

Summary
The droplet digital polymerase chain reaction (ddPCR) is a novel molecular technique that allows rapid quantification of rare target DNA sequences. Aim of this study was to explore the feasibility of the ddPCR technique to detect pathogen DNA in whole blood and to assess the diagnostic accuracy of ddPCR to detect bloodstream infections (BSIs), benchmarked against blood cultures. Broad‐range primers and probes were designed to detect bacterial 16S rRNA (and Gram stain for differentiation) and fungal 28S rRNA. To determine the detection limit of ddPCR, 10‐fold serial dilutions of E. coli and C. albicans were spiked in both PBS and whole blood. The diagnostic accuracy of ddPCR was tested in historically collected frozen blood samples from adult patients suspected of a BSI and compared with blood cultures. Analyses were independently performed by two research analysts. Outcomes included sensitivity and specificity of ddPCR. Within 4 h, blood samples were drawn, and DNA was isolated and analysed. The ddPCR detection limit was approximately 1–2 bacteria or fungi per ddPCR reaction. In total, 45 blood samples were collected from patients, of which 15 (33%) presented with positive blood cultures. The overall sensitivity of ddPCR was 80% (95% CI 52–96) and specificity 87% (95% CI 69–96). In conclusion, the ddPCR technique has considerable potential and is able to detect very low amounts of pathogen DNA in whole blood within 4 h. Currently, ddPCR has a reasonable sensitivity and specificity, but requires further optimization to make it more useful for clinical practice.
The droplet digital polymerase chain reaction (ddPCR) is a novel molecular technique that allows rapid quantification of rare target DNA sequences. Aim of this study was to explore the feasibility of the ddPCR technique to detect pathogen DNA in whole blood, and to assess the diagnostic accuracy of ddPCR to detect bloodstream infections, benchmarked against blood cultures. The ddPCR technique showed considerable potential and was able to detect very low amounts of pathogen DNA in whole blood within 4 h. Currently, ddPCR has a reasonable sensitivity and specificity, but requires further optimization to make it more useful for clinical use.

Introduction
Bloodstream infections (BSIs) are the foremost cause of death due to infections and significantly contribute to morbidity, prolonged hospital stay and increased healthcare expenditures (Kilgore and Brossette, 2008; Goto and Al‐Hasan, 2013; Rhee et al., 2017; Coopersmith et al., 2018). Currently, diagnosis of BSIs relies on blood cultures in order to identify the causative pathogen(s). Although blood cultures are considered the gold standard, these take ample time to provide guidance for antimicrobial treatment (Liesenfeld et al., 2014; Opota et al., 2015). Especially growth rates of daunting pathogens such as fungi may lead to delays of up to 5 days or more, compared to 1–2 days for most bacteria. This results in a significant time window before a correct diagnosis can be made. Since empirical broad‐spectrum antibiotic treatment is usually initiated immediately upon admission and later tailored based on culture results, development of microbial resistance is another important issue (Coopersmith et al., 2018). Because of its risk for metastatic infection with a possibly lethal course, a rapid diagnosis of BSIs is crucial to steer treatment and reduce the associated clinical adversities and cost (Kumar et al., 2006; Huang et al., 2013; Seymour et al., 2017; Martin‐Loeches et al., 2018).
The use of culture‐independent molecular techniques holds promise to improve BSI diagnostics, and several assays have become available that allow for a rapid detection of pathogens in whole blood (Tissari et al., 2010; Makristathis et al., 2014; Opota et al., 2015; Stevenson et al., 2016; Pilecky et al., 2018). If proven successful, the impact of any culture‐independent diagnostic test would be significant since early pathogen detection improves choice of therapy and narrows the spectrum of antimicrobial coverage, with a reduced risk for microbial resistance and potential side‐effects related to antimicrobial treatment (Stevenson et al., 2016; Seymour et al., 2017). Unfortunately, most molecular tests show acceptable specificity (90–95%), but only moderate sensitivity (65–85%) (Makristathis et al., 2014; Marco, 2017). This latter notion severely limits the feasibility of these tests for clinical use where false‐negative results may lead to poor outcomes.
The droplet digital polymerase chain reaction (ddPCR) is a novel molecular technique that has been developed to improve the sensitivity and quantification of rare target DNA sequences, for example in liquid biopsies for cancer monitoring, non‐invasive prenatal testing for genetic abnormalities and detection of DNA contaminants in bioprocessing (Hussain et al., 2016; Oellerich et al., 2017; Postel et al., 2018; Zhang et al., 2018; Wang et al., 2018; Tan et al., 2019; Galimberti et al., 2019). After DNA isolation, ddPCR generates approximately 20 000 miniscule droplets, which enriches target DNA sequences by reducing the competition with high‐copy templates (Fig. S1). Once PCR amplification has been carried out within each of these 20 000 droplets, fluorescent positive and negative droplets are measured and the concentration of target DNA is determined by a Poisson algorithm. Although ddPCR has shown to be a technique with potential and has been used for the detection of specific pathogens, its use in the setting of rapid BSI detection has not been explored yet (Yang et al., 2017; Li et al.,; Song et al., 2018; Wang et al., 2018). The use of broad‐range primers for the amplification of the highly conserved bacterial 16S rRNA and fungal 28S rRNA, and different fluorescence dye‐labelled probes, enables the detection of BSIs, discrimination between fungal and bacterial infections, and –in the latter case – specification of their Gram stain differentiation or genus (Fig. 1) (Klaschik et al., 2002; Yang et al., 2002; Vollmer et al., 2008; Wu et al., 2008; Horvath et al., 2013). Differentiation between these broad groups may be of interest in patients using central venous catheters, such as intestinal failure patients depending on lifelong home parenteral nutrition. Here, repeated catheter replacement due to catheter‐related BSIs may result in loss of venous access options. Rapid detection of (virulent) bacteria and fungi (mostly Candida) helps to decide what type of treatment should be started, but also whether a catheter can be salvaged or not (Pironi et al., 2016). A (rough) discrimination at the microbial genus level may also improve catheter salvage rates in patients with less virulent pathogens by tailoring antibiotic regimens.
Figure 1.
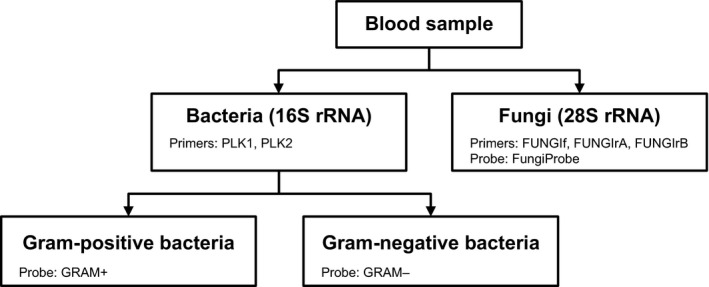
Detection of 16S rRNA and fungal 28S rRNA. Use of broad‐range primers and probes for bacterial 16S rRNA and fungal 28S rRNA enables the detection of bloodstream infections, and discrimination between fungal and bacterial infections. The fluorescence dye‐labelled probes GRAM + and GRAM– differentiate between Gram‐positive and Gram‐negative bacteria.
