

Abstract
Components of the renin–angiotensin system are well established targets for pharmacological intervention in a variety of disorders. Many such therapies abrogate the effects of the hypertensive and mitogenic peptide, angiotensin II, by antagonising its interaction with its receptor, or by inhibiting its formative enzyme, angiotensin-converting enzyme (ACE). At the turn of the millennium, a homologous enzyme, termed ACE2, was identified which increasingly shares the limelight with its better-known homologue. In common with ACE, ACE2 is a type I transmembrane metallopeptidase; however, unlike ACE, ACE2 functions as a carboxypeptidase, cleaving a single C-terminal residue from a distinct range of substrates. One such substrate is angiotensin II, which is hydrolysed by ACE2 to the vasodilatory peptide angiotensin 1–7. In this commentary we discuss the latest developments in the rapidly progressing study of the physiological and patho-physiological roles of ACE2 allied with an overview of the current understanding of its molecular and cell biology. We also discuss parallel developments in the study of collectrin, a catalytically inactive homologue of ACE2 with critical functions in the pancreas and kidney.
Abbreviations: ACE, angiotensin-converting enzyme; ADAM, a disintegrin and metalloproteinase; Ang, angiotensin; ARDS, acute respiratory distress syndrome; AT1, angiotensin receptor, type 1; CoV, coronavirus; HEK, human embryonic kidney; HNF, hepatocyte nuclear factor; mas, receptor for Ang (1-7); MDCK, Madin–Darby Canine Kidney; MODY, maturity-onset diabetes mellitus; RAS, renin–angiotensin system; SARS, severe acute respiratory syndrome; TACE, TNF-α converting enzyme; TNF, tumour necrosis factor
Keywords: ACE2, ACE, Angiotensin, ARDS, SARS, Diabetes
1. Introduction
Angiotensin-converting enzyme (ACE) is a critical regulator of the renin–angiotensin system and the target of a number of highly effective therapeutic agents used to treat cardiovascular and renal diseases. ACE is a metalloproteinase which converts the inactive decapeptide angiotensin I (Ang I) into the potent vasoconstrictor and mitogen angiotensin II (Ang II), while also metabolising the hypotensive peptide bradykinin. Although ACE inhibitors such as captopril and lisinopril have become mainstays in the treatment of cardiovascular disease, hypertension and congestive heart failure remain leading causes of death in the developed world. Due to this continuing morbidity and mortality, significant efforts have been made to identify new drug targets in the renin–angiotensin system (RAS).
2. A new member of the RAS
ACE2 was identified in 2000 by two groups simultaneously using distinct methodologies [1], [2]. It is an 805 amino acid type I transmembrane glycoprotein with an extracellular catalytic domain. ACE2 displays homology to two quite distinct proteins; its amino-terminal domain shares approximately 40% sequence identity with ACE, whereas its cytoplasmic and transmembrane domains display 48% homology to collectrin, a non-catalytic protein recently shown to have a critical role in amino acid absorption in the kidney [3], [4], pancreatic beta cell proliferation [5] and insulin exocytosis [6].
2.1. Cell biology and tissue distribution of ACE2
As predicted from analyses of its peptide sequence, heterologously expressed ACE2 localises predominantly to the plasma membrane [7]. Here, like ACE and collectrin [5], [8] it is subject to a juxtamembrane cleavage event (shedding) which releases the catalytically active ectodomain [7], [9]. This process is stimulated by phorbol ester, an event involving a promiscuous ‘sheddase’, ADAM17 (or TACE, TNF-α converting enzyme; Fig. 1 ) [9]. In common with ACE, the physiological role of ACE2 ectodomain shedding remains elusive as a role for circulating ACE2 has yet to be identified. Indeed, it is possible that shedding is a mechanism to regulate ACE2 activity at the cell surface. When expressed in polarised epithelial cells, ACE2 is predominantly trafficked to the apical surface; this is in contrast to ACE, which is evenly distributed between the apical and basolateral membranes [7]. The mechanisms underlying this difference are as yet unknown, but given the lack of homology between the cytoplasmic domains of the two enzymes, it is likely that this region may contain distinct targeting motifs.
Fig. 1.
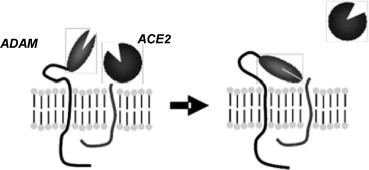
Illustration of the process of ectodomain shedding of ACE2. Upon stimulation by a variety of factors, a transmembrane proteinase (ADAM17) cleaves the extracellular juxtamembrane region of ACE2, releasing the catalytically active ectodomain into the extracellular milieu.
