

Abstract
The angiotensin converting enzyme 2 (ACE2) has been identified as a receptor for the severe acute respiratory syndrome associated coronavirus (SARS-CoV). Here we show that ACE2 expression on cell lines correlates with susceptibility to SARS-CoV S-driven infection, suggesting that ACE2 is a major receptor for SARS-CoV. The soluble ectodomain of ACE2 specifically abrogated S-mediated infection and might therefore be exploited for the generation of inhibitors. Deletion of a major portion of the cytoplasmic domain of ACE2 had no effect on S-driven infection, indicating that this domain is not important for receptor function. Our results point to a central role of ACE2 in SARS-CoV infection and suggest a minor contribution of the cytoplasmic domain to receptor function.
Keywords: Severe acute respiratory syndrome, Coronavirus, Angiotensin converting enzyme 2
A novel coronavirus (CoV) has been identified as the etiologic agent of severe acute respiratory syndrome (SARS) [1], [2], [3]. The SARS associated CoV (SARS-CoV) exhibits a genome organization similar to those of known CoVs [4], [5], [6], however, sequence analysis indicates that SARS-CoV does not cluster with known coronaviruses, but either constitutes a new branch [4], [5] or a subgroup of class II CoVs [7], [8]. Recent reports demonstrated that the spike protein (S) of SARS-CoV is sufficient to drive infection of cells and can be recognized by neutralizing antibodies in SARS patients [9], [10], [11]. Because of its essential role in viral entry, SARS-CoV S is an attractive target for vaccines and antiviral compounds. Indeed, immunization of mice with a S-based DNA vaccine triggered humoral and cellular immune responses and protected animals from SARS-CoV infection [12]. Moreover, a humanized monoclonal antibody specific for the S1 subunit of SARS-CoV S has been shown to potently inhibit SARS-CoV infection and could be employed for first line therapy [13].
Coronavirus infection of target cells depends on interactions of S with cellular receptors, such as CD13 for human CoV 229E and carcinoembryonic antigens (CEACAMs) for mouse hepatitis virus (MHV) [14]. Li et al. [15] identified the angiotensin converting enzyme 2 (ACE2) as a receptor for SARS-CoV. ACE2 expression on non-permissive cells confers susceptibility to SARS-CoV infection [11], indicating that ACE2 is sufficient to allow viral entry. The receptor-binding domain in SARS-CoV S has been mapped to amino acids 318–510, and efforts to identify regions in ACE2 contacted by S are underway [13], [16], [17], [18], [19]. However, it is unclear if ACE2 is the only receptor for SARS-CoV or if entry into certain tissues and organs like liver and kidney is facilitated by other receptors. Moreover, it is unknown whether the ACE2 ectodomain can be used to modulate S-driven infection; if so, the development of inhibitors mimicking the S-interaction domain in ACE2 might be justified. Finally, it needs to be determined if SARS-CoV S-binding to ACE2 triggers intracellular signaling events essential for infection. Here, we analyzed the interaction between S and ACE2 employing lentiviral particles bearing the SARS-CoV S-protein. These particles, so called pseudotypes, infect target cells in a S-dependent manner and can be used to analyze S-function under standard biosafety conditions [9], [10], [11].
Materials and methods
RT-PCR analysis of ACE2 mRNA expression. Total RNA was isolated from 1 × 106 cells using the RNeasy kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany). Five hundred nanograms of total RNA was treated with DNaseI followed by reverse transcription using oligo(dT) oligonucleotides and the AMV reverse transcriptase (Invitrogen). The resulting cDNAs were subjected to either amplification with oligonucleotides specific for GAPDH (GAPDH-5: 5′-ATGGGGAAGGTGAAGGTCGG-3′; GAPDH-3: 5′-ATACTTCTCATGGTTCACAC-3′) or ACE2 (ACE2-5: 5′-GTAAGGCCACTGCTCAACTAC-3′; ACE2-3: 5′-TTTTTCTAAAAGGAGGTCTGAACATC-3′). For detection of ACE2 transcripts, a nested PCR was performed using 5 μl of the first round PCR product as template and oligonucleotides ACE2nested-5 (5′-GATGGAGTACCGACTGGAGTCC-3′) and ACE2nested-3 (5′-CTAATATCGATGGAGGCATAAGG-3′) for amplification. As a control, nested PCRs were performed using the input RNA prior to reverse transcription.