The aim of this study was to evaluate the feasibility of ddPCR in the setting of rapid BSI diagnostics, by determining the ddPCR detection limit, and to evaluate the time to pathogen DNA detection in whole blood. A second aim was to test the diagnostic accuracy of the ddPCR by analysing historically collected whole blood samples from patients suspected of a BSI and compare the results with the current gold standard; blood cultures.
Results
Feasibility study
Pathogen DNA detection in water and whole blood
In a single PCR reaction, the ddPCR was able to detect pure bacterial and fungal DNA (Fig. 2), even after a 100 000 times dilution in water, correlating with a detection of approximately five bacteria per ddPCR reaction. As depicted in Fig. 2, in case of a positive result, a ‘cloud’ of positive droplets was observed above the threshold. Similar results were found after diluting pathogen DNA in human DNA (data not shown). The excess of human DNA did not increase the background signal ‘noise’ for the negative controls, suggesting that the primer/probe combinations were highly specific for bacterial and fungal DNA.
Figure 2.
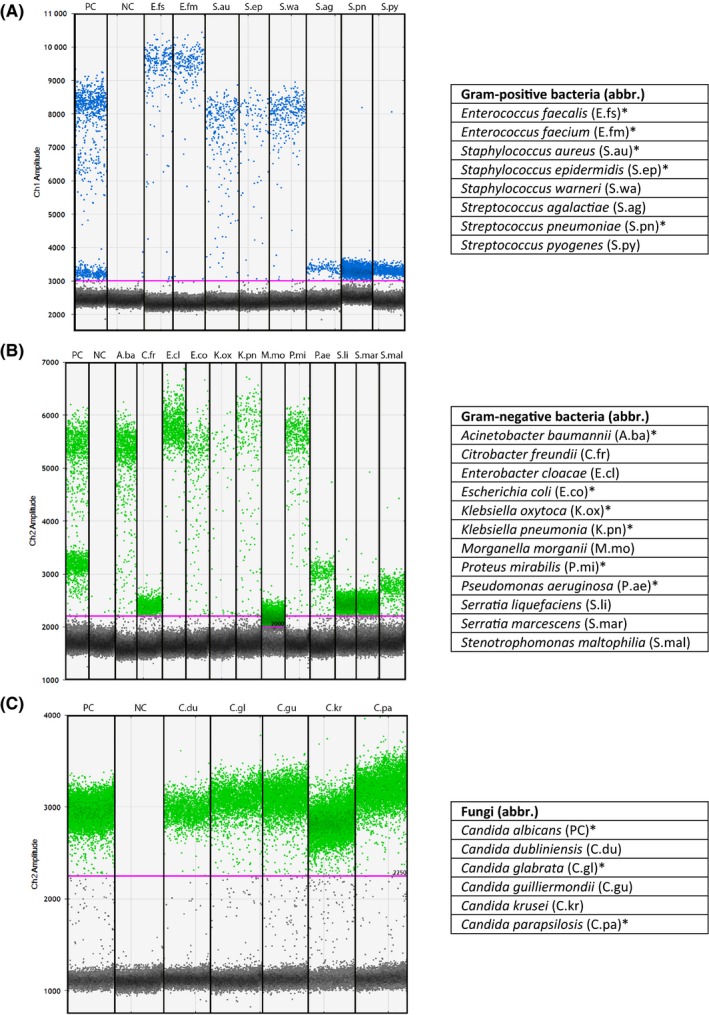
Detection of Gram‐positive bacteria, Gram‐negative bacteria and fungi using ddPCR. ddPCR, droplet digital polymerase chain reaction; NC, negative control (water); PC, positive control. All available 26 pathogen DNA samples were diluted in water. Positive controls included S. aureus and S. pneumoniae (A), E. coli and P. aeruginosa (B), and C. albicans (C). Abbreviations for pathogens are shown in the tables. *Most prevalent BSI‐causing microorganisms at our gastroenterology department.
To determine the ddPCR detection limit, E. coli and C. albicans were spiked in whole blood and subsequently isolated in a simple DNA isolation procedure, without pre‐isolating pathogens (Fig. 3 and Table S4). The ddPCR detection limit was below 5 (approximately 1–2) bacteria and 1–2 fungi per ddPCR reaction (approximately one copy of DNA per 40 000 human cells). The ddPCR detected E. coli in a 10‐ to 100‐fold lower concentration when compared to qPCR (Fig. 3A). For C. albicans, no difference was observed between the detection limits of ddPCR and qPCR (Fig. 3B).
Figure 3.
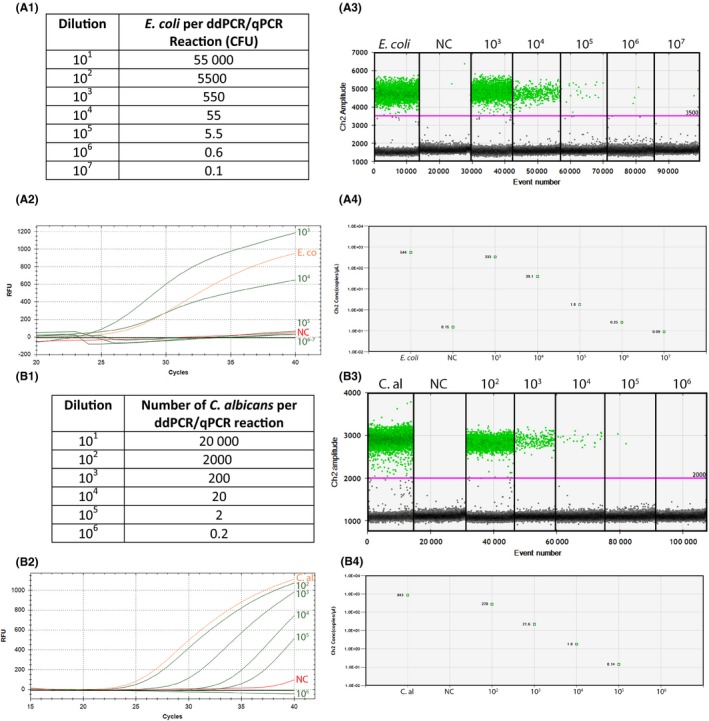
Detection limit of E. coli (A) and C. albicans (B) for ddPCR compared to qPCR. C.al, Candida albicans; CFU, colony‐forming units; ddPCR, droplet digital polymerase chain reaction; NC, negative control (whole blood); qPCR, quantitative polymerase chain reaction; RFU, relative fluorescence units. The amount of pathogens per ddPCR or qPCR reaction is shown in A1 and B1, respectively, determined by 10‐fold dilutions of E. coli and C. albicans in PBS (see also Table S4). qPCR results for both E. coli and C. albicans are shown in A2 and B2 respectively. A3 and B3 show ddPCR droplet results. The ddPCR concentrations per dilution in copies per µl are shown in A4 and B4. For qPCR, E. coli was not detectable after 104 dilutions, correlating with approximately 50 bacteria (A1, A2). The ddPCR detection limit was near 106 times dilution, resulting in approximately 1 to 2 bacteria per PCR reaction (A1, A3, A4). C. albicans was not detectable after 105 dilutions in qPCR, correlating with approximately 2 fungi (B1, B2). The ddPCR showed a similar detection limit (B1, B3, B4).
