

Abstract
In order to stimulate the development of drugs against severe acute respiratory syndrome (SARS), based on the atomic coordinates of the SARS coronavirus main proteinase determined recently [Science 13 (May) (2003) (online)], studies of docking KZ7088 (a derivative of AG7088) and the AVLQSGFR octapeptide to the enzyme were conducted. It has been observed that both the above compounds interact with the active site of the SARS enzyme through six hydrogen bonds. Also, a clear definition of the binding pocket for KZ7088 has been presented. These findings may provide a solid basis for subsite analysis and mutagenesis relative to rational design of highly selective inhibitors for therapeutic application. Meanwhile, the idea of how to develop inhibitors of the SARS enzyme based on the knowledge of its own peptide substrates (the so-called “distorted key” approach) was also briefly elucidated.
Keywords: SARS, Coronavirus proteinase, KZ7088, AG7088, Binding pocket, Octapeptide substrate, “Distorted key” mechanism
Severe acute respiratory syndrome (SARS) is a respiratory illness that has recently been reported in Asia, North America, and Europe. In general, SARS begins with a fever greater than 38 °C with symptoms such as headache, malaise, rigor, and body aches, followed by developing a dry cough and having trouble in breathing. The fatality rate for patients with SARS is presently around 15%. The primary way that the transmission of SARS occurs is mainly by close person-to-person contact, or by direct contact with infectious materials, such as respiratory secretions, from a person who has SARS. It is also possible that SARS can be spread more broadly through the air or by other ways that are currently not known. Since the spread of the new disease increases rapidly and so far no efficacious therapy is available, SARS has become a synonym of terror to human beings. Threatened by such a severe disease, scientists in all areas are facing a significant challenge, i.e., how to provide useful knowledge and technology that will lead to effective drugs against SARS. This is the need of the times.
Many evidences indicate that a previously unrecognised coronavirus exists in SARS patients. The newly-found virus, called SARS-coronavirus (SARS-CoV), is the leading hypothesis for the cause of SARS. It is also known that the process of cleaving the SARS-CoV polyproteins by a special proteinase, the so-called SARS coronavirus main proteinase (CoV Mpro), is a key step for the replication of SARS-CoV. The functional importance of the Mpro in the viral life cycle not only suggests that this proteinase is a culprit of SARS, but also makes it an attractive target for developing drugs directly against the new disease. However, to find effective compounds to inhibit the SARS-CoV Mpro, the first important thing is to understand the binding mechanism of the enzyme with its ligands. The present study was initiated in an attempt to gain some insight into this problem.
Materials and methods
The coordinates of SARS-coronavirus main proteinase, or SARS-CoV Mpro, recently determined by Anand et al. [1] were used as a targeted basis to conduct docking studies. The proteinase contains 304 amino acids, folded into three domains (Fig. 1 ): domain 1 (res. 8–99) is coloured magenta, domain 2 (res. 100–183) yellow, and domain 3 (res. 200–300) light blue. The N-terminal (res. 1–7) is coloured green, while the C-terminal (res. 301–304) red. The loop (res. 184–199) linking domain 2 and 3 is coloured purple. The binding cleft is located at the site between domains 1 and 2.
Fig. 1.
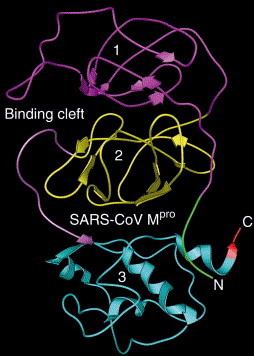
Ribbon illustration of the SARS-coronavirus main proteinase (SARS-CoV Mpro). The enzyme molecule consists of 304 amino acids. The entire peptide chain is folded into three domains: domain 1 (res. 8–99) is coloured magenta, domain 2 (res. 100–183) yellow, and domain 3 (res. 200–300) light blue. The N-terminal (res. 1–7) is coloured green, while the C-terminal (res. 301–304) red. The loop (res. 184–199) linking domain 2 and 3 is coloured purple. The binding cleft is located at the site between domains 1 and 2.