Plasmid construction and mutagenesis. Eukaryotic expression vectors for ACE2 were constructed by reverse transcription of total RNA derived from human (hu) 293T kidney cells and African green monkey (agm) Vero E6 cells using the SuperScript One-Step RT-PCR system (Invitrogen) and either oligonucleotides huACE2-5 (5′-CCCGGTACCCACCATGTCAAGCTCTTCCTGGCTCCTTCTC-3′) and huACE2-3 (5′-GGGCTCGAGTTTTTCTAAAAGGAGGTCTGAACATC-3′) or oligonucleotides agmACE2 (5′-CCCGGTACCCACCATGTCCAGCTCCTCCTGGCTCCTTCTC-3′) and agmACE2-3 (5′-GGGCTCGAGCTAAAAGGAAGTCTGAGCATCATCA-3′). The resulting amplificates were inserted into the pcDNA3.1 vector via KpnI and XhoI. Carboxy-terminal ACE2 deletion mutants were constructed by PCR using the huACE2-pcDNA3.1 construct as template and the huACE2-5 as 5′-oligonucleotide in combination with either ACE2-771 (5′-GAGCTCGAGTTATTCTCCTTTGCTAATATCGATGG-3′), ACE2-775 (5′-GAGCTCGAGTTAAGGATTTTCTCCACTTCTTGC-3′), ACE2-779 (5′-CAGCTCGAGTCTTGCTTTATTTTTCTTCTTCCG-3′) or ACE2-790 (5′-CAGCTCGAGTTATTTCTTCTTCCGATCTCTGATCC-3′) and cloned into the pcDNA3.1 vector via KpnI and XhoI. To obtain an expression plasmid encoding soluble ACE2, the huACE2 cDNA was amplified using the huACE2-5′ primer in combination with oligonucleotide ACE2sol-3 (5′-GGGCTCGAGTTAGGAAACAGGGGGCTGGTTAGGAG-3′) followed by insertion into pcDNA3.1 via KpnI and XhoI.
Cell culture, infection, and reporter assays. The lymphatic cell lines C8166 and BL41 were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum. 293T, Vero E6, HOS, Hep-2, and NIH3T3 cells were maintained in Dulbecco’s minimal essential medium (Gibco/BRL, Eggenstein, Germany) supplemented with 10% fetal calf serum; Huh-7 cells additionally received 1% amino acid cocktail (Invitrogen, San Diego, CA).
HIV-based pseudotypes were produced as described [9]. Briefly, the pNL4-3env*nef*-luc plasmid [20] was co-expressed in 293T cells together with an expression vector for the SARS-S protein or control envelope proteins (VSV-G, MLV). The supernatant was used for infection of target cells followed by determination of luciferase activity 72 h post infection using a commercially available kit according to the manufacturer’s protocol (Promega, Madison, WI).
Antibodies, Western blotting, and FACS analysis. 293T cells were transiently transfected with expression vectors encoding ACE2 and ACE2 deletion variants. After 48 h, cells were lysed in RIPA buffer (0.1% SDS, 1% NP40, 1% deoxycholate, 0.05 M Tris, pH 7.3, 0.15 M NaCl, and 150 mM PMSF) and separated by 10% PAGE followed by transfer to nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany). ACE2-proteins were detected using a monoclonal ACE2 antibody (R&D systems, Minneapolis). For FACS analysis, surface expression of ACE2 on transfected 293T cells was detected using a polyclonal ACE2 antibody (R&D systems, Minneapolis) in combination with a FITC-labeled secondary antibody.