Addition of higher volumes of isolated DNA, from 2 to 8 µl (approximately 100 to 400 ng), to the ddPCR reaction increased the number of positive droplets, without an increase in negative control droplets (Fig. S2). The quality of the droplet generation, however, decreased at a volume of 6 µl (approximately 300 ng) and higher; both the droplet’s sizes and amplitude of signals varied greatly.
Subsequently, all available 26 pathogen DNA samples were tested (Fig. 2). All pathogens were detected using ddPCR and discriminated by their Gram stain. Remarkably, the amplitude of droplet clouds between pathogens differed greatly, which made it possible to discriminate some bacteria at the genus level (Fig. 2A and 2B). Reason for these different amplitudes is a difference in binding efficiency of the probes on the pathogen DNA template; the better a probe fits to the binding site, the higher the amplitude of the droplet cloud becomes (Fig. S3).
An overload of DNA of two pathogens with different amplitudes resulted in two droplet ‘clouds’ with in between a third ‘cloud’ (Fig. S4). The latter ‘cloud’ consisted of droplets with signals positive for both pathogens. This effect disappeared in a 10–1000 dilution series (Fig. S4).
Time to diagnosis
The total time from drawing blood to detection of both bacteria and fungi amounted to approximately 4 h, including more than 1 h hands‐on time (60 min DNA isolation, 15 min PCR mixture and droplet generation), 2 h of ddPCR analysis and 45 min analysis of ddPCR results.
Diagnostic accuracy study
Patients and blood culture results
Patient demographics are shown in Table S5. In total, 45 blood samples were collected from patients suspected of a BSI. Of these, 15 (33%) presented with positive blood cultures (Table 1). Five (33%) blood cultures showed Gram‐positive bacteria, three (20%) Gram‐negative bacteria, three (20%) contained fungi and four (27%) blood cultures were polymicrobial.
Table 1.
Blood culture and ddPCR results of the patient cohort.
| Patient | Blood culture results | ddPCR | Bacteria | Gram‐positive bacteria | Gram‐negative bacteria | Fungi | Sequence results of blood samples |
|---|---|---|---|---|---|---|---|
| #1 | S. aureus (3fl) | – | – | – | – | – | Negative |
| #2 | Aeromonas sp. (3fl) | – | – | – | – | – | Negative |
| #3 | C. parapsilosis (1fl) | – | – | – | – | – | Negative |
| #4 | C. albicans (2fl) | + | + | + | – | + | C. albicans |
| #5 | C. parapsilosis (1fl) | + | + | – | + | + | C. parapsilosis |
| #6 | Diphtheroid rods (2fl) | + | + | – | + | – | Brevibacterium |
| #7 | CNS (2fl) | + | + | + | – | – | Negative |
| E. coli (2fl) | |||||||
| E. faecium (3fl) | |||||||
| S. viridans (1fl) | |||||||
| C. albicans (1fl) | |||||||
| #8 | E. faecalis (2fl) | + | + | + | – | – | Negative |
| #9 | CNS (4fl) | + | + | + | – | – | Negative |
| #10 | CNS (4fl) | + | + | + | – | – | Negative |
| E. faecium (1fl) | |||||||
| #11 | S. epidermidis (2fl) | + | + | + | – | – | Negative |
| #12 | K. pneumonia (3fl) | + | + | – | + | – | Negative |
| #13 | C. freundii (2fl) | + | + | – | + | – | S. marcescens |
| S. marcescens (3fl) | |||||||
| #14 | E. coli (2fl) | + | + | – | + | – | Negative |
| #15 | K. pneumoniae (1fl) | + | + | – | + | + | K. pneumoniae |
| P. mirabilis (1fl) | C. parapsilosis | ||||||
| C. parapsilosis (1fl) | |||||||
| #16 | Negative | + | + | + | – | – | Staphylococcus species |
| #17 | Negative | + | + | + | – | – | Negative |
| #18 | Negative | + | + | + | – | – | Negative |
| #19 | Negative | + | + | – | + | – | Negative |
| Other | 26 negative cases | – | – | – | – | – | N/A |
CNS, coagulase‐negative staphylococci; ddPCR, droplet digital polymerase chain reaction; fl, flask; N/A, not applicable.
Plus (+) is a positive result, minus (–) is a negative result. The flasks represent the number of blood flasks determined positive for a certain pathogen.
Blood culture versus ddPCR results
Blood culture and ddPCR results are summarized in Table 1. In three patients (#1–3), ddPCR was negative for pathogen DNA, while blood culture results were positive for either Gram‐positive or Gram‐negative bacteria, or a fungal pathogen (Table 1). DNA sequencing of these three blood samples gave negative results. In four patients (#16‐19), ddPCR was positive for pathogen DNA, while blood cultures remained negative. In these four cases, three blood samples gave negative DNA sequence results. Patient #16, however, had a DNA sequence positive for staphylococcus species, which was in line with ddPCR result. In three cases (patient #4–6), both blood cultures and ddPCR were positive, but ddPCR showed different results. For example, patient #4 and #5 had a fungal infection based on the blood culture results, while ddPCR detected additionally Gram‐positive and Gram‐negative pathogen DNA, respectively. Patient #6 was positive for Gram‐negative bacteria, while blood culture results showed Gram‐positive diphtheroid rods. DNA sequencing showed Brevibacterium DNA in the patient’s blood sample, which was in disagreement with both the blood culture and ddPCR result.
The remaining 35 blood culture results were in line with ddPCR results. A selection of relevant cases is shown in Fig. 4.
Figure 4.
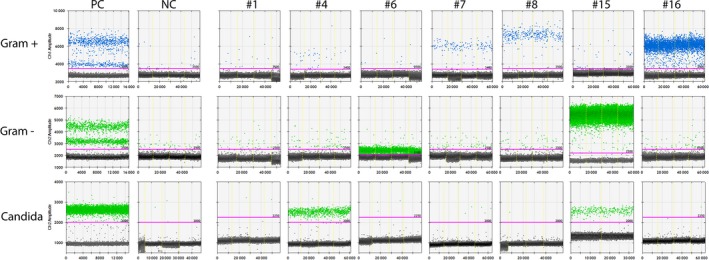
Selection of relevant patient results. ddPCR; droplet digital polymerase chain reaction, NC, negative control (whole blood), PC, positive control. PCs included S. aureus and S. pneumoniae for Gram‐positive bacteria, E. coli and P. aeruginosa for Gram‐negative bacteria, and C. albicans for Candida species. Note 1) patient samples may have been analysed on different days. Every patient sample has been compared with the NC and PC of that same day. In this figure, the most representative NCs/PCs for all patients have been selected for display purposes. Note 2) there is a ‘rain’ of droplets between the positive droplet clouds of the PCs. This ‘rain’ consists of droplets with signals positive for both positive controls (see also Fig. 4).
