

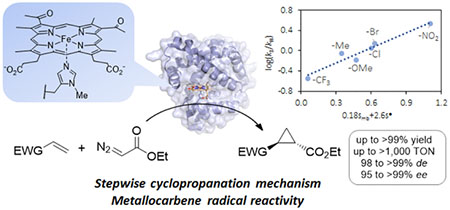
Abstract
Engineered myoglobins and other hemoproteins have recently emerged as promising catalysts for asymmetric olefin cyclopropanation reactions via carbene transfer chemistry. Despite this progress, the transformation of electron-poor alkenes has proven very challenging using these systems. Here, we describe the design of a myoglobin-based carbene transferase incorporating a non-native iron-porphyrin cofactor and axial ligand, as an efficient catalyst for the asymmetric cyclopropanation of electron-deficient alkenes. Using this metalloenzyme, a broad range of both electron-rich and electron-deficient alkenes are cyclopropanated with high efficiency and high diastereo- and enantioselectivity (up to >99% de and ee). Mechanistic studies revealed that the expanded reaction scope of this carbene transferase is dependent upon the acquisition of metallocarbene radical reactivity as a result of the reconfigured coordination environment around the metal center. The radical-based reactivity of this system diverges from the electrophilic reactivity of myoglobin and most of known organometallic carbene transfer catalysts. This work showcases the value of cofactor redesign toward tuning and expanding the reactivity of metalloproteins in abiological reactions and it provides a biocatalytic solution to the asymmetric cyclopropanation of electrodeficient alkenes. The metallocarbene radical reactivity exhibited by this biocatalyst is anticipated to prove useful in the context of a variety of other synthetic transformations.
Keywords: Cyclopropanation, electrondeficient olefins, myoglobin, carbene transfer catalysis, radical mechanism, Hammett
Graphical Abstract
Introduction
The transition metal-catalyzed cyclopropanation of alkenes with diazo compounds constitutes a convenient and direct strategy for the construction of cyclopropane rings,1–4 which are key structural motifs found in many drug molecules and biologically active natural products.5–8 While several transition metal complexes have proven effective for executing olefin cyclopropanations via carbene transfer chemistry,1–4 the development of biocatalytic strategies for realizing these transformations is highly desirable toward the implementation of sustainable methods for chemical synthesis. Over the past few years, iron-based heme-containing proteins and enzymes, such as myoglobins9–13 and cytochrome P450s14–18, respectively, have been identified as promising biocatalysts for promoting olefin cyclopropanation reactions with diazo compounds. In particular, our group previously reported the ability of engineered variants of sperm whale myoglobin (Mb) to catalyze the asymmetric cyclopropanation of vinylstyrenes in the presence of ethyl α-diazoacetate (EDA)9, 10 or other acceptor-only diazo compounds,11, 12 with high levels of diastereo- and enantioselectivity. Artificial metalloenzymes have also been developed and investigated for promoting abiological cyclopropanation reactions.19–25
Recent experimental and computational studies have provided insights into the mechanism of hemoprotein-catalyzed cyclopropanation reactions9, 26–28 and the role of the protein scaffold in controlling the stereoselectivity of these transformations.29 These studies support a mechanistic scenario in which an heme-carbenoid intermediate is initially formed upon reaction of the ferrous hemoprotein with the diazo reagent. This intermediate was shown to possess electrophilic reactivity9, 26 and be capable of engaging olefins and other nucleophiles30–32 in a carbene transfer reaction manifold. In the context of iron-porphyrin and hemoprotein-catalyzed intermolecular cyclopropanations with α-diazo esters, computational and experimental analyses support a catalytic mechanism involving a concerted, asynchronous insertion of the heme-bound carbenoids into the C=C bond of the olefin substrate.27, 29
Despite the progress made in developing biocatalytic strategies for olefin cyclopropanation, the scope of these transformations has remained largely restricted to aryl-substituted and aliphatic olefins, limiting the types of cyclopropane structures accessible using biocatalysis. In particular, the cyclopropanation of electron-deficient olefins has remained a largely unmet goal in this area. In the presence of diazoesters, these transformations can give access to electrophilic cyclopropanes carrying two electron-withdrawing groups, which represent valuable building blocks for medicinal chemistry as well as key intermediates for various synthetic applications.33–35 While currently lacking, biocatalytic approaches for realizing these transformations would be highly desirable.
Modification of the metal coordination environment can have a profound effect on the functional properties in metalloenzymes/proteins.18, 20, 21, 24, 36–48 In previous work, we demonstrated how alteration of the heme cofactor environment in myoglobin through substitution of the metal center,20, 49, 50 ‘proximal’ residue,20, 42 and/or porphyrin ligand,21 can alter the reactivity of this metalloprotein in the context of non-native nitrene and carbene transfer reactions. Here, we report the design of a cofactor-reconfigured myoglobin-based catalyst useful for the stereoselective cyclopropanation of electron-deficient olefins with diazoesters. Mechanistic studies show how the expanded reaction scope of this metalloprotein toward these challenging substrates is dependent upon the acquisition of radical carbene transfer reactivity and a dramatic change in the cyclopropanation mechanism, as a result of the combined effect of the non-native iron-porphyrin cofactor and axial ligand integrated into this metalloprotein.
Results and Discussion
Design and Generation of Cofactor Reconfigured Carbene Transferase
The reactivity of myoglobin (Mb) in carbene transfer reactions stems from its hemin cofactor (iron-protoporphyrin IX), which is coordinated at the iron center by the side-chain imidazolyl group of a conserved histidine residue (His93 in sperm whale myoglobin) as the ‘proximal’ axial ligand.51 The latter contributes to anchor the cofactor to the protein and participates in modulating both the redox and the oxygen binding properties of this metalloprotein.51 While engineered Mb have proven to be effective catalysts for the cyclopropanation of aryl-substituted olefins and aliphatic olefins,9, 10, 12, 29 the cyclopropanation of electron-deficient olefins has proven challenging using these systems. Notably, the cyclopropanation of electron-deficient alkenes has represented a notorious challenge also for synthetic carbene transfer catalysts,52–54 a phenomenon attributed to the electrophilic character of the metal-bound carbenes typically involved in these reactions.
To overcome these limitations, we envisioned the possibility of enhancing the electrophilic reactivity of the heme-carbene intermediate through modification of the heme cofactor and proximal ligand, as these are bound to directly influence the electronic properties of this reactive species in Mb-mediated cyclopropanation.27 As the parent scaffold for harboring these substitutions, we selected our previously reported variant Mb(H64V,V68A) (1, Figure 1a) due to its excellent stereoselectivity (>90–99% de and ee) and broad substrate scope in the cyclopropanation of vinylarenes with EDA.9, 10 Based on the considerations outlined above, we decided to substitute the native heme in Mb(H64V,V68A) (1) with iron-2,4-diacetyl deuteroporphyrin IX (Fe(DADP)) (Figure 1a), an electron-deficient heme analog amenable to incorporation into this hemoprotein.37 The incorporation of Fe(DADP) was previously shown to increase the Fe3+/Fe2+ reduction potential (Eo; couple) of Mb,37 a result consistent with the effect of the electronwithdrawing acetyl groups conjugated to the tetrapyrrole ring toward destabilizing the ferric form of the metalloprotein. This non-native cofactor was thus expected to potentially enhance the electrophilic character of the heme-carbene intermediate in the context of cyclopropanation reactions. Given the critical role of the proximal axial ligand in affecting the functional properties of myoglobin, we designed a second variant in which the Fe(DADP) cofactor is coordinated at the axial position by the non-canonical amino acid N-methyl-histidine (NMH, Figure 1a) in place of the native histidine ligand (His 93). This substitution was also reported to increase the Eo of Mb,46 an effect attributed to the loss of a hydrogen bond interaction between the proximal His ligand and a serine residue within the proximal pocket of the protein.55, 56 The aforementioned cofactor and axial ligand modifications were thus envisioned to potentially cooperate toward affecting the reactivity of the Mb variant as a carbene transferase, with the change in redox properties providing a proxy for detecting the occurrence of such additive or synergistic effect.
Figure 1.

a) High-resolution crystal structure29 of sperm whale Mb(H64V,V68A) (1) and structure of histidine- and N-methyl-histidine-ligated Fe(DADP) cofactor. b) Absorption spectra for ferric, ferrous and CO-bound form of Mb(H64V,V68A)[Fe(DADP)] (2). c) Absorption spectra for ferric, ferrous and CO-bound form of Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3). See Figure S1 for spectra corresponding to Mb(H64V,V68A) (1).
The corresponding Mb variants, Mb(H64V,V68A)[Fe(DADP)] (2) and Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3), could be successfully produced and isolated from E. coli in good yields (23 and 5 mg protein/L culture, respectively). Incorporation of the non-native Fe(DADP) cofactor was effectively achieved by adapting a protocol previously used for the recombinant production of cofactor-substituted variants of Mb20, 21 and other hemoproteins,57 whereas NHM was introduced at the axial position via amber stop codon suppression58 in E. coli cells co-expressing an orthogonal aminoacyl-tRNA/tRNA pair specifically evolved for genetic incorporation of this non-canonical amino acid.59
Characterization of Fe(DADP)-based Mb variants
Characterization of Mb(H64V,V68A)[Fe(DADP)] (2) and Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) by UV-vis spectroscopy showed a small blue-shift of the Soret band of the Fe(DADP) complex upon incorporation into these proteins (412–415 nm vs. 417 nm for Fe(DADP) alone; Figure 1b and 1c). Incubation of the Mb variants with sodium dithionite induces a shift of the Soret band from 415 nm to 434 and from 412 nm to 430 nm, respectively, corresponding to reduction of the protein from the ferric to the ferrous form. Further incubation with carbon monoxide resulted in a further shift of the Soret band to 428 and 422 nm, respectively, corresponding to CO-bound complex. Overall, the spectral features of the cofactor-substituted variants in their ferric, ferrous, and CO-bound form are similar, albeit not identical, to each other and compared to the heme-containing counterpart (Figure S1).
