

Graphical abstract
Keywords: IKKɛ, Inflammation, Cancer, NF-κB, IRF3
Abstract
The innate immune system forms our first line of defense against invading pathogens and relies for a major part on the activation of two transcription factors, NF-κB and IRF3. Signaling pathways that activate these transcription factors are intertwined at the level of the canonical IκB kinases (IKKα, IKKβ) and non-canonical IKK-related kinases (IKKɛ, TBK1). Recently, significant progress has been made in understanding the function and mechanism of action of IKKɛ in immune signaling. In addition, IKKɛ impacts on cell proliferation and transformation, and is thereby also classified as an oncogene. Studies with IKKɛ knockout mice have illustrated a key role for IKKɛ in inflammatory and metabolic diseases. In this review we will highlight the mechanisms by which IKKɛ impacts on signaling pathways involved in disease development and discuss its potential as a novel therapeutic target.
1. Introduction to canonical versus non-canonical IκB kinases (IKK)
Our first line of defense against viral and bacterial attacks relies on the innate immune system, which detects non-self products by means of specific surface, endosomal or cytosolic receptors. For example, bacterial lipopolysaccharide (LPS) is sensed by Toll-like receptor (TLR)-4 on the cell surface, whereas viral RNA is sensed by endosomal TLR3 or cytosolic retinoid acid-inducible gene (RIG-I) [1]. A major output from these receptors is the activation of transcription factors belonging to the nuclear factor-kappa B (NF-κB) and interferon (IFN) regulatory factor (IRF) family, which control the expression of multiple immune regulatory genes. In this review we will focus on p50/p65 NF-κB and IRF3, whose activation involves specific members of the inhibitor of κB (IκB) kinase (IKK) family. The canonical IKKs, IKKα and IKKβ, form a complex with the adaptor protein NEMO (also known as IKKγ), which has a regulatory role. This IKK complex is required for proper NF-κB signaling in response to multiple proinflammatory stimuli such as TNF and TLR ligands. IKKα and IKKβ are Ser/Thr kinases that phosphorylate the NF-κB inhibitor protein IκBα, resulting in its Lys48-linked polyubiquitination and subsequent proteasomal degradation. This allows NF-κB to translocate to the nucleus and bind to specific DNA elements [2]. IKKα and IKKβ contain an N-terminal catalytic kinase domain (KD), a more central leucine zipper (LZ) and helix loop helix (HLH) domain, and a C-terminal NEMO-binding domain (NBD) (Fig. 1 ). The major impact of the IKKs on NF-κB signaling inspired many researchers to search intensively for IKK-related kinases. Based on sequence similarities with IKKα and IKKβ, two IKK-related kinases, TANK binding kinase 1 (TBK1) and IKKɛ (also known as IKK-inducible or IKK-i), were discovered. IKKɛ and TBK1 are known as the non-canonical IKKs and expand the IKK family to four members [3], [4]. The kinase domain of IKKɛ shares 33% and 31% amino acid identity with the corresponding domains in IKKα and IKKβ, respectively, and 67% with TBK1 [5]. Furthermore, TBK1 and IKKɛ have a similar domain composition as the canonical IKKs. IKKβ and the non-canonical IKKs also share an ubiquitin-like domain (ULD), which is required for optimal kinase activity [6]. Importantly, IKKɛ and TBK1 lack a NBD and do not interact with NEMO, but each forms similar complexes with other specific scaffolding proteins (see Section 4).
Fig. 1.

Domain structure of the IKK family.
The IKK family can be divided in two groups: the classical or canonical IKKs (IKKα and IKKβ) and the non-canonical IKKs (TBK1 and IKKɛ). In addition, two splice variants of IKKɛ (IKKɛ-sv1 and IKKɛ-sv2), respectively missing 25 and 13 amino acids at their C-terminal end, have been described. The following domains are depicted: kinase domain (KD), leucine zipper (LZ), helix-loop-helix (HLH), NEMO-binding domain (NBD), ubiquitin-like domain (ULD). Numbers indicate kinase-activating phosphorylation sites.
Although IKKɛ and TBK1 were originally classified as IKKs based on their ability to phosphorylate IκBα upon overexpression, studies with IKKɛ and TBK1 deficient cells revealed that these kinases are dispensable for IκBα phosphorylation [7], [8]. Instead, both kinases were shown to contribute to LPS- and virus-induced phosphorylation of IRF3 and IRF7, allowing their homodimerization, nuclear import, and activation of type I IFN genes (IFN-α and IFN-β) [9]. IKKɛ and TBK1 are similar in their ability to activate IRF3 and IRF7 and their ability to phosphorylate the IκBα inhibitor of NF-κB, but they present some differences that may be of importance. For instance, deletion of the TBK1 gene leads to embryonic lethality at day 15 due to TNF-induced apoptosis in the liver [7], whereas IKKɛ deficient mice are viable [9]. The reason for these different phenotypes is still unclear but may in part reflect the differential expression and use of TBK1 and IKKɛ in various cell types (see also section 2). Moreover, TBK1 and IKKɛ may have nonredundant functions in other signaling pathways than those controlling IRF and canonical NF-κB activity. For example, recently published work demonstrated a specific role for TBK1 as a negative regulator of non-canonical NF-κB signaling in B cells stimulated with BAFF or anti-CD40, which was associated with the inducible TBK1 mediated phosphorylation of the kinase NIK, leading to its degradation [10]. Although B cells express both TBK1 and IKKɛ, NIK phosphorylation was highly specific for TBK1. Since TBK1 and IKKɛ share the same substrate consensus phosphorylation motif [11], substrate specificity is likely driven by recruitment of TBK1 or IKKɛ to discrete signaling complexes, possibly involving specific scaffolding proteins (see Section 4). Given the ever-growing number of TBK1 and IKKɛ interaction partners and substrates [12], localized kinase activation and substrate modification could be an effective strategy to confer TBK1 or IKKɛ specific functions.
2. Expression of IKKɛ and TBK1
While IKKα, IKKβ and TBK1 are constitutively expressed in most cell types, basal IKKɛ expression is only observed in specific tissues (pancreas, thymus and spleen) and cell types (T-cells and peripheral blood leukocytes). However, in other cell types (e.g. fibroblasts) IKKɛ is rapidly upregulated by cytokines (e.g. TNF, IL-1, IL-6, IFN-γ), microbial products (e.g. LPS, viral RNA), and phorbol esters (PMA), and has therefore also been called inducible IKK or IKK-i [12], [13]. The promoter of the IKKɛ gene contains two putative NF-κB binding sites, of which only one was shown to be functionally active [14], as well as seven STAT3-binding sites, of which two are active [15]. Whether the increase in IKKɛ expression induces kinase activation is currently still unclear. Recently, two IKKɛ splice variants, missing 25 amino acids (IKKɛ-sv1) or 13 amino acids (IKKɛ-sv2) at their C-terminal end, were detected in human peripheral blood mononuclear cells [16]. While these splice variants are ubiquitously expressed at the mRNA level, both protein isoforms are selectively upregulated by TNF stimulation or virus infection. IKKɛ is frequently overexpressed in a number of human cancers, in particular breast, ovarian and pancreatic cancer and has been implicated in tumorigenesis (see Section 6). Although IKKɛ and TBK1 show many overlapping activities that are relevant to innate immunity and inflammation, the more restricted expression of IKKɛ is of particular interest. In this review we will therefore focus on IKKɛ and mainly refer to TBK1 for reasons of comparison.