Diagnostic accuracy of ddPCR
The overall sensitivity and specificity of the ddPCR for detecting BSIs (either bacteria or fungi) was 80% (95% CI 52–96) and 87% (95% CI 69–96) respectively (Table 2). Similar results were found for the detection of bacteria. Differentiation between Gram‐positive and Gram‐negative bacteria resulted in a lower sensitivity (71% and 67%), but higher specificity (89% and 92%). For fungi detection, ddPCR sensitivity was 60% (95% CI 15–95) and specificity 100% (95% CI 91–100).
Table 2.
Diagnostic accuracy of the ddPCR.
| Bloodstream infection | Bacteria | Gram‐positive bacteria | Gram‐negative bacteria | Fungi | |
|---|---|---|---|---|---|
| Sensitivity (95% CI) | 80 (52–96) | 83 (52–98) | 71 (29–96) | 67 (22–96) | 60 (15–95) |
| Specificity (95% CI) | 87 (69–96) | 82 (65–93) | 89 (75–97) | 92 (79–98) | 100 (91–100) |
| LR+ (95% CI) | 6.00 (2.33–15.46) | 4.58 (2.13–9.87) | 6.79 (2.40–19.17) | 8.67 (2.54–29.52) | N/A |
| LR– (95% CI) | 0.23 (0.08–0.64) | 0.20 (0.06–0.73) | 0.32 (0.10–1.04) | 0.36 (0.12–1.12) | 0.40 (0.14–1.17) |
| PPV (95% CI) | 75 (54–89) | 63 (44–78) | 56 (31–78) | 57 (28–82) | N/A |
| NPV (95% CI) | 90 (76–96) | 93 (79–98) | 94 (84–98) | 95 (85–98) | 95 (85–99) |
CI, confidence interval; ddPCR, droplet digital polymerase chain reaction; LR−, negative likelihood ratio; LR+, positive likelihood ratiol; N/A, not applicable; NPV, negative predictive value; PPV, positive predictive value.
Discussion
In this study, we report the development of a novel diagnostic broad‐spectrum tool for rapid detection of bacteria and fungi in the setting of BSIs, based on the ddPCR technique. Besides its feasibility to detect pathogen DNA within a short time span, we also showed that ddPCR has a reasonable sensitivity and specificity to identify pathogens from whole blood.
The advantage of this ddPCR assay is its sensitive culture‐independent nature, based on pathogen DNA extraction directly from whole blood. It is a non‐challenging technique that can be implemented and performed in a simple laboratory setting. In comparison to other molecular tests, ddPCR assay does not require additional (time‐consuming and expensive) DNA enrichment steps prior to DNA extraction, such as centrifugation, filtration or microfluidic steps, to purify pathogens (Wellinghausen et al., 2009; Carrara et al., 2013; Loonen et al., 2014; Stevenson et al., 2016; Knabl et al., 2016; Pfaller et al., 2016; Pilecky et al., 2018). This simplification ultimately results in a rapid identification of pathogens within 4 h (including 1 h hands‐on time), whereas most molecular tests require 4–8 h, or longer (Pilecky et al., 2018). Especially in the setting of fungemia, rapid identification will likely have significant clinical impact. Here, accumulating evidence underscores the significance of early and appropriate antifungal treatment as an important driver of outcomes (Kullberg and Arendrup, 2015).
The detection limit of ddPCR was low with 1–2 bacteria and 1–2 fungi per PCR reaction. Similar ddPCR detection limits have been reported in other settings and mainly depend on the targets and primer–probe combinations (Wang et al., 2018). Since ddPCR assay does not include any enrichment steps prior to DNA extraction, pathogen DNA detection is based on a single PCR end reaction of 2 µl isolated DNA, including an overload of human DNA and 1–5 pathogen cells. This is in contrast to other systems, such as the SepsiTest (Molzym, Germany), which detect approximately 10–80 pathogen cells per millilitre in an already enriched PCR setting before a positive result is found, suggesting that ddPCR, at least in vitro, is a more sensitive method (Stevenson et al., 2016).
Due to the low amount of pathogen DNA in blood samples, we performed the ddPCR reaction at least in quadruple, as a single ddPCR reaction may not even contain pathogen DNA. It remains the question, however, whether such low pathogen DNA loads are clinically relevant. For some pathogens, such as S. aureus, very low pathogen loads may remain clinically relevant, whereas for other species this may not be the case. It is therefore important to set a lower limit of positive droplets, possibly per pathogen, where BSIs are determined positive. To lower the ddPCR detection limit even further, higher loads of DNA in a PCR reaction are required. Indeed, the addition of higher amounts of isolated DNA to the PCR reaction increased the number of positive droplets, but the quality of the droplets decreased at an amount of ≥ 300 ng. This is likely due to an increased viscosity, resulting in a wider range of droplet sizes and signals. Based on these results, we currently recommend to use a maximum of 200 ng of isolated DNA. According to the manufacturer, high amounts of human DNA per PCR reaction may indeed negatively affect the accuracy of DNA quantification (Bio‐Rad). A solution to this problem might be digestion of genomic DNA, which allows a PCR reaction to exceed 1 µg per 20 µl PCR reaction (containing approximately 25 pathogen cells per millilitre) without affecting DNA quantification. In addition, digestion may also decrease the scatter of droplets seen in ddPCR reactions with high loads of DNA.
Currently, based on our small retrospective cohort, the overall ddPCR sensitivity and specificity seems in line with other molecular tests on the market (Makristathis et al., 2014; Stevenson et al., 2016; Marco, 2017). Although the sensitivity and specificity of ddPCR is reasonable, it is obvious that a higher sensitivity and specificity is desirable in clinical practice. Especially for detection of fungemia, the sensitivity seems relatively low. More clinical (head‐to‐head) studies will need to be performed to establish the relevance of the ddPCR assay in clinical practice. Several factors may have affected the sensitivity and specificity of the ddPCR assay. For example, although blood cultures are considered the gold standard, contamination with other microbes, for instance during drawing of blood, may have occured (Coopersmith et al., 2018). In addition, up to half of suspected BSIs occur while blood cultures do not reveal microbial growth (Pilecky et al., 2018). This may be due to the presence of slow‐growing pathogens, or because of antibiotic therapy initiation before blood collection. The 10‐year‐old frozen blood samples used in this study may have affected the quality of the blood samples. In combination with retrospectively collected patient data, this could have had impact on the test results as well. Furthermore, we cannot rule out that the PCR assay may have been suppressed in certain patients, for example due to sample matrix components, such as heparin, immunoglobulins, or iron‐associated haemoglobin and lactoferrin (Coopersmith et al., 2018; Pilecky et al., 2018). Finally, it is yet unclear how extremely low pathogen DNA loads in the blood circulation (possibly due to previous infections) influence the sensitivity and specificity of the ddPCR (Coopersmith et al., 2018).
