

Abstract
Coronavirus 3C-like and Flavivirus NS2B-NS3 proteases utilize the chymotrypsin fold to harbor their catalytic machineries but also contain additional domains/co-factors. Over the past decade, we aimed to decipher how the extra domains/co-factors mediate the catalytic machineries of SARS 3C-like, Dengue and Zika NS2B-NS3 proteases by characterizing their folding, structures, dynamics and inhibition with NMR, X-ray crystallography and MD simulations, and the results revealed: 1) the chymotrypsin fold of the SARS 3C-like protease can independently fold, while, by contrast, those of Dengue and Zika proteases lack the intrinsic capacity to fold without co-factors. 2) Mutations on the extra domain of SARS 3C-like protease can transform the active catalytic machinery into the inactive collapsed state by structurally-driven allostery. 3) Amazingly, even without detectable structural changes, mutations on the extra domain are sufficient to either inactivate or enhance the catalytic machinery of SARS 3C-like protease by dynamically-driven allostery. 4) Global networks of correlated motions have been identified: for SARS 3C-like protease, N214A inactivates the catalytic machinery by decoupling the network, while STI/A and STIF/A enhance by altering the patterns of the network. The global networks of Dengue and Zika proteases are coordinated by their NS2B-cofactors. 5) Natural products were identified to allosterically inhibit Zika and Dengue proteases through binding a pocket on the back of the active site. Therefore, by introducing extra domains/cofactors, nature develops diverse strategies to regulate the catalytic machinery embedded on the chymotrypsin fold through folding, structurally- and dynamically-driven allostery, all of which might be exploited to develop antiviral drugs.
Keywords: SARS coronavirus, Flavivirus, 3C-like protease, NS2B-NS3 protease, Chymotrypsin fold, Enzymatic allostery, Correlated motions, Natural product
1. Introduction
In the last decades, several emerging and re-emerging viral diseases have broken out globally, which not only led to worldwide health hazards, but also caused significant damages to both the regional and global economies. While the severe acute respiratory syndrome (SARS) and the Middle-east respiratory syndrome (MERS) viruses represent paradigmatic examples of viruses completely new for humans, Dengue and Zika virus are considered to be the most important re-emerging arboviruses (Guzman and Harris, 2015, Ippolito and Rezza, 2017, Wikan and Smith, 2016). Noticeably, many emerging or re-emerging viruses are RNA viruses because they own high mutation rates for the viral enzymes, and consequently can quickly adapt to the varying conditions (Nichol et al., 2000).
SARS is a contagious disease with overall fatality rates of 14–15%, characterized by high fever, malaise, rigor, headache, and nonproductive cough, which suddenly appeared in 2002 in southern China and then rapidly spread to other countries through Hong Kong (Lee et al., 2003, Poutanen et al., 2003). To combat this unprecedented challenge, governmental agencies and scientists all over the world worked together intensely to identify its causative agent to be a novel coronavirus (Drosten et al., 2003, Ksiazek et al., 2003). Coronavirus belongs to the Coronaviridae, which are enveloped, positive-stranded RNA viruses with the largest single-stranded RNA genome (27–31 kilobases) among known RNA viruses (Drosten et al., 2003, Ksiazek et al., 2003, Lai and Cavanagh, 1997). Later, another coronavirus was identified to cause MERS. So far, neither an efficacious therapy nor a preventive treatment has been available despite tremendous efforts devoted to SARS and MERS related research.