3. Role of IKKɛ/TBK1 in NF-κB and IRF signaling
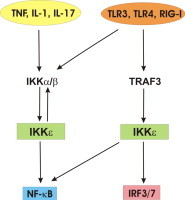
The identification of TBK1 and IKKɛ as IκBα kinases has always been very controversial and the source of many debates. Evidence is based primarily on the observation that overexpression of IKKɛ or TBK1 in cultured cells leads to phosphorylation of IκBα, be it only at one phosphoacceptor site (Ser36 or Ser32, respectively), driving increased IκBα turnover [13]. However, both IKKɛ and TBK1 deficient MEF cells display normal IκBα degradation in response to TNF, IL-1 or LPS, contradicting the overexpression data [7], [8]. Nevertheless, TNF-, IL-1- or LPS-induced NF-κB dependent gene expression is abrogated in the absence of IKKɛ [7], [8]. Therefore it is believed that IKKɛ influences NF-κB signaling and concomitant gene expression downstream of IκBα [12] (Fig. 2 ). In this context, several studies support a role for IKKɛ-mediated Ser468 and Ser536 phosphorylation of the p65 NF-κB subunit in the expression of a specific subset of NF-κB target genes in response to pro-inflammatory signals and viral infection [12], [17], [18], [19]. It has also been shown that activated p65 (phosphorylated on Ser468 or Ser536) serves as a docking site to bring IKKɛ-enzymatic activity to κB-containing inflammatory gene promoters in the nucleus, enabling IKKɛ to phosphorylate adjacent c-Jun and initiate nuclear receptor corepressor clearance [20]. These results suggest that p65-recruitment of IKKɛ to promoters that exhibit AP1 and κB sites in close proximity (e.g. inos, cxcl2, cxcl9, cxcl10, ccl4, tnfaip3) may regulate their activation by initiating corepressor turnover. Besides p65, IKKɛ/TBK1 also targets c-Rel NF-κB, leading to its nuclear accumulation and activation of NF-κB dependent gene expression [21].
Fig. 2.
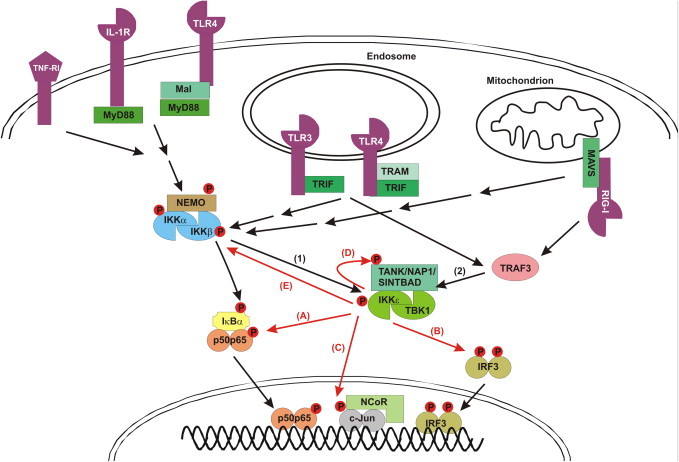
IKKɛ mediated signaling to NF-κB and IRF3 in response to specific receptors.
Non-canonical IKKs (IKKɛ and TBK1) can be activated by two signaling pathways: (1) IKKα/β mediated activation of IKKɛ/TBK1. Ligand binding to several receptors (TNF-R1, IL-1R, TLR4, RIG-I) initiates the recruitment of specific adaptor proteins (e.g. Mal, MyD88, TRAM, TRIF, MAVS), E3 ubiquitin ligases and kinases (not shown) to the receptor, eventually resulting in the activation of the canonical IKK (IKKα/IKKβ/NEMO) complex. This leads to the IKKβ-mediated phosphorylation and subsequent Lys48-linked polyubiquitination of IκBα, resulting in its proteasomal degradation and release of the p50–p65 NF-κB heterodimer, which then translocates to the nucleus. (2) IKKα/IKKβ-independent and TRAF3-dependent IKKɛ/TBK1 autoactivation. TRIF-dependent TLR3 and TLR4 signaling, as well as MAVS-dependent RIG-I signaling, induce IKKɛ/TBK1 autoactivation via TRAF3. This also requires the binding of IKKɛ/TBK1 to different scaffold proteins (TANK, NAP1, SINTBAD). IKKɛ/TBK1 then mediate different activities: (A) IKKɛ/TBK1 can phosphorylate NF-κB (p65), contributing to NF-κB dependent expression of specific genes. (B) IKKɛ/TBK1 can phosphorylate IRF3 (and IRF7; not shown), leading to its homodimerization and nuclear translocation. (C) IKKɛ/TBK1 can phosphorylate c-jun, leading to the release of the nuclear repressor complex (NCoR). (D) IKKɛ/TBK1 can phosphorylate their own scaffold proteins (TANK, NAP1 or SINTBAD), the function of which is still unclear. (E) IKKɛ/TBK1 is also able to phosphorylate the canonical IKKs, leading to their inactivation.
Next to their role in NF-κB signaling, TBK1 and IKKɛ are also implicated in IRF3 and IRF7 signaling in response to viral infection that is sensed by a diversity of receptors, such as TLR3 and RIG-I, leading to the production of type I IFNs [12] (Fig. 2). While IRF3 is constitutively expressed in the cytoplasm of many cells, IRF7 needs to be transcriptionally upregulated. Both IKKɛ and TBK1 directly phosphorylate IRF3 and IRF7 at their C-termini, resulting in their homo- and heterodimerization and translocation to the nucleus. Mass spectrometry pinpointed Ser386, Ser396 and Ser402 in IRF3 as redundant phosphorylation sites for IKKɛ upon viral infection of innate immune cells [22]. IKKɛ deficient MEF cells showed no change in IRF3 activation [9], whereas TBK1 deficient MEF cells showed reduced IRF3 activation upon TLR3 and TLR4 triggering [9], [23], [24]. However, IRF3 activation was completely abolished in TBK1 and IKKɛ double deficient MEFs [9]. IKKɛ overexpression, but not its kinase dead mutant, could restore IRF3 activation in TBK1 deficient cells, suggesting redundancy between both kinases [24]. Although IKKɛ and TBK1 seem to exert overlapping functions in IRF3 activation, mechanistic differences in the activation of TBK1 and IKKɛ following virus infection have been observed (e.g. mitochondrial localization of IKKɛ versus cytoplasmic localization of TBK1) [25]. Moreover, partially different substrates have been reported for both kinases. It is interesting to note that the TLR adaptor protein MyD88 was recently shown to abrogate TLR3-induced IFN production by preventing IKKɛ but not TBK1-mediated IRF3 phosphorylation [25], indicating that both kinases can also be differentially regulated.
4. Mechanism and regulation of IKKɛ/TBK1 activation
Although IKKɛ/TBK1 can be activated by several inflammatory stimuli, their activation by TLR3, TLR4 and RIG-I receptors has been best documented [26], [27]. Receptor stimulation induces the recruitment of specific adaptor proteins to the receptor (TRIF for TLR3 and TLR4, MAVS for RIG-I). Subsequently, these adaptors interact with the E3 ubiquitin ligase TRAF3 to activate IRF3, or with the E3 ubiquitin ligase TRAF6 and RIP1 kinase to activate NF-κB. Autoubiquitination of TRAF3 is needed for the recruitment and activation of IKKɛ and TBK1 [28]. At later time points, the presence of IKKɛ in the MAVS complex eventually leads to the release of TRAF3, with the shutdown of IFN signaling as a consequence [29].
