

Summary
Filoviruses, including Ebola and Marburg, cause fatal hemorrhagic fever in humans and primates. Understanding how these viruses enter host cells could help to develop effective therapeutics. An endosomal protein, Niemann-Pick C1 (NPC1), has been identified as a necessary entry receptor for this process, and priming of the viral glycoprotein (GP) to a fusion-competent state is a prerequisite for NPC1 binding. Here, we have determined the crystal structure of the primed GP (GPcl) of Ebola virus bound to domain C of NPC1 (NPC1-C) at a resolution of 2.3 Å. NPC1-C utilizes two protruding loops to engage a hydrophobic cavity on head of GPcl. Upon enzymatic cleavage and NPC1-C binding, conformational change in the GPcl further affects the state of the internal fusion loop, triggering membrane fusion. Our data therefore provide structural insights into filovirus entry in the late endosome and the molecular basis for design of therapeutic inhibitors of viral entry.
Graphical Abstract
Highlights
-
•
Structural basis of Ebola virus endosomal-receptor binding
-
•
NPC1 domain C (NPC1-C) displays a helical core structure with two protruding loops
-
•
NPC1-C binds to the primed Ebola virus GP (GPcl) protein with a low affinity
-
•
NPC1-C utilizes two protruding loops to engage a hydrophobic cavity on head of GPcl
The crystal structure of the primed Ebola virus glycoprotein in complex with domain C of the endosomal entry receptor Niemann-Pick C1 reveals structural insights into filovirus fusion to the late endosome and the molecular basis for designing therapeutic inhibitors of viral entry.
Introduction
The filovirus family Filoviradae, including the genera Ebolavirus and Marburgvirus, can cause a rapidly lethal hemorrhagic fever in humans, and at present, no clinically approved antiviral therapeutics are available. Since 1967, Marburg virus has emerged multiple times, with modern strains showing greater mortality (Geisbert et al., 2007, Malherbe and Strickland-Cholmley, 1968, Siegert et al., 1968, Towner et al., 2006). The Ebolavirus consists of five recognized species, including Zaire, Sudan, Reston, Bundibugyo and Tai Forest viruses, four of which (except the Reston virus) can infect humans (Kuhn et al., 2013, Kuhn and Jahrling, 2010). Among these filoviruses, Ebola virus (EBOV) and the Marburgvirus (MARV) Angola variant cause the most severe diseases, with case fatality rates reaching ∼90% (Feldmann and Geisbert, 2011, Sullivan et al., 2009). Recently, since December 2013, a historically unprecedented EBOV outbreak has occurred in the West Africa, causing more than 25,000 human infections and over 10,000 related deaths as of May 18th, 2015. Under this urgent situation, we are called for great efforts to develop the vaccines and antiviral therapeutics, which needs a comprehensive and decent understanding of the pathogenesis and molecular basis of EBOV infection.
Regarding the infection by filoviruses, including EBOV, the primarily infected cells are macrophages and dendritic cells. But the viruses exhibit a much broader cell tropism and can infect, upon primary infection, most of the cell types, including epithelial and non-epithelial cells, with the exception of lymphocytes and other non-adherent cells (Dube et al., 2010). Host cell attachment factors such as C-type lectins, including DC-SIGN (dendritic-cell-specific ICAM3-grabbing non-integrin; also known as CD209) and L-SIGN (liver and lymph node SIGN; also known as CLEC4M) and several cell-surface proteins such as integrins, T cell immunoglobulin and mucin domain-containing (TIM) proteins, and tyrosine protein kinase receptor 3 (TYRO3) family members have been shown to mediate the entry of filoviruses on the cell surface (Alvarez et al., 2002, Jemielity et al., 2013, Kondratowicz et al., 2011, Shimojima et al., 2007, Simmons et al., 2003, Takada et al., 2000, Takada et al., 2004, Wang et al., 2015). These attachment factors, however, do not function as authentic entry receptors (Brindley et al., 2011, Schornberg et al., 2009). Following binding to the cell surface, filoviruses are internalized by a macropinocytosis-like process and subsequently trafficked through early and late endosomes (Mulherkar et al., 2011, Nanbo et al., 2010, Saeed et al., 2010). The viral genome then penetrates into the cytoplasm after fusion of the viral envelope with the membrane of the late endosome. In the cytoplasm, the viral genome is replicated and transcribed, and new viral proteins are synthesized to assemble progeny virions, which bud from the cell surface (Bharat et al., 2011, Feldmann et al., 2013). Given its place at the initial step of virus infection, the entry process represents an ideal target for antiviral intervention: stopping the virus before it enters the cell.
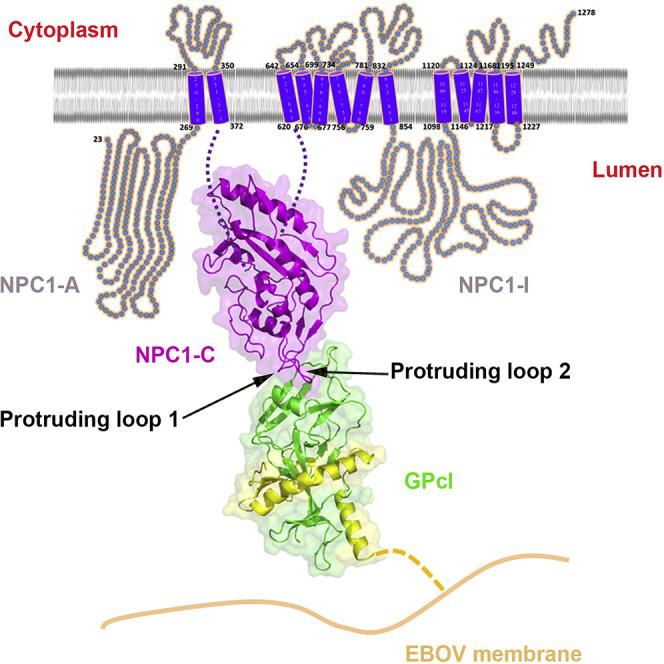
The trimeric glycoprotein (GP) spike on the envelope of filoviruses mediates all stages of virus entry, including attachment, entry, and fusion (Lee and Saphire, 2009). Like all the other Class I viral fusion proteins, the EBOV GP is synthesized as a single polypeptide that is subsequently cleaved by furin-like proteases into GP1 and GP2 subunits, which remain together through an inter-subunit disulfide bond and non-covalent interactions. In contrast to other Class I viral fusion proteins, however, the simple furin cleavage event is not sufficient to prime EBOV GP (Neumann et al., 2007, Wool-Lewis and Bates, 1999). After entering into the cell, the virus is eventually trafficked to late endosomes, where GP is further primed to remove some “cap” components, thereby triggering the induction of the crucial membrane fusion event, which leads to viral penetration. The functional EBOV GP priming is mediated by the cysteine proteases cathepsin B and cathepsin L (Chandran et al., 2005, Schornberg et al., 2006), which cleave GP1 within the β13-β14 loop (Dube et al., 2009, Hood et al., 2010, Lee et al., 2008). Cathepsin cleavage removes ∼60% of the amino acids from GP1, including the mucin-like domain, the glycan cap, and the outmost β strand of the proposed receptor binding region (Dube et al., 2009, Lee et al., 2008), resulting in a primed form of GP (named GPcl, the 19 kDa GP1 plus GP2) (Figure 1 A). Unlike the full-length GP, the primed GPcl could bind to an endosomal membrane protein Niemann-Pick C1 (NPC1) (Miller et al., 2012), which is an indispensable host entry factor for EBOV infection (Carette et al., 2011, Côté et al., 2011). NPC1 is a ubiquitously expressed protein with multiple transmembrane domains and resides primarily in the limiting membranes of late endosomes and lysosomes (Carstea et al., 1997, Davies and Ioannou, 2000). It functions to aid cholesterol egress out of late endosomes for re-distribution to cellular membranes (including endoplasmic reticulum and plasma membrane) in the assistance of a soluble NPC2 protein (Sleat et al., 2004). Further studies have revealed that the second luminal domain (Domain C) of NPC1 (NPC1-C) is responsible for the binding to GPcl (Figure 1A) (Miller et al., 2012). However, how GPcl binds to NPC1 remains elusive, and structural study should be carried out to investigate the molecular features of the binding interface, which will guide further development of small antiviral molecules and antibodies for prevention and/or treatment of EBOV infection.