To assess the impact of the cofactor and axial ligand substitution on the stability of the protein, their apparent melting temperature (Tm) was determined via thermal denaturation experiments using circular dichroism (CD) (Figure 2 and S2). These studies revealed that incorporation of the Fe(DADP) cofactor has no deleterious effect on the stability of the protein compared to heme-containing counterpart (Tm = 66.3°C for Mb(H64V,V68A)[Fe(DADP)] (2) vs. 66.0°C for Mb(H64V,V68A) (1)60). Although substitution of the proximal His with N-methyl-histidine causes a slight destabilization of the protein (ΔTm = −2.9°C), the Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) variant retains significant thermostability (Tm = 63.4°C; Figure 2), indicating that both structural modifications are well tolerated by the Mb scaffold.
Figure 2.

Thermal denaturation curves for Mb(H64V,V68A) (1), Mb(H64V,V68A)[Fe(DADP)] (2) and Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) as determined by circular dichroism (θ222).
The effect of these cofactor modifications on the Fe3+/Fe2+ reduction potential (E°) of the metalloprotein was then investigation using a UV-vis spectrochemical method.61 For comparison purposes, wild-type sperm whale Mb as well as a variant incorporating only the proximal ligand substitution, Mb(H64V,V68A,H93NMH), were included in these analyses. As expected, these experiments showed a shift of the redox potential toward more positive values for both the Fe(DADP)- and NMH-containing variant (E°Fe3+/Fe2+ = 59 ± 1 mV and 77 ± 6 mV, respectively; Figure S3b–c), compared to the parent protein Mb(H64V,V68A) (1) (E°Fe3+/Fe2+ = 54 ± 1 mV; Figure S3a). Using the same spectrochemical assay, wild-type Mb was determined to have an E° value of 47 ± 1 mV, which is very similar to those previously reported in the literature using other methods.37, 62 Notably, the combination of the Fe(DADP) cofactor with the NMH axial ligand leads to a dramatic increase of the redox potential, resulting in a E° value of 146 ± 3 mV for the Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) variant (Figure S3d). The nearly 100 mV increase in E° for the latter variant compared to the +6 to +23 mV increases for the Fe(DADP)- and NMH-containing variant, respectively, indicated a clear synergistic role of the non-native cofactor and axial ligand toward affecting the redox potential of the metalloprotein.
Cyclopropanation Activity
In order to evaluate the catalytic activity of Mb(H64V,V68A)[Fe(DADP)] (2) and Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) as cyclopropanation catalysts, these proteins were tested in a model reaction with styrene (4) and ethyl diazoacetate (EDA) under “standard reaction conditions” (10 mM styrene, 20 mM EDA, 10 mM sodium dithionite (Na2S2O4), 0.1 mol% protein, anaerobic conditions). As summarized in Table 1, both proteins produce the desired product 5a in quantitative yield and with excellent diastereo- and enantiomeric excess (>99% de and >98–99% ee; Table 1, Entry 2–3). The high cyclopropanation activity exhibited by Mb(H64V,V68A)[Fe(DADP)] (2) and Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) and the fact that both reactions proceed cleanly without the formation of carbene dimerization byproducts (diethyl maleate and fumarate) indicate that the [Fe(DADP)] cofactor can efficiently activate the diazo compound (EDA) promoting the carbene transfer reaction. In addition, both catalysts preserve the high stereoselectivity of the parent Mb(H64V,V68A) (1) variant, indicating that in either case the cofactor and cofactor/axial ligand substitution do not disrupt the active site structure of the protein.29
Table 1.
Catalytic activity of Mb(H64V,V68A) (1) and cofactor substituted variants thereof in the cyclopropanation of styrene with EDA.a
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Xb | Yield | TON | % de | % ee |
| 1 | 1 | -/red | >99% | >1000 | >99 | >99 |
| 2 | 2 | -/red | >99% | >1000 | >99 | >99 |
| 3 | 3 | -/red | >99% | >1000 | >99 | 98 |
| 4 | 1 | O2/red | 62% | 620 | >99 | >99 |
| 5 | 2 | O2/red | 39% | 390 | >99 | >99 |
| 6 | 3 | O2/red | 85% | 850 | >99 | 98 |
| 7 | 1 | −/− | 10% | 150 | >99 | >99 |
| 8 | 2 | −/− | 86% | 865 | >99 | >99 |
| 9 | 3 | −/− | >99% | >1000 | >99 | >99 |
| 10 | 3 | O2/- | 12% | 125 | >99 | >99 |
| 11c | 3 | −/− | >99% | 2155 | >99 | >99 |
| 12d | 3 | −/− | 40% | 4010 | >99 | >99 |
Reaction conditions: 10 mM styrene (4), 20 mM EDA, 10 μM purified Mb variant in KPi 50 mM (pH 7), r.t., 16 hours. Yield, diastereomeric and enantiomeric excess determined by chiral GC-FID analysis using 1,3-benzodioxole as internal standard.
Variable parameter (X): “-/red”=anaerobic, 10 mM Na2S2O4; “−/−“=anaerobic, no reductant; “O2/red”=aerobic, 10 mM Na2S2O4; “O2/-“=aerobic, no reductant.
Reaction performed using 5 μM protein.
Reaction performed using 1 μM protein.
Since modification of the porphyrin cofactor and/or proximal ligand can affect the sensitivity of Mb-based carbene transferases to oxygen and/or non-reducing conditions,20, 21, 42, 47 the cyclopropanation activity of the [Fe(DADP)]-based variants was also tested in the presence and in the absence of oxygen and/or reductant (Na2S2O4) (Table 1 and Table S1). While the catalytic activity of Mb(H64V,V68A) (1) decreases in the presence of oxygen (62% vs. >99% yield), Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) maintains high cyclopropanation activity (85% yield) under aerobic and reducing conditions (Table 1, Entry 6 vs. 4). This activity is higher than in the presence of the Fe(DADP) cofactor or the His93NMH substitution alone (Table S1, Entry 3), indicating a combined effect of these substitutions toward increasing the oxygen tolerance of the biocatalyst. Unlike Mb(H64V,V68A) (1), both [Fe(DADP)]-based variants and in particular Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) exhibit high cyclopropanation activity (86–100% yield) also under non-reducing (and anaerobic) conditions (Table 1, Entry 7–9).
As the [Fe(DADP)]-containing variants are prepared as ferric proteins, the catalytic activity under non-reducing conditions can be explained based on the ability of these variants to undergo in situ reduction by the EDA reagent to produce the catalytically active ferrous form42 or, alternatively, by their ability to catalyze this reaction also in their ferric form, as previously observed for an iron-chlorin e6 containing Mb variant.21 To gain insights into this aspect, the same reactions were carried out in the presence of CO, which binds to the ferrous but not the ferric form these proteins as confirmed by UV-vis spectroscopy (Figure 1b–c and Figure S4). CO binding to the iron center is expected to inhibit cyclopropanase activity. Under these conditions, the cyclopropanation product 5a was obtained only in trace amounts (Table S1, Entry 8). Control experiments ruled out that CO-mediated reduction63 of this protein occurs within a timescale relevant for observation of cyclopropanation activity (i.e., <30–45 min) (Figure S4). These results suggest that in situ generated ferrous form may contribute to the observed catalytic activity of the Fe(DADP)-containing variants under non-reducing conditions. Given the lower activity of Mb(H64V,V68A) (1) under these conditions, it is possible that the Fe(DADP) cofactor makes these variants susceptible to reduction by EDA, which can act as a mild reducing agent,64–67 a behavior that may be at least in part attributed to their increased redox potential (Eo) compared to the heme-containing parent protein.
Reactivity toward Aryl- and Heteroatom-Substituted Olefins
Based on the positive results from the studies above, we then assessed the catalytic performance of the Fe(DADP)-based variants toward the cyclopropanation of aryl-substituted olefins under non-reducing and anaerobic conditions. As shown in Scheme 1, a diverse panel of para-, ortho-, meta-, and α- substituted styrene derivatives were efficiently converted to the desired cyclopropanation products 5b-h in good to excellent yield, and excellent trans-diastereoselectivity (98 to >99% de) and (1S,2S)-enantioselectivity (98% to >99% ee). Both electron-donating (5b-e) and electron-withdrawing groups (5f-h) in the phenyl ring were well tolerated. In addition, the sterically more demanding α-methylstyrene and 2-vinyl-naphtalene were also efficiently converted to the corresponding cyclopropane 5i and 5j with high diastereo- and enantioselectivity. Moreover, unlike Mb(H64V,V68A)[Fe(DADP)] (2), Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) showed also good activity toward non-styrenyl substrates such as phenylvinylether and phenylvinylsulfane, producing 5k, and 5l, respectively, in good to high yield and with high stereoselectivity. The number of catalytic turnovers (TON) observed with these substrates are 2- to 3-fold higher than those obtained with Mb(H64V,V68A) (1) under reducing conditions. The unactivated olefin substrate (allyloxy)benzene was also converted by Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) into the cyclopropane product 5m. In this case, the Fe(DADP)-based catalyst also offers improved stereoselectivity (>99% de and ee) compared to the parent enzyme (95% de, 86% ee).
Scheme 1.

Substrate scope of Mb(H64V,V68A)[Fe(DADP)] (2) and Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3).a
a Reaction conditions: 10 mM alkene, 20 mM EDA, 10 μM Mb variant 2 or 3 in KPi buffer (50 mM, pH 7), r.t., 16 hrs, anaerobic conditions. Yield and diastereomeric excess were determined by chiral GC-FID analysis. Enantiomeric excess was determined by chiral GC-FID or SFC analysis. b 50 mM alkene, 20 mM EDA, 20 μM catalyst 3.
Stereoselective Cyclopropanation of Electron-Deficient Olefins
The high cyclopropanation activity of Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) toward the monohalogenated styrenes (products 5f, 5g, and 5h in Scheme 1) was in line with our goal of enhancing the reactivity of Mb-based systems toward electron-poor olefins and encouraged us to assess its scope toward more challenging substrates such as trifluoromethyl-, nitro-, and pentafluoro-styrenes (Scheme 2). Gratifyingly, the corresponding cyclopropanation products 5n-q could be obtained in high (69–85%) to quantitative yield (5o), along with high diastereo- and enantioselectivity (98 to >99% de; 86 to >99% ee). Notably, 4-vinylbenzensulfonamide, whose cyclopropanation failed using Mb(H64V,V68A) (1) or commonly adopted organometallic carbene transfer catalysts such as Rh2(OAc)4 and Fe(TPP)Cl (Table S2), was successfully cyclopropanated in the presence of Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) to afford 5r in 48% yield and >99% de and 84% ee (Scheme 2).