At the protein level, IKKɛ (and TBK1) activity is regulated by phosphorylation at a single site (Ser172) in its activation loop [30], in contrast to IKKα and IKKβ, which require phosphorylation at two sites (IKKα: Ser176 and Ser180; IKKβ: Ser177 and Ser181) for their function. Although overexpression studies indicated a role for autophosphorylation of IKKɛ/TBK1 at Ser172 [13], [31], activation of the endogenous kinases does not only involve autophosphorylation. Indeed, cells treated with the TBK1 and IKKɛ inhibitor BX795 still showed phosphorylation of TBK1 and IKKɛ at Ser172 in response to poly(I:C), LPS, TNF and IL-1 [32], suggesting the involvement of other kinases. Clark and co-workers could show that Ser172 phosphorylation of IKKɛ/TBK1 is impaired in IKKα deficient cells treated with an IKKɛ inhibitor [31], indicating IKKα as a potential IKKɛ kinase. In line with these observations, TNF-induced activation of IKKɛ/TBK1 seems to be mediated solely by the canonical IKKs [31]. Interestingly, IKKɛ/TBK1 activation by the canonical IKK pathway only results in NF-κB activation and not IRF3 activation.
Similar to the canonical kinases IKKα and IKKβ, which require the formation of a complex with the adaptor protein NEMO, also TBK1 and IKKɛ activation involves specific adaptor proteins. So far, three scaffold proteins have been shown to bind IKKɛ/TBK1 and to promote IKKɛ/TBK1-mediated phosphorylation of IRF3 and IRF7: NAK associated protein 1 (NAP1), TNF receptor-associated factor (TRAF) family member-associated NF-κB activator (TANK) and similar to NAP and TBK1 adaptor (SINTBAD) [30], [33], [34], [35]. Therefore, it is an attractive idea that different scaffold proteins might assemble distinct TBK1 and IKKɛ complexes under distinct conditions, providing signaling specificity. Whether or not these scaffold proteins assemble hetero- or homodimers of IKKɛ and TBK1 is currently still unknown.
In contrast to the positive regulation of NF-κB dependent gene expression by IKKɛ/TBK1, the latter have also been described to negatively regulate NF-κB activation in response to TNF or IL-1 stimulation. More specifically, IKKɛ/TBK1 was shown to directly phosphorylate the canonical IKKs outside their activation loop, restricting their activity [31] (Fig. 2). Interestingly, this negative regulation of the canonical IKKs by IKKɛ/TBK1 was impaired in TANK-deficient macrophages [36]. Using these cells it was also shown that TANK is required for IKKɛ/TBK1 and NF-κB activation in response to LPS via both the MyD88- and TRIF-dependent pathways, and mediates the interaction of the non-canonical IKKs with the canonical IKKs. The latter most likely involves the binding of TANK to the canonical IKK adaptor protein NEMO [37]. Together, these findings demonstrate a key role for the IKKɛ/TBK1 adaptor protein TANK in enabling the canonical and non-canonical IKKs to regulate each other.
In analogy to NEMO, one can expect that the mechanisms driving IKKɛ/TBK1 activation most likely require different posttranslational modifications of their scaffold proteins. Indeed, IKKɛ/TBK1 dependent phosphorylation and Lys63-linked polyubiquitination of TANK have already been described in response to LPS stimulation [33], but their significance is still unclear.
Multiple negative regulatory mechanisms are in place to avoid excessive IKKɛ/TBK1 activity and IFN production. For example, the deubiquitinase DUBA reverses TRAF3 ubiquitination, disconnecting TRAF3 from its substrates TBK1 and IKKɛ [38]. Also the deubiquitinase CYLD negatively regulates RIG-I induced interferon production by deubiquitinating RIG-I and TBK1/IKKɛ [39], [40]. Conversely, IKKɛ directly phosphorylates CYLD at Ser418, hereby decreasing its deubiquitinating potential but increasing the IKKɛ-induced cell transformation [41] (Fig. 3 ) (see Section 6). Others have shown that the ubiquitin-editing enzyme A20, in conjunction with Tax1 binding protein 1 (TAX1BP1), antagonizes Lys63-polyubiquitination of TBK1 and IKKɛ. Surprisingly, A20-mediated inhibition of TBK1/IKKɛ Lys63-polyubiquitination was independent of its DUB function [42]. Recently, the E3 ubiquitin ligases TRIP and DTX4 have been shown to negatively regulate IRF3 activation by inducing TBK1 Lys48-polyubiquitination and proteosomal degradation [43], [44]. DTX4 showed weak or no activity against IKKɛ, while the activity of TRIP against IKKɛ was not studied. Interestingly, also many viruses themselves interfere with the IRF pathway and antiviral IFN production at the level of IKKɛ and/or TBK1. For example, specific viral components disturb the interaction of IKKɛ/TBK1 with other signaling proteins (e.g. Ebola virus VP35 protein [45], M protein of Corona virus [46], Hepatitis B or C virus [47], [48]). Alternatively, viral proteins may act as alternative substrates for IKKɛ and TBK1, thereby targeting these kinases for degradation (e.g. Borna disease virus P protein, paramyxovirus V proteins [49], [50]). These virus adaptations further emphasize the importance of the non-canonical kinases in antiviral signaling.
Fig. 3.
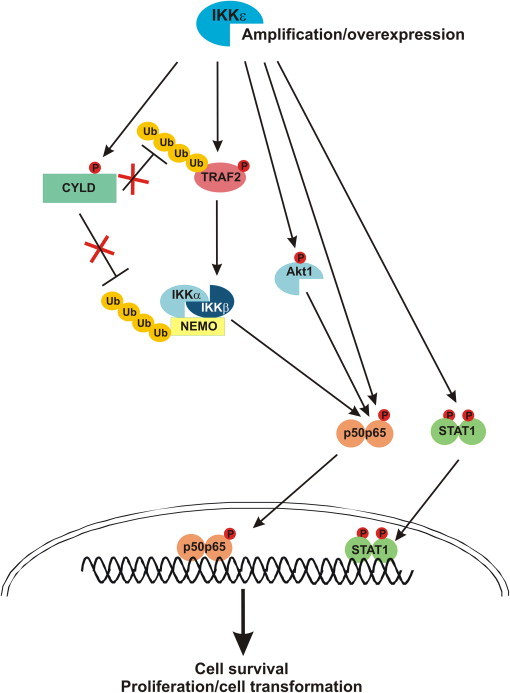
Mechanisms contributing to the oncogenic potential of IKKɛ.
Overexpression of IKKɛ in tumor cells induces cell survival, cell transformation and proliferation by different mechanisms involving IKKɛ mediated phosphorylation of specific substrates. IKKɛ can either directly or indirectly (via Akt phosphorylation and activation) phosphorylate NF-κB (p65), leading to increased NF-κB dependent gene expression. IKKɛ also phosphorylates and inactivates the tumor suppressor CYLD, preventing CYLD from deubiquitinating specific substrates in the NF-κB signaling pathway. In addition, phosphorylation of TRAF2 activates its E3 ubiquitin ligase activity. Both CYLD and TRAF2 phosphorylation thus increase ubiquitin-dependent NF-κB signaling. IKKɛ also directly phosphorylates STAT1, increasing its gene activating potential.
5. Role of IKKɛ in inflammatory and metabolic diseases
IKKɛ activity has been linked to the pathology of inflammatory diseases such as rheumatoid arthritis (RA) [51], [52], [53]. For instance, IKKɛ is constitutively expressed and phosphorylated in synovial intimal lining of RA patients, resulting in uncontrolled IRF3-driven production of proinflammatory mediators such as IFN-β, matrix metalloproteinases and chemokines [52]. Further supporting a major contribution of IKKɛ to the pathogenesis of RA is the finding that mice deficient in IKKɛ show less synovial inflammation in a passive K/BxN arthritis model due to lower expression of inflammatory mediators [51]. Furthermore, IKKɛ single nucleotide polymorphisms have been associated with the early stages of RA [54], and genome wide associated studies revealed IKKɛ as a susceptibility locus for systemic lupus erythematosus (SLE), in which type I IFNs play a crucial role [55]. Computational analysis of predicted protein-protein interactions also pinpointed IKKɛ as a potential therapeutic target in psoriasis [56].