On the other hand, Dengue is the most prevalent mosquito-borne viral disease with >500 million human infections annually and 2.5 billion people at risk, particularly in tropical and subtropical regions (Bhatt et al., 2013, Guzman et al., 2010). The disease is caused by Dengue virus (DENV) of the Flaviviridae family, which also includes several other human pathogens including the West Nile virus, Japanese encephalitis virus, yellow fever virus and Zika virus (Bhatt et al., 2013, Guzman et al., 2010, Chan et al., 2016a, Chan et al., 2016b, Ramos da Silva and Gao, 2016, Dick et al., 1952; Wikan and Smith, 2016). Zika virus (ZIKV) was one of Flavivirus that reemerged in Brazil in 2015. Previously, Zika is a neglected, mosquito-borne flavivirus because of its relatively small geographical spread and mild clinical symptoms. In fact, the first biological ZIKV sample was isolated from a sentinel rhesus monkey in the Zika Forest of Uganda in 1947 (Dick et al., 1952), and it was later found that ZIKV is transmitted to humans by Aedes mosquitoes. However since 2007, large epidemics of Asian genotype ZIKV have been reported around the world (Wikan and Smith, 2016; Baronti et al., 2014, Bogoch et al., 2016). Recently it was predicted that one-third of the world population might be at risk of infection (Bogoch et al., 2016). The spread of ZIKA infection is facilitated by the ease of vertical (Mlakar et al., 2016) and sexual human-to-human transmissions (D'Ortenzio et al., 2016). Most seriously, recent studies have revealed that ZIKV infection is associated with many other diseases: Guillain-Barre syndrome and microcephaly in newborn infants of mothers infected with ZIKV during pregnancy (Mlakar et al., 2016, Li et al., 2016a, Cugola et al., 2016), thrombocytopenia (Sharp et al., 2016), multipleorgan failures (Sikka et al., 2016), and possibly male infertility (Chan et al., 2016a). On the other hand, currently there are neither vaccines nor other effective treatments available to fight Dengue and Zika diseases.
Both Coronavirus and Flavivirus are positive-stranded RNA viruses. SARS-CoV has a large RNA genome (Lai and Cavanagh, 1997), which encodes two polyproteins, namely pp1a (∼450 kDa) and pp1ab (∼750 kDa) required for genome replication and transcription. On the other hand, Flavivirus including Dengue and Zika viruses have RNA genomes with a size of approximately 11 kb, which encode a single polyprotein (∼340 kDa) (Perera and Kuhn, 2008, Lu and Gong, 2017). Intriguingly, the translation products of the genomes of SARS CoV and Flavivirus are very large replicative polyproteins, which thus need subsequent cleavage by host and virus encoded proteinases to release the functional subunits. Briefly, two polyproteins of SARS-CoV are usually processed by two proteases, one with a chymotrypsin fold and the other with a papain-like topology. The 33-kDa viral protease with the catalytic machinery hosted by the chymotrypsin fold (Fig. 1 A) was called “main proteinase” or, alternatively, the “3C-like protease (3CLp)” to indicate its similarity to the picornavirus 3C protease in sharing the chymotrypsin fold and cleavage specificity (Anand et al., 2002, Yang et al., 2003, Allaire et al., 1994, Tong, 2002, Hilgenfeld, 2014, Mesters et al., 2006).
Fig. 1.

Three-dimensional structures of the proteases of SARS-CoV, Dengue and Zika viruses. (A)The dimeric structure of the wild-type 3C-like protease of SARS-CoV. The chymotrypsin folds hosting the catalytic machinery are colored in blue and the extra helical domain in purple. (B) The NS2B-NS3 two component proteases in open and closed forms of Dengue and Zika viruses. The chymotrypsin folds adopted by NS3 protease domain to host the catalytic machinery are colored in blue and the co-factor NS2B in purple.
On the other hand, the polyproteins encoded by Flavivirus including Dengue and Zika are cleaved into 10 proteins, including three structural proteins (capsid, membrane, and envelope) and seven nonstructural proteins (NS1, NS2A/B, NS3, NS4A/B, and NS5) (Chambers et al., 1990). The cleavage is carried out by host cell proteases including furin and signalaseas, as well as a virus-encoded serine protease NS2B-NS3 protease. The NS3 protease domain located within the N-terminal part of NS3 also adopts a chymotrypsin-like fold to host the catalytic machinery (Fig. 1B). Amazingly, the flavivirus catalytic machinery completely embedded on the NS3 protease domain only becomes active upon being complexed with a stretch of approximately 40 amino acids from the cytosolic domain of NS2B, thus called two-component protease (Erbel et al., 2006, Noble and Shi, 2012, Noble et al., 2012, Lei et al., 2016, Luo et al., 2015).