Strengths of this study include its new insights in the practical abilities of ddPCR, the systematic and broad set‐up of analyses, and the combination of both testing the feasibility and diagnostic accuracy of this method. In addition, we focused on clinically relevant outcomes, such as a sensitive detection of pathogens in whole blood within a short time frame. The choice for broad‐range primers/probes for bacteria and fungi resulted in potential clinically relevant decision‐making steps. Another strength is the primer–probe combination for Gram‐positive bacteria and fungi, which had a very low background signal ‘noise’ for the negative controls.
On the other hand, further improvement seems desirable as there is still a substantial background noise for Gram‐negative bacteria for the lower amplitudes (2000–3000). In the event of very low pathogen DNA loads, this may limit the detection of these pathogens. Unfortunately, it is yet unclear what causes this background noise. In contrast to some molecular systems, the current ddPCR assay only discriminates between certain bacteria on genus level, but not on species level (Wellinghausen et al., 2009; Carrara et al., 2013; Liesenfeld et al., 2014). As some pathogens have droplet clouds that overlap, it is sometimes impossible to differentiate between monobacterial and polymicrobial blood samples (e.g. two Gram‐positive bacteria droplet clouds with similar amplitudes) or between bacteria (e.g. between CNS and E. faecium, as in patient #10), while this is important for clinical decision‐making. Thus, modifications of the ddPCR system seem necessary for clinical practice, for example by adding extra channels to the device for additional primer–probe combinations (whether or not on 16S/28S rRNA) to detect pathogen‐specific sequences (e.g. nuc or eap genes for S. aureus species), but also to allow random access when the system is already running (Hussain et al., 2008; Wang et al., 2018). Another limitation is that, in line with most molecular tests, the ddPCR set‐up does not provide information regarding microbial susceptibility to antibiotics (Coopersmith et al., 2018; Pilecky et al., 2018). Finally, automation of the ddPCR technique seems desirable for clinical practice, as 1 h hands‐on time requires dedicated personnel, also in the weekends and nights.
Future research should focus on establishing larger prospective patient cohorts in various clinical settings, such as haemodialysis, oncology or home parenteral nutrition, but also the emergency department and intensive care unit. These studies may help to determine the relevance of low amounts of positive droplets in blood samples and define the ddPCR detection limit in clinical practice. In addition, studies may investigate the contribution of patient monitoring during bloodstream infection treatment, which is particularly helpful in patients with disseminated infections (Savilampi et al., ClinicalTrials.gov). It may be interesting to investigate correlations between the pathogen load (copies µl−1) and the severity of sepsis as well. Finally, in this study, total DNA was extracted in the presence of either lysozyme or lyticase. Reason for this separated step of DNA extraction was the presence of Gram‐negative bacterial DNA in the lyticase formulation. Hence, combining lysozyme and lyticase results in false‐positive outcomes for Gram‐negative bacteria. It would be interesting to investigate whether adding DNAse to a mixture of lysozyme and lyticase prevents this problem. This will likely result in faster, more efficient and less expensive analyses.
In conclusion, this study shows that the ddPCR technique has considerable potential and that it is able to detect pathogen DNA in whole blood within a relative short time span of 4 h. The technique enabled differentiation between Gram‐positive bacteria, Gram‐negative bacteria and fungi, but also discrimination between certain bacteria on genus level. Although our relatively small retrospective cohort showed a reasonable sensitivity and specificity, additional prospective research is mandatory to optimize the ddPCR assay and to determine the role of the ddPCR technique in the clinical setting.
Experimental procedures
Feasibility study
Pathogen samples
Twenty‐six DNA samples of pathogens in various concentrations (range 3.6–424 ng µl−1) were obtained from the Medical Microbiology department of the Radboud university medical centre in Nijmegen, the Netherlands.
DNA isolation
Total DNA was extracted from 200 µl whole blood with or without pathogens using the High Pure PCR Template Preparation Kit, version 20 (Roche Diagnostics GmbH, Mannheim, Germany) in the presence of lysozyme (Sigma‐Aldrich, St Louis, MO, USA) for the detection of bacteria (16S rRNA), or lyticase (Sigma‐Aldrich) for the detection of fungi (28S rRNA). DNA was isolated according to the manufacturer’s protocol, except for an elution step of 100 µl instead of 200 µl DNA. DNA samples with an approximate concentration of 50 ng µl−1 were stored at 4–8°C until further use.
Detection and spiking of pathogen (DNA)
To determine whether it was possible to detect pathogen DNA in water, pathogen DNA (10 ng µl−1) was added to water in a ratio of 1:10–1:100 000. To determine the detection limit of the ddPCR and qPCR, 10‐fold serial dilutions (in duplo) of E. coli and C. albicans were spiked in both 200 µl phosphate‐buffered saline (PBS; for counting) and 200 µl whole blood (for ddPCR and qPCR). For counting, E. coli was dispersed on lysogeny broth (LB) agar plates and colonies were counted the next day. For counting C. albicans, microorganisms were manually counted using a Bürker‐Türk counting chamber.
Primers and probes
Two sets of primers (Sigma‐Aldrich) and probes (Sigma‐Aldrich, or IDT, Coralville, IA, USA) were designed for the detection of bacterial 16S rRNA (including Gram differentiation) and fungal 28S rRNA (Fig. 1). Primers and probes were designed based on the most prevalent BSI‐causing microorganisms at our department of Gastroenterology and Hepatology over the last 40 years, but likely detect additional bacterial and fungal species (Fig. 2 and Table S1). The primers and probes were later tested on a total of 26 available pathogen DNA samples (Fig. 2 and Table S2). The primers used for the amplification of bacterial 16S rRNA were forward PLK1 and reverse PLK2 (Table S3) (Klaschik et al., 2002). Two internal hybridization probes were adapted from Klaschik et al. for Gram‐positive bacteria (GRAM+) and for Gram‐negative bacteria (GRAM−); both probes binding the 16s rRNA template (Klaschik et al., 2002). The 6‐fluorescein amidite (6‐FAM) and hexachloro‐fluorescein amidite (HEX) were covalently bound to the 5′‐end of the GRAM + and GRAM‐ probe, respectively, for simultaneous detection of bacterial species.
Amplification of the fungal 28S rRNA was performed using a forward primer (FUNGIf) and two reverse primers (FUNGIrA and FUNGIrB; 50% each). The internal hybridization probe for fungi was FungiProbe (Table S3). These primers and probes were adapted from Vollmer et al. (2008).