IKKɛ has also been implicated in pulmonary inflammation. In this context, IKKɛ deficient mice showed fewer infiltrating neutrophils and diminished expression of proinflammatory cytokines and chemokines after intranasal administration of IL-17. Surprisingly, in this model IKKɛ deficiency did not affect NF-κB activation, but did reduce MAP kinase signaling [57]. More specifically, IKKɛ was shown to be responsible for phosphorylation of the IL-17 receptor adaptor Act-1, thereby activating the TRAF2/TRAF5 pathway leading to increased MAP kinase activation and leaving the TRAF6/NF-κB axis emanating from Act-1 undisturbed. Increased MAP kinase activation led to increased chemokine mRNA stability, contributing to increased chemokine production and neutrophil infiltration into the lungs. A role for IKKɛ in the regulation of IL-17 responses is also illustrated by the recent finding that IKKɛ can promote the AKT-mTOR signaling pathway, which mediates IL-1 induced Th17 maintenance, by phosphorylating and inactivating GSK3α [58]. Taken together, many studies indicate an important role of IKKɛ in the pathophysiology of inflammatory diseases, mainly by regulating NF-κB, IFN and IL-17 responses.
IKKɛ might also be a potential target to treat inflammatory pain. In inflammatory pain models (hind paw inflammation evoked by injection of zymosan or formalin), IKKɛ-deficient mice exhibited a significantly reduced nociceptive behavior in comparison with wild type mice, indicating that IKKɛ contributes to the development of inflammatory hyperalgesia [59]. At the same time, the provoked NF-κB activation in nociceptive neurons (neurons involved in perception of pain) was reduced, as reflected by lower inflammatory gene expression. Of interest, this process is independent of type I IFN responses, suggesting that IKKɛ is promoting inflammatory hyperalgesia merely by activating the NF-κB pathway.
Finally, IKKɛ has been reported as an important link between inflammation and obesity. Obese mice have major risks to develop inflammatory or metabolic diseases such as type 2 diabetes [60]. In fact, obesity mimics a permanent low-grade inflammatory condition due to dietary fatty acid recognition by TLR4 [61] or hypoxia [62]. Elevated NF-κB activity in obese mice drives increased IKKɛ expression in the liver, adipocytes and adipose tissue macrophages [63], [64]. Furthermore, mice deficient in IKKɛ were found to be protected from high fat diet-induced obesity and showed less chronic liver inflammation, hepatic steatosis and insulin resistance. These events are regulated by changes in the expression of regulatory proteins and enzymes that are involved in glucose and lipid metabolism, and by a decrease in the production of proinflammatory cytokines and proteins involved in insulin resistance. Transfection of cultured adipocytes and hepatoma cells with IKKɛ induced similar changes, suggesting a direct role for IKKɛ in the regulation of hepatic inflammation. However, contradictory results were reported by another group, showing no difference between IKKɛ knockout and wild type mice in a model of high fat diet-induced obesity and insulin resistance [65]. Therefore, more work is needed to define the precise role of IKKɛ in the development of metabolic diseases.
6. Role of IKKɛ in cancer
IKKɛ has been associated with the initiation and progression of multiple cancers and might function as an oncogene for malignant transformation. For instance, several breast cancer cell lines and ∼30% of primary human breast tumors express high levels of IKKɛ [66]. Increased IKKɛ expression is due to an up till now unknown mutation regulating IKKɛ transcript levels or to an amplification of the 1q32 region comprising the IKBKE locus [67], [68]. Ectopic expression of IKKɛ in immortalized mammary epithelial cells at levels found in human cancer cells renders them tumorigenic, confirming that the allele amplified in breast cancer specimens is transforming [67]. High IKKɛ expression is often associated with the accumulation of c-Rel and p65 NF-κB subunits in the nucleus (e.g. in primary breast tumors), and IKKɛ silencing in several breast cancer cell lines was shown to reduce NF-κB activation and cell proliferation [69]. Similarly, introducing a kinase inactive IKKɛ mutant (IKKɛ K38A) in breast cancer cells, which exerts a dominant negative effect on endogenous IKKɛ, reduced NF-κB dependent gene expression (e.g. cyclin D1 and RelB) [70], demonstrating the necessity of IKKɛ catalytic activity. Recently, this was further confirmed by demonstrating the requirement of IKKɛ-mediated phosphorylation at Ser418 of the tumor suppressor CYLD, which prevents its deubiquitinating activity on NF-κB signaling proteins such as TRAF2 and NEMO [41], [71] (Fig. 3). Moreover, IKKɛ also activates the E3 ubiquitin ligase TRAF2 by direct phosphorylation at Ser11, resulting in increased TRAF2 ubiquitination [71]. Together these events lead to enhanced NF-κB activation, which promotes survival, transformation and proliferation of mammary epithelial cells (Fig. 3). Furthermore, IKKɛ (as well as TBK1) may contribute to enhanced NF-κB activity and tumorigenesis by directly phosphorylating NF-κB p65 (as described above) or by phosphorylating Akt, which then phosphorylates and activates p65 [72], [73]. In addition, elevated IKKɛ levels are also associated with STAT1 activation in different primary tumors and cell lines derived from a diversity of cancers, like lung and breast carcinoma [15], [74], [75], which may also contribute to the oncogenic activities of IKKɛ. As IRF3 suppression did not affect IKKɛ-induced cell transformation, the oncogenic potential of IKKɛ seems to be independent of its IRF3 signaling function [67].
More recently, human ovarian cancer cell lines and primary tumors were also shown to have elevated levels and activity of IKKɛ, which was associated with a lower overall survival rate [76], [77]. Furthermore, alterations of IKKɛ were associated with late-stage and high-grade tumors, suggesting a role of IKKɛ in ovarian tumor progression rather than in tumor initiation. Finally, IKKɛ was also described as an oncogene in prostate and oesophageal squamous cell carcinoma with increased levels of different NF-κB family members [78], [79], [80], [81], and in clear cell renal cell carcinoma [82].
High levels of IKKɛ coincide with resistance to well established chemotherapeutics [83]. This is also the case for elevated NF-κB activation, which is known to contribute to cancer cell survival and to reduce sensitivity to chemotherapeutic agents and ionizing radiation [84]. Unfortunately, breast cancer cells in which IKKɛ was inactivated retained their resistance to chemotherapeutics despite a lowered NF-κB activation, suggesting that additional mechanisms must be involved in the regulation of cell death [69], [77], [85]. Finally, IKKɛ also accumulates in subnuclear promyelocytic leukemia (PML) bodies upon genotoxic stress, where it undergoes SUMOylation, leading to its activation and ultimately resulting in NF-κB mediated anti-apoptotic responses [86]. In this respect, it is worthwhile to mention that several glioma cell lines and human primary glioma tissues exhibit elevated levels of IKKɛ and are less sensitive to DNA damage-induced apoptosis [87]. Suppression of IKKɛ, however, renders the cells more sensitive, confirming its prosurvival function.