Figure 1.

Model of NPC1 Bound to the Primed EBOV GP and Overall Structure of NPC1-C
(A) The full-length EBOV GP was primed by cathepsin L and B in the late endosome, and the highly glycosylated 130 kDa GP1 subunit was cleaved into a 19 kDa GP1, generating a primed form of GP (GPcl). Then, the GPcl binds to the domain C of NPC1 molecule (NPC1-C), which is an endosomal 13-transmembrane protein with three large luminal domains (domains A, C, and I).
(B) Crystal structure of NPC1-C. The helices (α and η) are colored in cyan, the β strands are colored in magenta, and the loops are colored in light pink. The disulfide bonds are colored in yellow. NPC1-C displays a helical core structure surrounded by several β strands with two extended loops.
(C) Topological secondary structure of NPC1-C. Secondary elements are shown as cylinders and arrows for helices and β strands.
See also Figures S1, S2, and S3 and Table S1.
Here, we have determined the crystal structures of free NPC1-C and its complex with GPcl. The results revealed that NPC1-C displays a helical structure core surrounded by several β strands and contains two extended loops protruding outward for ligand interactions. Further GPcl/NPC1-C complex structure indeed shows that NPC1-C utilizes the two protruding loops to engage a hydrophobic cavity on the head of GPcl. Despite a low affinity between NPC1-C and GPcl as revealed by the biophysical analyses, upon enzymatic cleavage and NPC1-C binding, several conformational changes are shown to be induced in GPcl. After conformational changes, the uplift of the short 310 helix in the β3-α1 loop likely helps to release the N-terminal portion of the internal fusion loop (IFL), thereby triggering the membrane fusion. The mutagenesis work in NPC1-C further confirmed that the protruding loop 2 plays a more important role in the binding of NPC1-C to GPcl.
Results
Structure of NPC1-C
The 1,278 residue human NPC1 contains 13 predicted transmembrane-spanning helices and 3 large luminal domains, domains A, C, and I (Figure 1A), the overall structural features of which remain to be illustrated. The crystal structure of the first luminal domain (domain A, also called N-terminal domain, NTD) is previously reported, showing a helical structural fold that contains a deep pocket for binding of sterols (Kwon et al., 2009). The structure information of the second and third luminal domains (domains C and I, respectively) of NPC1, however, are uncharacterized. Here, we used the pET21a expression vector to produce human NPC1-C (residues 374 to 620) in E. coli BL21 (DE3), and the protein exists as insoluble inclusion bodies. Then the inclusion bodies were refolded to obtain soluble NPC1-C, which behaves as a single monomeric peak in gel filtration (Figure S1 A). Some droplet-like crystals were obtained using the sitting drop vapor diffusion crystallization method, and the best crystal diffracted to a high resolution of 2.0 Å. As there is no homology structural model, the crystal structure of NPC1-C was determined by using single wavelength anomalous dispersion (SAD) phasing method with the help of a gold derivative. After several rounds of refinement, the final Rfree and Rwork reached 21.9 and 18.2, respectively (Table S1).
Figure S1.

Characterization of Refolded NPC1-C Protein and Analysis of Potential N-Link Glycosylation Sites, Related to Figure 1
(A) The refolded NPC1-C protein was eluted as a single sharp peak in the gel filtration on Superdex 200® column (GE Healthcare). The NPC1-C protein shows perfect purity, which is ran as a single band in the SDS-PAGE without any extra bands. Irrelevant lanes were removed during preparation of the figure.
(B) The NPC1-C has seven potential N-link glycosylation sites, six of which locate in the loops except one (N452) at the tip of α4 helix. The sites are shown in red spheres in the NPC1-C structure.
Overall, NPC1-C is mainly composed of seven α helices in the center as a core and seven β strands surrounding the core, with two protruding loops (Figure 1B). The first protruding loop (loop 1) is localized between β2 and β3 strands, while the second protruding loop (loop 2) is between α4 and α5 helices (Figure 1B). NPC1-C contains four cysteine residues, all of which were involved in forming disulfide bonds (C468-C479, C516-C533) (Figure 1C). Through analysis of primary protein sequence, NPC1-C has seven potential N-linked glycosylation sites, six of which locate in the loops, while one locates at the tip of α4 helix (Figure S1B). No glycans are observed in the crystal structure because the NPC1-C protein is expressed in bacterial cells that lack post-translational glycosylation modifications. However, the surface exposure of the seven sites indicates their accessibility for N-glyco-transferases.
The intact luminal NPC1-C consists of residues 372 to 640. In our crystal structure, only residues 384 to 624 are visible (Figure S2 ). We cannot see the electron density for the N-terminal 12 amino acids and C-terminal 16 amino acids (Figure S2), indicating that these regions might be flexible loops. It is assumed that, with these flexible N-terminal and C-terminal loops, the domain C can protrude from the endosomal membrane (Figure S3 ) and be easily bound by the primed GP molecule.
Figure S2.

Sequence Alignment of NPC1-C and NPC1, Related to Figure 1
The intact luminal domain C consists of residues 372 to 640. In our crystal structure, only residues 384 to 624 are visible, and the secondary structures are labeled. We cannot see the electron density for the N-terminal 12 amino acids and C-terminal 16 amino acids, indicating these disordered regions might be flexible loops.
Figure S3.

Protruding Model of NPC1-C, Related to Figure 1
We cannot see the density for both ends immediately adjacent to the TM segments. The missing residues (in dash lines) of both the N-terminal and C-terminal could be flexible loops, which will not make the visible domain C structure to be tightly attached to the endosomal membrane. It is assumed that, with these flexible N-terminal and C-terminal loops, the domain C can protrude from the endosomal membrane and be easily bound by the primed GP molecule.
NPC1-C Bound to the GPcl with a Low Affinity
The mucin-deleted ectodomain of EBOV GP (GP-ectoΔmucin) was expressed in insect cells using baculovirus expression system and purified as a single homotrimeric peak by gel filtration (Figure S4 A) following the previously reported method (Lee et al., 2008). Subsequently, thermolysin was used to cleave the GP-ecto protein to obtain GPcl, which also exists as a similar homotrimeric peak with relatively smaller size in the gel filtration (Figure S4A), following the previously developed method (Hashiguchi et al., 2015). The thermolysin, a protease that functions at neutral pH, can substitute for the cathepsin enzymes in priming GP (Schornberg et al., 2006). As expected, GP1 is cleaved into a 19 kDa protein moiety after thermolysin digestion (Figure S4A), resembling the version of GP component responsible for receptor binding in the endosome.
Figure S4.