In vivo, ACE2 is predominantly expressed in the heart, kidneys and testes, and at a lower level in a wide variety of tissues, particularly the colon and lung [2]. In the heart, ACE2 is essentially confined to the endothelia, in kidney to the luminal surface of the tubular epithelial cells [1], [2] and, in testes, to the adult Leydig cells [10].
2.2. Substrate specificity of ACE2
Unlike ACE, ACE2 functions as a carboxypeptidase, cleaving a single carboxy-terminal residue from its peptide substrates [2]. A wide variety of potential substrates have been identified for ACE2, both within the RAS and elsewhere [11]. It is able to cleave both Ang I and Ang II, to Ang (1–9) and Ang (1–7), respectively. Whilst the affinity for the former is poor in comparison with ACE, making it unlikely to be a physiological substrate (except under conditions in which ACE activity is inhibited), ACE2 efficiently cleaves Ang II to Ang (1–7) [12]. Ang (1–7) is increasingly becoming recognised as a key peptide within the RAS [13]. Ang (1–7) is known to potentiate the vasodilatory effects of bradykinin [14], stimulate NO and prostaglandin release [15], and antagonise the actions of Ang II [16] (Fig. 2 ). Hence, the ability of ACE2 to degrade Ang II and simultaneously increase Ang (1–7) would effectively oppose the actions of ACE, suggesting the balance of the levels of the two enzymes would be critical in pathologies in the aetiologies of which Ang II is implicated (Fig. 2A).
Fig. 2.
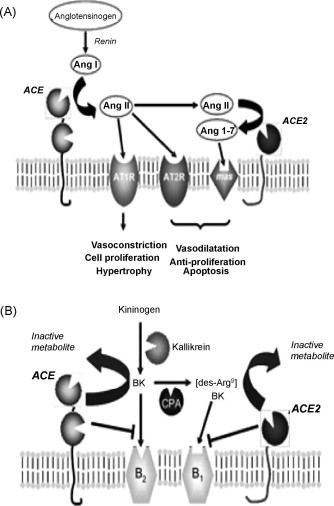
Schematics to illustrate the involvement of ACE and ACE2 in the regulation of the functions of the RAS (A) and the kinin-kininogen system (B). See text for details. CPA, carboxypeptidase A.
In addition to its activity on Ang II, ACE2 is also able to hydrolyse other non-RAS peptides which have roles in maintaining cardiovascular homeostasis such as [des-Arg9] bradykinin (Fig. 2B), a member of the kininogen-kinin system [17]. [des-Arg9] bradykinin is formed from bradykinin by the action of carboxypeptidases and is an agonist for the B1 receptor, which is induced upon tissue injury [18]. Bradykinin, a vasodilator which acts through the B2 receptor, is produced from its precursor kininogen by kallikrein and is degraded by ACE [17] (Fig. 2B). Whilst the degradation of bradykinin by ACE is known to be an important aspect of blood pressure regulation, the significance of the degradation of [des-Arg9] bradykinin by ACE2 remains to be established. In addition to [des-Arg9] bradykinin, ACE2 is also able to degrade apelin-13, a peptide proposed to cause vasoconstriction and known to regulate fluid homeostasis, and other non-RAS peptides such as kinetensin, dynorphin A and neurotensin [11].
2.3. Structural considerations
ACE and ACE2 belong to the gluzincin clan of metalloproteinases, all of which catalyse reactions by utilising zinc, coordinated by conserved histidines within the active site, to facilitate nucleophilic attack on the carbonyl bond of the substrate by a water molecule, forming a non-covalently bound intermediate. In addition to the two histidines (located within the HEXXH motif), a further glutamate residue is involved in coordinating the zinc ion; this is located 23 amino acids C-terminally to the HEXXH motif in both ACE and ACE2 [19], [20]. Despite these similarities, however, ACE and ACE2 function differently (the former normally releasing a C-terminal dipeptide from its substrate; peptidyl dipeptidase action) and the latter releasing a single amino acid (a strict carboxypeptidase). In addition, the activity of ACE2 is unaffected by a range of ACE inhibitors, such as captopril and lisinopril [21], but is sensitive to inhibition by the dipeptide Pro-Phe [12] and by the peptide analogues DX600 [22] and MLN 4760 ((S,S)-2-[1-carboxy-2-[3-(3,5-dichlorobenzyl)-3H-imidazol4-yl]-ethylamino]-4-methylpentanoicacid) [23]. MLN 4760 was the first rationally designed inhibitor of ACE2, based on the C-terminal dipeptide of Ang I (His-Leu), and has high potency (K i = 0.44 nM) and specificity. Molecular modelling studies [12] and the resolution of the ACE2 crystal structure [20] revealed that these substrate specificity differences are a result of the smaller binding pocket in ACE2 due to arginine-273 making a salt-bridge with the C-terminus of the substrate, whereas in ACE this residue is substituted by the smaller glutamine. Comparison of the structure of ACE2 in the presence and absence of MLN 4760 revealed a large ‘hinge-bending’ motion, in which movement of the catalytic subdomains induced by inhibitor binding repositioned key residues for catalysis [20].