7. Development and characterization of IKKɛ inhibitors
The role of IKKɛ in the development of inflammatory and metabolic diseases, as well as cancer, indicate the potential of IKKɛ as a therapeutic target. So far, only few small molecule inhibitors of IKKɛ have been described. BX-795 was the first IKKɛ inhibitor on the market and suppresses TBK1 and IKKɛ activity at nanomolar concentrations in vitro [32]. The specificity of BX-795 is however an issue since it was originally developed as an inhibitor of 3-phosphoinositide-dependent protein kinase 1 (PDK1) and only later found to also inhibit IKKɛ [88]. Moreover, BX-795 shows off-target effects towards several other kinases, including JNK and p38 MAP kinases [31], [32]. A modified version of BX-795, MRT67307, no longer inhibits JNK or p38 MAP kinases, but still interferes with the activity of TBK1 and PDK1 [31], [32]. More recently, a series of azabenzimidole derivatives and 2,4-diamino-5-cyclopropyl pyrimidines with improved kinase selectivity and drug-like properties were described [88], [89]. However, these compounds still inhibit both IKKɛ and TBK1, with a slightly higher potency against TBK1. In mice, the pyrimidines significantly inhibited LPS-induced release of IFN-β, although toxicity was observed at higher doses [88], [89]. It is worth mentioning that also a number of naturally occurring compounds (polyphenoles such as (−)-epigallocatechin-3-gallate, lutolin, quercetin) with anti-inflammatory properties have been shown to target TBK1 [90], [91]. Activities against IKKɛ have not been described, but in general the specificity of these polyphenoles is low and most of them have multiple targets.
The development of more specific and better IKKɛ inhibitors may be enabled by the recent elucidation of the substrate specificity of IKKɛ and TBK1 [11]. These studies demonstrated that the consensus phosphorylation motif of IKKɛ differs from that of IKKβ, but is identical to that of TBK1, suggesting that the development of IKKɛ inhibitors that do not target TBK1 may be very difficult. Nevertheless, high-throughput screening using a specific IKKɛ/TBK1 substrate peptide resulted in several lead molecules that showed selectivity for either IKKɛ or TBK1 [11]. However, as IKKɛ and TBK1 show many overlapping functions in oncogenic and inflammatory signaling pathways, it is likely that the therapeutic effectiveness of IKKɛ specific inhibitors may be hampered by the redundant activity of TBK1. Therefore, at least in some cases inhibition of the activity of both kinases may be a better therapeutic approach. Finally, as overexpression of IKKɛ is linked with tumorigenesis, therapeutic approaches that would reduce IKKɛ expression to normal levels may be a valid alternative. Therefore, more fundamental research on the molecular mechanisms that regulate IKKɛ expression may further boost the development of IKKɛ targeting drugs.
8. Future perspectives
The potential value of IKKɛ as therapeutic target for anti-inflammatory or anti-cancer therapies requires further investigation into the mechanisms and pathways involved. The pharmaceutical industry has been very active in the development of IKKα/IKKβ inhibitors as novel anti-inflammatory agents, but so far with limited success. Since NF-κB mediates a number of physiological functions, non-selective and complete inhibition of the NF-κB pathway may lead to serious side-effects. This is also illustrated by the fact that p65-, IKKα-, IKKβ-, or NEMO-deficient mice die during embryonic development or perinatally. Compounds that more selectively repress the activation of NF-κB in response to specific receptors or the expression of only a specific subset of NF-κB dependent genes would be associated with fewer side effects. Therefore, IKKɛ could well be such a target with great clinical value. This is supported by the fact that IKKɛ knockout mice, in contrast to IKKα or IKKβ knockout mice, are viable and fertile. Since TBK1 knockout mice also die embryonically, the challenge will be to develop IKKɛ specific inhibitors that do not target TBK1. In this context, it will also be important to further determine the relative contribution of IKKɛ and TBK1 in different pathways (e.g. NF-κB versus IRF) and pathophysiological processes. Modulation of IKKɛ or TBK1 expression and activity in distinct cell types or tissues by means of conditional knockout mice will therefore be of high value. In addition, knowledge of the different IKKɛ substrates and the physiological relevance of their phosphorylation may help to predict efficiency or side effects of IKKɛ inhibitors.
The relationship between the canonical and non-canonical IKKs, and other signaling pathways, is also an open line of investigation. In particular the negative regulatory effect of IKKɛ/TBK1 on the canonical IKKs could suggest that inhibition of IKKɛ/TBK1 may actually have a pro-inflammatory effect. In this context, a more detailed understanding of the molecular mechanisms involved in the counter-regulation in the IKK family could be helpful. Many of the players that are involved (e.g. TANK, NEMO) in the connection between IKK and IKK-related kinases are phosphorylated or modified by different types of ubiquitin chains, but the consequence of these modifications is not fully clear. IKKɛ is also believed to form distinct complexes with different adaptor proteins (TANK, NAP1, SINTBAD), which could perform different functions. Future studies on the existence and function of IKKɛ complexes consisting of distinct IKKɛ adaptor proteins are therefore of high interest as it may allow more selective IKKɛ targeting. Such studies may also help to explain why some stimuli (e.g. TNF, IL-1, IL-17) activate IKKɛ/TBK1 without inducing the phosphorylation of IRF3, whereas other stimuli (e.g. LPS, viral RNA) do activate this pathway via IKKɛ/TBK1. The strong inducibility of IKKɛ expression and its overexpression in multiple tumors is also worthwhile to examine. What are the upstream signals that can regulate IKKɛ expression? What determines IKKɛ stability? Is IKKɛ overexpression sufficient to be oncogenic? In this context, generating (e.g. mammary) tissue specific IKKɛ transgenic mice may be very informative.
In summary, the generation and exploitation of IKKɛ specific inhibitors and conditional knockout/knockin/transgenic mice of IKKɛ and its regulators or substrates is likely to give answers to many of the above mentioned questions. Eventually, this could lead to the use of IKKɛ specific inhibitors for the successful treatment of autoimmunity, obesity, diabetes and certain cancers.
Acknowledgements
Research in the authors’ lab is supported by grants from the ‘Interuniversity Attraction Poles (IAP-VII, contract P7/32)’, the Fund for Scientific Research (FWO)-Flanders (grants G0619.10, G0089.10, 3G023611, G028712N, G046612N, G016413N, G027413N), the ‘Foundation Against Cancer’, the ‘Strategic Basic Research’ programme of the IWT, and the ‘Hercules’, ‘GOA’, and ‘Group-ID MRP’ initiatives of Ghent University. LV holds a FWO postdoctoral fellowship.