Two ligands were selected for the docking studies. One was KZ7088 (Fig. 2A ) and the other the octapeptide AVLQSGFR. As shown in Fig. 2A, KZ7088 is a derivative by modifying AG7088 (Fig. 2B). The latter was developed by Pfizer and is currently in clinical trials for the treatment of rhinovirus, a pathogen that can cause the common cold. Although it is a putative candidate for docking studies related to SARS drug finding, AG7088 has a p-fluorophenylalanine side chain (p-fluorobenzyl), which might be too long (or bulky) to fit into the relevant binding pocket [1]. Accordingly, KZ7088 with a modified side chain by removing -CH2 (Fig. 2) could well serve as a starting point for modification that will quickly lead to effective drug candidates for the treatment of SARS. The reason to select the octapeptide AVLQSGFR is as follows. (1) The protease-susceptible sites in proteins usually extend to an octapeptide, as generally formulated by P4P3P2P1P1′P2′P3′P4′ with the scissile bond located between the subsites P1 and P1′, as generally expressed by P1↓P1′ [2], [3], [4]. (2) The SARS coronavirus enzyme and several viral proteinases exhibit Gln ↓ (Ser, Ala, and Gly) specificity [1]. (3) According to the “lock-and-key” mechanism in enzymology, the octapeptide cleavable by the SARS proteinase must have a good fit for binding to the active site. However, such a peptide, after a modification of its scissile bond with some simple routine procedure, will completely lose its cleavability but it can still bind to the active site. Actually, the molecule thus modified can be compared to a “distorted key” that can be inserted into a lock but can neither open the lock nor be pulled out from it, spontaneously becoming an ideal competitive inhibitor against the SARS proteinase. Therefore, it would provide us with useful insights for drug design by studying the binding mechanism of SARS proteinase with octapeptides that are cleavable by the enzyme itself. For a detailed discussion about the rationale of “distorted key,” see a review by Chou [5] and Fig. 2 therein.
Fig. 2.
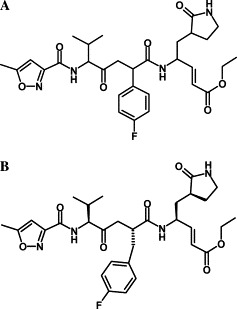
Chemical structure of KZ7088 (A), a derivative of AG7088 (B) by removing –CH2 from its fluorophenylalanine side chain.
The software package MOE (Chemical Computing Ltd.) was used to conduct the docking of the ligands to the SARS-coronavirus main proteinase. A similar approach was successfully used to investigate the binding interaction of the Cdk5–Nck5a*–ATP [6], [7] and that of caspase-9 with Ac-DVAD-fmk [8].
Results and discussion
The overall structure obtained by docking KZ7088 to SARS-CoV Mpro is given in Fig. 3 , where the SARS-coronavirus main proteinase is in ribbon drawing, the KZ7088 in ball-and-stick drawing, and the white mesh represents the binding pocket for KZ7088. The constituents of the binding pocket are defined by those residues that have at least one heavy atom (i.e., other than hydrogen) with a distance ⩽5 Å from a heavy atom of KZ7088 (a similar binding pocket was defined for ATP in the Cdk5–Nck5a*–ATP complex [6] that has later proved quite useful in identifying functional domains and stimulating the relevant truncation experiments [9]). The pocket thus defined consists of 23 residues, as listed in Table 1 . It would be intriguing to probe the binding pocket by site-directed mutagenesis of single amino acids as an avenue to find out which of the 23 residues is the most sensitive for the catalytic activity. A close view of the binding interaction is given in Fig. 4 . As shown from the figure, KZ7088 is tethered to the enzyme by six hydrogen bonds (blue dotted lines). The detailed atoms in forming the hydrogen bonds are given in Table 2 .
Fig. 3.