Characterization of GP-ectoΔmucin and GPcl Proteins and Binding Affinities of 2G4 Bound to GP and GPcl, Related to Figure 2
(A) The GPectoΔmucin protein was eluted as a single peak (the elution volume represents a molecular weight of ∼180 kDa, homotrimer) in the gel filtration of Superdex® 200 column (GE Healthcare). The primed GP protein (GPcl) was also eluted as a single peak (the elution volume represents a molecular weight of ∼120 kDa). In the SDS-PAGE, after thermolycin digestion, the GP1 subunit was cleaved into a 19 kDa band, the molecular weight of which is similar to the GP2 subunit.
(B) The GP-ectoΔmucin and GPcl proteins were immobilized on the CM5 chip with about 2000 response units. Then serial concentrations of 2G4 Fab were flow through the chip. The BIAcore® diagrams reveal that 2G4 Fab binds to GP or GPcl with slow on and off rates. Moreover, 2G4 Fab binds to GP or GPcl with similar binding affinities (8.2 nM versus 3.8 nM). The KD values were calculated by the BIAcore® 3000 analysis software (BIAevaluation Version 4.1).
In order to test the active interactions between purified NPC1-C and GPcl proteins, we performed surface plasmon resonance (SPR) experiments to explore the binding affinity of these two proteins using a BIAcore 3000 instrument. The GP-ectoΔmucin or GPcl protein was immobilized on a CM5 chip, which was then flown through with the NPC1-C protein. Before we tested the NPC1-C protein, we first used 2G4 antibody (a member of the ZMapp cocktail (Qiu et al., 2014), which shows active binding capacity to the stem region of EBOV GP protein) as a positive control, confirming that the GP-ectoΔmucin and GPcl protein on the chip remain in the native prefusion state (Figure S4B). We found that 2G4 binds to GP-ectoΔmucin and GPcl protein with similar binding affinities (8.2 nM versus 3.8 nM) (Figure S4B). As expected, GP-ectoΔmucin does not bind to NPC1-C (Figure 2 A), while GPcl binds to NPC1-C with a low affinity of 158 micromolar (μM) (Figures 2B and 2C). This binding affinity is much lower than that of some other highly pathogenic viruses such as MERS-CoV and SARS-CoV, which is among the nanomolar (nM) range (Lu et al., 2013).
Figure 2.

Binding Affinity of GPcl Bound to NPC1-C
(A) BIAcore diagram of refolded E. coli-expressed NPC1-C protein bound to the GP-ectoΔmucin protein. The refolded NPC1-C shows no binding to the GP-ectoΔmucin.
(B and C) BIAcore diagram and saturation curve of refolded NPC1-C protein bound to the GPcl protein. The refolded NPC1-C binds to the GPcl with a low affinity and a fast kinetics.
(D) BIAcore diagram of glycosylated 293T-expressed NPC1-C protein bound to the GP-ectoΔmucin protein. The glycosylated NPC1-C also shows no binding to the GP-ectoΔmucin.
(E and F) BIAcore diagram and saturation curve of glycosylated NPC1-C protein bound to the GPcl protein. The glycosylated NPC1-C binds to the GPcl with a similar low affinity and a fast kinetics. Response units were plotted against protein concentrations. The KD values were calculated by the BIAcore 3000 analysis software (BIAevaluation Version 4.1).
See also Figures S4 and S5.
In order to evaluate effect of the glycosylations in NPC1-C protein on binding of GPcl, we expressed glycosylated NPC1-C protein in 293T mammalian cells. This glycosylated NPC1-C protein exists as a dispersing band in the SDS-PAGE with an average molecular weight of about 55 kDa (Figure S5 ). Further SPR experiments revealed that the glycosylated NPC1-C does not bind to GP-ectoΔmucin (Figure 2D) but binds to GPcl with a comparable low affinity of 140 μM (Figures 2E and 2F) to the refolded NPC1-C protein (158 μM). It indicates that the glycosylations in NPC1-C have no effect on the binding of GPcl.
Figure S5.

Characterization of Glycosylated NPC1-C Protein Expressed in Mammalian 293T Cells, Related to Figure 2
The NPC1-C protein was expressed in 293T mammalian cells as secreted protein. After purification with affinity chromatography using HisTrap FF column (GE Healthcare), the glycosylated NPC1-C protein was eluted as a single peak in the gel filtration on Superdex 200 column (GE Healthcare). The NPC1-C protein shows good purity, which is run as a dispersing single band in the SDS-PAGE, with an average molecular weight of 55 kDa. The protein was further confirmed by Western blot assay with anti-histag antibody. Irrelevant lanes were removed during preparation of the figure.
Complex Structure of NPC1-C and GPcl
The NPC1-C and GPcl were incubated together at a molar ratio of 1:1, and then crystallization screen was performed with the mixed protein complex. Initially, low-resolution-diffractive crystals were obtained, and after optimization of crystallization condition, a high-resolution-diffractive dataset was finally collected at the synchrotron facility. The complex structure was determined by molecular replacement at a resolution of 2.3 Å.
Structural analysis of the bound GPcl demonstrates that the cathepsin enzymes indeed cleave the GP protein within the β13-β14 loop (Figure 3 A), and only residues 31 to 188 of GP1 subunit and residues 509 to 598 of GP2 subunit are visible in the crystal structure. After cathepsin cleavage, the glycan cap and mucin domain components would be removed (Figure 3A).
Figure 3.

Complex Structure of NPC1-C Bound to GPcl
(A) The bound GPcl structure. The visible portions of GP1 and GP2 subunits are colored in green and yellow, respectively, and the other invisible or cleavage-removed portions are colored in gray.
(B) Overall complex structure. The NPC1-C uses its two protruding loops to bind to the head of GPcl at a perpendicular angle.
(C) Surface representations of interacting residues in the NPC1-C and GPcl. The residues responsible for the binding in the NPC1-C (top) are colored in blue, and the interacting residues in the GPcl (bottom) are colored in orange. The NPC1-C mainly utilizes aromatic residues to engage the cavity of the GPcl, which mainly consists of hydrophobic residues.
See also Table S1.
The solved complex structure reveals that NPC1-C binds the membrane-distal head of GPcl with a perpendicular angle (Figure 3B). NPC1-C mainly uses its two protruding loops to engage a hydrophobic cavity in the head of primed GP1 (Figure 3B). The hydrophobic cavity was formed by residues from the α1 helix, the β4, β7, β9, and β10 strands, and the β9-β10 and β12-β13 loops (Figure 3B). The protruding loop 1 of NPC1-C contacts one side of the cavity, while the protruding loop 2 inserts into the cavity (Figure 3B). In the protruding loop 1, seven residues (Y420, Q421, Y423, P424, S425, G426, and D428) are involved in the interaction (Figure 3C, top), among which residues Y423 and P424 contribute most of the atom-to-atom contacts (Table 1 ). In the protruding loop 2, six residues (D501, D502, F503, F504, V505, and Y506) participate in the binding process (Figure 3C, top), and residues F503, F504, and Y506 play the major role by contributing the majority of the tight hydrophobic interactions with the head cavity of GPcl (Table 1). Moreover, residue D501 of NPC1-C forms a hydrogen bond with residue T83 of GPcl in the cavity (Figure 3C and Table 1). Reciprocally, the hydrophobic cavity of GPcl consists of residues V79, P80, T83, W86, G87, F88, L111, E112, I113, V141, G145, P146, C147, A152, and I170, while the other residues of K114, G118, S142, G143, and T144 form a hydrophilic patch contacting the protruding loop 1 with polar interactions (Figure 3C, bottom). Of these polar contacts, the NPC1-C residue Q421 forms a hydrogen bond with the GPcl residue T144, and the NPC1-C residue D428 could form a potential salt bridge with GPcl residue K114 in the hydrophilic patch (Figure 3C and Table 1).