Scheme 2.

Mb(H64V,V68A,H93NMH)[Fe(DADP)]-catalyzed cyclopropanation of electrondeficient olefins.a
a Reaction conditions: 10 mM alkene, 20 mM EDA, 10 μM catalyst 3 in KPi buffer (50 mM, pH 7), r.t., 16 hrs, anaerobic conditions. Yield and diastereomeric excess were determined by chiral GC-FID analysis. Enantiomeric excess was determined by chiral GC-FID or SFC analysis. b 20 mM alkene, 10 mM EDA, 20 μM 3. c 50 mM alkene, 20 mM EDA, 20 μM 3. d 2.5 mM alkene, 2.5 mM EDA, 5 μM 3.
Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) could be successfully applied to the stereoselective cyclopropanation of a variety of other electron-deficient non-styrenyl olefins. While acrylate esters are notoriously challenging substrates for transition metal-catalyzed cyclopropanation,52, 68 phenyl and benzyl acrylate could be converted to the desired products 5s and 5t, respectively, in good to high yields (49–89%) and high enantiopurity (98–99% de; 99% ee). Vinyl benzoate, which is unreactive in the presence of Fe(TPP)Cl and Mb(H64V,V68A) (1) and only poorly reactive toward Rh2(OAc)4 (Table S3), was also efficiently cyclopropanated by Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) to give enantiopure 5u in quantitative yield with 99% de and 97% ee. Time-course experiments showed this reaction proceeds rapidly, with >90% of the final product being formed within the first 10 minutes and that it is faster than the cyclopropanation of styrene (Figure S5). It is also worth noting that this transformation can provide an expedite route (i.e., via hydrolysis of 5u) to cyclopropanols, which are valuable building blocks for drug synthesis. Finally, the Mb(H64V,V68A,H93NMH)[Fe(DADP)]-catalyzed cyclopropanation of N-vinyl-amides and succinamides was also successful, affording the corresponding cyclopropanes 5v and 5w in good to moderate yields (75–33%) and high stereoselectivity. Comparable selectivity but somewhat reduced yield (by 20–30%) were obtained for the Mb(H64V,V68A,H93NMH)[Fe(DADP)]-catalyzed reactions with the electrondeficient olefins in the presence of sodium dithionite, indicating an optimal performance of this catalyst under non-reducing conditions.
In general, Mb(H64V,V68A)[Fe(DADP)] (2) and Mb(H64V,V68A,H93NMH) proved to be inferior catalysts for the cyclopropanation of the electrodeficient alkenes compared to Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3). For example, cyclopropane 5r and 5u were obtained in <20% yields using either of these catalysts compared to 50–80% yields obtained with Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) under the same reaction conditions (Tables S2 and S3). Mirroring the observed effect on other functional properties (i.e. E0, sensitivity to reductant/oxygen), these results point at a synergistic effect of the Fe(DADP) cofactor and axial N-methyl-histidine ligand toward conferring high reactivity toward these challenging substrates.
Mechanistic Studies
To gain insights into the mechanistic basis for the divergent reactivity of Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) compared to His-ligated heme counterpart, we measured the relative rates of these catalysts for the cyclopropanation of a series of para-substituted styrenes vs. styrene via competition experiments. Consistent with our previous observations,9 styrene derivatives carrying electrondonating groups (i.e., -Me, -OMe) showed higher reactivity than styrene in the Mb(H64V,V68A)-catalyzed reactions, whereas electron-deficient styrenes (e.g., p-CF3, p-NO2) exhibited reduced cyclopropanation rates compared to styrene (Table 2, Figure S6). Plots of the corresponding log(kX/kH) values against the Hammett constants69 σp and σ+ gave relatively good linear correlations, yielding a ρp value of −1.20 (R2 = 0.84, Figure 3a) and a ρ+ value of −0.77 (R2 = 0.77), respectively. Similar plots generated after excluding p-bromo-styrene, which showed the largest deviation from the trend, yielded even stronger linear correlations (R2 = 0.88–0.93) without affecting the ρ values (ρp = −1.20; ρ+ = −0.79). The large negative ρ values is consistent with the electrophilic nature of the metal carbene intermediate involved in these reactions.9, 27
Table 2.
Relative rates scales for the cyclopropanation of para-substituted styrene catalyzed by Mb(H64V,V68A) (1) and Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) and sigma and sigma-dot scales for para substituents.a
 | ||||||
|---|---|---|---|---|---|---|
| Substituent | kX / kH (catalyst 1) |
kX / kH (catalyst 3) |
σp | σ+ | σmb | σ•JJ |
| -OMe | 1.04 | 0.65 | −0.27 | −0.778 | −0.77 | 0.23 |
| -Me | 0.61 | 0.89 | −0.17 | −0.31 | −0.29 | 0.15 |
| -Cl | 0.39 | 1.12 | 0.23 | 0.11 | 0.11 | 0.22 |
| -Br | 0.66 | 1.39 | 0.23 | 0.15 | 0.13 | 0.23 |
| -CF3 | 0.16 | 0.28 | 0.54 | 0.61 | 0.49 | −0.01 |
| -NO2 | 0.04 | 3.43 | 0.78 | 0.79 | 0.86 | 0.36 |
Figure 3.

Hammett plots. (a) Plot of log(kX/kH) for Mb(H64V,V68A) (1) vs. σ constant. (b-d) Plots of log(kX/kH) for Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) vs. σ constant (b), σ•JJ constant (c) and 0.18σmb+2.6σ•JJ (d).
In stark contrast to the reactivity trend exhibited by Mb(H64V,V68A) (1), the rate of Mb(H64V,V68A,H93NMH)[Fe(DADP)]-catalyzed cyclopropanation is accelerated by the presence of electron-withdrawing groups in the styrene substrates, whereas reduced cyclopropanation rates are observed with the electron-rich styrenes relative to unsubstituted styrene (Table 2, Figure S7). The corresponding log(kX/kH) values showed no correlation with the polar substituent constant σp or σ+ (R2 = 0.07–0.10) (Figure 3b). The non-linearity of these plots, along with our previous observations concerning the radical mechanism of hemoprotein-catalyzed nitrene transfer reactions,70 prompted to us to consider the potential involvement of intermediates with significant radical character in Mb(H64V,V68A,H93NMH)[Fe(DADP)]-catalyzed cyclopropanation.
Supporting this hypothesis, a very good linear correlation (R2 = 0.86) was obtained between the log(kX/kH) values and Jiang’s spin-delocalization substituent constants σ•JJ72 (Figure 3c; Table 3). An even stronger correlation (R2 = 0.93) was achieved using the dual-parameter equation log(kX/kH) = ρXσX + ρ•σ•, which takes into consideration of both the polar (σX) and spin-delocalization effects (σ•) of the substituents.73–75 Fitting of this equation to the data via multiple linear regression using Jiang’s σmb71 polar scale and σ•JJ72 radical scale yielded a ρmb and ρ•JJ value of 0.18 and 2.60, respectively (Figure 3d, Table 3). While the improved correlation achieved with the extended equation compared to the radical scale alone suggests the participation of both polar and spin effects in affecting the rate of the reaction, the small ρmb/ρ•JJ ratio of 0.06 indicates a dominance of the spin-delocalization effect over the polar effect.75 In stark contrast, dual-parameter fitting of the Mb(H64V,V68A) (1) data yielded a large negative σmb (−0.79 vs. +0.18 for 3; Table 3) and a ρmb/ρ•JJ above the unity (= 1.5), i.e. features which have been attributed to reactions dominated by polar effects.75
Table 3.
Rho and R2 values for single- and dual-parameters correlations of log(kX/kH) values for Mb(H64V,V68A) (1) and Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) with sigma and sigma-dot parameters. N.a. = not applicable due to non-linear correlation.
| Catalyst | Parameters | ρX | ρmb | ρ•JJ | |ρmb/ρ•JJ| | R2 |
|---|---|---|---|---|---|---|
| 1 | σp | −1.20 | - | - | - | 0.84 |
| 1 | σ+ | −0.77 | - | - | - | 0.77 |
| 1 | σ•JJ | - | - | n.a. | - | 0.05 |
| 1 | σmb/ρ•JJ | - | −0.79 | −0.52 | 1.50 | 0.84 |
| 3 | σp | n.a. | - | - | - | 0.10 |
| 3 | σ+ | n.a | - | - | - | 0.07 |
| 3 | σ•JJ | - | - | 2.70 | - | 0.86 |
| 3 | σmb/σ•JJ | - | 0.18 | 2.60 | 0.06 | 0.93 |
To further probe the occurrence of a radical mechanism in the Mb(H64V,V68A,H93NMH)[Fe(DADP)]-catalyzed cyclopropanation process, reactions were carried out using EDA and the isotopically labelled substrate cis-β-deuterostyrene (d-4a) (see Experimental details), where a (partial) loss of stereospecificity would reveal the occurrence of a radical intermediate. Insightfully, this reaction led to the formation of d-5a in low yield as a 1.2:1 mixture of cis and trans isomers with respect to the -D and -Ph groups, as determined based on 1H and 2H NMR spectra (Scheme 4). In contrast, the same reaction in the presence of Mb(H64V,V68A) (1) proceeds stereospecifically with no scrambling of the deuterium label. In comparison, ~40% isomerization was observed for the same reaction using Co(TPP) as the catalyst,27 a result in line with the radical cyclopropanation mechanism previously established for this system.76, 77 Altogether, these experiments provided strong evidence for the intermediacy of a radical intermediate in the Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) reaction.
Scheme 4.

Enzymatic cyclopropanation reactions with cis-β-deuterostyrene (d-4a).