Inhibition of S-mediated entry into target cells by soluble ACE2. Soluble ACE2 protein was harvested from the supernatant of transiently transfected 293T cells followed by concentration of the protein using a CentriconPlus ultrafilter with a 30 kDa molecular weight cut-off (Millipore, Eschborn, Germany). For inhibition of S-mediated infection, S- or control pseudotypes standardized for equal luciferase activity upon infection of target cells were pre-incubated with different dilutions of soluble ACE2 for 1 h at 37 °C followed by infection of 293T target cells. Luciferase activity was determined in cell extracts after 72 h.
Results and discussion
ACE2 expression correlates with permissiveness to SARS-CoV S-driven infection
SARS-CoV engages ACE2 for entry into permissive Vero E6 cells [15]; however, it is unclear if SARS-CoV can use alternative receptors for infection of other target cells. Therefore, we investigated if ACE2 expression correlates with permissiveness to SARS-CoV S-mediated infection. To this end, total RNA from a panel of cell lines of known permissiveness to SARS-CoV S-driven infection was isolated, reverse transcribed, and the ACE2 sequence was amplified by nested PCR (Fig. 1A ). As a control, the GAPDH cDNA was also amplified. In a first round of PCR employing outer primers, GAPDH expression was readily detected in cDNA from all cell lines tested, whereas no signal for ACE2 was observed. In a second round of PCR with inner primers specific for ACE2, amplificates were obtained using cDNA from Huh-7, Hep-2, Vero E6, and 293T cells, which are susceptible to S-driven infection [1], [2], [9], [10]. In contrast, ACE2 sequences were not amplified from cDNA derived from HeLa, HOS, C8166, BL41, and NIH3T3 cell lines, which are largely refractory to S-mediated infection [9], [10], [11]. Thus, ACE2 expression in cell lines correlates with susceptibility to SARS-CoV S-driven infection, indicating that ACE2 plays an important role in SARS-CoV replication.
Fig. 1.
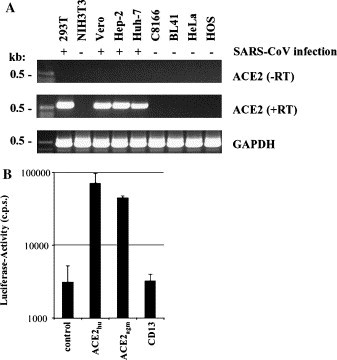
(A) Expression of ACE2 in cell culture cell lines susceptible (+) or refractory (−) to SARS-CoV infection. Total RNA was isolated from the indicated cell lines followed by reverse transcription. Subsequently, a nested PCR with ACE2-specific oligonucleotides was performed using either the resulting cDNAs as templates (middle panel, +RT) or employing the input RNA (upper panel, −RT). As a control, all cDNAs were subjected to a PCR with GAPDH-specific oligonucleotides (lower panel). (B) Enhanced SARS-CoV S-mediated entry into 293T cells transiently over-expressing ACE2. ACE2 of human (hu) and African green monkey (agm) origin or human CD13 were transiently expressed in 293T cells followed by infection with SARS-CoV S-pseudotypes carrying a luciferase reporter gene. After 72 h, cells were lysed and luciferase activity was determined in the cell extracts. Each experiment was performed in quadruplicate and repeated at least three times with independent virus stocks.
We next analyzed if high amounts of ACE2 can augment infection of permissive cells by lentiviral pseudotypes harboring SARS-CoV S as described previously [9], [10], [11]. Plasmids encoding ACE2 and CD13, the receptor for human CoV 229E [14], were transiently transfected into 293T cells followed by infection with pseudotypes harboring S. Both ACE2 and CD13 were efficiently expressed on the cell surface (Fig. 3B and data not shown); however, only expression of ACE2 but not of CD13 enhanced S-driven infection compared to infection of control cells, confirming the specific interaction of SARS-CoV S with ACE2 (Fig. 1B).
Fig. 3.