References
- 1.Olive C. Pattern recognition receptors: sentinels in innate immunity and targets of new vaccine adjuvants. Expert Rev Vaccines. 2012;11:237–256. doi: 10.1586/erv.11.189. [DOI] [PubMed] [Google Scholar]
- 2.Gilmore T.D., Wolenski F.S. NF-kappaB: where did it come from and why. Immunol Rev. 2012;246:14–35. doi: 10.1111/j.1600-065X.2012.01096.x. [DOI] [PubMed] [Google Scholar]
- 3.Peters R.T., Liao S.M., Maniatis T. IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol Cell. 2000;5:513–522. doi: 10.1016/s1097-2765(00)80445-1. [DOI] [PubMed] [Google Scholar]
- 4.Tojima Y., Fujimoto A., Delhase M., Chen Y., Hatakeyama S., Nakayama K. NAK is an IkappaB kinase-activating kinase. Nature. 2000;404:778–782. doi: 10.1038/35008109. [DOI] [PubMed] [Google Scholar]
- 5.Chien Y., Kim S., Bumeister R., Loo Y.M., Kwon S.W., Johnson C.L. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127:157–170. doi: 10.1016/j.cell.2006.08.034. [DOI] [PubMed] [Google Scholar]
- 6.Ikeda F., Hecker C.M., Rozenknop A., Nordmeier R.D., Rogov V., Hofmann K. Involvement of the ubiquitin-like domain of TBK1/IKK-i kinases in regulation of IFN-inducible genes. EMBO J. 2007;26:3451–3462. doi: 10.1038/sj.emboj.7601773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonnard M., Mirtsos C., Suzuki S., Graham K., Huang J., Ng M. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 2000;19:4976–4985. doi: 10.1093/emboj/19.18.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kravchenko V.V., Mathison J.C., Schwamborn K., Mercurio F., Ulevitch R.J. IKKi/IKKepsilon plays a key role in integrating signals induced by pro-inflammatory stimuli. J Biol Chem. 2003;278:26612–26619. doi: 10.1074/jbc.M303001200. [DOI] [PubMed] [Google Scholar]
- 9.Hemmi H., Takeuchi O., Sato S., Yamamoto M., Kaisho T., Sanjo H. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med. 2004;199:1641–1650. doi: 10.1084/jem.20040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin J., Xiao Y., Chang J.H., Yu J., Hu H., Starr R. The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-kappaB signaling. Nat Immunol. 2012;13:1101–1109. doi: 10.1038/ni.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hutti J.E., Porter M.A., Cheely A.W., Cantley L.C., Wang X., Kireev D. Development of a high-throughput assay for identifying inhibitors of TBK1 and IKKepsilon. PLoS ONE. 2012;7:e41494. doi: 10.1371/journal.pone.0041494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clement J.F., Meloche S., Servant M.J. The IKK-related kinases: from innate immunity to oncogenesis. Cell Res. 2008;18:889–899. doi: 10.1038/cr.2008.273. [DOI] [PubMed] [Google Scholar]
- 13.Shimada T., Kawai T., Takeda K., Matsumoto M., Inoue J., Tatsumi Y. IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IkappaB kinases. Int Immunol. 1999;11:1357–1362. doi: 10.1093/intimm/11.8.1357. [DOI] [PubMed] [Google Scholar]
- 14.Wang N., Ahmed S., Haqqi T.M. Genomic structure and functional characterization of the promoter region of human IkappaB kinase-related kinase IKKi/IKKvarepsilon gene. Gene. 2005;353:118–133. doi: 10.1016/j.gene.2005.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo J., Kim D., Gao J., Kurtyka C., Chen H., Yu C. IKBKE is induced by STAT3 and tobacco carcinogen and determines chemosensitivity in non-small cell lung cancer. Oncogene. 2012 doi: 10.1038/onc.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Koop A., Lepenies I., Braum O., Davarnia P., Scherer G., Fickenscher H. Novel splice variants of human IKKepsilon negatively regulate IKKepsilon-induced IRF3 and NF-kB activation. Eur J Immunol. 2011;41:224–234. doi: 10.1002/eji.201040814. [DOI] [PubMed] [Google Scholar]
- 17.Bao X., Indukuri H., Liu T., Liao S.L., Tian B., Brasier A.R. IKKepsilon modulates RSV-induced NF-kappaB-dependent gene transcription. Virology. 2010;408:224–231. doi: 10.1016/j.virol.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geng H., Wittwer T., Dittrich-Breiholz O., Kracht M., Schmitz M.L. Phosphorylation of NF-kappaB p65 at Ser468 controls its COMMD1-dependent ubiquitination and target gene-specific proteasomal elimination. EMBO Rep. 2009;10:381–386. doi: 10.1038/embor.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreno R., Sobotzik J.M., Schultz C., Schmitz M.L. Specification of the NF-kappaB transcriptional response by p65 phosphorylation and TNF-induced nuclear translocation of IKK epsilon. Nucleic Acids Res. 2010;38:6029–6044. doi: 10.1093/nar/gkq439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang W., Ghisletti S., Perissi V., Rosenfeld M.G., Glass C.K. Transcriptional integration of TLR2 and TLR4 signaling at the NCoR derepression checkpoint. Mol Cell. 2009;35:48–57. doi: 10.1016/j.molcel.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris J., Oliere S., Sharma S., Sun Q., Lin R., Hiscott J. Nuclear accumulation of cRel following C-terminal phosphorylation by TBK1/IKK epsilon. J Immunol. 2006;177:2527–2535. doi: 10.4049/jimmunol.177.4.2527. [DOI] [PubMed] [Google Scholar]
- 22.Fujii K., Nakamura S., Takahashi K., Inagaki F. Systematic characterization by mass spectrometric analysis of phosphorylation sites in IRF-3 regulatory domain activated by IKK-i. J Proteomics. 2010;73:1196–1203. doi: 10.1016/j.jprot.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 23.McWhirter S.M., Fitzgerald K.A., Rosains J., Rowe D.C., Golenbock D.T., Maniatis T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci USA. 2004;101:233–238. doi: 10.1073/pnas.2237236100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perry A.K., Chow E.K., Goodnough J.B., Yeh W.C., Cheng G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J Exp Med. 2004;199:1651–1658. doi: 10.1084/jem.20040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siednienko J., Gajanayake T., Fitzgerald K.A., Moynagh P., Miggin S.M. Absence of MyD88 Results in Enhanced TLR3-Dependent Phosphorylation of IRF3 and Increased IFN-{beta} and RANTES Production. J Immunol. 2011 doi: 10.4049/jimmunol.1003093. [DOI] [PubMed] [Google Scholar]
- 26.Kumar H., Kawai T., Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 27.Maelfait J., Beyaert R. Emerging role of ubiquitination in antiviral RIG-I signaling. Microbiol Mol Biol Rev. 2012;76:33–45. doi: 10.1128/MMBR.05012-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verstrepen L., Verhelst K., Carpentier I., Beyaert R.T.A.X1B.P1. a ubiquitin-binding adaptor protein in innate immunity and beyond. Trends Biochem Sci. 2011;36:347–354. doi: 10.1016/j.tibs.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 29.Paz S., Vilasco M., Werden S.J., Arguello M., Joseph-Pillai D., Zhao T. A functional C-terminal TRAF3-binding site in MAVS participates in positive and negative regulation of the IFN antiviral response. Cell Res. 2011 doi: 10.1038/cr.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chau T.L., Gioia R., Gatot J.S., Patrascu F., Carpentier I., Chapelle J.P. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated. Trends Biochem Sci. 2008;33:171–180. doi: 10.1016/j.tibs.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 31.Clark K., Peggie M., Plater L., Sorcek R.J., Young E.R., Madwed J.B. Novel cross-talk within the IKK family controls innate immunity. Biochem J. 2011;434:93–104. doi: 10.1042/BJ20101701. [DOI] [PubMed] [Google Scholar]