To further corroborate this conclusion, the Mb(H64V,V68A,H93NMH)[Fe(DADP)]-catalyzed reaction with styrene and EDA was carried out in the presence and in absence of the radical spin trapping agents 5,5-dimethyl-1-pyrroline N-oxide (DMPO) (Table 4). In the presence of DMPO, a significant reduction (by 58%) in the yield of the cyclopropane product 5a was observed under reducing conditions compared to a parallel reaction lacking the spin trapping agent (Entry 7 vs. 6). A similar effect but with a higher degree of inhibition (77% reduction in yield) was observed under non-reducing conditions. Very similar results were obtained from parallel experiments involving 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPOL) in place of DMPO as the spin trapping agent (Table 4, Entries 3 and 8). In contrast, the Mb(H64V,V68A)-catalyzed cyclopropanation reaction showed only a small reduction in yield upon addition of DMPO or TEMPOL (Table 4, Entries 2–3 vs. 1). In line with these results, kinetic experiments showed a drastic reduction of the cyclopropanation rate of Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) in the presence of DMPO compared to a reaction without it (38 vs. 670 turnovers min−1), whereas the cyclopropanation rate of Mb(H64V,V68A) (1) was only minimally affected by the spin trapping agent (510 vs. 550 turnovers min−1; Figure S10).
Table 4.
Inhibition experiments with spin trapping agents in styrene cyclopropanation reaction catalyzed by Mb(H64V,V68A) (1) or Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3).a
| Entry | Catalyst | Styrene | Inhibitor | Yield | % de | % ee |
|---|---|---|---|---|---|---|
| 1 | 1 | Yes | None | >99% | 99 | >99 |
| 2 | 1 | Yes | DMPO | 89% | 99 | >99 |
| 3 | 1 | Yes | TEMPOL | 90% | >99 | 99 |
| 4 | 1 | No | None | 95%b | - | - |
| 5 | 1 | No | DMPO | 80%b | - | - |
| 6 | 3 | Yes | None | >99% | >99 | >99 |
| 7 | 3 | Yes | DMPO | 42% | >99 | 99 |
| 8 | 3 | Yes | TEMPOL | 40% | >99 | 99 |
| 9 | 3 | No | None | 88%b | - | - |
| 10 | 3 | No | DMPO | 8%b | - | - |
Reaction conditions: 10 mM styrene, 20 mM EDA, 100 mM DMPO (or TEMPOL), 20 μM Mb variant, 10 mM sodium dithionite in 50 mM potassium phosphate buffer (pH 7) containing 10% DMF, r.t., 16 hours. Yield, diastereomeric and enantiomeric excess were determined by chiral GC-FID analysis.
Yield of diethyl fumarate and maleate determined by chiral GC-FID analysis.
To probe the step at which DMPO-mediated inhibition occurs, inhibition experiments were performed in the presence of EDA but in the absence of styrene, under which conditions the carbene dimerization products diethyl fumarate/maleate are produced. Insightfully, nearly full suppression (>95%) of the carbene dimerization product was observed in the presence of DMPO with Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) (Table 4, Entry 10 vs. 9), whereas only little inhibition (<20%) was observed with Mb(H64V,V68A) (1) (Table 4, Entry 5 vs. 4). Taken together, the Hammett analyses and radical rearrangement/trapping experiments converged in supporting (a) the occurrence of a step-wise radical-based mechanism for the Mb(H64V,V68A,H93NMH)[Fe(DADP)]-catalyzed olefin cyclopropanation reaction and (b) the intermediacy of a metallocarbene species with significant radical character, as evidenced by its susceptibility to inhibition by spin trapping agents.
On the basis of these results, a plausible mechanism for this reaction is proposed in Scheme 5. Under reducing conditions, the ferrous form of the protein (I) reacts with the diazo reagent to form an iron-bound carbene intermediate (II). While the structural and electronic features of this species awaits further elucidation, this intermediate clearly possesses distinct reactivity compared to the heme-bound carbene intermediate27 and possesses significant radical character, as evidenced by its reactivity toward the radical trapping agent. Upon generation of this species, metallocarbene-radical activation of the olefin substrates is envisioned to ensue, leading to a carbon-centered radical intermediate (III). Ring closure leads to the cyclopropane product and regenerates the catalyst.
Scheme 5.

Proposed mechanism for Mb(H64V,V68A,H93NMH)[Fe(DADP)]-catalyzed cyclopropanation reaction.
Under non-reducing conditions, a similar mechanism may be operative after generation of the catalytically competent ferrous form via in situ reduction of the ferric protein by the diazo compound. This reductive process could be facilitated by the increase in the redox potential associated with the incorporation of NMH-ligated Fe(DADP) cofactor compared to the histidine-ligated heme counterpart. While the CO inhibition experiments would support this hypothesis, a cyclopropanation pathway mediated by the ferric form of this protein is also considered viable. Fe(III)-based catalysts capable of promoting cyclopropanation reactions have been described67, 78–81 and we previously reported a Mb variant functioning as cyclopropanase in its ferric state.21 This alternative scenario is suggested by various pieces of experimental data, including the higher cyclopropanation activity of Mb(H64V,V68A,H93NMH)[Fe(DADP)] (3) with the electrondeficient olefins and the higher degree of DMPO-induced inhibition observed under non-reducing vs. reducing conditions. Future studies are warranted to elucidate these aspects in more detail.
Further Discussion
The radical-based stepwise mechanism operating in the present system is reminiscent of that reported for olefin cyclopropanation catalyzed by Zhang and de Bruin’s Co-porphyrins76, 77 and an open-shell iron alkylidene complex recently reported by Deng and coworkers,82 both of which systems were found to be reactive toward electron-deficient alkenes.52, 82 The reactivity of the Fe(DADP)-based biocatalyst 3 toward electron-poor olefins (Scheme 2) sets it apart from the electrophilic reactivity of Fisher-type metallocarbenes involved in cyclopropanation reactions catalyzed by Mb (ρ = − 1.20; Figure 3a) and various organometallic catalysts, including iron- (ρ = −0.6865; ρ+ = −0.4183), ruthenium- (ρ+ = −0.4484), and iridium-porphyrins (ρ+ = −0.2285), mononuclear Rh (ρ+ = −0.4086) and copper complexes (ρ = − 0.8587; ρ+ = −0.7686; ρ+ = −0.7488). Furthermore, the positive ρmb value (0.18), along with the large ρ•JJ (2.60), obtained for the Fe(DADP)-based catalyst suggest the involvement of a metallocarbene radical intermediate with some nucleophilic character. Such characteristics, which arise from a synergistic role of the Fe(DADP) cofactor and NMH axial ligand, can explain the expanded substrate scope of the cofactor engineered biocatalyst toward electron-poor (5n-5w; Scheme 2) and unactivated olefins (5m; Scheme 1) in addition to electron-rich alkenes (5a-5l; Scheme 1).
As supported by our results, reconfiguration of the first coordination sphere around the iron center via the porphyrin and axial ligand substitutions induces a dramatic change in the mechanism of the intermolecular olefin cyclopropanation investigated here, going from a (predominantly) concerted carbene transfer process dominated by polar effects and mediated by an electrophilic heme-carbene intermediate,27 to a stepwise radical mechanism dominated by spin delocalization effects and mediated by a metallocarbene intermediate with radical-type, dual nucleophilic/electrophilic character. While ligand-induced changes in reaction mechanisms have been in some cases observed for transition metal-catalyzed transformations,89, 90 this is the first instance, to the best of our knowledge, where this effect is achieved in the context of a metalloprotein.
It should be further noted that we previously observed a latent radical reactivity of free hemin in intermolecular cyclopropanation reactions with EDA (2% isomerization with β-deuterostyrene)27, whereas more recent studies from our laboratory showed evidence for the occurrence of radical-mediated rearrangements in Mb-catalyzed intramolecular cyclopropanation reactions.91 Based on this knowledge and the present work, we set forth the hypothesis that hemoproteins may possess a dual (or multistate) reactivity in carbene transfer reactions, possibly involving distinct intermediates and reaction pathways, akin to that proposed for metal- and metalloenzyme-catalyzed oxo transfer processes.92, 93 Importantly, the present studies demonstrate how modification of the first coordination sphere around the iron can provide an effective strategy for enhancing the radical-type carbene transfer reactivity of this system, enabling cyclopropanation reactions not readily accessible using their native cofactor-containing counterparts. Further studies are ongoing to elucidate the nature of the catalytic intermediates involved in these reactions and better understand the role played by the cofactor/axial ligands in favouring the observed radical-type reactivity, which is only rarely found among organometallic carbene transfer catalysts.76, 82
Conclusion
In summary, we have described the design and development of a metalloprotein catalyst useful for the stereoselective cyclopropanation of both electron-rich and electron-poor alkenes. A variety of electrondeficient styrenes and alkenes, which are either poorly reactive or unreactive toward heme-based protein catalysts, were effectively cyclopropanated by this system with high catalytic activity as well as high diastereo- and enantioselectivity. Our mechanistic studies show that the expanded reaction scope of this biocatalyst compared to the heme-based counterpart is dependent upon the acquisition of radical-type carbene transfer reactivity as a result of the reconfigured primary coordination sphere around the iron center through a N-methyl-histidine-ligated Fe(DADP) cofactor. Hammett analyses and radical rearrangement/trapping experiments provide evidence for the involvement of a radical-based, stepwise mechanism in these reactions, which differs from the concerted nonsynchronous carbene transfer mechanism previously found to dominate Mb- and iron-porphyrin-catalyzed intermolecular cyclopropanations with diazoesters.94, 95 This study highlights the value of cofactor engineering for tuning the reactivity and expanding the reaction scope of metalloproteins in the context of abiological reactions. Furthermore, in view of the unique and attractive features of radical-mediated reactions,96–99 the metallocarbene radical reactivity exhibited by the present system is expected to prove useful in the context of other synthetically valuable transformations.
EXPERIMENTAL DETAILS
General Information.
All chemicals and reagents were purchased from commercial suppliers (Sigma-Aldrich, Alfa Aesar, Frontier Scientific Inc, Chem-Impex Int.) and use without any further purification, unless otherwise stated. All dry reactions were carried out under argon pressure in oven-dried glassware with magnetic stirring using standard gas-tight syringes, cannula, and septa. 1H and 13C NMR spectra were measured on a Bruker DPX-400 instrument (operating at 400MHz for 1H, 100 MHz for 13C) or a Bruker DPX- instrument (operating at 500 MHz for 1H and 125 MHz for 13C). Column chromatography purification was carried out using AMD Silica Gel 60 230–400 mesh. Thin Layer Chromatography (TLC) was carried out using Merck Millipore TLC silica gel 60 F254 glass paltes. Gas chromatography (GC) analyses were carried out using a Shimadzu GC-2010 gas chromatograph equipped with a FID detector, and a Cyclosil-B column (30 m × 0.25 mm × 0.25μm film). Supercritical Fluid Chromatography (SFC) analysis were performed using a JASCO Analytical and Semi-Preparative SFC instrument equipped with a column oven (35 °C), photodiode array detector, a backpressure regulator (12.0 MPa), a carbon dioxide pump and a sample injection volume of 3 μL.