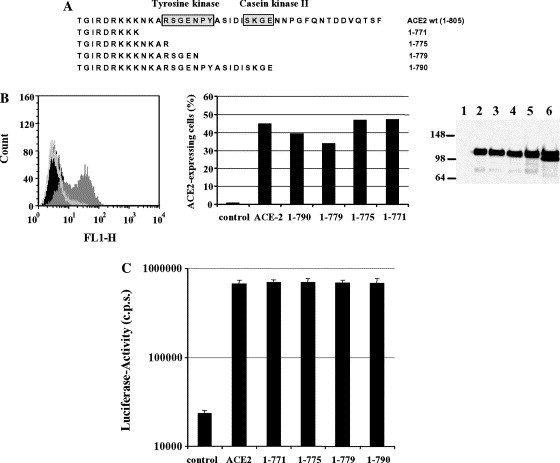
Analysis of the contribution of the ACE2 cytoplasmic domain to receptor function. (A) Schematic overview depicting the C-terminal ACE2 mutants analyzed. Putative tyrosine and casein kinase motifs are boxed. (B) Surface expression of ACE2 and the ACE2 deletion mutants. ACE2 was transiently expressed in 293T cells and analyzed by FACS using a polyclonal ACE2 antiserum followed by incubation with a polyclonal FITC-labeled secondary antibody (left panel, dark grey). As controls, pcDNA3-transfected cells were incubated with the secondary antibody (black line) or with both the ACE2-specific antiserum in combination with the secondary antibody (light grey). Similarly, the indicated ACE2 deletion mutants were subjected to FACS analysis; the percentage of ACE2 expressing cells is shown (middle panel). In parallel, expression of wild type ACE2 (lane 2) and all ACE2 mutants was examined by Western blot analysis (right panel: lane 1, pcDNA3; lane 3, mutant 1–790; lane 4, mutant 1–779; lane 5, mutant 1–775; and lane 6, mutant 1–771). (C) Role of the cytoplasmic domain within ACE2 for SARS-CoV S-mediated infection of target cells. ACE2 and the indicated deletion mutants were transiently expressed in 293T cells followed by infection with S-pseudotypes carrying luciferase as reporter gene. After 72 h, the luciferase activity was determined. Each experiment was performed in quadruplicate and repeated at least three times with independent virus preparations.
The soluble ACE2 ectodomain inhibits SARS-CoV S-driven infection
Next, we investigated if pre-incubation of SARS-CoV S-bearing pseudotypes with the soluble ACE2 ectodomain modulates viral infectivity. We anticipated that pre-incubation of virions with soluble ACE2 might block binding of SARS-CoV S to membrane bound ACE2, since it has been shown that soluble ACE2 blocks the attachment of a S-immunoglobulin fusion protein to permissive Vero E6 cells [15]. Alternatively, pre-coating of virions with soluble receptor could activate the fusogenic activity in SARS-CoV S and facilitate infection of target cells. To obtain soluble ACE2, a stop codon was introduced at position 740 of the ACE2 coding sequence, resulting in a truncated reading frame encoding the entire ectodomain as previously described [21]. Transfection of 293T cells with the plasmid encoding soluble ACE2 resulted in efficient release of the protein into the supernatant, while no soluble protein was detected in supernatants of cells transfected with wild type ACE2 (Fig. 2A ). The supernatant containing soluble ACE2 was concentrated by filtration through an ultrafilter with a 30 kDa molecular weight cut-off (CentriconPLUS80) and employed for inhibition studies (Fig. 2B). Viral pseudotypes bearing SARS-CoV S or VSV-G were standardized for comparable production of luciferase upon infection of target cells, incubated with the indicated dilutions of supernatant containing soluble ACE2, and used for infection of 293T cells. The soluble ACE2 containing supernatant blocked SARS-CoV S-driven infection in a dose-dependent manner, but did not appreciably modulate VSV-G- and MLV-glycoprotein-mediated infection (Fig. 2B and data not shown), indicating that the ACE2 ectodomain and possibly portions thereof can be used to specifically block SARS-CoV S-driven infection.