Table 1.
Interaction between GPcl and NPC1-C
| GPcl | Contactsa | NPC1-C |
|---|---|---|
| V79 | 3, 2, 4 | Y423, F504, Y506 |
| P80 | 2, 19, 1 | D501, Y506, L528 |
| T83 | 7, 9, 3 | D501, F503, Y506 |
| W86 | 11 | F503 |
| G87 | 4 | F503 |
| F88 | 3, 14 | D502, F503 |
| L111 | 2, 3 | F503, F504 |
| E112 | 3 | F504 |
| I113 | 6, 6 | F503, F504 |
| K114 | 8, 1 | Q421, D428 |
| G118 | 1 | Q421 |
| V141 | 10, 5, 4 | Y423, P424, F504 |
| S142 | 6, 16, 8, 12, 3 | Y423, P424, S425, G426, F504 |
| G143 | 6, 1, 2, 4 | Q421, Y423, G426, F504 |
| T144 | 4, 13, 2, 5, 6 | Y420, Q421, F503, F504, V505 |
| G145 | 4, 2, 1 | F503, F504, V505 |
| P146 | 3, 5, 4 | D501, D502, F503 |
| C147 | 1 | D502 |
| A152 | 2 | F503 |
| I170 | 1 | F503 |
Numbers represent the number of atom-to-atom contacts between the GPcl residues and the NPC1-C residues, which were analyzed by the Contact program in CCP4 suite (the distance cutoff is 4.5 Å).
Structural Comparison between GP and GPcl
Before GP is cleaved by the cathepsin enzymes, the binding site for NPC1-C is shielded by the glycan cap (Figure 4 A). The β14 strand forms a hydrogen bond network with the β9 strand, and two aromatic residues F225 and Y232 insert into the hydrophobic cavity in the head of GPcl. By contrast, NPC1-C takes advantage of four aromatic residues (Y423, F503, F504, and Y506) and one hydrophobic residue P424 to interact with the hydrophobic cavity. Of special importance, residue F503 inserts into the cavity at a deeper position (Figure 4B). Thus, the binding of NPC1-C mimics the interaction between the glycan cap and the cavity but likely with a better binding capacity by providing more aromatic residues. Interestingly, a recent study revealed that a similar hydrophobic cavity is found in the primed Marburg virus GP protein, and the cavity can be targeted by a neutralizing antibody MR78 (Hashiguchi et al., 2015). MR78 binds to the cavity mainly through two aromatic residues F111.2 and Y112.2 (Figure 4C), with a similar hydrophobic interaction in the binding of NPC1-C to GPcl.
Figure 4.

NPC1-C Mimics Similar but Stronger Interactions like the Glycan Cap in the Unprimed GP
(A) In the unprimed GP structure, the glycan cap utilizes two aromatic residues, F225 and Y232, to insert into the hydrophobic receptor binding cavity in the head of GPcl.
(B) NPC1-C takes advantage of four aromatic residues (Y423, F503, F504, and Y506) and one hydrophobic residue P424 to interact with the hydrophobic cavity. In particular, the residue F503 inserts into the cavity at a deeper position. Thus, the binding of NPC1-C mimics the interaction like the glycan cap but likely with a better binding capacity by providing more aromatic residues.
(C) An MARV-neutralizing antibody MR79 targets the hydrophobic binding cavity of MARV GP through aromatic residues (F111.2 and Y112.2).
Through comparison between the unbound GP and the NPC1-C-bound GPcl, several conformational changes are observed. Two most dominant changes occur in the binding site of NPC1-C, including the β7-β8 loop and the α1 helix. In reference to that in the unbound GP, the β7-β8 loop moves downward, whereas the α1 helix goes upward upon engagement with NPC1-C (Figure 5 A). With the movement of the α1 helix, the short 310 helix in the β3-α1 loop raises upward correspondingly (Figure 5A). In the unbound GP, residue N73 in the beta turn forms a hydrogen bond with the main-chain carboxyl oxygen of residue K510, which is in the N-terminal portion of the internal fusion loop (IFL) (Figure 5B). The interaction can compromise the electric repulsion between K510 and R559 (in the HR1 helix of GP2) (Figure 5B). In the bound GPcl, due to the uplift of the short 310 helix, residue N73 does not interact with residue K510, and the side chain of K510 points inward and easily generates an electric repulsion with residue R559 (Figure 5C). The unfavorable interaction likely helps to release the IFL and therefore make ready to trigger the membrane fusion in the late endosomes.
Figure 5.

Conformational Changes in the GPcl upon Enzymatic Cleavage and NPC1-C Engagement
(A) Comparison of bound GPcl (GP1 in green and GP2 in yellow) and unbound GP-ectoΔmucin (gray glycan cap is not shown here). Three conformational changes are observed in the bound GPcl, including sites I, II, and III. At site I, the β7-β8 loop moves downward; at site II, the α1 helix raises upward. Accompanied with the raising α1 helix, an upward movement of the 310 helix in the β3-α1 loop occurs at site III.
(B) Zoomed view of the site III in the unbound GP-ectoΔmucin. The residue N73 in the 310 helix could form a hydrogen bond with the residue K510 in the N-terminal portion of the IFL. This interaction could separate the electric repulsion between K510 and R559 (in the HR1 helix of GP2).
(C) Zoomed view of the site III in the bound GPcl. The upward-moved residue N73 does not form a hydrogen bond with the residue K510, and the electric repulsion between the K510 and R539 may help to release the IFL and trigger the membrane fusion.
Mutagenesis Confirms the Key Interaction Residues
In order to clarify the interactions between the NPC1-C and GPcl, we performed mutagenesis work with NPC1-C and used the SPR experiment to test the binding. The structural analyses in the above section revealed that residues Y423, P424, F503, F504, and Y506 play the major role by contributing most of the interactions. Thus, we substituted these five sites with alanine (A) or glycine (G) residues, including quintuple substitution, double substitution (Y423&P424 or F503&F504), and single substitution. Unfortunately, the quintuple-A mutant (Y423A&P424A&F503A&F504A&Y506A), double mutant (Y423A&P424A), and two of the single mutants (Y423G and Y506G) cannot be refolded (Figures S6 and S7 ). As expected, the quintuple-G substitution (Y423G&P424G&F503G&F504G&Y506G) in NPC1-C abolishes its binding to GPcl (Figure 6 A). The double mutant in loop 1 (Y423G&P424G) dramatically reduces the binding affinity, and the double mutants (both F503A&F504A and F503G&F504G) abolish the binding (Figures 6B–6D). The single mutants in loop 2 (F503G, F504G, F503A, F504A, and Y506A) similarly abolish the binding to GPcl, as observed for the quintuple mutant (Figures 6E–6I). However, the single mutants in loop 1 (P424G, Y423A and P424A) can keep the binding capacity to GPcl but with slightly lower affinities (Figures 6J–6L), compared with the wild-type NPC1-C. Taken together, the mutagenesis work points out that the protruding loop 2 contributes more to the GPcl binding.
Figure S6.