Synthetic Procedures.
Detailed procedures for the synthesis of Fe(DADP)Cl, cis-β-deuterostyrene and cyclopropanes 5a-w are provided in the Supporting Information.
Growth Media.
Cell cultures were grown in enriched M9 medium which was prepared follows. For 1 L, 779 mL deionized H2O were mixed with 200 mL M9 salts (5X) solution, 10 mL casamino acids (20% m/v), 10 mL glycol, 1 mL MgSO4 (2M), and 100 μL CaCl2 (1M). The M9 (5X) solution was prepared by dissolving 15 g Na2HPO4, 7.5 g K2HPO4, 2.5 g NaCl, 5 g NH4Cl in 1 L deionized H2O and sterilized by autoclaving. The casamino acids, glycol and MgSO4 solutions were autoclaved separately. The CaCl2 solution was sterilized by filtration. Enriched M9 agar plates were prepared by adding 17 g agar to 1 L of enriched M9 media containing all of the aforementioned components at the specified concentrations with the exception of CaCl2, which was added immediately prior to plating. To media and plates, ampicillin was added to a final concentration of 100 mg/L and chloramphenicol was added to a final concentration of 34 mg/L.
Protein Expression.
Cloning of MbH64V,V68A was described previously.9 Mb(H64V,V68A) was expressed in E.coli C41(DE3) cells as follows. After transformation, cells were grown in TB medium (ampicillin, 100 mg/L) at 37°C (200 rpm) until OD600 reached 0.6. Cells were then induced with 0.25 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG) and 0.3 mM γ-aminolevulinic acid (ALA). After induction, cultures were shaken at 180 rpm and 27 °C and harvested after 20 h by centrifugation at 4,000 rpm at 4°C. The cells were resuspended in 20 mL of Ni-NTA Lysis Buffer (50 mM KPi, 250 mM NaCl, 10 mM histidine, pH 8.0). Resuspended cells were frozen and stored at −80°C until purification.
Expression of Cofactor-Substituted Myoglobin variants.
Mb(H64V,V68A)Fe(DADP) was expressed in E.coli C41(DE3) cells using pET22_Mb(H64V,V68A) as follows. The cells were plated on a lysogeny broth agar plate containing ampicillin and a single colony of freshly transformed cells was cultured in 5 mL of lysogeny broth medium containing ampicillin. The overnight cultures were transferred to 1 L enriched M9 medium containing ampicillin, followed by incubation at 37°C with shaking (180 rpm). At OD600 of 1.4, cells were induced with 0.25 mM IPTG and the induced culture was shaken at 27°C and rpm 180 for additional 16 to 20 hours. After harvesting, the cells were pelleted by centrifugation, resuspended in 20 mL of Ni- NTA Lysis Buffer (50 mM KPi, 250 mM NaCl, 10 mM histidine, pH 8.0) and 20 μL of a cofactor solution (30 mg/mL in DMF) were added. Cells were lysed by sonication and the cell lysate was clarified by centrifugation (14,00 rpm, 4°C, 30 min). For expression of Mb(H64V,V68A,H93NMH)Fe(DADP), pET29b_Mb(H64V,V68A,H93NMH) and pEVOL-PyIRS(NMH) were used to transform E.coli BL21(DE3) and the cells were plated on a lysogeny broth agar plate containing ampicillin and chloramphenicol. A single colony of freshly transformed cells was cultured overnight in 5 mL of lysogeny broth medium containing ampicillin and chloramphenicol. The overnight cultures were transferred to 1 L enriched M9 medium containing ampicillin and chloramphenicol, followed by incubation at 37°C with shaking (180 rpm). At OD600 of 0.4, cell cultures were condensed by centrifugation (4,000 rpm, 4°C, 30 min) followed by resuspension of the cell pellets in 200 mL of enriched M9 medium. To the condensed expression cultures, N-3-methyl-L-histidine (NMH) and arabinose were added to a final concentration of X mM and X mM, respectively, and then cultures were shaken at 24°C and 180 rpm for 20 minutes before induction with 0.25 mM IPTG. The induced culture was shaken at 24°C and rpm 180 for additional 16 to 20 hours. After harvesting, the cells were pelleted by centrifugation, resuspended in 20 mL of Ni- NTA Lysis Buffer (50 mM KPi, 250 mM NaCl, 10 mM histidine, pH 8.0) and 20 μL of a cofactor solution (30 mg/mL in DMF) were added. Cells were lysed by sonication and the cell lysate was clarified by centrifugation (14,00 rpm, 4°C, 30 min).
Protein Purification.
The clarified lysate was transferred to a Ni-NTA column equilibrated with Ni-NTA Lysis Buffer. The resin was washed with 50 mL of Ni-NTA Lysis Buffer and then 50 mL of Ni-NTA Wash Buffer (50 mM KPi, 250 mM NaCl, 20 mM histidine, pH 8.0). Proteins were eluted with Ni-NTA Elution Buffer (50 mM KPi, 250 mM NaCl, 250 mM histidine, pH 7.0). After elution from the Ni-NTA column, the protein was buffer exchanged against 50 mM KPi buffer (pH 7.0) using 10 kDa Centricon filters. The concentration of the Mb variants (ferric form) was determined using the following extinction coefficients: ε410=157 mM−1cm−1 for Fe(ppIX)-containing variants and ε470=60 mM−1cm−1 for Fe(DADP)-containing variants, which was determined using the pyridine hemocrome assay (Analyt. Biochem. 1987, 161, 1).
Tm determination.
Thermal denaturation experiments were carried out using a JASCO J-1100 CD spectrophotometer equipped with variable temperature/wavelength denaturation analysis software and samples of purified Mb variant at 3 μM in 50 mM potassium phosphate buffer (pH 7.0). Thermal denaturation curves were measured by monitoring the change in molar ellipticity at 222 or 228 nm over a temperature range from 13°C to 100°C. The temperature increase was set to 0.5°C per minute with an equilibration time of 10 seconds. Data integration time for the melt curve was set to 4 seconds with a bandwidth of 1 nm. Linear baselines for the folded (θf) and unfolded state (θu) were generated using the low temperature (θf = mf T + bf) and high temperature (θu = mu T + bu) equations fitted to the experimental data before and after global unfolding, respectively Using these equations, the melt data were converted to fraction of folded protein (Ff) vs. temperature plots and the resulting curve was fitted to a sigmoidal equation (θfit) via nonlinear regression analysis in SigmaPlot from which apparent melting temperatures were derived. The reported mean values and standard errors were derived from experiments performed at least in duplicate.
Reduction potential determination.
Reactions were carried out on a 1 mL scale in a solution of KPi (50 mM, pH 7) containing xanthine (30 mM stock solution); protein; dye; catalase (10 mg/mL stock solution); a mixture of glucose (1 M stock solution)/glucose oxidase (175 μM stock solution), and xanthine oxidase (175 μM stock solution). In a sealed vial, a solution of buffer containing glucose (5 mM final concentration), glucose oxidase (50 μg/mL final concentration), and xanthine (300 μM final concentration) was degassed by bubbling argon for 3 min. A buffered solution containing the myoglobin variant and dye was carefully degassed in a similar manner in a sealed cuvette (the concentration of the dye was adjusted by titration to give an absorbance which is approximately equal to that of the highest absorbance band in the protein spectra). The two solutions were then mixed together via cannula, and then catalase (5 μg/mL final concentration) and xanthine oxidase (50 nM final concentration) were added to initiate the two-electron oxidation of xanthine to uric acid and the corresponding reduction of protein and dye. The reactions were monitored by UV–Vis spectrophotometry and the data were plotted. The reduction potential was determined by adding the standard reduction potential of dye to the value of the y intercept obtained by fitting the data to the Nernst equation (Eq. 1).
| (Eq. 1) |
The absorbance values corresponding to the protein (based on the Soret band of the oxidised form) and the dye (Figure S3) were used to determine the ratio of concentrations of oxidized (ox) to reduced (red) form of both protein and dye at each stage of the experiment (Eq. 2).
| (Eq. 2) |
Cyclopropanation reactions.
General cyclopropanation reactions were carried out at a 400 μL-scale using 10 μM Mb variant, 10 mM styrene and 20 mM EDA. In a typical procedure, potassium phosphate buffer (50 mM, pH 7.0) was degassed by bubbling argon into the mixture for 3 min in a sealed vial. A buffered solution containing the myoglobin variant was carefully degassed in a similar manner in a separate vial. The two solutions were then mixed together via cannula. Reactions were initiated by addition of 10 μL of styrene (from a 0.4 M stock solution in ethanol), followed by the addition of 10 μL of EDA (from a 0.8 M stock solution in ethanol) with a syringe, and the reaction mixture was stirred for 16 hours at room temperature, under positive argon pressure. Aerobic reactions were carried out without degassing the solution with argon and in open reaction vessels. Reactions with reductant were performed in a similar manner adding to the potassium phosphate buffer 40 μL of sodium dithionite (100 mM stock solution in buffer).
Product analysis.
The reactions were analyzed by adding 20 μL of internal standard (benzodioxole, 100 mM in methanol) to the reaction mixture, followed by extraction with 400 μL of dichloromethane (DCM) and analyzed by GC–FID (see the Analytical Methods in the Supporting Information). Calibration curves of the different cyclopropane products were constructed using synthetically produced authentic standards (see the Synthetic Procedures in the Supporting Information). All measurements were performed at least in duplicate. For each experiment, negative control samples containing either no enzyme or no reductant were included. For stereoselectivity determination, the samples were analyzed by chiral GC–FID or chiral SFC as described in the Supporting Information
Kinetic experiments.