Pathogen DNA detection
The presence of bacterial 16S rRNA or fungal 28S rRNA was detected separately using the QX200 Droplet Digital PCR system (Bio‐Rad, Hercules, CA, USA) according to the manufacturer’s protocol. The ddPCR mastermix had a final volume of 22 µl and contained 1× ddPCR Supermix for Residual DNA Quantification (Bio‐Rad), 900 nM of forward and reverse primer, 250 nM probe, 2 µl (approximately 50 ng µl−1) of isolated DNA and DNase‐free water. In total, 20 µl of the total mixture was used to generate droplets using the QX200 Droplet Generator (Bio‐Rad). Subsequently, the droplet suspension was transferred into a 96‐wells plate and PCR amplification was performed on a C1000 Terminal Cycler (Bio‐Rad) with the following cycling parameters: 95°C for 10 min, 40 cycles (with ramp rate 2°C s−1) of 95°C for 30 s and 61°C for 1 min, and a final step at 98°C for 10 min. The droplets were analysed on the QX200 Droplet Reader (Bio‐Rad), and results were visualized with QuantaSoft software version 1.7.4 (Bio‐Rad). Thresholds were set manually above the negative (signal‐arm) droplets. Droplets above the threshold were determined positive (signal‐rich). A sample was defined positive when a higher number of droplets/concentration (copies µl−1) was found, when compared to background control samples. The latter included pooled whole blood samples of healthy volunteers or water.
ddPCR and qPCR detection limit
A quantitative PCR (qPCR) analysis was performed to compare the detection limit of the ddPCR with qPCR. The reaction mix was produced as described above. A 10‐fold dilution series of E. coli and C. albicans in PBS was performed. Subsequently, the presence of bacterial or fungal DNA was detected using the CFX 96 real‐time PCR detection system and analysed with CFX Manager 3.0 (Bio‐Rad). The final dilution where pathogen DNA could be detected by either the ddPCR or qPCR was considered the detection limit of the PCR reaction.
Sanger sequencing
To verify bacterial 16S rRNA and fungal 28S rRNA, DNA fragments amplified by qPCR were run on an agarose gel and isolated using the QIAEXII Gel Extraction Kit (Qiagen, Hilden, Germany) and sequenced with the BigDye Terminator Kit and ABI3730 capillary sequencer (Perkin‐Elmer Applied Biosystems, Boston, MA, USA) at the department of Genetics of the Radboud university medical centre.
Diagnostic accuracy study
Historical patient cohort
To test the diagnostic accuracy of the ddPCR for detecting BSIs, we analysed historically collected blood samples (between 2008 and 2010) of adult patients suspected of a BSI and compared these samples with blood cultures drawn on the same day combined with retrospectively collected clinical data. All included patients were admitted to our department of Gastroenterology and Hepatology. A BSI was suspected in patients with ≥ 2 SIRS criteria and/or other clinical signs, such as chills and/or hypotension (Bone et al., 1992).
Blood culture and blood sample analyses
In case a patient was suspected of having a BSI, at least two sets of blood cultures (aerobe and anaerobe; BD BACTEC, BD medical, NJ, USA) were routinely collected and transported to the department of Medical Microbiology for cultivation at 37°C for at least 5 days or until positive. Isolates were identified at species level using standard microbiological procedures.
After collecting blood for cultures, 1 ml residual EDTA blood was frozen (−20°C) in a DNA‐free tube (Sarstedt, Nümbrecht, Germany) until further use. DNA was isolated with lysozyme for the 16S rRNA ddPCR reaction, or with lyticase for the 28S rRNA ddPCR reaction. Each sample was measured in quadruple using ddPCR and compared with background– negative and positive controls. Next, the ddPCR results were analysed independently by two research analysts, both of whom were blinded for the diagnosis and treatment of the patients. Finally, their results were compared with the outcomes of the blood cultures. In case the ddPCR detected a blood sample positive for a pathogen, the sample was sequenced and compared with blood culture results to verify identical pathogens.
Patient data collection and outcomes
The following baseline characteristics were collected: gender, age, main underlying disease, presence of diabetes, parenteral nutrition use and infectious symptoms. Main outcomes of the diagnostic accuracy study included sensitivity, specificity, positive– and negative predictive value (PPV, NNV), and positive – and negative likelihood ratio (LR+, LR−) of the ddPCR test.
Ethics statement
The ethics committee of the Radboud university medical centre approved the use of residual whole blood and retrospectively collected data and waived the need for informed consent (reference number 2018‐4582).
Statistical analysis
Baseline characteristics were presented as medians with interquartile ranges. Outcomes, such as sensitivity, specificity, PPV, NNV, LR+, and LR– were descriptive in nature and expressed in percentages with 95% confidence intervals (95% CIs) or ratio’s with 95% CIs. All statistical analyses were performed with SPSS statistical software package version 22.0 (SPSS Inc., Chicago, IL, USA).
Conflict of Interest
None declared.
Supporting information
Fig. S1. ddPCR droplet generation. ddPCR, droplet digital polymerase chain reaction. The ddPCR generates 20000 droplets from a blood sample. PCR amplification is carried out within each of the 20000 droplets. As the amount of pathogen DNA in a single droplet is relatively increased compared to the background of human DNA, the chance for detecting pathogen DNA is increased as well.
Fig. S2. Addition of higher volumes of E. coli DNA to the PCR reaction.
Fig. S3. Overview of all 26 tested pathogens and their probe binding sites with ddPCR.
Fig. S4. Dilution series of DNA from patient #13 and a DNA overload of E. coli and S. marcescens (PC).
Table S1. GenBank numbers of the most prevalent BSI‐causing microorganisms at our department of Gastroenterology and Hepatology.
Table S2. Available ATCC numbers of the 26 tested pathogen DNA samples.
Table S3. Primers and probes.
Table S4. Counting results of E. coli and C. albicans.
Table S5. Patient demographics.
Acknowledgements
We are grateful to Amelieke Cremers and Arjan de Jong for providing the pathogen DNA samples and their assistance.
Microbial Biotechnology (2020) 13(3), 657–668
Funding Information
This study was funded by the Dutch governmental Organization for Health Research and Development (ZonMw; project number 427002002). The funder had no involvement in the study design and production of the protocol, collection of data, analyses and interpretation of data, writing of the report, and the decision to submit the article for publication.
References
- Bio-Rad H, CA, USA . Droplet digital PCR application guide. URL https://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6407.pdf
- Bone, R.C. , Balk, R.A. , Cerra, F.B. , Dellinger, R.P. , Fein, A.M. , Knaus, W.A. , et al. (1992) Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 101: 1644–1655. [DOI] [PubMed] [Google Scholar]
- Carrara, L. , Navarro, F. , Turbau, M. , Seres, M. , Moran, I. , Quintana, I. , et al. (2013) Molecular diagnosis of bloodstream infections with a new dual‐priming oligonucleotide‐based multiplex PCR assay. J Med Microbiol 62(Pt 11): 1673–1679. 10.1099/jmm.0.064758-0 [DOI] [PubMed] [Google Scholar]
- ClinicalTrials.gov . URL https://clinicaltrials.gov/ct2/show/NCT03782454?cond=NCT03782454.