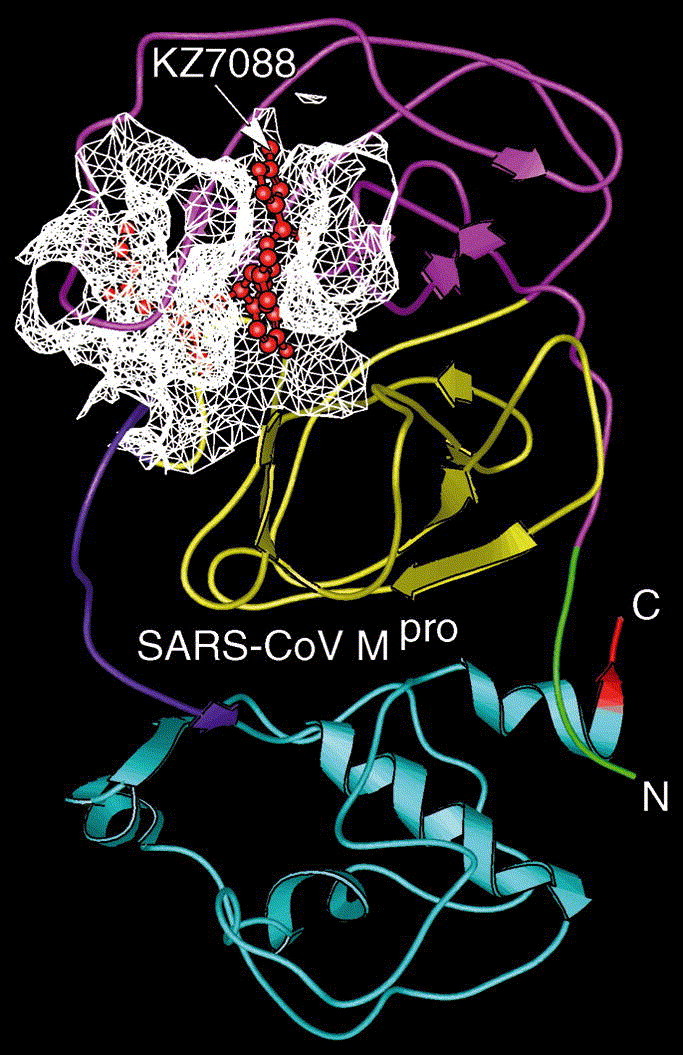
An overall view of the complex obtained by docking KZ7088 to SARS-CoV Mpro, where the SARS enzyme is in ribbon drawing, the KZ7088 in ball-and-stick drawing, and the white mesh represents the binding pocket for KZ7088. The 23 constituent residues of the binding pocket are listed in Table 1.
Table 1.
Binding pocket of the SARS proteinase for KZ7088
| Cys-22 | Gly-23 | Thr-24 | Thr-25 | Leu-27 | His-41 | Val-42 | Cys-44 |
| Thr-45 | Ala-46 | Glu-47 | Asp-48 | Met-49 | Leu-50 | Asn-51 | Pro-52 |
| Tyr-54 | Cys-145 | His-164 | Met-165 | Asp-187 | Arg-188 | Gln-189 |
Fig. 4.
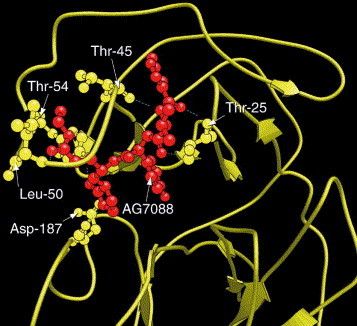
A close view of the binding interaction between KZ7088 and the SARS enzyme. As shown in the figure, KZ7088 is tethered to the enzyme by six hydrogen bonds (blue dotted lines). The detailed atoms in forming the hydrogen bonds are given in Table 2.
Table 2.