Fig. 2.
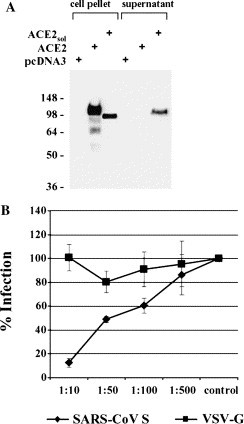
Expression of soluble ACE2 protein and inhibition of SARS-CoV S-driven infection. (A) Expression of the soluble ACE2 ectodomain. Either a pcDNA3 control vector (lane 1), wild type ACE2 (lane 2) or an ACE2 variant comprising only the ectodomain (lane 3) was transiently expressed in 293T cells. After 48 h, cells and culture supernatants (lanes 4–6) were harvested and analyzed for ACE2 expression via Western blot. (B) Inhibition of S-mediated entry into 293T cells by soluble ACE2. S-bearing pseudotypes and VSV-G pseudotypes normalized for equal luciferase activity (104 c.p.s.) upon infection of target cells were pre-incubated with the indicated dilutions of concentrated soluble ACE2 and used for infection of 293T cells. Luciferase activity was determined in cell extracts after 72 h. The relative luciferase units obtained after infection in the absence of soluble ACE2 was set as 100%. Each experiment was performed in quadruplicate and repeated three times; similar results were obtained with a different soluble ACE2 preparation and with independent virus stocks.
Consensus kinase motifs in the ACE2 cytoplasmic domain are not required for receptor function
The ACE2 cytoplasmic domain harbors consensus sites for tyrosine kinases and casein kinase II [21], and an interaction of this domain with cellular proteins might be important for receptor function. The contribution of the ACE2 cytoplasmic domain to receptor function was assessed by analyzing ACE2 variants in which portions of this domain were deleted. The largest deletion removed the kinase consensus sites and reduced the tail length from 42 to 9 amino acids (Fig. 3A ). All ACE2 variants were expressed to comparable degrees on the surface and in lysates of transiently transfected 293T cells (Fig. 3B) and efficiently enhanced S-driven infection (Fig. 3C), indicating that amino acids 771–805 in the ACE2 cytoplasmic tail are dispensable for enhancement of SARS-CoV S-mediated infection of 293T cells.
ACE2: a promising target and tool for antiviral therapy
The identification of ACE2 as a receptor for SARS-CoV greatly advanced the understanding of viral cell tropism and opened new opportunities for therapeutic intervention. Thus, the development of compounds that block binding of SARS-CoV S to ACE2 without inhibiting the natural function of ACE2 is now a desirable goal. For these strategies to be successful, however, it needs to be determined if ACE2 is the only receptor for SARS-CoV. This is especially important in the light of recent findings demonstrating that SARS-CoV cell tropism is broader than initially appreciated [9], [10]. Here, we analyzed several cell lines of known permissiveness to SARS-CoV S-mediated infection for expression of ACE2 messenger RNA (mRNA). Expression of ACE2 mRNA correlated with permissiveness to infection for all cell lines examined, indicating that ACE2 plays a central role in SARS-CoV infection. Nevertheless, it cannot be excluded that alternative receptors promote viral entry into certain cell types and tissues.
Evidence obtained with other viruses indicates that incubation with soluble receptors can modulate infection. For example, a tetrameric form of the HIV receptor CD4 potently abrogates HIV-1 infection, and this effect is recapitulated to some degree by a 27 amino acid comprising CD4 mimicking synthetic peptide [22], [23]. In our hands, the soluble ACE2 ectodomain specifically blocked infection by SARS-CoV S-bearing pseudotypes. This validates the receptor-binding site in S as a therapeutic target, although the development of therapeutics based on the ACE2 ectodomain is going to be challenging. Nevertheless, it will be interesting to examine if smaller portions of the ACE2 ectodomain exhibit inhibitory activity. Finally, the solution of the structure of S bound to ACE2 must provide the basis for the rational design of compounds that target residues in S contacted by ACE2.