3. Physiological and patho-physiological roles of ACE2
3.1. Cardiovascular disease
The high level of expression of ACE2 in the heart together with its ability to hydrolyse angiotensin peptides have suggested a role for ACE2 in maintaining cardiovascular physiology from the outset, a hypothesis subsequently supported by experimental data. Crackower et al. [24] showed that deletion of ACE2 in mice resulted in elevated cardiac and plasma Ang II together with impaired cardiac contractility which increased with age. These changes were associated with an upregulation of hypoxia-induced genes, consistent with a role for Ang II in the ACE2 null phenotype. This hypothesis is given further credence by the observation that the defects in the Ace2-null mice were reversed by deletion of ACE in the same mice [24], an intervention which also abrogated the increased Ang II levels. Furthermore, a recent study by the same group showed that the cardiomyopathy observed in Ace2-null mice involves Ang II acting through the AT1 receptor to activate PI3Kγ-mediated pathways [25]. It should be noted, however, that a similar study by Gurley et al. [26] failed to detect any changes in cardiac function in a distinct strain of Ace2-null mice but instead observed enhanced susceptibility to Ang II-induced hypertension. Furthermore, deletion of ACE2 did not cause abnormalities which might be expected as a result of increased Ang II, such as cardiac hypertrophy, fibrosis and altered conductance [24], suggesting that ACE2 may not exert its effects in the heart solely via Ang II. Interestingly, overexpression of ACE2 in cardiac myocytes did cause changes in cardiac conductivity, resulting in arrest and sudden death [27]. These contradictory data suggest that the role of ACE2 in the cardiovascular system may be more complex than is immediately apparent.
Further evidence for a role of ACE2 in maintaining cardiovascular homeostasis via Ang II regulation is provided by studies conducted by Zisman et al. [28] which detected increased ACE2 and Ang (1–7) forming activity in failing human hearts. Increases in ACE2 expression and activity have been observed in rats following arterial ligation and AT1 receptor blockade [29], [30]. In a separate study, arterial ligation of rats resulted in increased Ang II and Ang (1–7) levels alongside cardiac hypertrophy, an effect reversed by treatment with AT1 receptor antagonists, which also increased cardiac ACE2 levels [31]. In this study, however, no change was observed in ACE2 levels following arterial ligation. Whilst these studies again highlight some conflicting data, it is likely that ACE2 may play a protective role in the early stages of heart failure by elevating Ang 1–7 levels. In addition to its postulated roles in the heart, the ace2 gene which is present on the X chromosome maps to a quantitative trait locus (QTL) in a number of rat models of hypertension. Furthermore, salt-sensitive hypertensive rats have reduced ACE2 transcript and protein expression [24]. In humans, single nucleotide polymorphisms associated with increased risk of cardiovascular disease have been identified within the ACE2 gene locus [32].
3.2. ARDS and SARS
A role for the RAS in the development of lung disease has been suggested by studies in rodents showing AT1 receptor antagonists protect against experimentally induced pulmonary fibrosis [33], and an increased mortality rate in acute respiratory distress syndrome (ARDS) patients carrying the ACE DD polymorphism [34]. ARDS can be triggered by a variety of insults including acid aspiration, peritoneal sepsis and acute pancreatitis, and has a high mortality rate due to associated pulmonary oedema, inflammation and hypoxia. A recent study by Kuba et al. [35] identified a protective role for ACE2 in mice in which ARDS had been experimentally induced by distinct insults. In this study, Ace2 knockout mice developed more severe ARDS than wild-type littermates, an effect which was reversed in mice in which the ace gene had also been disrupted, or upon treatment with recombinant ACE2 protein. In addition, at1R knockout mice also had less severe disease and both Ace2 −/y and wild-type mice responded positively to AT1 receptor antagonists, suggesting the involvement of Ang II in ARDS progression. It can therefore be postulated that ACE2 exerts its protective role by metabolising Ang II and thereby abrogating its deleterious effects.