Hydrogen bond interactions between the SARS proteinase and KZ7088
| Atom in SARS proteinase | Atom in KZ7088 |
|---|---|
| Thr-25 N | O-9 |
| Thr-25 O-γ1 | O-20 |
| Thr-45 O | O-15 |
| Leu-50 N | O-68 |
| Tyr-54 O-η | O-67 |
| Asp-187 O | O-72 |
The overall structure obtained by docking the octapeptide AVLQSGFR to SARS-CoV Mpro is given in Fig. 5 , where the octapeptide is coloured white (in ball-and-stick drawing). As shown in Fig. 6A , there are six hydrogen bonds (blue dotted lines) formed between the octapeptide and the SARS proteinase. Those residues involved in forming the hydrogen bonds from the enzyme are: Arg-40, His-41, Phe-185, Asp-187, and Gln-189. The detailed atoms in forming the hydrogen bonds are given in Table 3 , which may provide useful information for in-depth understanding of the mechanism of the nucleophilic attack of the active site on the carbonyl carbon atom of the scissile peptide bond [10]. It is interesting to see that, after a single amino acid mutation for the octapeptide at the position P1, i.e., substitution of Q by E, the interaction of hydrogen bonding between the enzyme and the mutated octapeptides AVLESGFR would be reduced to only three hydrogen bonds (Fig. 6B) from the original six (Fig. 6A). This finding has further supported the observations by Anand et al. [1] that Gln at the position P 1 of the octapeptide plays a key role for the specificity of the SARS proteinase.
Fig. 5.
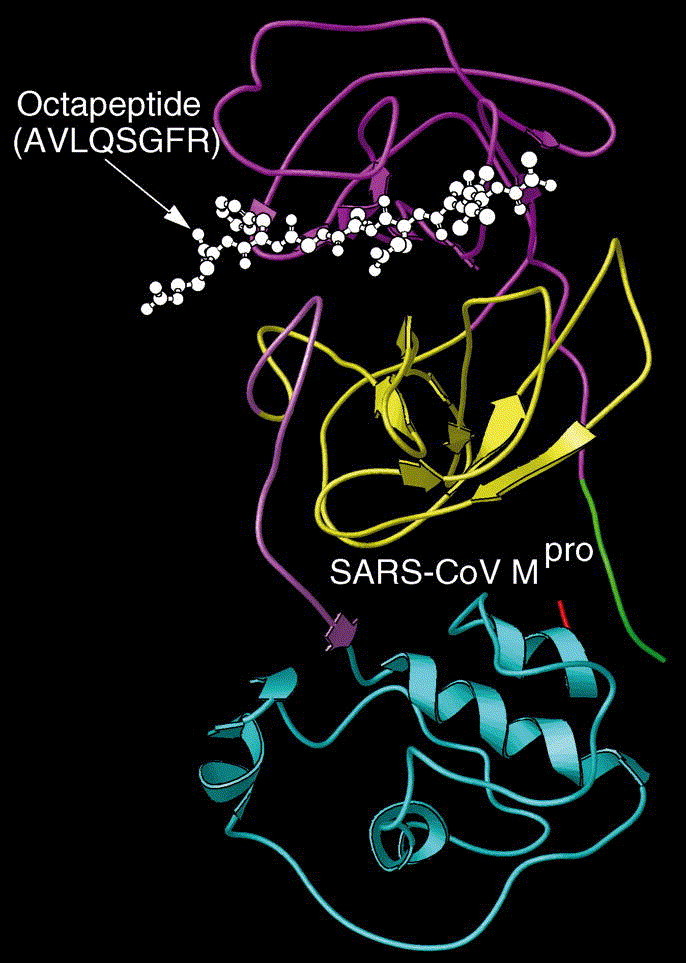
An overall view of the complex obtained by docking the octapeptide AVLQSGFR to SARS-coronavirus main proteinase, where the octapeptide is coloured white (in ball-and-stick drawing), and the SARS enzyme is in ribbon drawing. As shown in the figure, the octapeptide is inserted into the cleft between domains 1 and 2 as expected.
Fig. 6.
