis. Acta Pharmacol. Sin. 2003;24:505–511. [PubMed] [Google Scholar]
- 8.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 10.Binns M.M., Boursnell M.E., Cavanagh D., Pappin D.J., Brown T.D. Cloning and sequencing of the gene encoding the spike protein of the coronavirus IBV. J. Gen. Virol. 1985;66(Pt. 4):719–726. doi: 10.1099/0022-1317-66-4-719. [DOI] [PubMed] [Google Scholar]
- 11.Available from 〈http://www.ncbi.nlm.nih.gov/〉
- 12.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 14.Jones D.T. GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J. Mol. Biol. 1999;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- 15.Available from 〈http://www.rcsb.org/pdb/〉
- 16.Available from 〈http://us.expasy.org/sprot/〉
- 17.Murzin A.G., Brenner S.E., Hubbard T., Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 18.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 19.Pearlman D.A., Case D.A., Caldwell J.W., Ross W.S., Cheatman III T.E., Ferguson D.M., Seibel G.L., Chandra Singh U., Weiner P.K., Kollman P.A. University of California; San Francisco: 1995. AMBER 4.1. [Google Scholar]
- 20.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 21.Koradi R., Billeter M., Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. 29–32. [DOI] [PubMed] [Google Scholar]
- 22.Fariselli P., Riccobelli P., Casadio R. Role of evolutionary information in predicting the disulfide-bonding state of cysteine in proteins. Proteins. 1999;36:340–346. [PubMed] [Google Scholar]
- 23.Goodford P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985;28:849–857. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- 24.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 25.Klein-Seetharaman J., Oikawa M., Grimshaw S.B., Wirmer J., Duchardt E., Ueda T., Imoto T., Smith L.J., Dobson C.M., Schwalbe H. Long-range interactions within a nonnative protein. Science. 2002;295:1719–1722. doi: 10.1126/science.1067680. [DOI] [PubMed] [Google Scholar]