Gel Filtration Curves of Refolded Alanine(A)-Substituted NPC1-C Mutant Proteins, Related to Figure 6
Different groups of A-substituted NPC1-C mutant proteins were design at the five sites (Y423, P424, F503, F504 and F506), which contribute most of interaction between NPC1-C and GPcl. The quintuple-A and Y423A&P424A mutants cannot be refolded, as there are only aggregates peaks in the gel filtration curves on Superdex 200 (GE Healthcare). For other mutant proteins, we can see monomer peak with different refolding efficiencies.
Figure S7.

Gel Filtration Curves of Refolded Glycine(G)-Substituted NPC1-C Mutant Proteins, Related to Figure 6
Different groups of A-substituted NPC1-C mutant proteins were design at the five sites (Y423, P424, F503, F504 and F506), which contribute most of interaction between NPC1-C and GPcl. The Y423G and Y506G mutants cannot be refolded, as there are only aggregates peaks in the gel filtration curves on Superdex 200 (GE Healthcare). For other mutant proteins, we can see monomer peak with different refolding efficiencies.
Figure 6.

Alanine/Glycine-Scanning Mutagenesis Experiments on NPC1-C
(A–I) BIAcore diagrams of quintuple-G, Y423G&P424G, F503G&F504G, F503A&F504A, F503G, F504G, F503A, F504A, and Y506A mutants of NPC1-C binding to the GPcl. These nine mutants abolish the binding to the GPcl.
(J) BIAcore diagram and saturation curve of P424G mutant of NPC1-C binding to the GPcl. The P424G mutant can keep binding to the GPcl with an affinity of 213 μM, which is slightly lower than that of wild-type NPC1-C.
(K) BIAcore diagram and saturation curve of Y423A mutant of NPC1-C binding to the GPcl. The Y423A mutant can keep binding to the GPcl with an affinity of 166 μM, which is slightly lower than that of wild-type NPC1-C.
(L) BIAcore diagram and saturation curve of P424A mutant of NPC1-C binding to the GPcl. The P424A mutant can keep binding to the GPcl with an affinity of 281 μM, which is slightly lower than that of wild-type NPC1-C. Response units were plotted against protein concentrations. The KD values were calculated by the BIAcore 3000 analysis software (BIAevaluation Version 4.1).
See also Figures S6 and S7.
Discussion
Virus-cell membrane fusion is the means by which all enveloped viruses, including devastating human pathogens such as filovirus, influenza virus, and human immunodeficiency virus (HIV), enter cells and initiate the life cycle of virus infection (Backovic and Rey, 2012). This membrane fusion process is executed by one or more viral envelope glycoproteins, including one that is generally defined as the fusion protein. As viral fusion proteins can mediate virus entry and genome release, leading to infection, they are increasingly exploited as targets for antiviral interventions (Harrison, 2015). The fusion can occur either on the cell plasma membrane or endosomal membrane (Harrison, 2015).
With major breakthroughs in our understanding of the proteins or protein complexes that mediate membrane fusion between enveloped viruses and their host cells, the fusion trigger can be classified into four major types: (1) interaction of the fusion protein with a cell-surface receptor; (2) sequential interactions with a cell-surface receptor and co-receptor/s; (3) exposure to low pH environment in endosomes after entering the cell by a general “swallow” process (e.g., endocytosis); and (4) sequential interactions with a cell-surface receptor followed by exposure to low pH environment in endosomes (White et al., 2008). After fusion trigger, the fusion protein would undergo a series of marked conformational changes (White et al., 2008). A key early event is the exposure of the fusion loop (or fusion peptide), which is otherwise either buried or fastened in the native fusion protein (Harrison, 2015, White et al., 2008). When the fusion loop or fusion peptide is released, it binds to the target membrane, forming a rod-shaped intermediate, which connects the viral and target membranes. Next, the fusion protein bends approximately in half by conformational changes and draws the viral and target membranes into close proximity, finally resulting in the two membranes to merge (Harrison, 2015, Wang et al., 2005, White et al., 2008).
It seems that EBOV fusion does not follow the above-mentioned four types of fusion-trigger manners, and the viral GP needs first to be primed inside the endosome, after entering the cell by cell-surface receptor or co-factor induction, to expose its endosome-receptor binding domain for its fusion trigger (White et al., 2008). There is an EBOV receptor residing in the endosome, and therefore a low pH environment is needed to facilitate conformational changes in the primed GP after its endosomal receptor binding (Brecher et al., 2012, Gregory et al., 2011, Schornberg et al., 2006). It has been identified that the endosome-residing NPC1 is the endosome-receptor to bind to the primed GP (Carette et al., 2011, Côté et al., 2011). It has been shown that GP is processed by cathepsin L and B to yield a primed GP (GPcl) for its full function to trigger the membrane fusion (Chandran et al., 2005, Schornberg et al., 2006). NPC1-C domain has been proposed to be the binding partner of the primed GP (Miller et al., 2012).
In this study, we present the structural evidence that NPC1-C indeed binds to the primed EBOV GP, and SPR experiments reveal that the binding affinity is low. Since the binding affinity between individual NPC1-C and the primed GP is low, an obvious question raised here would be how the primed GP can efficiently bind to the endosomal receptor NPC. We can see that there are two possibilities: either the confinement in the late endosomes makes the local concentration of NPC1 to be very high, in which case a dissociation constant in the order of 100 μM would be sufficient, or that additional domains of NPC1 also contribute to the binding affinity. These possibilities or beyond should be pursued in the future.
The receptor binding region for NPC1-C is mainly a hydrophobic cavity in the head of the primed GP and is contacted by the two protruding loops of NPC1-C. Upon enzymatic cleavage and NPC1-C binding, we have observed several conformational changes in the GPcl, of which the upward movement of a short 310 helix in the β3-α1 loop likely helps to release the N-terminal portion of the IFL in the GP2 subunit, which is probably triggering the membrane fusion. As the crystal structure of free GPcl is not resolved yet, we do not know if it is cathepsin cleavage that induces a membrane-fusion-priming conformational change in GPcl or the binding of GPcl by NPC1-C. However, our data support the notion that there should be a fifth fusion triggering type, i.e., exposure to a low pH environment in the late endosome and interaction with an endosomal receptor. The Marburgvirus has a similar hydrophobic cavity in its head of the primed GP (Hashiguchi et al., 2015) and also needs NPC1 for viral fusion (Carette et al., 2011, Côté et al., 2011). This fusion-triggering mechanism likely represents a unique feature for all the filoviruses.
The structural data of the GPcl/NPC1-C complex have immediately pointed toward the development of therapeutic agents that target their binding interface as the strategies to treat infections by EBOV and other filoviruses. Previously, compounds have been found to potentially inhibit EBOV entry by targeting the NPC1 molecule (Côté et al., 2011). There are two classes of NPC1-interacting compounds that can hamper the filovirus entry and infection, typified by compound 3.47 and compound U18666A (Côté et al., 2011). The former compound was shown to competitively block the primed GP binding to the membrane (Côté et al., 2011), and according to our structural information, this compound probably binds to the two protruding loops of NPC1-C, which should be explored in the future. By contrast, the latter compound cannot block the binding of primed GP to the NPC1-C but rather binding to a different site on NPC1 and causing endosomal calcium depletion, similar to other calcium channel inhibitors targeting the two-pore channels (TPCs) (Sakurai et al., 2015). A full-length structure of NPC1 molecule is ideal to illuminate the true nature of the molecular basis of these inhibitors’ effect, which should be pursued in the future, though we solved the NPC1-C domain structure in this study. In addition to designing inhibitors targeting NPC1, our data also provide another important direction for inhibitor design. By targeting the hydrophobic binding cavity in the head of the primed GP, some therapeutic agents, such as peptide inhibitors or small molecules, which can easily penetrate the cell membrane and reach the primed GP in the late endosomes, can be developed. Recently, a cross-reactive human antibody MR78 was identified to target this hydrophobic binding cavity of both MARV and EBOV (Hashiguchi et al., 2015). MR78 can neutralize the MARV but cannot neutralize the EBOV (Flyak et al., 2015), which might result from the fact that the hydrophobic binding cavity of MARV GP is partially exposed, whereas the cavity of EBOV GP is fully shielded by glycan cap before cathepsin cleavage in the endosome. The antibody cannot easily penetrate the cell membrane and reach the primed GP in the late endosomes, whereas the peptide inhibitors and small molecules can execute it. These previous studies, together with our study, provide a map by which therapy with cross-reactive antibodies and small inhibitors of entry could be developed. Otherwise, recent research advances in the nucleoprotein and its chaperoned VP35 protein also provide key targets for therapeutic intervention (Dong et al., 2015, Kirchdoerfer et al., 2015, Leung et al., 2015).