Due to the essential roles of the SARS 3C-like protease, as well as Dengue and Zika NS2B-NS3 proteases in their replication, they have been extensively validated to be promising targets for design of therapeutic inhibitors for treating SARS, Dengue and Zika. Over the past decade, we have been mainly focused on deciphering how the extra domains/co-factors of the SARS 3C-like, Dengue and Zika NS2B-NS3 proteases mediate their catalytic machineries by characterizing their folding, structures, dynamics and inhibition with NMR spectroscopy, X-ray crystallography and molecular dynamics (MD) simulations. Here, we summarize our results with a particular emphasis on the structurally- and dynamically-driven mechanisms by which the extra domains/co-factors mediate the enzymatic machineries engineered within the chymotrypsin fold.
2. The 3C-like protease of SARS coronavirus (SARS-CoV)
2.1. The catalytic machinery is under extensive regulation by the extra domain
Before the SARS outbreak, in 2002 the main proteinase of transmissible gastroenteritis coronavirus (TGEV) associated with severe diarrhoea in young pigs was reported (Anand et al., 2002). Strikingly, this proteinase shares with the picornavirus 3C protease in utilizing the chymotrypsin fold to host the catalytic machinery, thus called “3C-like protease”. Nevertheless, two distinguishable differences exist: 1) the TGEV 3C-like protease gains an extra C-terminal α-helical domain of >100 residues during the evolution, and 2) it exists in a dimeric form. However, the roles of these two features in the enzymatic catalysis remained largely elusive.
In order to fight SARS, immediately after the successful identification of a new coronavirus as the etiological agent of SARS on March 2003, we decided to focus on the 3C-like protease of SARS-CoV by exploring the functional roles of the two unique properties. Therefore, upon the release of the genome sequence of SARS-CoV, we immediately identified the sequence of the SARS 3C-like protease and further dissected it into the chymotrypsin fold hosting the catalytic machinery and the extra domain based on the sequence alignment between SARS-CoV and TGEV (Shi et al., 2004). Later, the crystal structure of the SARS 3C-like protease was reported (Yang et al., 2003), indicating that exactly like the TGEV one, the SARS 3C-like protease also has a helical extra domain and exists in a dimeric form (Fig. 2 A). As such, our previous dissection properly separated the chymotrypsin fold (Fig. 2B) from the extra domain (Fig. 2C). The well-dispersed NMR 1H-15N HSQC spectra for both chymotrypsin fold (Fig. 2D) and the extra domain (Fig. 2E) suggested that they are independently folded. However, the isolated chymotrypsin fold existed in a monomeric form and showed no catalytic activity although it hosts the entire catalytic machinery. On the other hand, the extra domain existed in a dimeric form. As such, the results led two major conclusions: 1) the SARS 3C-like protease is only active in the dimeric form; and 2) the extra domain appears to play a key role in maintaining the catalytic activity and dimerization (Shi et al., 2004). Later studies indicated that the extra domain was sufficient to maintain the dimerization of the SARS protease even with the N-finger deleted (Zhong et al., 2008).
Fig. 2.