In 2003, the surprising discovery was made that ACE2 is a functional receptor for the causative agent of severe acute respiratory syndrome (SARS), the SARS coronavirus [36]. SARS achieved widespread notoriety for its high mortality rate (approaching 10%) in infected individuals due to an atypical pneumonia resulting in respiratory failure due to ARDS [37]. ACE2 was serendipitously identified as a receptor for SARS-CoV in vitro using co-immunoprecipitation techniques [36] and has subsequently been shown to be essential for SARS infection in vivo [35]. Interaction of SARS-CoV with ACE2 occurs via trimers of the SARS spike protein, a loop of which extends into a hydrophobic pocket of ACE2; here a methyl group of threonine 487 of the spike interacts with lysine 353 of ACE2 [38]. The acquisition of a threonine at position 487 appears to have been critical in the adaptation of the SARS virus to humans; in the civet (believed to be the source of the outbreak), this position is occupied by a serine residue which renders it unable to bind human ACE2 [39].
In the first study to demonstrate that ACE2 functions as the SARS receptor in vivo [35], infection of mice with SARS resulted in downregulation of ACE2 and a concomitant increase in Ang II, and increased severity of lung damage which could be improved by treatment with AT1 antagonists. This downregulation of ACE2 in response to SARS infection, leading to increased disease severity resulting from increases in Ang II, may be a significant contributor to the high mortality rates resulting from SARS. It has therefore been suggested that administration of recombinant ACE2 may be an effective therapy [40].
3.3. Renal disease
The renal RAS plays a vital role in maintaining electrolyte and fluid homeostasis and the kidney is host to high levels of RAS components, including renin [41], ACE [42], AT receptors [43] and ACE2 [1]. Alterations of ACE levels are associated with differing susceptibility to experimental renal damage and elevated Ang II levels have been detected in diseased kidneys in humans [44], suggesting that regulating Ang II expression may be critical for maintaining renal function. ACE2 is expressed on the luminal surface of proximal and distal tubules and at a lower level in glomeruli [7], [45], [46]. In this regard it displays a similar pattern of expression to ACE, and may, like ACE, regulate the interstitial levels of angiotensin peptides. Evidence for this is provided by Li et al. [46], who showed that isolated proximal tubules, when incubated with Ang I, generated Ang (1–7), a process which was blocked by addition of the ACE2 inhibitor DX600. The results of this study are surprising as they indicate that ACE2 in kidney is generating Ang (1–7) indirectly from Ang I (ACE2 is able to convert Ang I to Ang (1–9) which can then be converted to Ang (1–7) by ACE, but the kinetics for this are not favourable [21]) rather than directly from Ang II, but still indicate a role for ACE2 in generating interstitial Ang (1–7). In addition, ACE2 levels are elevated in the kidneys of young diabetic mice in parallel with reduced ACE expression [47]. This is in contrast with a separate study showing markedly reduced levels of ACE2 in a model of diabetic nephropathy [48]; the reasons for this difference are not immediately apparent, but may indicate that ACE2 is protective in the early stages of the disease but then a decline in its expression allows elevated Ang II levels to contribute to the development of nephropathy. This is supported in part by the observation that some Ace2 deficient mice develop Ang II dependent glomerulosclerosis with aging [25]. Furthermore, infusion of the specific ACE2 inhibitor MLN 4760 resulted in marked albuminuria in diabetic mice [49]. ACE2 levels are also known to rise together with Ang (1–7) during gestation in rats [50]; this may be a protective mechanism to obviate the effects of hypertension in the kidney during pregnancy.