For the kinetic measurement, reactions were carried out on a 400 μL-scale in KPi buffer (pH 7.0) using 5 μM Mb variant, 2.5 mM vinyl benzoate (from a 0.2 M stock solution in ethanol) and 2.5 mM EDA (from a 0.2 M stock solution in ethanol). At regular intervals 20 μL of solution were collected and quenched with 50 μL of 0.2 M HCl. Aliquots were analysed by adding 20 μL of internal standard (benzodioxole, 50 mM in ethanol) followed by extraction with 400 μL of dichloromethane and analysis by gas chromatography (GC). For the Hammett analyses, reactions were carried out on a 400 μL scale in KPi buffer (pH 7.0) and 10% ethanol using 1 μM or 2 μM Mb variant (see Figure S6 and S7 for details), 1.25 mM substituted styrene (from a 0.1 M stock solution in ethanol), 1.25 mM styrene (from a 0.1 M stock solution in ethanol) and 2.5 mM EDA (from a 0.2 M stock solution in ethanol). The reactions were quenched after 2, 4 and 5 min by adding 100 μL of 0.2 M HCl and 20 μL of internal standard (benzodioxole, 50 mM in ethanol) to the reaction mixture. The products were extracted with 400 μL of dichloromethane and analysed by gas chromatography (GC). Representative kinetic plots corresponding to these experiments are reported in Figure S6 and S7.
Radical Spin Trap Experiments.
Reactions were carried out on a 400 μL-scale using 10 μM Mb variant, 10 mM styrene, 20 mM EDA with or without 100 mM 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPOL) or 5,5-Dimethyl-1-pyrroline N-oxide (DMPO), and 10 mM sodium dithionite. In a typical reaction, a solution containing sodium dithionite (100 mM stock solution) in potassium phosphate buffer (50 mM, pH 7.0, 10% dimethylformamide (DMF)) was degassed by bubbling argon into the mixture for 3 min in a septum-capped vial. A buffered solution containing the myoglobin variant was carefully degassed in a similar manner in a separate vial. The two solutions were then mixed together via cannula. Reactions were initiated by addition of 10 μL of styrene (from a 0.4 M stock solution in DMF), followed by the addition of radical trap reagent (40 μL, 1M stock solution in DMF) and 10 μL of EDA (from a 0.8 M stock solution in DMF) with a syringe. The reaction was stirred for 16 hours at room temperature, under positive argon pressure. For product analysis, 20 μL of internal standard (benzodioxole, 50 mM in ethanol) were added to the reaction mixture followed by extraction with 400 μL of dichloromethane and analysis by gas chromatography (GC).
Reactions with cis-β-deutero-styrene.
Reactions were carried out at 2 mL-scale using 60 μM Mb variant, 0.2 M cis-β-d1-styrene, 0.4 M ethyl diazoacetate (EDA). In a typical reaction, potassium phosphate buffer (50 mM, pH 7.0, 10% DMF) was degassed by bubbling argon into the mixture for 3 min in a sealed vial. A buffered solution containing the myoglobin variant was carefully degassed in a similar manner in a separate vial. The two solutions were then mixed together via cannula. Reactions were initiated by addition of cis-β-d1-styrene (42.1 mg, 0.40 mmol), followed by the addition of EDA (84.0 mL, 0.80 mmol) with a syringe. The reaction mixture was stirred for 16 hours at room temperature, under positive argon pressure. The reaction products were extracted with dichloromethane (2 mL, three rimes), and the organic layers were collected and dried over MgSO4. The organic solvent was removed by rotary evaporation, and the crude was purified via flash chromatography (silica gel, hexane:AcOEt 99:1). The analytical data for 3a-d and 3b-d are in accord with those reported in literature. Enantiopure d-5acis: 1H NMR (400 MHz, CDCl3): δ 7.30 (t, J = 7.2 Hz, 2H), 7.22 (t, J = 7.2 Hz, 1H), 7.11 (d, J = 7.2 Hz, 2H), 4.20 (q, J = 14.4 Hz, 7.2 Hz, 2H), 2.53 (dd, J = 9.2 Hz, 4.0 Hz, 1H), 1.91 (t (=dd), J = 4.0 Hz, 1H), 1.61 (dd, J = 8.9 Hz, 5.2 Hz, 1H), 1.30 (t, J = 7.2 Hz, 3H) ppm. 2H NMR (60 MHz, CDCl3): δ 1.32 ppm (s, 1H). Enantiopure d-5atrans: 1H NMR (400 MHz, CDCl3): δ 7.28–7.21 (m, 2H), 7.16 (t, J = 7.1 Hz, 1H), 7.06 (d, J = 7.2 Hz, 2H), 3.83 (q, J = 7.1 Hz, 2H), 2.54 (t, J = 9.0 Hz, 1H), 2.03 (t, J = 8.5 Hz, 1H), 1.28 (d, J = 7.0 Hz, 1H), 0.93 ppm (t, J = 7.1 Hz, 3H). 2H NMR (60 MHz, CDCl3): δ 1.72 ppm (s, 1H).
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the U.S. National Institute of Health grant GM098628. MS instrumentation was supported by the U.S. NSF grant CHE-0946653.
Footnotes
Supporting Information
Supporting information includes supplementary Tables and Figures, synthetic procedures, analytical methods, and compound characterization data.
REFERENCES
- (1).Doyle MP, and Forbes DC Recent Advances in Asymmetric Catalytic Metal Carbene Transformations, Chem. Rev 1998, 98, 911–936. [DOI] [PubMed] [Google Scholar]
- (2).Lebel H, Marcoux JF, Molinaro C, and Charette AB Stereoselective cyclopropanation reactions, Chem. Rev 2003, 103, 977–1050. [DOI] [PubMed] [Google Scholar]
- (3).Davies HML, and Hedley SJ Intermolecular reactions of electron-rich heterocycles with copper and rhodium carbenoids, Chem. Soc. Rev 2007, 36, 1109–1119. [DOI] [PubMed] [Google Scholar]
- (4).Pellissier H Recent developments in asymmetric cyclopropanation, Tetrahedron 2008, 64, 7041–7095. [Google Scholar]
- (5).Davies HML, and Denton JR Application of donor/acceptor-carbenoids to the synthesis of natural products, Chem. Soc. Rev 2009, 38, 3061–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Reichelt A, and Martin SF Synthesis and properties of cyclopropane-derived peptidomimetics, Acc. Chem. Res 2006, 39, 433–442. [DOI] [PubMed] [Google Scholar]
- (7).Talele TT The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules, J. Med. Chem 2016, 59, 8712–8756. [DOI] [PubMed] [Google Scholar]
- (8).Ebner C, and Carreira EM Cyclopropanation Strategies in Recent Total Syntheses, Chem. Rev 2017, 117, 11651–11679. [DOI] [PubMed] [Google Scholar]
- (9).Bordeaux M, Tyagi V, and Fasan R Highly Diastereoselective and Enantioselective Olefin Cyclopropanation Using Engineered Myoglobin-Based Catalysts, Angew. Chem. Int. Ed 2015, 54, 1744–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bajaj P, Sreenilayam G, Tyagi V, and Fasan R Gram-Scale Synthesis of Chiral Cyclopropane-Containing Drugs and Drug Precursors with Engineered Myoglobin Catalysts Featuring Complementary Stereoselectivity, Angew. Chem. Int. Ed 2016, 55, 16110–16114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tinoco A, Steck V, Tyagi V, and Fasan R Highly Diastereo- and Enantioselective Synthesis of Trifluoromethyl-Substituted Cyclopropanes via Myoglobin-Catalyzed Transfer of Trifluoromethylcarbene, J. Am. Chem. Soc 2017, 139, 5293–5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chandgude AL, and Fasan R Highly Diastereo- and Enantioselective Synthesis of Nitrile-Substituted Cyclopropanes by Myoglobin-Mediated Carbene Transfer Catalysis, Angew. Chem. Int. Ed 2018, 57, 15852–15856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Vargas D, Khade R, Zhang Y, and Fasan R Biocatalytic strategy for highly diastereo- and enantioselective synthesis of 2,3-dihydrobenzofuran based tricyclic scaffolds., Angew. Chem. Int. Ed 2019, 58, 10148–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Coelho PS, Brustad EM, Kannan A, and Arnold FH Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes, Science 2013, 339, 307–310. [DOI] [PubMed] [Google Scholar]
- (15).Gober JG, Rydeen AE, Gibson-O’Grady EJ, Leuthaeuser JB, Fetrow JS, and Brustad EM Mutating a Highly Conserved Residue in Diverse Cytochrome P450s Facilitates Diastereoselective Olefin Cyclopropanation, Chembiochem 2016, 17, 394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Knight AM, Kan SBJ, Lewis RD, Brandenberg OF, Chen K, and Arnold FH Diverse Engineered Heme Proteins Enable Stereodivergent Cyclopropanation of Unactivated Alkenes, ACS Central Sci. 2018, 4, 372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Brandenberg OF, Prier CK, Chen K, Knight AM, Wu Z, and Arnold FH Stereoselective Enzymatic Synthesis of Heteroatom-Substituted Cyclopropanes, ACS Catal. 2018, 8, 2629–2634. [Google Scholar]
- (18).Chen K, Zhang SQ, Brandenberg OF, Hong X, and Arnold FH Alternate Heme Ligation Steers Activity and Selectivity in Engineered Cytochrome P450-Catalyzed Carbene-Transfer Reactions, J. Am. Chem. Soc 2018, 140, 16402–16407. [DOI] [PubMed] [Google Scholar]
- (19).Srivastava P, Yang H, Ellis-Guardiola K, and Lewis JC Engineering a dirhodium artificial metalloenzyme for selective olefin cyclopropanation, Nat. Commun 2015, 6, 7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sreenilayam G, Moore EJ, Steck V, and Fasan R Metal substitution modulates the reactivity and extends the reaction scope of myoglobin carbene transfer catalysts, Adv. Synth. Cat 2017, 359, 2076–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sreenilayam G, Moore EJ, Steck V, and Fasan R Stereoselective olefin cyclopropanation under aerobic conditions with an artificial enzyme incorporating an iron-chlorin e6 cofactor, ACS Catal. 2017, 7, 7629–7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Dydio P, Key HM, Nazarenko A, Rha JYE, Seyedkazemi V, Clark DS, and Hartwig JF An artificial metalloenzyme with the kinetics of native enzymes, Science 2016, 354, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wolf MW, Vargas DA, and Lehnert N Engineering of RuMb: Toward a Green Catalyst for Carbene Insertion Reactions, Inorg. Chem 2017, 56, 5623–5635. [DOI] [PubMed] [Google Scholar]