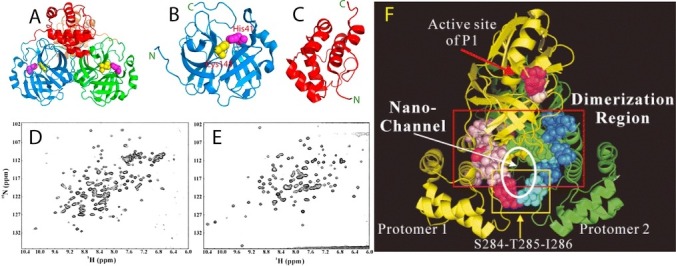
The catalytic machinery of the SARS 3C-like protease is under extensive regulation by the extra domain. (A)The dimeric structure of the wild-type 3C-like protease of SARS-CoV with the catalytic dyad His41-Cys145 displayed in spheres. Dissected chymotrypsin fold hosting the catalytic machinery (B) and extra helical domain (C). Two-dimensional 1H-15N NMR HSQC spectra of the dissected chymotrypsin fold (D), and extra helical domain (E). (F) Ribbon representation of the dimeric SARS 3C-like protease with the residues we previously identified to be critical for dimerization and catalytic activity in the sphere mode. The N-terminal five residues of protomer 1 are shown in yellow while those of protomer 2 are shown in green. Dimerization-critical residues on the extra domain are in pink for protomer 1 and blue for protomer 2. The hotpink spheres are used to indicate residues Ser284–Thr285–Ile286 and Phe291 for protomer 1 and cyan for protomer 2. The proposed nano-channel is indicated. The active site residues His41 and Cys145 are shown in red and light-brown, respectively.
To systematically map out the extra-domain residues critical for dimerization and catalytic activity, we mutated all 15 extra-domain residues to Ala one by one, which have close contacts (≤7 Å) with any residues on another protomer. Moreover, we also constructed two multiple-mutation mutants (STI/A and STIF/A), and five deletion mutants (Shi and Song, 2006). The obtained results led to the identification of the four regions critical for dimerization. In addition to the N terminus within the chymotrypsin fold which was well-recognized from its three-dimensional structure (Yang et al., 2003), other three regions within the extra domain are also critical for enzyme dimerization: namely the region around Asn214, region over Glu288–Asp289–Glu290, and the region over Arg298–Gln299 on the C-terminal last helix. Another unexpected finding is that the reduction of the side-chain volumes by Ala-mutation of the residues of two regions over Ser284–Thr285–Ile286 and Phe291 significantly boosted the enzymatic activity. As a result, two super-active mutants were constructed by mutating Ser284–Thr285–Ile286 or Ser284–Thr285–Ile286 and Phe291 to Ala, which showed significant enhancement of proteolytic activity but reduction of the enzymatic specificity (Shi and Song, 2006).
Most strikingly, the majority of the critical residues for dimerization and catalysis are clustered together to form a nano-scale channel passing through the central region of the dimeric enzyme (Fig. 2F). We thus speculated that this nano-channel plays a key role in allosterically relaying regulatory effects from the extra domain to the catalytic machinery located on the chymotrypsin fold (Shi and Song, 2006). Remarkably, this speculation has been strongly supported by our further structural and dynamic studies of the SARS 3C-like protease with both experimental and computational approaches (Shi et al., 2008, Shi et al., 2011, Lim et al., 2014).
2.2. Inactivation of the catalytic machinery by structurally-driven allostery
To understand the underlying mechanism for the extra domain to allosterically control the catalytic machinery by dimerization required a high-resolution structure of the monomeric extra-domain mutant of the SARS 3C-like protease. Therefore we have attempted to crystallize several monomer-like mutants we identified (Shi and Song, 2006), and successfully obtained the high-quality single crystals of the R298A mutant. Subsequently we determined its crystal structure at a resolution of 1.75 Å (Shi et al., 2008). Very unexpectedly, except for a 40-degree reorientation between the chymotrypsin fold and the extra domain (Fig. 3 A), the backbones and side chains of the separated chymotrypsin fold and extra domain are highly superimposable between R298A mutant and wild-type form. Detailed examination of the residue-specific root mean square deviations (RMSD) revealed that while the extra domain only has a backbone root mean square deviation of only 0.41 Å, relatively large conformational variations could be recognized within the chymotrypsin fold, in particular over several loops constituting the catalytic machinery, including residues 117 to 125, 133 to 144, and 166 to 169.
Fig. 3.