3.4. Chronic liver injury
A role for ACE2 has also been proposed in the development of liver fibrosis and subsequent cirrhosis. Paizis et al. [51] identified an increase in ACE2 levels in response to bile-duct ligation in rats, and in human cirrhotic liver. In both cases, ACE2 levels were low in corresponding controls, with expression confined to endothelial cells and perivenular hepatocytes, but were widespread in the parenchymal tissue of diseased livers. Intriguingly, ACE2 expression was high in cells exposed to low oxygen levels, and ACE2 could be induced by hypoxia in vitro. This is in keeping with studies showing increased ACE2 levels in myocardial ischaemia, and raises the possibility that ACE2 may exert a protective effect in tissue injury by increasing local oxygenation levels by abrogating Ang II-mediated vasoconstriction. This hypothesis has been further strengthened by a recent study showing that the increase in ACE2 levels following bile-duct ligation is accompanied by increases in Ang 1–7 and its receptor, mas [52]. This study, however, did not detect any direct protective role for Ang 1–7, suggesting that ACE2 may exert positive effects in liver injury predominantly by reducing Ang II levels.
4. Collectrin
The ACE2 homologue, collectrin (also known as Tmem27), was first identified as a 27 kDa kidney-specific developmentally regulated transmembrane protein of unknown function shortly after the discovery of ACE2 [53]. Recently collectrin has been implicated as a critical regulator of amino acid uptake in the proximal tubules of the kidney [3], [4]. Disruption of collectrin in mice results in downregulation of a variety of renal apical amino acid transporters and associated aminoaciduria. A recent study by Zhang et al. [54] provided evidence of a role for hepatocyte nuclear factor 1β (HNF-1β) in regulating renal collectrin expression, and showed collectrin is essential for the maintenance of primary cilia and polarity of collecting duct cells. Mutations in HNF-1β are associated with polycystic kidney disease and MODY5 (maturity-onset diabetes mellitus of the young, type 5), sufferers of which also develop renal disease. These findings provide a link with studies of the role of collectrin in the pancreas, where it has recently been shown to regulate pancreatic islet cell growth and insulin exocytosis. Akpinar et al. [5] showed that collectrin levels were reduced in mice in which the related hepatocyte nuclear factor 1α (HNF-1α) had been deleted; mutations in this transcription factor are associated with the development of MODY. This reduction in expression correlated with decreased islet mass; in keeping with this, overexpression of collectrin resulted in increased islet mass. Similar results were obtained by Fukui et al. [6], however this study also provided evidence of a role for collectrin in regulating insulin secretion by facilitating SNARE complex formation. Additionally, Akpinar et al. [5] showed that collectrin, like ACE2, is subject to ectodomain shedding. In the case of collectrin, however, this shedding event appeared to occur exclusively in pancreatic β-cells, suggesting the involvement of a sheddase with very restricted distribution and therefore presumably via a mechanism distinct from that of ACE2. Whilst there is much work to be done elucidating the mechanisms underlying the interaction of collectrin with renal amino acid transporters and its actions in the pancreas, given the range of diseases in which collectrin is now implicated, such as Hartnup disease and diabetes, it is clear that these discoveries are likely to provide new opportunities for therapeutic intervention.
5. Perspectives and concluding remarks
The importance of ACE2 as a regulator of the local RAS is becoming increasingly apparent. Through its ability to metabolise Ang II to Ang (1–7) it is able to regulate local Ang II levels thereby modulating its effects. Disturbance of the balance of expression of ACE2 and its homologue ACE could alter the levels of Ang II and contribute to the development of a range of pathologies, from myocardial infarction to SARS. Furthermore, the recent explosion of research into the ACE2 homologue, collectrin, has revealed new possible physiological functions for ACE2 independent of its catalytic activity. While there still exists a myriad of opportunities for further research, recent findings have revealed intriguing possibilities for novel avenues of pharmacological intervention, most probably involving strategies to upregulate ACE2 activity.
Acknowledgement
We would like to acknowledge the financial support provided by the Biotechnology and Biological Sciences Research Council (UK).