- (24).Ohora K, Meichin H, Zhao LM, Wolf MW, Nakayama A, Hasegawa J, Lehnert N, and Hayashi T Catalytic Cyclopropanation by Myoglobin Reconstituted with Iron Porphycene: Acceleration of Catalysis due to Rapid Formation of the Carbene Species, J. Am. Chem. Soc 2017, 139, 17265–17268. [DOI] [PubMed] [Google Scholar]
- (25).Villarino L, Splan KE, Reddem E, Alonso-Cotchico L, de Souza CG, Lledos A, Marechal JD, Thunnissen AMWH, and Roelfes G An Artificial Heme Enzyme for Cyclopropanation Reactions, Angew. Chem. Int. Ed 2018, 57, 7785–7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Khade RL, and Zhang Y Catalytic and Biocatalytic Iron Porphyrin Carbene Formation: Effects of Binding Mode, Carbene Substituent, Porphyrin Substituent, and Protein Axial Ligand, J. Am. Chem. Soc 2015, 137, 7560–7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wei Y, Tinoco A, Steck V, Fasan R, and Zhang Y Cyclopropanations via Heme Carbenes: Basic Mechanism and Effects of Carbene Substituent, Protein Axial Ligand, and Porphyrin Substitution, J. Am. Chem. Soc 2018, 140, 1649–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lewis RD, Garcia-Borras M, Chalkley MJ, Buller AR, Houk KN, Kan SBJ, and Arnold FH Catalytic iron-carbene intermediate revealed in a cytochrome c carbene transferase, Proc. Natl. Acad. Sci. USA 2018, 115, 7308–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tinoco A, Wei Y, Bacik J-P, Carminati DM, Moore EJ, Ando N, Zhang Y, and Fasan R Origin of High Stereocontrol in Olefin Cyclopropanation Catalyzed by an Engineered Carbene Transferase, ACS Catal. 2019, 9 1514–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tyagi V, Sreenilayam G, Bajaj P, Tinoco A, and Fasan R Biocatalytic Synthesis of Allylic and Allenyl Sulfides through a Myoglobin-Catalyzed Doyle-Kirmse Reaction, Angew. Chem. Int. Ed 2016, 55, 13562–13566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tyagi V, and Fasan R Myoglobin-Catalyzed Olefination of Aldehydes, Angew. Chem. Int. Ed 2016, 55, 2512–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Vargas DA, Tinoco A, Tyagi V, and Fasan R Myoglobin-Catalyzed C-H Functionalization of Unprotected Indoles, Angew. Chem. Int. Ed 2018, 57, 9911–9915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gnad F, and Reiser O Synthesis and applications of beta-aminocarboxylic acids containing a cyclopropane ring, Chem. Rev 2003, 103, 1603–1623. [DOI] [PubMed] [Google Scholar]
- (34).Wong HNC, Hon MY, Tse CW, Yip YC, Tanko J, and Hudlicky T Use of Cyclopropanes and Their Derivatives in Organic-Synthesis, Chem. Rev 1989, 89, 165–198. [Google Scholar]
- (35).Danishefsky S Electrophilic Cyclopropanes in Organic-Synthesis, Acc. Chem. Res 1979, 12, 66–72. [Google Scholar]
- (36).Marshall NM, Garner DK, Wilson TD, Gao YG, Robinson H, Nilges MJ, and Lu Y Rationally tuning the reduction potential of a single cupredoxin beyond the natural range, Nature 2009, 462, 113–U127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bhagi-Damodaran A, Petrik ID, Marshall NM, Robinson H, and Lu Y Systematic Tuning of Heme Redox Potentials and Its Effects on O-2 Reduction Rates in a Designed Oxidase in Myoglobin, J. Am. Chem. Soc 2014, 136, 11882–11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mirts EN, Bhagi-Damodaran A, and Lu Y Understanding and Modulating Metalloenzymes with Unnatural Amino Acids, Non-Native Metal Ions, and Non-Native Metallocofactors, Acc. Chem. Res 2019, 52, 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Oohora K, Kihira Y, Mizohata E, Inoue T, and Hayashi T C(sp(3))-H Bond Hydroxylation Catalyzed by Myoglobin Reconstituted with Manganese Porphycene, J. Am. Chem. Soc 2013, 135, 17282–17285. [DOI] [PubMed] [Google Scholar]
- (40).Oohora K, Onoda A, and Hayashi T Hemoproteins Reconstituted with Artificial Metal Complexes as Biohybrid Catalysts, Acc. Chem. Res 2019, 52, 945–954. [DOI] [PubMed] [Google Scholar]
- (41).Onderko EL, Silakov A, Yosca TH, and Green MT Characterization of a selenocysteine-ligated P450 compound I reveals direct link between electron donation and reactivity, Nat. Chem 2017, 9, 623–628. [DOI] [PubMed] [Google Scholar]
- (42).Moore EJ, and Fasan R Effect of proximal ligand substitutions on the carbene and nitrene transferase activity of myoglobin, Tetrahedron 2019, 75, 2357–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Coelho PS, Wang ZJ, Ener ME, Baril SA, Kannan A, Arnold FH, and Brustad EM A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo, Nat. Chem. Biol 2013, 9, 485–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Hyster TK, Farwell CC, Buller AR, McIntosh JA, and Arnold FH Enzyme-Controlled Nitrogen-Atom Transfer Enables Regiodivergent C-H Amination, J. Am. Chem. Soc 2014, 136, 15505–15508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Green AP, Hayashi T, Mittl PRE, and Hilyert D A Chemically Programmed Proximal Ligand Enhances the Catalytic Properties of a Heme Enzyme, J. Am. Chem. Soc 2016, 138, 11344–11352. [DOI] [PubMed] [Google Scholar]
- (46).Pott M, Hayashi T, Mori T, Mittl PRE, Green AP, and Hilvert D A Noncanonical Proximal Heme Ligand Affords an Efficient Peroxidase in a Globin Fold, J. Am. Chem. Soc 2018, 140, 1535–1543. [DOI] [PubMed] [Google Scholar]
- (47).Hayashi T, Tinzl M, Mori T, Krengel U, Proppe J, Soetbeer J, Klose D, Jeschke G, Reiher M, and Hilvert D Capture and characterization of a reactive haem-carbenoid complex in an artificial metalloenzyme, Nat. Catal 2018, 1, 578–584. [Google Scholar]
- (48).Neya S, Nagai M, Nagatomo S, Hoshino T, Yoneda T, and Kawaguchi AT Utility of heme analogues to intentionally modify heme-globin interactions in myoglobin, Biochim. Biophys. Acta 2016, 1857, 582–588. [DOI] [PubMed] [Google Scholar]
- (49).Bordeaux M, Singh R, and Fasan R Intramolecular C(sp(3))H amination of arylsulfonyl azides with engineered and artificial myoglobin-based catalysts, Bioorg. Med. Chem 2014, 22, 5697–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Moore EJ, Steck V, Bajaj P, and Fasan R Chemoselective Cyclopropanation over Carbene Y-H Insertion Catalyzed by an Engineered Carbene Transferase, J. Org. Chem 2018, 83, 7480–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Springer BA, Sligar SG, Olson JS, and Phillips GN Mechanisms of Ligand Recognition in Myoglobin, Chem. Rev 1994, 94, 699–714. [Google Scholar]
- (52).Chen Y, Ruppel JV, and Zhang XP Cobalt-catalyzed asymmetric cyclopropanation of electron-deficient olefins, J. Am. Chem. Soc 2007, 129, 12074–+. [DOI] [PubMed] [Google Scholar]
- (53).Wang HB, Guptill DM, Varela-Alvarez A, Musaev DG, and Davies HML Rhodium-catalyzed enantioselective cyclopropanation of electron-deficient alkenes, Chem. Sci 2013, 4, 2844–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Lindsay VNG, Fiset D, Gritsch PJ, Azzi S, and Charette AB Stereoselective Rh-2(S-IBAZ)(4)-Catalyzed Cyclopropanation of Alkenes, Alkynes, and Allenes: Asymmetric Synthesis of Diacceptor Cyclopropylphosphonates and Alkylidenecyclopropanes, J. Am. Chem. Soc 2013, 135, 1463–1470. [DOI] [PubMed] [Google Scholar]
- (55).Shiro Y, Iizuka T, Marubayashi K, Ogura T, Kitagawa T, Balasubramanian S, and Boxer SG Spectroscopic Study of Ser92 Mutants of Human Myoglobin - Hydrogen-Bonding Effect of Ser92 to Proximal His93 on Structure and Property of Myoglobin, Biochemistry 1994, 33, 14986–14992. [DOI] [PubMed] [Google Scholar]
- (56).Decatur SM, Belcher KL, Rickert PK, Franzen S, and Boxer SG Hydrogen bonding modulates binding of exogenous ligands in a myoglobin proximal cavity mutant, Biochemistry 1999, 38, 11086–11092. [DOI] [PubMed] [Google Scholar]
- (57).Kawakami N, Shoji O, and Watanabe Y Single-Step Reconstitution of Apo-Hemoproteins at the Disruption Stage of Escherichia coli Cells, Chembiochem 2012, 13, 2045–2047. [DOI] [PubMed] [Google Scholar]
- (58).Wang L, and Schultz PG Expanding the genetic code, Angew. Chem. Int. Ed 2004, 44, 34–66. [DOI] [PubMed] [Google Scholar]