Structurally-driven allostery of the SARS 3C-like protease. (A) The monomeric mutant R298A (blue) and a protomer of the wild-type SARS 3C-like protease (red) superimposed over the catalytic fold (12–180). (B) Conformations over the residues Ile136-Lys137-Gly138-Ser139-Phe140-Leu141-Asn142-Gly143-Ser144-Cys145 in different crystal structures. Red, catalytically inactive R298A; green, catalytically inactive protomer of 1UJ1; yellow, catalytically inactive 2BX4; violet, catalytically inactive 2BX3; cyan, catalytically inactive 2GT8; blue, catalytically active 2H2Z. (C) The interaction network proposed for maintaining the dimeric structure, with one protomer in purple and another in cyan. Hydrogen bonds are indicated by cyan dashed lines, hydrophobic interactions by red dashed lines, and salt bridges by blue dashed lines. The active-site Cys145 residues of both protomers are displayed as spheres. Interestingly, it appears that Tyr126 is utilized for maintaining dimerization via an aromatic-hydrophobic interaction with Met6 of the opposite protomer, as well as for stabilizing the catalytic machinery via an aromatic stack interaction with Phe140 of the same protomer. (D) Superimposition of three monomeric mutants: Gly11Ala (yellow), Ser139Ala (cyan) and Arg298Ala (red).
The most distinguishable characteristic in the inactivated R298A catalytic machinery is the chameleon formation of a short 310-helix by residues Ser139-Phe140-Leu141, which instead assume a dynamic loop conformation in the active wild-type enzyme structure. This transformation appears to abolish the key stack interaction between the aromatic rings of Phe140 and His163, as well as significantly twists the conformation of the residues Gly143-Ser144-Cys145, thus leading to the collapsed substrate binding pocket and oxyanion hole (Fig. 3B). As inspired by this observation, we therefore examined all available crystal structures of the SARS-CoV 3CL protease, and to our great surprise, in the dimeric structures, this characteristic is absent in the active protomers but can be found in the collapsed protomers. Therefore, we proposed that even in the dimeric enzyme, due to the high conformational dynamics required for the catalytic actions, the catalytic machinery can be trapped into the collapsed conformations by a variety of unfavorable factors. Nevertheless, in the context of dimeric form, the collapsed enzyme still has the potential to be reactivated once the unfavorable factors are removed. By a sharp contrast, if dimerization is abolished, the catalytic machinery will be permanently frozen in the collapsed state, thus leading to an irreversible inactivation of the monomeric enzyme as observed on the R298A mutant. Strikingly, it has been recently demonstrated that by introducing mutations into the short region to disrupt the characteristic 310-helix, the monomeric and inactive mutants of the SARS 3C-like protease could become reactivated (Li et al., 2016b).
It is also surprised to find that although the dimeric SARS-CoV 3C-like protease has a very large dimeric interface area of over 1000 Å2, one single interfacial mutation such as Gl1A within the chymotrypsin fold (Chen et al., 2016; Hu et al., 2009), and R298A within the extra domain is sufficient to eliminate the dimerization. Most amazingly, it has been also shown that the residues for controlling dimerization are not just limited to the interfacial ones, because residues located away from the dimerization interface was also identified to be crucial for dimerization (Barrila et al., 2006, Barrila et al., 2010). To rationalize these observations, we thus proposed that in the SARS 3C-like protease, there exist two interaction networks: one responsible for maintaining the dimeric structure and another for implementing catalytic actions (Fig. 3C). By use of overlapped residues such as Tyr126, dimerization becomes elegantly coupled to catalysis of the SARS-CoV 3C-like protease. Indeed, despite having mutation residues at very different locations, three monomeric mutants G11A (Chen et al., 2008), S139A (Hu et al., 2009) and R298A (Shi et al., 2008) available so far all share highly-superimposable backbone structures, in which the characteristic short 310-helix forms within the region Ile136-Gly143 of all three mutants (Fig. 3D).