References
- 1.Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N. A novel angiotensin converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87(5):E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 2.Tipnis S.R., Hooper N.M., Hyde R., Karran E., Christie G., Turner A.J. A human homolog of angiotensin converting enzyme: cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275(43):33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 3.Danilczyk U., Sarao R., Remy C., Benabbas C., Stange G., Richter A. Essential role for collectrin in renal amino acid transport. Nature. 2006;444(7122):1088–1091. doi: 10.1038/nature05475. [DOI] [PubMed] [Google Scholar]
- 4.Malakauskas S.M., Quan H., Fields T.A., McCall S.J., Yu M.J., Kourany W.M. Aminoaciduria and altered renal expression of luminal amino acid transporters in mice lacking novel gene collectrin. Am J Physiol Renal Physiol. 2007;292(2):F533–F544. doi: 10.1152/ajprenal.00325.2006. [DOI] [PubMed] [Google Scholar]
- 5.Akpinar P., Kuwajima S., Krutzfeldt J., Stoffel M. Tmem27: a cleaved and shed plasma membrane protein that stimulates pancreatic beta cell proliferation. Cell Metab. 2005;2(6):385–397. doi: 10.1016/j.cmet.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Fukui K., Yang Q., Cao Y., Takahashi N., Hatakeyama H., Wang H. The HNF-1 target collectrin controls insulin exocytosis by SNARE complex formation. Cell Metab. 2005;2(6):373–384. doi: 10.1016/j.cmet.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Warner F.J., Lew R.A., Smith A.I., Lambert D.W., Hooper N.M., Turner A.J. Angiotensin-converting enzyme 2 (ACE2), but not ACE, is preferentially localized to the apical surface of polarized kidney cells. J Biol Chem. 2005;280(47):39353–39362. doi: 10.1074/jbc.M508914200. [DOI] [PubMed] [Google Scholar]
- 8.Turner A.J. Exploring the structure and function of zinc metallopeptidases: old enzymes and new discoveries. Biochem Soc Trans. 2003;31(Pt 3):723–727. doi: 10.1042/bst0310723. [DOI] [PubMed] [Google Scholar]
- 9.Lambert D.W., Yarski M., Warner F.J., Thornhill P., Parkin E.T., Smith A.I. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2) J Biol Chem. 2005;280(34):30113–30119. doi: 10.1074/jbc.M505111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Douglas G.C., O’Bryan M.K., Hedger M.P., Lee D.K., Yarski M.A., Smith A.I. The novel angiotensin-converting enzyme (ACE) homolog, ACE2, is selectively expressed by adult Leydig cells of the testis. Endocrinology. 2004;145(10):4703–4711. doi: 10.1210/en.2004-0443. [DOI] [PubMed] [Google Scholar]
- 11.Vickers C., Hales P., Kaushik V., Dick L., Gavin J., Tang J. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277(17):14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 12.Guy J.L., Jackson R.M., Acharya K.R., Sturrock E.D., Hooper N.M., Turner A.J. Angiotensin-converting enzyme-2 (ACE2): comparative modeling of the active site, specificity requirements, and chloride dependence. Biochemistry. 2003;42(45):13185–13192. doi: 10.1021/bi035268s. [DOI] [PubMed] [Google Scholar]
- 13.Iyer S.N., Averill D.B., Chappell M.C., Yamada K., Allred A.J., Ferrario C.M. Contribution of angiotensin-(1–7) to blood pressure regulation in salt-depleted hypertensive rats. Hypertension. 2000;36(3):417–422. doi: 10.1161/01.hyp.36.3.417. [DOI] [PubMed] [Google Scholar]
- 14.Greco A.J., Master R.G., Fokin A., Jr., Baber S.R., Kadowitz P.J. Angiotensin-(1–7) potentiates responses to bradykinin but does not change responses to angiotensin I. Can J Physiol Pharmacol. 2006;84(11):1163–1175. doi: 10.1139/y06-053. [DOI] [PubMed] [Google Scholar]
- 15.Rajendran S., Chirkov Y.Y., Campbell D.J., Horowitz J.D. Angiotensin-(1–7) enhances anti-aggregatory effects of the nitric oxide donor sodium nitroprusside. J Cardiovasc Pharmacol. 2005;46(4):459–463. doi: 10.1097/01.fjc.0000176729.51819.a6. [DOI] [PubMed] [Google Scholar]
- 16.Grobe J.L., Mecca A.P., Lingis M., Shenoy V., Bolton T.A., Machado J.M. Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1–7) Am J Physiol Heart Circ Physiol. 2007;292(2):H736–H742. doi: 10.1152/ajpheart.00937.2006. [DOI] [PubMed] [Google Scholar]