- (59).Xiao H, Peters FB, Yang PY, Reed S, Chittuluru JR, and Schultz PG Genetic Incorporation of Histidine Derivatives Using an Engineered Pyrrolysyl-tRNA Synthetase, ACS Chem. Biol 2014, 9, 1092–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Moore EJ, Zorine D, Hansen WA, Khare SD, and Fasan R Enzyme stabilization via computationally guided protein stapling, Proc. Natl. Acad. Sci. USA 2017, 114, 12472–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Efimov I, Parkin G, Millett ES, Glenday J, Chan CK, Weedon H, Randhawa H, Basran J, and Raven EL A simple method for the determination of reduction potentials in heme proteins, FEBS Lett. 2014, 588, 701–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Battistuzzi G, Bellei M, Casella L, Bortolotti CA, Roncone R, Monzani E, and Sola M Redox reactivity of the heme Fe3+/Fe2+ couple in native myoglobins and mutants with peroxidase-like activity, J. Biol. Inorg. Chem 2007, 12, 951–958. [DOI] [PubMed] [Google Scholar]
- (63).Bickar D, Bonaventura C, and Bonaventura J Carbon-Monoxide Driven Reduction of Ferric Heme and Heme-Proteins, J. Biol. Chem 1984, 259, 777–783. [PubMed] [Google Scholar]
- (64).Salomon RG, and Kochi JK Copper(I) Catalysis in Cyclopropanations with Diazo-Compounds - Role of Olefin Coordination, J. Am. Chem. Soc 1973, 95, 3300–3310. [Google Scholar]
- (65).Wolf JR, Hamaker CG, Djukic JP, Kodadek T, and Woo LK Shape and Stereoselective Cyclopropanation of Alkenes Catalyzed by Iron Porphyrins, J. Am. Chem. Soc 1995, 117, 9194–9199. [Google Scholar]
- (66).Lai TS, Chan FY, So PK, Ma DL, Wong KY, and Che CM Alkene cyclopropanation catalyzed by Halterman iron porphyrin: participation of organic bases as axial ligands, Dalton T. 2006, 4845–4851. [DOI] [PubMed] [Google Scholar]
- (67).Simkhovich L, Mahammed A, Goldberg I, and Gross Z Synthesis and characterization of germanium, tin, phosphorus, iron, and rhodium complexes of tris(pentafluorophenyl)corrole, and the utilization of the iron and rhodium corroles as cyclopropanation catalysts, Chem. Eur. J 2001, 7, 1041–1055. [DOI] [PubMed] [Google Scholar]
- (68).Renata H, Wang ZJ, Kitto RZ, and Arnold FH P450-catalyzed asymmetric cyclopropanation of electron-deficient olefins under aerobic conditions, Catal. Sci. Technol 2014, 4, 3640–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Chapman NB, and Shorter J (1982) Correlation analysis of organic reactivity, Plenum Press, New York. [Google Scholar]
- (70).Singh R, Kolev JN, Sutera PA, and Fasan R Enzymatic C(sp(3))-H Amination: P450-Catalyzed Conversion of Carbonazidates into Oxazolidinones, ACS Catal. 2015, 5, 1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Ji GZ, Jiang XK, Zhang YH, Yuan SG, Yu CX, Shi YQ, Zhang XL, and Shi WT The Spin Delocalization Substitutent Parameter Sigma-Jj .5. Correlation-Analysis of F-19 Chemical-Shifts of Substituted Trifluorostyrenes - the Unresolved Polar Substituent Parameter Sigma-Mb, J. Phys. Org. Chem 1990, 3, 643–650. [Google Scholar]
- (72).Jiang XK, and Ji GZ A Self-Consistent and Cross-Checked Scale of Spin-Delocalization Substituent Constants, the Sigma.(Jj) Scale, J. Org. Chem 1992, 57, 6051–6056. [Google Scholar]
- (73).Dincturk S, and Jackson RA Free-Radical Reactions in Solution .7. Substituent Effects on Free-Radical Reactions - Comparison of the Sigma-Scale with Other Measures of Radical Stabilization, J. Chem. Soc. Perk. T 1981, 1127–1131. [Google Scholar]
- (74).Kim SS, Zhu Y, and Lee KH Thermal isomerizations of ketenimines to nitriles: Evaluations of sigma-dot (sigma(center dot)) constants for spin-delocalizations, J. Org. Chem 2000, 65, 2919–2923. [DOI] [PubMed] [Google Scholar]
- (75).Jiang XK Establishment and successful application of the sigma(JJ)center dot scale of spin-delocalization substituent constants, Acc. Chem. Res 1997, 30, 283–289. [Google Scholar]
- (76).Dzik WI, Xu X, Zhang XP, Reek JNH, and de Bruin B ‘Carbene Radicals’ in CoII(por)-Catalyzed Olefin Cyclopropanation, J. Am. Chem. Soc 2010, 132, 10891–10902. [DOI] [PubMed] [Google Scholar]
- (77).Lu H, Dzik WI, Xu X, Wojtas L, de Bruin B, and Zhang XP Experimental Evidence for Cobalt(III)-Carbene Radicals: Key Intermediates in Cobalt(II)-Based Metalloradical Cyclopropanation, J. Am. Chem. Soc 2011, 133, 8518–8521. [DOI] [PubMed] [Google Scholar]
- (78).Edulji SK, and Nguyen ST Catalytic olefin cyclopropanation using mu-oxo-bis[(salen)iron(III)] complexes, Organometallics 2003, 22, 3374–3381. [Google Scholar]
- (79).Intrieri D, Le Gac S, Caselli A, Rose E, Boitrel B, and Gallo E Highly diastereoselective cyclopropanation of alpha-methylstyrene catalysed by a C-2-symmetrical chiral iron porphyrin complex, Chem. Commun 2014, 50, 1811–1813. [DOI] [PubMed] [Google Scholar]
- (80).Carminati DM, Intrieri D, Caselli A, Le Gac S, Boitrel B, Toma L, Legnani L, and Gallo E Designing ‘Totem’ C2 -Symmetrical Iron Porphyrin Catalysts for Stereoselective Cyclopropanations, Chemistry 2016, 22, 13599–135612. [DOI] [PubMed] [Google Scholar]
- (81).Morandi B, and Carreira EM Iron-Catalyzed Cyclopropanation in 6 M KOH with in Situ Generation of Diazomethane, Science 2012, 335, 1471–1474. [DOI] [PubMed] [Google Scholar]
- (82).Liu J, Hu L, Wang L, Chen H, and Deng L An Iron(II) Ylide Complex as a Masked Open-Shell Iron Alkylidene Species in Its Alkylidene-Transfer Reactions with Alkenes, J. Am. Chem. Soc 2017, 139, 3876–3888. [DOI] [PubMed] [Google Scholar]
- (83).Li Y, Huang JS, Zhou ZY, Che CM, and You XZ Remarkably stable iron porphyrins bearing nonheteroatom-stabilized carbene or (Alkoxycarbonyl) carbenes: Isolation, X-ray crystal structures, and carbon atom transfer reactions with hydrocarbons, J. Am. Chem. Soc 2002, 124, 13185–13193. [DOI] [PubMed] [Google Scholar]
- (84).Che CM, Huang JS, Lee FW, Li Y, Lai TS, Kwong HL, Teng PF, Lee WS, Lo WC, Peng SM, and Zhou ZY Asymmetric inter- and intramolecular cyclopropanation of alkenes catalyzed by chiral ruthenium porphyrins. Synthesis and crystal structure of a chiral metalloporphyrin carbene complex, J. Am. Chem. Soc 2001, 123, 4119–4129. [DOI] [PubMed] [Google Scholar]
- (85).Anding BJ, Ellern A, and Woo LK Olefin Cyclopropanation Catalyzed by Iridium(III) Porphyrin Complexes, Organometallics 2012, 31, 3628–3635. [Google Scholar]
- (86).Kwong HL, Wong WL, Lee WS, Cheng LS, and Wong WT New chiral 2,2 ′: 6 ′,2 ″-terpyridine ligands from the chiral pool: synthesis, crystal structure of a rhodium complex and uses in copper- and rhodium-catalyzed enantioselective cyclopropanation of styrene, Tetrahedron-Asymmetr. 2001, 12, 2683–2694. [Google Scholar]
- (87).Diaz-Requejo MM, Perez PJ, Brookhart M, and Templeton JL Substituent effects on the reaction rates of copper-catalyzed cyclopropanation and aziridination of para-substituted styrenes, Organometallics 1997, 16, 4399–4402. [Google Scholar]
- (88).Kwong HL, Cheng LS, Lee WS, Wong WL, and Wong WT Novel chiral di-2-pyridyl ketone ligands: Crystal structure of a copper complex and its activity in the copper-catalyzed enantioselective cyclopropanation of styrene, Eur. J. Inorg. Chem 2000, 1997–2002. [Google Scholar]
- (89).Mahandru GM, Liu G, and Montgomery J Ligand-dependent scope and divergent mechanistic behavior in nickel-catalyzed reductive couplings of aldehydes and alkynes, J. Am. Chem. Soc 2004, 126, 3698–3699. [DOI] [PubMed] [Google Scholar]
- (90).Lee YC, Patil S, Golz C, Strohmann C, Ziegler S, Kumar K, and Waldmann H A ligand-directed divergent catalytic approach to establish structural and functional scaffold diversity, Nat. Commun 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Chandgude AL, Ren X, and Fasan R Stereodivergent Intramolecular Cyclopropanation Enabled by Engineered Carbene Transferases, J. Am. Chem. Soc 2019, 141, 9145–9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Shaik S, Hirao H, and Kumar D Reactivity of high-valent iron-oxo species in enzymes and synthetic reagents: A tale of many states, Acc. Chem. Res 2007, 40, 532–542. [DOI] [PubMed] [Google Scholar]
- (93).Meunier B, de Visser SP, and Shaik S Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes, Chem. Rev 2004, 104, 3947–3980. [DOI] [PubMed] [Google Scholar]
- (94).Tinoco A, Wei Y, Bacik J-P, Carminati DM, Moore EJ, Ando N, Zhang Y, and Fasan R Origin of High Stereocontrol in Olefin Cyclopropanation Catalyzed by an Engineered Carbene Transferase, ACS Catal. 2019, 9, 1514–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Wei Y, Tinoco A, Steck V, Fasan R, and Zhang Y Cyclopropanations via Heme Carbenes: Basic Mechanism and Effects of Carbene Substituent, Protein Axial Ligand, and Porphyrin Substitution, J. Am. Chem. Soc 2018, 140, 1649–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Prier CK, Rankic DA, and MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Lu HJ, and Zhang XP Catalytic C-H functionalization by metalloporphyrins: recent developments and future directions, Chem. Soc. Rev 2011, 40, 1899–1909. [DOI] [PubMed] [Google Scholar]
- (98).Studer A, and Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective, Angew. Chem. Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]
- (99).Kuijpers PF, van der Vlugt JI, Schneider S, and de Bruin B Nitrene Radical Intermediates in Catalytic Synthesis, Chem. Eur. J 2017, 23, 13819–13829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.