- Coopersmith, C.M. , De Backer, D. , Deutschman, C.S. , Ferrer, R. , Lat, I. , Machado, F.R. , et al. (2018) Surviving sepsis campaign: research priorities for sepsis and septic shock. Intensive Care Med 44: 1400–1426. 10.1007/s00134-018-5175-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimberti, S. , Genuardi, E. , Mazziotta, F. , Iovino, L. , Morabito, F. , Grassi, S. , et al. (2019) The minimal residual disease in Non‐Hodgkin's lymphomas: from the laboratory to the clinical practice. Front Oncol 9: 528 10.3389/fonc.2019.00528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto, M. , and Al‐Hasan, M.N. (2013) Overall burden of bloodstream infection and nosocomial bloodstream infection in North America and Europe. Clin Microbiol Infect 19: 501–509. 10.1111/1469-0691.12195 [DOI] [PubMed] [Google Scholar]
- Horvath, A. , Peto, Z. , Urban, E. , Vágvölgyi, C. , and Somogyvári, F. (2013) A novel, multiplex, real‐time PCR‐based approach for the detection of the commonly occurring pathogenic fungi and bacteria. BMC Microbiol 13: 300 10.1186/1471-2180-13-300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, A.M. , Newton, D. , Kunapuli, A. , Gandhi, T.N. , Washer, L.L. , Isip, J. , et al. (2013) Impact of rapid organism identification via matrix‐assisted laser desorption/ionization time‐of‐flight combined with antimicrobial stewardship team intervention in adult patients with bacteremia and candidemia. Clin Infect Dis 57: 1237–1245. 10.1093/cid/cit498 [DOI] [PubMed] [Google Scholar]
- Hussain, M. , von Eiff, C. , Sinha, B. , Joost, I. , Herrmann, M. , Peters, G. , et al. (2008) eap Gene as novel target for specific identification of Staphylococcus aureus . J Clin Microbiol 46: 470–476. 10.1128/jcm.01425-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain, M. , Fantuzzo, R. , Mercorelli, S. , and Cullen, C. (2016) A direct droplet digital PCR method for quantification of residual DNA in protein drugs produced in yeast cells. J Pharm Biomed Anal 123: 128–131. 10.1016/j.jpba.2016.01.050 [DOI] [PubMed] [Google Scholar]
- Kilgore, M. , and Brossette, S. (2008) Cost of bloodstream infections. Am J Infect Control 36: S172e1–3. 10.1016/j.ajic.2008.10.004 [DOI] [PubMed] [Google Scholar]
- Klaschik, S. , Lehmann, L.E. , Raadts, A. , Book, M. , Hoeft, A. , and Stuber, F. (2002) Real‐time PCR for detection and differentiation of gram‐positive and gram‐negative bacteria. J Clin Microbiol 40: 4304–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knabl, L. , Mutschlechner, W. , and Orth‐Holler, D. (2016) Evaluation of a multiplex OnSpot Primer‐Extension PCR assay in the diagnosis of sepsis. J Microbiol Methods 120: 91–93. 10.1016/j.mimet.2015.12.001 [DOI] [PubMed] [Google Scholar]
- Kullberg, B.J. , and Arendrup, M.C. (2015) Invasive candidiasis. N Engl J Med 373: 1445–1456. 10.1056/NEJMra1315399 [DOI] [PubMed] [Google Scholar]
- Kumar, A. , Roberts, D. , Wood, K.E. , Light, B. , Parrillo, J.E. , Sharma, S. , et al. (2006) Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med 34: 1589–1596. 10.1097/01.Ccm.0000217961.75225.E9 [DOI] [PubMed] [Google Scholar]
- Li, H. , Bai, R. , Zhao, Z. , Tao, L. , Ma, M. , Ji, Z. , et al. (2018) Application of droplet digital PCR to detect the pathogens of infectious diseases. Biosci Rep 38: BSR20181170 10.1042/BSR20181170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesenfeld, O. , Lehman, L. , Hunfeld, K.P. , and Kost, G. (2014) Molecular diagnosis of sepsis: new aspects and recent developments. Eur J Microbiol Immunol 4: 1–25. 10.1556/EuJMI.4.2014.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loonen, A.J. , de Jager, C.P. , Tosserams, J. , Kusters, R. , Hilbink, M. , Wever, P.C. , et al. (2014) Biomarkers and molecular analysis to improve bloodstream infection diagnostics in an emergency care unit. PLoS ONE 9: e87315 10.1371/journal.pone.0087315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makristathis, A. , Riss, S. , and Hirschl, A.M. (2014) A novel fluorescence in situ hybridization test for rapid pathogen identification in positive blood cultures. Clin Microbiol Infect 20: O760–O763. 10.1111/1469-0691.12561 [DOI] [PubMed] [Google Scholar]
- Marco, F. (2017) Molecular methods for septicemia diagnosis. Enferm Infecc Microbiol Clin 35: 586–592. 10.1016/j.eimc.2017.03.002 [DOI] [PubMed] [Google Scholar]
- Martin‐Loeches, I. , Forster, R. , and Prina‐Mello, A. (2018) Intensive care medicine in 2050: nanotechnology. Emerging technologies and approaches and their impact on critical care. Intensive Care Med 44: 1299–1301. 10.1007/s00134-017-5002-y [DOI] [PubMed] [Google Scholar]
- Oellerich, M. , Schutz, E. , Beck, J. , Kanzow, P. , Plowman, P.N. , Weiss, G.J. , and Walson, P.D. (2017) Using circulating cell‐free DNA to monitor personalized cancer therapy. Crit Rev Clin Lab Sci 54: 205–218. 10.1080/10408363.2017.1299683 [DOI] [PubMed] [Google Scholar]
- Opota, O. , Croxatto, A. , Prod'hom, G. , and Greub, G. (2015) Blood culture‐based diagnosis of bacteraemia: state of the art. Clin Microbiol Infect 21: 313–322. 10.1016/j.cmi.2015.01.003 [DOI] [PubMed] [Google Scholar]
- Pfaller, M.A. , Wolk, D.M. , and Lowery, T.J. (2016) T2MR and T2Candida: novel technology for the rapid diagnosis of candidemia and invasive candidiasis. Future Microbiol 11: 103–107. 10.2217/fmb.15.111 [DOI] [PubMed] [Google Scholar]
- Pilecky, M. , Schildberger, A. , Orth‐Holler, D. , and Weber, V. (2018) Pathogen enrichment from human whole blood for the diagnosis of bloodstream infection: Prospects and limitations. Diagn Microbiol Infect Dis 94: 7–14. 10.1016/j.diagmicrobio.2018.11.015 [DOI] [PubMed] [Google Scholar]
- Pironi, L. , Arends, J. , Bozzetti, F. , Cuerda, C. , Gillanders, L. , Jeppesen, P.B. , et al. (2016) ESPEN guidelines on chronic intestinal failure in adults. Clin Nutr 35: 247–307. 10.1016/j.clnu.2016.01.020 [DOI] [PubMed] [Google Scholar]