In conclusion, our results presented in this study for the structural basis of primed EBOV GP bound to its endosome-residing receptor NPC1 expand our understanding in the fusion trigger mechanism of enveloped viruses and provide valuable information for the design of therapeutics against infections by filoviruses.
Experimental Procedures
Protein Production and Crystallization
The cDNAs encoding domain C residues 374–620 of NPC1 were cloned into pET21a vector with a C-terminal His6-tag. The NPC1-C mutants were constructed by site-directed mutagenesis kit. The NPC1 domain C proteins (NPC1-C) were expressed in Escherichia coli strain BL21 (DE3) as inclusion bodies and then refolded in vitro using the method as previously described (Shi et al., 2011). The refolded NPC1-C proteins were then concentrated and purified by gel filtration on a HiLoad 16/60 Superdex 75 PG column (GE Healthcare). The fully glycosylated NPC1-C protein was expressed in 293T cells using the pCDNA4.0 vector and purified by metal affinity chromatography using a HisTrap HP 5 ml column (GE Healthcare) and then further purified by gel filtration on a HiLoad 16/60 Superdex 200 PG column (GE Healthcare). The EBOV transmembrane-and-mucin-domains-removed GP (GP-ectoΔmucin) protein was constructed as previously described (Lee et al., 2008) and expressed in baculovirus expression system using Sf9 cells (Shi et al., 2013, Stevens et al., 2004, Zhang et al., 2010). Soluble GP-ectoΔmucin protein was harvested from the culture supernatants by metal affinity chromatography and purified by gel filtration on a HiLoad 16/60 Superdex 200 PG column (GE Healthcare). To mimic endosomal protease cleavage and produce primed EBOV GP (GPcl), which was capable of NPC1 binding, 1 mg of EBOV GP-ectoΔmucin protein was incubated with 5 μg thermolysin overnight and purified on a Superdex 200 column.
All the crystals were obtained by using the sitting drop vapor diffusion method with 1 μl protein mixing with 1 μl reservoir solution. Diffractive crystals of free NPC1-C protein were obtained in a condition consisting of 0.07 M sodium acetate trihydrate (pH 4.6), 5.6% (w/v) polyethylene glycol 4,000, and 30% (v/v) glycerol at the protein concentration of 5 mg/ml. Derivative crystals were obtained by soaking NPC1-C crystals overnight in mother liquor containing 1 mM KAuCl4.
To obtain the complex crystals of NPC1-C bound to GPcl, the individual proteins were in vitro mixed at a molar ratio of 1:1 and incubated at 4°C for about 2 hr before concentrating and being used for crystallization. The high-quality complex crystals were obtained in a reservoir solution of 15% (v/v) pentaerythritolpropoxylate, 0.2 M sodium chloride (pH 5.5), and 0.1 M MES-NaOH with a protein concentration of 10 mg/ml.
Data Collection and Structure Determination
For data collection, all crystals were cryo-protected by briefly soaking in reservoir solution supplemented with 20% (v/v) glycerol before flash-cooling in liquid nitrogen. All the datasets were processed with HKL2000 software (Otwinowski and Minor, 1997). The structure of NPC1-C was determined by the SAD method with anomalous signal method using Au derivative with SHELXD (Usón and Sheldrick, 1999) and Phaser (Read, 2001). The complex structure was solved by molecular replacement method using Phaser with the solved NPC1-C structure and previously reported GP structure (PDB code, 3CSY) as the search models. The atomic models were completed with Coot (Emsley and Cowtan, 2004) and refined with phenix.refine in Phenix (Adams et al., 2010), and the stereochemical qualities of the final models were assessed with PROCHECK (Laskowski et al., 1993). Data collection, processing, and refinement statistics are summarized in Table S1.
SPR Analysis
The SPR analysis was performed using a BIAcore 3000 machine with CM5 chips (GE Healthcare) at room temperature (25°C). All the proteins using in SPR analysis were exchanged to BIAcore buffer, consisting of 10 mM HEPES (pH 7.5), 150 mM NaCl, and 0.005% (v/v) Tween-20, via gel filtration. The WT and mutant NPC1-C proteins or antibody 2G4 were serially diluted to a serial of concentrations. The analytes were then used to flow over the chip surface with the response units measured. The binding kinetics was analyzed with the software BIAevaluation Version 4.1 using 1:1 Langmuir binding model.
Author Contributions
G.F.G. and Y.S. designed and supervised the study. H.W. and J.S. conducted the experiments. J.Q. collected the datasets and solved the structures. Y.S. and G.F.G. analyzed the data and wrote the manuscript. G.L. and J.Y. participated in the manuscript-editing and discussion.
Acknowledgments
This work was supported by the special project of Ebola virus research from the President Foundation of Chinese Academy of Sciences (CAS), the National Natural Science Foundation of China (NSFC, Grant No. 81590761), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB08020100), and China Ministry of Science and Technology National 973 Project (Grant numbers 2013CB531502 and 2014CB542503). We acknowledge Zheng Fan and Yiwei Liu for assistance with SPR experiments in Core Facility in Institute of Microbiology, CAS. We thank the staff of BL19U1 beamline at National Center for Protein Sciences Shanghai and Shanghai Synchrotron Radiation Facility (Shanghai, People’s Republic of China) and BL17U beamline at Shanghai Synchrotron Radiation Facility for assistance during data collection. Xu Yang, Xiaoying Xu, Wenqian Duan, Yan Li, and Qihui Wang were involved in protein preparation and cell culture work during the revision stage of the manuscript. Y.S. is supported by the Excellent Young Scientist Program of the Chinese Academy of Sciences and the Youth Innovation Promotion Association CAS (2015078). G.F.G. is a leading principal investigator of the NSFC Innovative Research Group (Grant number 81321063).
Published: January 14, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2015.12.044.
Accession Numbers
The crystal structures of NPC1-C and NPC1-C/GPcl complex have been submitted to Protein Data Bank with accession numbers PDB: 5F18 and PDB: 5F1B, respectively.