- 17.Turner A.J., Hooper N.M. The angiotensin-converting enzyme gene family: genomics and pharmacology. Trends Pharmacol Sci. 2002;23(4):177–183. doi: 10.1016/s0165-6147(00)01994-5. [DOI] [PubMed] [Google Scholar]
- 18.Kakoki M., McGarrah R.W., Kim H.S., Smithies O. Bradykinin B1 and B2 receptors both have protective roles in renal ischemia/reperfusion injury. Proc Natl Acad Sci USA. 2007;104(18):7576–7581. doi: 10.1073/pnas.0701617104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guy J.L., Jackson R.M., Jensen H.A., Hooper N.M., Turner A.J. Identification of critical active-site residues in angiotensin-converting enzyme-2 (ACE2) by site-directed mutagenesis. FEBS J. 2005;272(14):3512–3520. doi: 10.1111/j.1742-4658.2005.04756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Towler P., Staker B., Prasad S.G., Menon S., Tang J., Parsons T. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem. 2004;279(17):17996–18007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rice G.I., Thomas D.A., Grant P.J., Turner A.J., Hooper N.M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J. 2004;383(Pt 1):45–51. doi: 10.1042/BJ20040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang L., Sexton D.J., Skogerson K., Devlin M., Smith R., Sanyal I. Novel peptide inhibitors of angiotensin-converting enzyme 2. J Biol Chem. 2003;278(18):15532–15540. doi: 10.1074/jbc.M212934200. [DOI] [PubMed] [Google Scholar]
- 23.Dales N.A., Gould A.E., Brown J.A., Calderwood E.F., Guan B., Minor C.A. Substrate-based design of the first class of angiotensin-converting enzyme-related carboxypeptidase (ACE2) inhibitors. J Am Chem Soc. 2002;124(40):11852–11853. doi: 10.1021/ja0277226. [DOI] [PubMed] [Google Scholar]
- 24.Crackower M.A., Sarao R., Oudit G.Y., Yagil C., Kozieradzki I., Scanga S.E. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 25.Oudit G.Y., Herzenberg A.M., Kassiri Z., Wong D., Reich H., Khokha R. Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin II-dependent glomerulosclerosis. Am J Pathol. 2006;168(6):1808–1820. doi: 10.2353/ajpath.2006.051091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gurley S.B., Allred A., Le T.H., Griffiths R., Mao L., Philip N. Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J Clin Invest. 2006;116(8):2218–2225. doi: 10.1172/JCI16980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donoghue M., Wakimoto H., Maguire C.T., Acton S., Hales P., Stagliano N. Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins. J Mol Cell Cardiol. 2003;35(9):1043–1053. doi: 10.1016/s0022-2828(03)00177-9. [DOI] [PubMed] [Google Scholar]
- 28.Zisman L.S., Keller R.S., Weaver B., Lin Q., Speth R., Bristow M.R. Increased angiotensin-(1–7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme homologue ACE2. Circulation. 2003;108(14):1707–1712. doi: 10.1161/01.CIR.0000094734.67990.99. [DOI] [PubMed] [Google Scholar]
- 29.Ferrario C.M., Trask A.J., Jessup J.A. Advances in biochemical and functional roles of angiotensin-converting enzyme 2 and angiotensin-(1–7) in regulation of cardiovascular function. Am J Physiol Heart Circ Physiol. 2005;289(6):H2281–H2290. doi: 10.1152/ajpheart.00618.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burrell L.M., Risvanis J., Kubota E., Dean R.G., MacDonald P.S., Lu S. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J. 2005;26(4):369–375. doi: 10.1093/eurheartj/ehi114. discussion 322–4. [DOI] [PubMed] [Google Scholar]
- 31.Ishiyama Y., Gallagher P.E., Averill D.B., Tallant E.A., Brosnihan K.B., Ferrario C.M. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension. 2004;43(5):970–976. doi: 10.1161/01.HYP.0000124667.34652.1a. [DOI] [PubMed] [Google Scholar]
- 32.Yang W., Huang W., Su S., Li B., Zhao W., Chen S. Association study of ACE2 (angiotensin I-converting enzyme 2) gene polymorphisms with coronary heart disease and myocardial infarction in a Chinese Han population. Clin Sci (Lond) 2006;111(5):333–340. doi: 10.1042/CS20060020. [DOI] [PubMed] [Google Scholar]
- 33.Li X., Rayford H., Uhal B.D. Essential roles for angiotensin receptor AT1a in bleomycin-induced apoptosis and lung fibrosis in mice. Am J Pathol. 2003;163(6):2523–2530. doi: 10.1016/S0002-9440(10)63607-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marshall R.P., Webb S., Bellingan G.J., Montgomery H.E., Chaudhari B., McAnulty R.J. Angiotensin-converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2002;166(5):646–650. doi: 10.1164/rccm.2108086. [DOI] [PubMed] [Google Scholar]