Binding of SARS-CoV S to ACE2 could induce intracellular signaling which might be required for receptor function. Indeed, the cytoplasmic domain of ACE2 contains consensus sites for kinases, suggesting an interaction of this domain with cellular proteins involved in signal transduction. Moreover, the ACE2 cytoplasmic domain could be involved in internalization, which seems to be required for S-driven infection [9], [10]. However, large deletions in the ACE2 cytoplasmic tail did not appreciably modulate expression and receptor function, at least in the context of 293T cells over-expressing receptor (Fig. 3C). These data argue against an important role of the ACE2 cytoplasmic domain in mediating SARS-CoV S-driven infection, albeit different cellular backgrounds need to be examined. Moreover, it cannot be excluded that the minimal sequences still remaining in the cytoplasmic tail of the ACE2 deletion-variants examined play a role in signaling or internalization. However, the introduction of larger deletions abrogated surface but not intracellular expression (data not shown) of the receptor, suggesting that sequences between amino acids 766 and 771 are required for the insertion of ACE2 into the plasma membrane.
In summary, our results argue for a crucial role of ACE2 in SARS-CoV S infection and underline that the interaction of SARS-CoV S with ACE2 is an attractive target for therapeutic intervention.
Acknowledgements
We thank R. Kammerer for reagents, G. Simmons for sharing unpublished results, and B. Fleckenstein for constant encouragement and support. This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB466 to H.H., M.G., A.M., M.K., T.G., and S.P.; Fe140/5-2 to G.H.F.) and the Wilhelm Sander Foundation (1996.047.3 to G.H.F.).
References
- 1.Drosten C, Gunther S, Preiser W, van der W.S, Brodt H.R, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier R.A, Berger A, Burguiere A.M, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra J.C, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk H.D, Osterhaus A.D, Schmitz H, Doerr H.W. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Ksiazek T.G, Erdman D, Goldsmith C.S, Zaki S.R, Peret T, Emery S, Tong S, Urbani C, Comer J.A, Lim W, Rollin P.E, Dowell S.F, Ling A.E, Humphrey C.D, Shieh W.J, Guarner J, Paddock C.D, Rota P, Fields B, DeRisi J, Yang J.Y, Cox N, Hughes J.M, LeDuc J.W, Bellini W.J, Anderson L.J. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 3.Peiris J.S, Lai S.T, Poon L.L, Guan Y, Yam L.Y, Lim W, Nicholls J, Yee W.K, Yan W.W, Cheung M.T, Cheng V.C, Chan K.H, Tsang D.N, Yung R.W, Ng T.K, Yuen K.Y. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marra M.A, Jones S.J, Astell C.R, Holt R.A, Brooks-Wilson A, Butterfield Y.S, Khattra J, Asano J.K, Barber S.A, Chan S.Y, Cloutier A, Coughlin S.M, Freeman D, Girn N, Griffith O.L, Leach S.R, Mayo M, McDonald H, Montgomery S.B, Pandoh P.K, Petrescu A.S, Robertson A.G, Schein J.E, Siddiqui A, Smailus D.E, Stott J.M, Yang G.S, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth T.F, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples G.A, Tyler S, Vogrig R, Ward D, Watson B, Brunham R.C, Krajden M, Petric M, Skowronski D.M, Upton C, Roper R.L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 5.Rota P.A, Oberste M.S, Monroe S.S, Nix W.A, Campagnoli R, Icenogle J.P, Penaranda S, Bankamp B, Maher K, Chen M.H, Tong S, Tamin A, Lowe L, Frace M, DeRisi J.L, Chen Q, Wang D, Erdman D.D, Peret T.C, Burns C, Ksiazek T.G, Rollin P.E, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus A.D, Drosten C, Pallansch M.A, Anderson L.J, Bellini W.J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 6.Stadler K, Masignani V, Eickmann M, Becker S, Abrignani S, Klenk H.D, Rappuoli R. Nat. Rev. Microbiol. 