Supplemental Information
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez C.P., Lasala F., Carrillo J., Muñiz O., Corbí A.L., Delgado R. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 2002;76:6841–6844. doi: 10.1128/JVI.76.13.6841-6844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backovic M., Rey F.A. Virus entry: old viruses, new receptors. Curr. Opin. Virol. 2012;2:4–13. doi: 10.1016/j.coviro.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharat T.A., Riches J.D., Kolesnikova L., Welsch S., Krähling V., Davey N., Parsy M.L., Becker S., Briggs J.A. Cryo-electron tomography of Marburg virus particles and their morphogenesis within infected cells. PLoS Biol. 2011;9:e1001196. doi: 10.1371/journal.pbio.1001196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecher M., Schornberg K.L., Delos S.E., Fusco M.L., Saphire E.O., White J.M. Cathepsin cleavage potentiates the Ebola virus glycoprotein to undergo a subsequent fusion-relevant conformational change. J. Virol. 2012;86:364–372. doi: 10.1128/JVI.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindley M.A., Hunt C.L., Kondratowicz A.S., Bowman J., Sinn P.L., McCray P.B., Jr., Quinn K., Weller M.L., Chiorini J.A., Maury W. Tyrosine kinase receptor Axl enhances entry of Zaire ebolavirus without direct interactions with the viral glycoprotein. Virology. 2011;415:83–94. doi: 10.1016/j.virol.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette J.E., Raaben M., Wong A.C., Herbert A.S., Obernosterer G., Mulherkar N., Kuehne A.I., Kranzusch P.J., Griffin A.M., Ruthel G. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstea E.D., Morris J.A., Coleman K.G., Loftus S.K., Zhang D., Cummings C., Gu J., Rosenfeld M.A., Pavan W.J., Krizman D.B. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- Chandran K., Sullivan N.J., Felbor U., Whelan S.P., Cunningham J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté M., Misasi J., Ren T., Bruchez A., Lee K., Filone C.M., Hensley L., Li Q., Ory D., Chandran K., Cunningham J. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature. 2011;477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J.P., Ioannou Y.A. Topological analysis of Niemann-Pick C1 protein reveals that the membrane orientation of the putative sterol-sensing domain is identical to those of 3-hydroxy-3-methylglutaryl-CoA reductase and sterol regulatory element binding protein cleavage-activating protein. J. Biol. Chem. 2000;275:24367–24374. doi: 10.1074/jbc.M002184200. [DOI] [PubMed] [Google Scholar]
- Dong S., Yang P., Li G., Liu B., Wang W., Liu X., Xia B., Yang C., Lou Z., Guo Y., Rao Z. Insight into the Ebola virus nucleocapsid assembly mechanism: crystal structure of Ebola virus nucleoprotein core domain at 1.8 Å resolution. Protein Cell. 2015;6:351–362. doi: 10.1007/s13238-015-0163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube D., Brecher M.B., Delos S.E., Rose S.C., Park E.W., Schornberg K.L., Kuhn J.H., White J.M. The primed ebolavirus glycoprotein (19-kilodalton GP1,2): sequence and residues critical for host cell binding. J. Virol. 2009;83:2883–2891. doi: 10.1128/JVI.01956-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube D., Schornberg K.L., Shoemaker C.J., Delos S.E., Stantchev T.S., Clouse K.A., Broder C.C., White J.M. Cell adhesion-dependent membrane trafficking of a binding partner for the ebolavirus glycoprotein is a determinant of viral entry. Proc. Natl. Acad. Sci. USA. 2010;107:16637–16642. doi: 10.1073/pnas.1008509107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Feldmann H., Geisbert T.W. Ebola haemorrhagic fever. Lancet. 2011;377:849–862. doi: 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann H., Sanchez A., Geisbert T.W. Filoviridae: Marburg and Ebola Viruses. In: Knipe D.M., Howley P.M., editors. Fields Vriology. Lippincott, Williams & Wilkins; Philadelphia: 2013. pp. 923–956. [Google Scholar]
- Flyak A.I., Ilinykh P.A., Murin C.D., Garron T., Shen X., Fusco M.L., Hashiguchi T., Bornholdt Z.A., Slaughter J.C., Sapparapu G. Mechanism of human antibody-mediated neutralization of Marburg virus. Cell. 2015;160:893–903. doi: 10.1016/j.cell.2015.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbert T.W., Daddario-DiCaprio K.M., Geisbert J.B., Young H.A., Formenty P., Fritz E.A., Larsen T., Hensley L.E. Marburg virus Angola infection of rhesus macaques: pathogenesis and treatment with recombinant nematode anticoagulant protein c2. J. Infect. Dis. 2007;196(Suppl 2):S372–S381. doi: 10.1086/520608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory S.M., Harada E., Liang B., Delos S.E., White J.M., Tamm L.K. Structure and function of the complete internal fusion loop from Ebolavirus glycoprotein 2. Proc. Natl. Acad. Sci. USA. 2011;108:11211–11216. doi: 10.1073/pnas.1104760108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison S.C. Viral membrane fusion. Virology. 2015;479-480:498–507. doi: 10.1016/j.virol.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiguchi T., Fusco M.L., Bornholdt Z.A., Lee J.E., Flyak A.I., Matsuoka R., Kohda D., Yanagi Y., Hammel M., Crowe J.E., Jr., Saphire E.O. Structural basis for Marburg virus neutralization by a cross-reactive human antibody. Cell. 2015;160:904–912. doi: 10.1016/j.cell.2015.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood C.L., Abraham J., Boyington J.C., Leung K., Kwong P.D., Nabel G.J. Biochemical and structural characterization of cathepsin L-processed Ebola virus glycoprotein: implications for viral entry and immunogenicity. J. Virol. 2010;84:2972–2982. doi: 10.1128/JVI.02151-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemielity S., Wang J.J., Chan Y.K., Ahmed A.A., Li W., Monahan S., Bu X., Farzan M., Freeman G.J., Umetsu D.T. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog. 2013;9:e1003232. doi: 10.1371/journal.ppat.1003232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchdoerfer R.N., Abelson D.M., Li S., Wood M.R., Saphire E.O. Assembly of the Ebola Virus Nucleoprotein from a Chaperoned VP35 Complex. Cell Rep. 2015;12:140–149. doi: 10.1016/j.celrep.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratowicz A.S., Lennemann N.J., Sinn P.L., Davey R.A., Hunt C.L., Moller-Tank S., Meyerholz D.K., Rennert P., Mullins R.F., Brindley M. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. USA. 2011;108:8426–8431. doi: 10.1073/pnas.1019030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn J.H., Jahrling P.B. Clarification and guidance on the proper usage of virus and virus species names. Arch. Virol. 2010;155:445–453. doi: 10.1007/s00705-010-0600-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn J.H., Bao Y., Bavari S., Becker S., Bradfute S., Brister J.R., Bukreyev A.A., Chandran K., Davey R.A., Dolnik O. Virus nomenclature below the species level: a standardized nomenclature for natural variants of viruses assigned to the family Filoviridae. Arch. Virol. 2013;158:301–311. doi: 10.1007/s00705-012-1454-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H.J., Abi-Mosleh L., Wang M.L., Deisenhofer J., Goldstein J.L., Brown M.S., Infante R.E. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137:1213–1224. doi: 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R.A., Macarthur M.W., Moss D.S., Thornton J.M. Procheck - a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- Lee J.E., Saphire E.O. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009;4:621–635. doi: 10.2217/fvl.09.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.E., Fusco M.L., Hessell A.J., Oswald W.B., Burton D.R., Saphire E.O. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008;454:177–182. doi: 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D.W., Borek D., Luthra P., Binning J.M., Anantpadma M., Liu G., Harvey I.B., Su Z., Endlich-Frazier A., Pan J. An Intrinsically Disordered Peptide from Ebola Virus VP35 Controls Viral RNA Synthesis by Modulating Nucleoprotein-RNA Interactions. Cell Rep. 2015;11:376–389. doi: 10.1016/j.celrep.2015.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G., Hu Y., Wang Q., Qi J., Gao F., Li Y., Zhang Y., Zhang W., Yuan Y., Bao J. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature. 2013;500:227–231. doi: 10.1038/nature12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malherbe H., Strickland-Cholmley M. Human disease from monkeys (Marburg virus) Lancet. 1968;1:1434. doi: 10.1016/s0140-6736(68)92023-0. [DOI] [PubMed] [Google Scholar]