- 32.Clark K., Plater L., Peggie M., Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J Biol Chem. 2009;284:14136–14146. doi: 10.1074/jbc.M109.000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gatot J.S., Gioia R., Chau T.L., Patrascu F., Warnier M., Close P. Lipopolysaccharide-mediated interferon regulatory factor activation involves TBK1-IKKepsilon-dependent Lys(63)-linked polyubiquitination and phosphorylation of TANK/I-TRAF. J Biol Chem. 2007;282:31131–31146. doi: 10.1074/jbc.M701690200. [DOI] [PubMed] [Google Scholar]
- 34.Kawai T., Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 35.Ryzhakov G., Randow F.S.I.N.T.B.A.D. a novel component of innate antiviral immunity, shares a TBK1-binding domain with NAP1 and TANK. EMBO J. 2007;26:3180–3190. doi: 10.1038/sj.emboj.7601743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clark K., Takeuchi O., Akira S., Cohen P. The TRAF-associated protein TANK facilitates cross-talk within the IkappaB kinase family during Toll-like receptor signaling. Proc Natl Acad Sci USA. 2011;108:17093–17098. doi: 10.1073/pnas.1114194108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chariot A., Leonardi A., Muller J., Bonif M., Brown K., Siebenlist U. Association of the adaptor TANK with the I kappa B kinase (IKK) regulator NEMO connects IKK complexes with IKK epsilon and TBK1 kinases. J Biol Chem. 2002;277:37029–37036. doi: 10.1074/jbc.M205069200. [DOI] [PubMed] [Google Scholar]
- 38.Kayagaki N., Phung Q., Chan S., Chaudhari R., Quan C., O’Rourke K.M. DUBA: a deubiquitinase that regulates type I interferon production. Science. 2007;318:1628–1632. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- 39.Friedman C.S., O’Donnell M.A., Legarda-Addison D., Ng A., Cardenas W.B., Yount J.S. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008;9:930–936. doi: 10.1038/embor.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang M., Wu X., Lee A.J., Jin W., Chang M., Wright A. Regulation of IkappaB kinase-related kinases and antiviral responses by tumor suppressor CYLD. J Biol Chem. 2008;283:18621–18626. doi: 10.1074/jbc.M801451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hutti J.E., Shen R.R., Abbott D.W., Zhou A.Y., Sprott K.M., Asara J.M. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKepsilon promotes cell transformation. Mol Cell. 2009;34:461–472. doi: 10.1016/j.molcel.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parvatiyar K., Barber G.N., Harhaj E.W. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem. 2010;285:14999–15009. doi: 10.1074/jbc.M110.109819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cui J., Li Y., Zhu L., Liu D., Songyang Z., Wang H.Y. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol. 2012;13:387–395. doi: 10.1038/ni.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang M., Wang L., Zhao X., Zhao K., Meng H., Zhao W. TRAF-interacting protein (TRIP) negatively regulates IFN-beta production and antiviral response by promoting proteasomal degradation of TANK-binding kinase 1. J Exp Med. 2012;209:1703–1711. doi: 10.1084/jem.20120024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prins K.C., Cardenas W.B., Basler C.F. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J Virol. 2009;83:3069–3077. doi: 10.1128/JVI.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siu K.L., Kok K.H., Ng M.H., Poon V.K., Yuen K.Y., Zheng B.J. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J Biol Chem. 2009;284:16202–16209. doi: 10.1074/jbc.M109.008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaukinen P., Sillanpaa M., Nousiainen L., Melen K., Hepatitis Julkunen I. C virus NS2 protease inhibits host cell antiviral response by inhibiting IKKepsilon and TBK1 functions. J Med Virol. 2013;85:71–82. doi: 10.1002/jmv.23442. [DOI] [PubMed] [Google Scholar]
- 48.Yu S., Chen J., Wu M., Chen H., Kato N., Hepatitis Yuan Z. B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J Gen Virol. 2010;91:2080–2090. doi: 10.1099/vir.0.020552-0. [DOI] [PubMed] [Google Scholar]
- 49.Lu L.L., Puri M., Horvath C.M., Sen G.C. Select paramyxoviral V proteins inhibit IRF3 activation by acting as alternative substrates for inhibitor of kappaB kinase epsilon (IKKe)/TBK1. J Biol Chem. 2008;283:14269–14276. doi: 10.1074/jbc.M710089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Unterstab G., Ludwig S., Anton A., Planz O., Dauber B., Krappmann D. Viral targeting of the interferon-{beta}-inducing Traf family member-associated NF-{kappa}B activator (TANK)-binding kinase-1. Proc Natl Acad Sci USA. 2005;102:13640–13645. doi: 10.1073/pnas.0502883102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corr M., Boyle D.L., Ronacher L., Flores N., Firestein G.S. Synergistic benefit in inflammatory arthritis by targeting I kappaB kinase epsilon and interferon beta. Ann Rheum Dis. 2009;68:257–263. doi: 10.1136/ard.2008.095356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sweeney S.E., Mo L., Firestein G.S. Antiviral gene expression in rheumatoid arthritis: role of IKKepsilon and interferon regulatory factor 3. Arthritis Rheum. 2007;56:743–752. doi: 10.1002/art.22421. [DOI] [PubMed] [Google Scholar]
- 53.Vervoordeldonk M.J., Aalbers C.J., Tak P.P. Interferon beta for rheumatoid arthritis: new clothes for an old kid on the block. Ann Rheum Dis. 2009;68:157–158. doi: 10.1136/ard.2008.097899. [DOI] [PubMed] [Google Scholar]
- 54.Dieguez-Gonzalez R., Akar S., Calaza M., Perez-Pampin E., Costas J., Torres M. Genetic variation in the nuclear factor kappaB pathway in relation to susceptibility to rheumatoid arthritis. Ann Rheum Dis. 2009;68:579–583. doi: 10.1136/ard.2007.087304. [DOI] [PubMed] [Google Scholar]
- 55.Sandling J.K., Garnier S., Sigurdsson S., Wang C., Nordmark G., Gunnarsson I. A candidate gene study of the type I interferon pathway implicates IKBKE and IL8 as risk loci for SLE. Eur J Hum Genet. 2011;19:479–484. doi: 10.1038/ejhg.2010.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Park D., Jeong H.O., Kim B.C., Ha Y.M., Young Chung H. Computational approach to identify enzymes that are potential therapeutic candidates for psoriasis. Enzyme Res. 2011;2011:826784. doi: 10.4061/2011/826784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bulek K., Liu C., Swaidani S., Wang L., Page R.C., Gulen M.F. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat Immunol. 2011;12:844–852. doi: 10.1038/ni.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gulen M.F., Bulek K., Xiao H., Yu M., Gao J., Sun L. Inactivation of the Enzyme GSK3alpha by the Kinase IKKi Promotes AKT-mTOR Signaling Pathway that Mediates Interleukin-1-Induced Th17 Cell Maintenance. Immunity. 2012;37:800–812. doi: 10.1016/j.immuni.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moser C.V., Kynast K., Baatz K., Russe O.Q., Ferreiros N., Costiuk H. The protein kinase IKKepsilon is a potential target for the treatment of inflammatory hyperalgesia. J Immunol. 2011;187:2617–2625. doi: 10.4049/jimmunol.1004088. [DOI] [PubMed] [Google Scholar]
- 60.Shoelson S.E., Herrero L., Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132:2169–2180. doi: 10.1053/j.gastro.2007.03.059. [DOI] [PubMed] [Google Scholar]
- 61.Tsukumo D.M., Carvalho-Filho M.A., Carvalheira J.B., Prada P.O., Hirabara S.M., Schenka A.A. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56:1986–1998. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- 62.Ye J., Gao Z., Yin J., He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab. 2007;293:E1118–E1128. doi: 10.1152/ajpendo.00435.2007. [DOI] [PubMed] [Google Scholar]