- Postel, M. , Roosen, A. , Laurent‐Puig, P. , Taly, V. , and Wang‐Renault, S.‐F. (2018) Droplet‐based digital PCR and next generation sequencing for monitoring circulating tumor DNA: a cancer diagnostic perspective. Exp Rev Mol Diagn 18: 7–17. 10.1080/14737159.2018.1400384 [DOI] [PubMed] [Google Scholar]
- Rhee, C. , Dantes, R. , Epstein, L. , Murphy, D.J. , Seymour, C.W. , Iwashyna, T.J. , et al. (2017) Incidence and trends of sepsis in US hospitals using clinical vs claims data, 2009–2014. JAMA 318: 1241–1249. 10.1001/jama.2017.13836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour, C.W. , Gesten, F. , Prescott, H.C. , Friedrich, M.E. , Iwashyna, T.J. , Phillips, G.S. , et al. (2017) Time to treatment and mortality during mandated emergency care for sepsis. N Engl J Med 376: 2235–2244. 10.1056/NEJMoa1703058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, N. , Tan, Y. , Zhang, L. , Wei, L. , Qing, G. , Ming‐zhe, Y. et al. (2018) Detection of circulating Mycobacterium tuberculosis‐specific DNA by droplet digital PCR for vaccine evaluation in challenged monkeys and TB diagnosis. Emerg Microb Infect 7: 78 10.1038/s41426-018-0076-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson, M. , Pandor, A. , Martyn‐St James, M. , Rachid, R. , Lesley, U. , John, S. , et al. (2016) Sepsis: the LightCycler SeptiFast Test MGRADE(R), SepsiTest and IRIDICA BAC BSI assay for rapidly identifying bloodstream bacteria and fungi ‐ a systematic review and economic evaluation. Health Technol Assess (Winchester, England) 20: 1–246. 10.3310/hta20460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, C. , Chen, X. , Wang, F. , Wang, D. , Cao, Z. , Zhu, X. , et al. (2019) A multiplex droplet digital PCR assay for non‐invasive prenatal testing of fetal aneuploidies. The Analyst 144: 2239–2247. 10.1039/c8an02018c [DOI] [PubMed] [Google Scholar]
- Tissari, P. , Zumla, A. , Tarkka, E. , Mero, S. , Savolainen, L. , Vaara, M. , et al. (2010) Accurate and rapid identification of bacterial species from positive blood cultures with a DNA‐based microarray platform: an observational study. Lancet (London, England) 375: 224–2230. 10.1016/s0140-6736(09)61569-5 [DOI] [PubMed] [Google Scholar]
- Vollmer, T. , Stormer, M. , Kleesiek, K. , and Dreier, J. (2008) Evaluation of novel broad‐range real‐time PCR assay for rapid detection of human pathogenic fungi in various clinical specimens. J Clin Microbiol 46: 1919–1926. 10.1128/jcm.02178-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Yan, W. , Fu, S. , Wang, Y. , Xu, J. , and Ye, C. (2018) Multiple cross displacement amplification coupled with nanoparticles‐based lateral flow biosensor for detection of Staphylococcus aureus and identification of methicillin‐resistant S. aureus . Front Microbiol 9: 907 10.3389/fmicb.2018.00907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Bergelson, S. , and Feschenko, M. (2018) Determination of lentiviral infectious titer by a novel droplet digital PCR method. Hum Gene Ther Meth 29: 96–103. 10.1089/hgtb.2017.198 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Cooper, R. , Bergelson, S. , and Feschenko, M. (2018) Quantification of residual BHK DNA by a novel droplet digital PCR technology. J Pharm Biomed Anal 159: 477–482. 10.1016/j.jpba.2018.07.022 [DOI] [PubMed] [Google Scholar]
- Wellinghausen, N. , Kochem, A.J. , Disque, C. , Muhl, H. , Gebert, S. , Winter, J. , and Sakka, S.G. (2009) Diagnosis of bacteremia in whole‐blood samples by use of a commercial universal 16S rRNA gene‐based PCR and sequence analysis. J Clin Microbiol 47: 2759–2765. 10.1128/jcm.00567-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y.D. , Chen, L.H. , Wu, X.J. , Shang, S.‐q. , Lou, J.‐t. , Du, L.‐z. , and Zhao, Z.‐y. (2008) Gram stain‐specific‐probe‐based real‐time PCR for diagnosis and discrimination of bacterial neonatal sepsis. J Clin Microbiol 46: 2613–2619. 10.1128/jcm.02237-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, S. , Lin, S. , Kelen, G.D. , Quinn, T. C. , Dick, J.D. , Gaydos, C.A. , and Rothman, R.E. (2002) Quantitative multiprobe PCR assay for simultaneous detection and identification to species level of bacterial pathogens. J Clin Microbiol 40: 3449–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Han, X. , Liu, A. , Bai, X. , Xu, C. , Bao, F. , et al. (2017) Use of digital droplet PCR to detect Mycobacterium tuberculosis DNA in whole blood‐derived DNA samples from patients with pulmonary and extrapulmonary tuberculosis. Front Cell Infect Microbiol 7: 369 10.3389/fcimb.2017.00369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Gong, X. , He, Y. , Huang, L. , Zhang, Q. , Liu, Y. et al. (2018) [Non‐invasive prenatal diagnosis for beta‐thalassemia by detecting paternal CD41‐42 mutation in cell‐free DNA derived from maternal plasma with droplet digital PCR]. Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi. Chin J Med Genet 35: 787–790. 10.3760/cma.j.issn.1003-9406.2018.06.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. ddPCR droplet generation. ddPCR, droplet digital polymerase chain reaction. The ddPCR generates 20000 droplets from a blood sample. PCR amplification is carried out within each of the 20000 droplets. As the amount of pathogen DNA in a single droplet is relatively increased compared to the background of human DNA, the chance for detecting pathogen DNA is increased as well.
Fig. S2. Addition of higher volumes of E. coli DNA to the PCR reaction.
Fig. S3. Overview of all 26 tested pathogens and their probe binding sites with ddPCR.
Fig. S4. Dilution series of DNA from patient #13 and a DNA overload of E. coli and S. marcescens (PC).
Table S1. GenBank numbers of the most prevalent BSI‐causing microorganisms at our department of Gastroenterology and Hepatology.
Table S2. Available ATCC numbers of the 26 tested pathogen DNA samples.
Table S3. Primers and probes.
Table S4. Counting results of E. coli and C. albicans.
Table S5. Patient demographics.