- 35.Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ware L.B., Matthay M.A. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 38.Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309(5742):1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 39.Li W., Zhang C., Sui J., Kuhn J.H., Moore M.J., Luo S. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24(8):1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuba K., Imai Y., Rao S., Jiang C., Penninger J.M. Lessons from SARS: control of acute lung failure by the SARS receptor ACE2. J Mol Med. 2006;84(10):814–820. doi: 10.1007/s00109-006-0094-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fasciolo J.C. The renin content of kidney. Acta Physiol Lat Am. 1950;1(1):7–19. [PubMed] [Google Scholar]
- 42.Lieberman J., Sastre A. Angiotensin-converting enzyme activity in postmortem human tissues. Lab Invest. 1983;48(6):711–717. [PubMed] [Google Scholar]
- 43.Brown C.A., Zusman R.M., Haber E. Identification of an angiotensin receptor in rabbit renomedullary interstitial cells in tissue culture: correlation with prostaglandin biosynthesis. Circ Res. 1980;46(6):802–807. doi: 10.1161/01.res.46.6.802. [DOI] [PubMed] [Google Scholar]
- 44.Mezzano S.A., Ruiz-Ortega M., Egido J. Angiotensin II and renal fibrosis. Hypertension. 2001;38(3 Pt 2):635–638. doi: 10.1161/hy09t1.094234. [DOI] [PubMed] [Google Scholar]
- 45.Ye M., Wysocki J., William J., Soler M.J., Cokic I., Batlle D. Glomerular localization and expression of angiotensin-converting enzyme 2 and angiotensin-converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol. 2006;17(11):3067–3075. doi: 10.1681/ASN.2006050423. [DOI] [PubMed] [Google Scholar]
- 46.Li N., Zimpelmann J., Cheng K., Wilkins J.A., Burns K.D. The role of angiotensin converting enzyme 2 in the generation of angiotensin 1–7 by rat proximal tubules. Am J Physiol Renal Physiol. 2005;288(2):F353–F362. doi: 10.1152/ajprenal.00144.2004. [DOI] [PubMed] [Google Scholar]
- 47.Wysocki J., Ye M., Soler M.J., Gurley S.B., Xiao H.D., Bernstein K.E. ACE and ACE2 activity in diabetic mice. Diabetes. 2006;55(7):2132–2139. doi: 10.2337/db06-0033. [DOI] [PubMed] [Google Scholar]
- 48.Tikellis C., Johnston C.I., Forbes J.M., Burns W.C., Burrell L.M., Risvanis J. Characterization of renal angiotensin-converting enzyme 2 in diabetic nephropathy. Hypertension. 2003;41(3):392–397. doi: 10.1161/01.HYP.0000060689.38912.CB. [DOI] [PubMed] [Google Scholar]
- 49.Soler MJ, Wysocki J, Ye M, Lloveras J, Kanwar Y, Batlle D. ACE2 inhibition worsens glomerular injury associated with increased ACE expression in streptozotocin-induced diabetic mice. Kidney Int; 2007. [DOI] [PubMed]
- 50.Brosnihan K.B., Neves L.A., Joyner J., Averill D.B., Chappell M.C., Sarao R. Enhanced renal immunocytochemical expression of ANG-(1–7) and ACE2 during pregnancy. Hypertension. 2003;42(4):749–753. doi: 10.1161/01.HYP.0000085220.53285.11. [DOI] [PubMed] [Google Scholar]
- 51.Paizis G., Tikellis C., Cooper M.E., Schembri J.M., Lew R.A., Smith A.I. Chronic liver injury in rats and humans upregulates the novel enzyme angiotensin converting enzyme 2. Gut. 2005;54(12):1790–1796. doi: 10.1136/gut.2004.062398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herath CB, Warner FJ, Lubel JS, Dean RG, Jia Z, Lew RA, et al. Upregulation of hepatic angiotensin-converting enzyme 2 (ACE2) and angiotensin-(1–7) levels in experimental biliary fibrosis. J Hepatol; 2007. [DOI] [PMC free article] [PubMed]
- 53.Zhang H., Wada J., Hida K., Tsuchiyama Y., Hiragushi K., Shikata K. Collectrin, a collecting duct-specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. J Biol Chem. 2001;276(20):17132–17139. doi: 10.1074/jbc.M006723200. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y., Wada J., Yasuhara A., Iseda I., Eguchi J., Fukui K. The role for HNF-1beta-targeted collectrin in maintenance of primary cilia and cell polarity in collecting duct cells. PLoS ONE. 2007;2:e414. doi: 10.1371/journal.pone.0000414. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]