2.3. Inactivation of the catalytic machinery by dynamically-driven allostery
In our systematic mapping, we also identified one mutant N214A, which showed a dramatically reduced activity but still remained dimeric (Shi and Song, 2006). In order to understand the underlying mechanism, we determined its crystal structure, and indeed it still adopted a dimeric structure (Shi et al., 2011). Most unexpectedly, N214A not only has the overall backbone structure (Fig. 4 A and B), but also the side chains of the key residues of the catalytic machinery (Fig. 4C) almost identical to those of the WT protease. So we hypothesized that the N214A mutation inactivated the catalytic machinery of the SARS 3C-like protease by triggering dynamical changes which were not reflected by the average crystal structure.
Fig. 4.

Dynamically-driven inactivation of the catalytic machinery by the N214A mutation on the extra domain. (A)Overall superimposition of the dimeric Asn214Ala (violet) and WT (cyan) structures. (B) Dimeric structure of the SARS 3C-like protease showing the catalytic dyad His41-Cys145 located on the cleft of domain I (brown) and domain II (blue) of the chymotrypsin fold, as well as Asn214 on the extra domain. Superimpositions of the catalytically critical residues of WT (violet) and Asn214Ala (cyan) for protomers A (C) and B (D) respectively. (E) Dynamic behavior of the catalytic dyad as reflected by the time-trajectories of the distance between NE2 of His41 and SG of Cys145 atoms, of the Chi1 dihedral angle of His41 and of the Chi2 dihedral angle of His41 of WT; R298A and N214A.
In order to explore this possibility, we utilized molecular dynamics (MD) simulations, which is a powerful tool to gain insights into the dynamic mechanism that underlies protein function (Karplus and McCammon, 1983, Hammes-Schiffer and Benkovic, 2006, Dodson et al., 2008, Ma and Nussinov, 2010). We initiated MD simulations for the WT, N214A and R298A enzymes, as well as two artificial monomers derived from the dimeric WT and N214A structures (Shi et al., 2011). Remarkably, three forms of the SARS 3C-like proteases display very distinct dynamic behaviors. Among them the WT enzyme has the largest conformational rigidity with the lowest root-mean-square deviations (RMSD) values averaged over three simulations (1.75 and 1.76 Å respectively for two protomers). By contrast, R298A shows the highest overall conformational flexibility, with the largest average RMSD (2.50 Å). Interestingly, despite having the crystal structures very similar to those of WT, both N214A protomers have much larger conformational flexibility than the WT ones, with average RMSD values of 2.20 and 2.34 Å respectively for two protomers (Shi et al., 2011).
Subsequently, we exhaustively analyzed the conformational changes of individual residues of different forms in the MD simulations. The most dramatic and relevant changes are located within the catalytic machinery composed of the catalytic dyad and substrate binding subsite S1. In all previously-determined crystal structures of SARS 3C-like protease, the distance between NE2 of His41 and SG of Cys145 ranges from 3.6 to 3.9 Å. Furthermore, previous MD simulations also revealed that the dynamic stability of this distance is extremely critical for the maintaining the catalytic competency of the SARS 3C-like protease (Pang, 2004, Tan et al., 2005, Chen et al., 2006, Yin et al., 2007). As shown in the time-trajectories of this distance in our simulations for WT, R298A and N214A (Fig. 4E), for the WT enzyme, the average value of the distance is 3.83 Å, while is 4.18 Å for R298A. Intriguingly, N214A has the largest average value of this distance (4.26 Å). In particular, in the trajectory of one simulation, there are several very large fluctuations. We also analyzed the time-trajectories of the angles Chi1 and Chi2 of His41 for WT, R298A and N214A (Fig. 4E). Interestingly in both WT protomers, the Chi1 and Chi2 trajectories are dynamically stable. For R298A, the Chi1 and Chi2 trajectories are also relatively stable. However, Chi1 and Chi2 trajectories of both protomers of N214A are dynamically very unstable and jumping among several conformational clusters.