2003;1:209–218. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eickmann M, Becker S, Klenk H.D, Doerr H.W, Stadler K, Censini S, Guidotti S, Masignani V, Scarselli M, Mora M, Donati C, Han J.H, Song H.C, Abrignani S, Covacci A, Rappuoli R. Science. 2003;302:1504–1505. doi: 10.1126/science.302.5650.1504b. [DOI] [PubMed] [Google Scholar]
- 8.Snijder E.J, Bredenbeek P.J, Dobbe J.C, Thiel V, Ziebuhr J, Poon L.L, Guan Y, Rozanov M, Spaan W.J, Gorbalenya A.E. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofmann H, Hattermann K, Marzi A, Gramberg T, Geier M, Krumbiegel M, Kuate S, Überla K, Niedrig M, Pöhlmann S. The S protein of severe acute respiratory syndrome (SARS) associated coronavirus mediates entry into hepatoma cell lines and is targeted by neutralizing antibodies in infected patients. J. Virol. 2004;78:6134–6142. doi: 10.1128/JVI.78.12.6134-6142.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simmons G, Reeves J.D, Rennekamp A.J, Amberg S.M, Piefer A.J, Bates P. Proc. Natl. Acad. Sci. USA. 2004;101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang P, Chen J, Zheng A, Nie Y, Shi X, Wang W, Wang G, Luo M, Liu H, Tan L, Song X, Wang Z, Yin X, Qu X, Wang X, Qing T, Ding M, Deng H. Biochem. Biophys. Res. Commun. 2004;315:439–444. doi: 10.1016/j.bbrc.2004.01.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Z.Y, Kong W.P, Huang Y, Roberts A, Murphy B.R, Subbarao K, Nabel G.J. Nature. 2004;428:561–564. doi: 10.1038/nature02463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sui J, Li W, Murakami A, Tamin A, Matthews L.J, Wong S.K, Moore M.J, Tallarico A.S, Olurinde M, Choe H, Anderson L.J, Bellini W.J, Farzan M, Marasco W.A. Proc. Natl. Acad. Sci. USA. 2004;101:2536–2541. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gallagher T.M, Buchmeier M.J. Virology. 2001;279:371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li W, Moore M.J, Vasilieva N, Sui J, Wong S.K, Berne M.A, Somasundaran M, Sullivan J.L, Luzuriaga K, Greenough T.C, Choe H, Farzan M. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Babcock G.J, Esshaki D.J, Thomas W.D, Jr., Ambrosino D.M. J. Virol. 2004;78:4552–4560. doi: 10.1128/JVI.78.9.4552-4560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prabakaran P, Xiao X, Dimitrov D.S. Biochem. Biophys. Res. Commun. 2004;314:235–241. doi: 10.1016/j.bbrc.2003.12.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong S.K, Li W, Moore M.J, Choe H, Farzan M. J. Biol. Chem. 2004;279:3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao X, Chakraborti S, Dimitrov A.S, Gramatikoff K, Dimitrov D.S. Biochem. Biophys. Res. Commun. 2003;312:1159–1164. doi: 10.1016/j.bbrc.2003.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Connor R.I, Chen B.K, Choe S, Landau N.R. Virology. 1995;206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- 21.Tipnis S.R, Hooper N.M, Hyde R, Karran E, Christie G, Turner A.J. J. Biol. Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 22.Martin L, Stricher F, Misse D, Sironi F, Pugniere M, Barthe P, Prado-Gotor R, Freulon I, Magne X, Roumestand C, Menez A, Lusso P, Veas F, Vita C. Nat. Biotechnol. 2003;21:71–76. doi: 10.1038/nbt768. [DOI] [PubMed] [Google Scholar]
- 23.Pohlmann S, Doms R.W. Curr. Drug Targets Infect. Disord. 2002;2:9–16. doi: 10.2174/1568005024605864. [DOI] [PubMed] [Google Scholar]