- 63.Chiang S.H., Bazuine M., Lumeng C.N., Geletka L.M., Mowers J., White N.M. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell. 2009;138:961–975. doi: 10.1016/j.cell.2009.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Olefsky J.M. IKKepsilon: a bridge between obesity and inflammation. Cell. 2009;138:834–836. doi: 10.1016/j.cell.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 65.Scheja L., Heese B., Seedorf K. Beneficial effects of IKKepsilon-deficiency on body weight and insulin sensitivity are lost in high fat diet-induced obesity in mice. Biochem Biophys Res Commun. 2011;407:288–294. doi: 10.1016/j.bbrc.2011.02.137. [DOI] [PubMed] [Google Scholar]
- 66.Shen R.R., Hahn W.C. Emerging roles for the non-canonical IKKs in cancer. Oncogene. 2011;30:631–641. doi: 10.1038/onc.2010.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boehm J.S., Zhao J.J., Yao J., Kim S.Y., Firestein R., Dunn I.F. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 68.Krishnamurthy S., Basu A. Regulation of IKKepsilon Expression by Akt2 Isoform. Genes Cancer. 2011;2:1044–1050. doi: 10.1177/1947601912444604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qin B., Cheng K. Silencing of the IKKepsilon gene by siRNA inhibits invasiveness and growth of breast cancer cells. Breast Cancer Res. 2010;12:R74. doi: 10.1186/bcr2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eddy S.F., Guo S., Demicco E.G., Romieu-Mourez R., Landesman-Bollag E., Seldin D.C. Inducible IkappaB kinase/IkappaB kinase epsilon expression is induced by CK2 and promotes aberrant nuclear factor-kappaB activation in breast cancer cells. Cancer Res. 2005;65:11375–11383. doi: 10.1158/0008-5472.CAN-05-1602. [DOI] [PubMed] [Google Scholar]
- 71.Shen R.R., Zhou A.Y., Kim E., Lim E., Habelhah H., Hahn W.C. IκB kinase ɛ phosphorylates TRAF2 to promote mammary epithelial cell transformation. Mol Cell Biol. 2012;32:4756–4768. doi: 10.1128/MCB.00468-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guo J.P., Coppola D., Cheng J.Q. IKBKE protein activates Akt independent of phosphatidylinositol 3-kinase/PDK1/mTORC2 and the pleckstrin homology domain to sustain malignant transformation. J Biol Chem. 2011;286:37389–37398. doi: 10.1074/jbc.M111.287433. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 73.Xie X., Zhang D., Zhao B., Lu M.K., You M., Condorelli G. IkappaB kinase epsilon and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci USA. 2011;108:6474–6479. doi: 10.1073/pnas.1016132108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tenoever B.R., Ng S.L., Chua M.A., McWhirter S.M., Garcia-Sastre A., Maniatis T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science. 2007;315:1274–1278. doi: 10.1126/science.1136567. [DOI] [PubMed] [Google Scholar]
- 75.Watson C.J., Neoh K. The Stat family of transcription factors have diverse roles in mammary gland development. Semin Cell Dev Biol. 2008;19:401–406. doi: 10.1016/j.semcdb.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 76.Hsu S., Kim M., Hernandez L., Grajales V., Noonan A., Anver M. IKK-ɛ coordinates invasion and metastasis of ovarian cancer. Cancer Res. 2012;72:5494–5504. doi: 10.1158/0008-5472.CAN-11-3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo J.P., Shu S.K., He L., Lee Y.C., Kruk P.A., Grenman S. Deregulation of IKBKE is associated with tumor progression, poor prognosis, and cisplatin resistance in ovarian cancer. Am J Pathol. 2009;175:324–333. doi: 10.2353/ajpath.2009.080767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kang M.R., Kim M.S., Kim S.S., Ahn C.H., Yoo N.J., Lee S.H. NF-kappaB signalling proteins p50/p105, p52/p100, RelA, and IKKepsilon are over-expressed in oesophageal squamous cell carcinomas. Pathology. 2009;41:622–625. doi: 10.3109/00313020903257756. [DOI] [PubMed] [Google Scholar]
- 79.Peant B., Diallo J.S., Dufour F., Le Page C., Delvoye N., Saad F. Over-expression of IkappaB-kinase-epsilon (IKKepsilon/IKKi) induces secretion of inflammatory cytokines in prostate cancer cell lines. Prostate. 2009;69:706–718. doi: 10.1002/pros.20912. [DOI] [PubMed] [Google Scholar]
- 80.Peant B., Forest V., Trudeau V., Latour M., Mes-Masson A.M., Saad F. IkappaB-Kinase-epsilon (IKKepsilon/IKKi/IkappaBKepsilon) expression and localization in prostate cancer tissues. Prostate. 2011;71:1131–1138. doi: 10.1002/pros.21329. [DOI] [PubMed] [Google Scholar]
- 81.Seo S.I., Song S.Y., Kang M.R., Kim M.S., Oh J.E., Kim Y.R. Immunohistochemical analysis of NF-kappaB signaling proteins IKKepsilon, p50/p105, p52/p100 and RelA in prostate cancers. APMIS. 2009;117:623–628. doi: 10.1111/j.1600-0463.2009.02506.x. [DOI] [PubMed] [Google Scholar]
- 82.Hildebrandt M.A., Tan W., Tamboli P., Huang M., Ye Y., Lin J. Kinome expression profiling identifies IKBKE as a predictor of overall survival in clear cell renal cell carcinoma patients. Carcinogenesis. 2012;33:799–803. doi: 10.1093/carcin/bgs018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guo J.P., Shu S.K., Esposito N.N., Coppola D., Koomen J.M., Cheng J.Q. IKKepsilon phosphorylation of ERalpha-Ser167 and contribution to tamoxifen resistance in breast cancer. J Biol Chem. 2010;285:3676–3684. doi: 10.1074/jbc.M109.078212. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 84.Arlt A., Schafer H. NFkappaB-dependent chemoresistance in solid tumors. Int J Clin Pharmacol Ther. 2002;40:336–347. doi: 10.5414/cpp40336. [DOI] [PubMed] [Google Scholar]
- 85.Tapia M.A., Gonzalez-Navarrete I., Dalmases A., Bosch M., Rodriguez-Fanjul V., Rolfe M. Inhibition of the canonical IKK/NF kappa B pathway sensitizes human cancer cells to doxorubicin. Cell Cycle. 2007;6:2284–2292. doi: 10.4161/cc.6.18.4721. [DOI] [PubMed] [Google Scholar]
- 86.Renner F., Moreno R., Schmitz M.L. SUMOylation-Dependent Localization of IKK epsilon in PML Nuclear Bodies Is Essential for Protection against DNA-Damage-Triggered Cell Death. Mol Cell. 2010;37:503–515. doi: 10.1016/j.molcel.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 87.Guan H., Zhang H., Cai J., Wu J., Yuan J., Li J. IKBKE is over-expressed in glioma and contributes to resistance of glioma cells to apoptosis via activating NF-kappaB. J Pathol. 2011;223:436–445. doi: 10.1002/path.2815. [DOI] [PubMed] [Google Scholar]
- 88.Wang T., Block M.A., Cowen S., Davies A.M., Devereaux E., Gingipalli L. Discovery of azabenzimidazole derivatives as potent, selective inhibitors of TBK1/IKKepsilon kinases. Bioorg Med Chem Lett. 2012;22:2063–2069. doi: 10.1016/j.bmcl.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 89.McIver E.G., Bryans J., Birchall K., Chugh J., Drake T., Lewis S.J. Synthesis and structure-activity relationships of a novel series of pyrimidines as potent inhibitors of TBK1/IKKepsilon kinases. Bioorg Med Chem Lett. 2012;22:7169–7173. doi: 10.1016/j.bmcl.2012.09.063. [DOI] [PubMed] [Google Scholar]
- 90.Lee J.K., Kim S.Y., Kim Y.S., Lee W.H., Hwang D.H., Lee J.Y. Suppression of the TRIF-dependent signaling pathway of Toll-like receptors by luteolin. Biochem Pharmacol. 2009;77:1391–1400. doi: 10.1016/j.bcp.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 91.Youn H.S., Lee J.Y., Saitoh S.I., Miyake K., Kang K.W., Choi Y.J. Suppression of MyD88- and TRIF-dependent signaling pathways of Toll-like receptor by (−)-epigallocatechin-3-gallate, a polyphenol component of green tea. Biochem Pharmacol. 2006;72:850–859. doi: 10.1016/j.bcp.2006.06.021. [DOI] [PubMed] [Google Scholar]