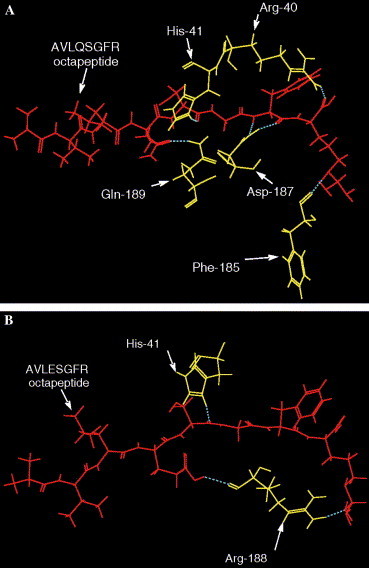
Illustration to show the hydrogen bonding interaction of the SARS proteinase with (A) the octapeptide AVLQSGFR and (B) its mutated octapeptide AVLESGFR. As shown in the figure, after a single amino acid mutation for the octapeptide at the position P1, i.e., substitution of Q by E, the relevant hydrogen bonds are reduced from the original six (A) to only three (B).
Table 3.
Hydrogen bond interactions between the SARS proteinase and AVLQSGFR octapeptide
| Atom in SARS proteinase | Atom in AVLQSGFR octapeptide |
|---|---|
| Arg-40 N-η2 | Arg-8 O |
| His-41 N-δ1 | Ser-5 O |
| Phe-185 O | Arg-8 N-ε |
| Asp-187 O-δ2 | Phe-7 O |
| Asp-187 O-δ2 | Arg-8 O |
| Gln-189 N-ε2 | Gln-4 O-ε1 |
Conclusions
In addition to the atomic coordinates of the SARS-CoV main proteinase recently provided by Anand et al. [1], the detailed binding interactions of the SARS enzyme with KZ7088 (a derivative of AG7088) and the octapeptide AVLQSGFR obtained through the present docking studies will further provide a solid footing for conducting structure-based drug design against SARS.
Footnotes
Abbreviations: SARS, severe acute respiratory syndrome; CoV, coronavirus; Mpro, main proteinase.
References
- 1.K. Anand, J. Ziebuhr, P. Wadhwani, J.R. Mesters, R. Hilgenfeld, Science, www.scienceexpress.org, 13 May 2003 (online) [DOI] [PubMed]
- 2.Schechter I., Berger A. Biochem. Biophys. Res. Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 3.Miller M., Schneider J., Sathyanarayana B.K., Toth M.V., Marshall G.R., Clawson L., Selk L., Kent S.B., Wlodawer A. Science. 1989;246:1149–1152. doi: 10.1126/science.2686029. [DOI] [PubMed] [Google Scholar]
- 4.Chou K.C. J. Biol. Chem. 1993;268:16938–16948. [PubMed] [Google Scholar]
- 5.Chou K.C. Anal. Biochem. 1996;233:1–14. doi: 10.1006/abio.1996.0001. [DOI] [PubMed] [Google Scholar]
- 6.Chou K.C., Watenpaugh K.D., Heinrikson R.L. Biochem. Biophys. Res. Commun. 1999;259:420–428. doi: 10.1006/bbrc.1999.0792. [DOI] [PubMed] [Google Scholar]
- 7.Tarricone C., Dhavan R., Peng J., Areces L.B., Tsai L.H., Musacchio A. Mol. Cell. 2001;8:657–669. doi: 10.1016/s1097-2765(01)00343-4. [DOI] [PubMed] [Google Scholar]
- 8.Chou K.C., Tomasselli A.G., Heinrikson R.L. FEBS Lett. 2000;470:249–256. doi: 10.1016/s0014-5793(00)01333-8. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J., Luan C.H., Chou K.C., Johnson G.V.W. Proteins. 2002;48:447–453. doi: 10.1002/prot.10173. [DOI] [PubMed] [Google Scholar]
- 10.Chou K.C., Howe W.J. Biochem. Biophys. Res. Commun. 2002;292:702–708. doi: 10.1006/bbrc.2002.6686. [DOI] [PubMed] [Google Scholar]