- Miller E.H., Obernosterer G., Raaben M., Herbert A.S., Deffieu M.S., Krishnan A., Ndungo E., Sandesara R.G., Carette J.E., Kuehne A.I. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 2012;31:1947–1960. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulherkar N., Raaben M., de la Torre J.C., Whelan S.P., Chandran K. The Ebola virus glycoprotein mediates entry via a non-classical dynamin-dependent macropinocytic pathway. Virology. 2011;419:72–83. doi: 10.1016/j.virol.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanbo A., Imai M., Watanabe S., Noda T., Takahashi K., Neumann G., Halfmann P., Kawaoka Y. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog. 2010;6:e1001121. doi: 10.1371/journal.ppat.1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann G., Geisbert T.W., Ebihara H., Geisbert J.B., Daddario-DiCaprio K.M., Feldmann H., Kawaoka Y. Proteolytic processing of the Ebola virus glycoprotein is not critical for Ebola virus replication in nonhuman primates. J. Virol. 2007;81:2995–2998. doi: 10.1128/JVI.02486-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Qiu X., Wong G., Audet J., Bello A., Fernando L., Alimonti J.B., Fausther-Bovendo H., Wei H., Aviles J., Hiatt E. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature. 2014;514:47–53. doi: 10.1038/nature13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read R.J. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr. D Biol. Crystallogr. 2001;57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- Saeed M.F., Kolokoltsov A.A., Albrecht T., Davey R.A. Cellular entry of ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 2010;6:e1001110. doi: 10.1371/journal.ppat.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai Y., Kolokoltsov A.A., Chen C.C., Tidwell M.W., Bauta W.E., Klugbauer N., Grimm C., Wahl-Schott C., Biel M., Davey R.A. Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science. 2015;347:995–998. doi: 10.1126/science.1258758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schornberg K., Matsuyama S., Kabsch K., Delos S., Bouton A., White J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006;80:4174–4178. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schornberg K.L., Shoemaker C.J., Dube D., Abshire M.Y., Delos S.E., Bouton A.H., White J.M. Alpha5beta1-integrin controls ebolavirus entry by regulating endosomal cathepsins. Proc. Natl. Acad. Sci. USA. 2009;106:8003–8008. doi: 10.1073/pnas.0807578106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Qi J., Iwamoto A., Gao G.F. Plasticity of human CD8αα binding to peptide-HLA-A∗2402. Mol. Immunol. 2011;48:2198–2202. doi: 10.1016/j.molimm.2011.05.009. [DOI] [PubMed] [Google Scholar]
- Shi Y., Zhang W., Wang F., Qi J., Wu Y., Song H., Gao F., Bi Y., Zhang Y., Fan Z. Structures and receptor binding of hemagglutinins from human-infecting H7N9 influenza viruses. Science. 2013;342:243–247. doi: 10.1126/science.1242917. [DOI] [PubMed] [Google Scholar]
- Shimojima M., Ikeda Y., Kawaoka Y. The mechanism of Axl-mediated Ebola virus infection. J. Infect. Dis. 2007;196(Suppl 2):S259–S263. doi: 10.1086/520594. [DOI] [PubMed] [Google Scholar]
- Siegert R., Shu H.L., Slenczka W. [Isolation and identification of the “Marburg virus”] Dtsch. Med. Wochenschr. 1968;93:604–612. doi: 10.1055/s-0028-1105103. [DOI] [PubMed] [Google Scholar]
- Simmons G., Reeves J.D., Grogan C.C., Vandenberghe L.H., Baribaud F., Whitbeck J.C., Burke E., Buchmeier M.J., Soilleux E.J., Riley J.L. DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology. 2003;305:115–123. doi: 10.1006/viro.2002.1730. [DOI] [PubMed] [Google Scholar]
- Sleat D.E., Wiseman J.A., El-Banna M., Price S.M., Verot L., Shen M.M., Tint G.S., Vanier M.T., Walkley S.U., Lobel P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. USA. 2004;101:5886–5891. doi: 10.1073/pnas.0308456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens J., Corper A.L., Basler C.F., Taubenberger J.K., Palese P., Wilson I.A. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science. 2004;303:1866–1870. doi: 10.1126/science.1093373. [DOI] [PubMed] [Google Scholar]
- Sullivan N.J., Martin J.E., Graham B.S., Nabel G.J. Correlates of protective immunity for Ebola vaccines: implications for regulatory approval by the animal rule. Nat. Rev. Microbiol. 2009;7:393–400. doi: 10.1038/nrmicro2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada A., Watanabe S., Ito H., Okazaki K., Kida H., Kawaoka Y. Downregulation of beta1 integrins by Ebola virus glycoprotein: implication for virus entry. Virology. 2000;278:20–26. doi: 10.1006/viro.2000.0601. [DOI] [PubMed] [Google Scholar]
- Takada A., Fujioka K., Tsuiji M., Morikawa A., Higashi N., Ebihara H., Kobasa D., Feldmann H., Irimura T., Kawaoka Y. Human macrophage C-type lectin specific for galactose and N-acetylgalactosamine promotes filovirus entry. J. Virol. 2004;78:2943–2947. doi: 10.1128/JVI.78.6.2943-2947.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towner J.S., Khristova M.L., Sealy T.K., Vincent M.J., Erickson B.R., Bawiec D.A., Hartman A.L., Comer J.A., Zaki S.R., Ströher U. Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J. Virol. 2006;80:6497–6516. doi: 10.1128/JVI.00069-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usón I., Sheldrick G.M. Advances in direct methods for protein crystallography. Curr. Opin. Struct. Biol. 1999;9:643–648. doi: 10.1016/s0959-440x(99)00020-2. [DOI] [PubMed] [Google Scholar]
- Wang X.J., Bai Y.D., Zhang G.Z., Zhao J.X., Wang M., Gao G.F. Structure and function study of paramyxovirus fusion protein heptad repeat peptides. Arch. Biochem. Biophys. 2005;436:316–322. doi: 10.1016/j.abb.2005.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Qi J., Liu N., Li Y., Gao J., Zhang T., Chai Y., Gao F., Zhang H., Li X. Crystal structures of human TIM members: Ebolavirus entry-enhancing receptors. Chin. Sci. Bull. 2015;60:3438–3453. [Google Scholar]
- White J.M., Delos S.E., Brecher M., Schornberg K. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008;43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wool-Lewis R.J., Bates P. Endoproteolytic processing of the ebola virus envelope glycoprotein: cleavage is not required for function. J. Virol. 1999;73:1419–1426. doi: 10.1128/jvi.73.2.1419-1426.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Qi J., Shi Y., Li Q., Gao F., Sun Y., Lu X., Lu Q., Vavricka C.J., Liu D. Crystal structure of the swine-origin A (H1N1)-2009 influenza A virus hemagglutinin (HA) reveals similar antigenicity to that of the 1918 pandemic virus. Protein Cell. 2010;1:459–467. doi: 10.1007/s13238-010-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.