All results together revealed that while in WT, the catalytic machinery stably retains in the active state over the whole simulations; in R298A it remains largely collapsed in the inactivated state. This led to the conclusion that the active and collapsed states of the catalytic machinery of the SARS 3C-like protease are not only structurally very distinguishable as revealed by the crystal structures (Fig. 3), but also dynamically well separated as decoded by MD simulations. Most surprisingly, although in the crystal structure, the catalytic machinery of N214A adopted the active state as WT, after several nanoseconds of simulations, it became dynamically unstable and many residues constituting the catalytic machinery jump to sample the conformations highly resembling those of R298A. Therefore, we concluded that despite being located on the helical extra domain, the N214A mutation is able to triggers the changes of the enzyme dynamics even in the context of the dimeric form, which ultimately inactivates the catalytic machinery located on the chymotrypsin fold. The MD simulations thus unveiled that the catalysis of the SARS 3C-like protease is critically dependent on the protein dynamics, which can be amazingly modulated by the perturbation on the extra domain. Consequently, mediating the dynamics may offer a potential avenue to inhibit the SARS-CoV 3C-like protease.
2.4. Enhancement of the catalytic machinery by dynamically-driven allostery
In our previous systematic mapping, we have also engineered two multiple mutants with S284-T285-I286 and S284-T285-I286-Phe291 (Fig. 5 A) replaced by Ala, designated as STI/A and STIF/A respectively, which have 3.6- and 6.2-fold activity increases but reduced substrate-specificity (Shi and Song, 2006). In order to understand the underlying mechanisms, we also determined their crystal structures (Lim et al., 2014), which still adopt the dimeric structure almost identical to that of the wild-type (WT), except for slightly tighter packing between two extra domains (Fig. 5B–D). This inspired us again to hypothesize that the enhancing effects on the catalytic machinery of these mutations located on the extra domain are mostly dynamically driven, as we previously observed on the N214A mutant which dramatically inactivated the catalytic machinery, even without significant perturbation of the average three-dimensional structure (Shi et al., 2011).
Fig. 5.

Dynamically-driven enhancement of the catalytic machinery by the STI/A and STIF/A mutations on the extra domain. (A) The dimeric structure of the SARS 3C-like protease showing the locations of catalytic dyad, Phe291, Ser284-Thr285-Ile286. The cavity of the nano-channel is represented by the mesh for STI/A (B), WT (C) and STIF/A (D). (E) The cavity volumes of the nano-channel of STI/A in the first simulation at 0, 25, 50, 75 and 100 ns. (F) The cavity volumes of the nano-channel of STIF/A in the first simulation at 0, 25, 50, 75 and 100 ns. (G) The crystal structure of STI/A with key residues having relevant dynamical changes displayed and labeled. The cavity is represented by the violet mesh.
As such, we subsequently conducted 100-ns molecular dynamics (MD) simulations for WT, N214A, STI/A and STIF/A (Lim et al., 2014). Interestingly, even in the context of the dimeric forms, STI/A showed larger overall RMSD than WT over 100-ns simulations. Nevertheless, an exhaustive analysis revealed that the larger RMSD values for the dimeric STI/A are mostly resulting from the motions of the extra domains (Lim et al., 2014). Most distinguishably, in the simulations of STI/A (Fig. 5E) and STIF/A (Fig. 5F), two extra domains become further tightly packed, thus leading to a significant volume reduction of the nano-channel constituted by residues from both catalytic and extra domains, which we previously proposed to relay the regulatory effect from the extra domains to the catalytic machinery (Shi and Song, 2006). The enhanced packing leads to a slight increase of the dynamic stability of the N-finger and the first helix residues, which subsequently triggers the redistribution of dynamics over residues directly contacting them. These changes ultimately lead to the enhanced dynamic stability of residues constituting the catalytic machinery including Asn28, Thr25 and Cys45; Glu166 and His172; as well as the catalytic dyad His41-145. For example, the distance between NE2 of His41 and SG of Cys145, whose dynamic stability of is extremely critical for the stable formation of a hydrogen bond pivotal for maintaining the catalytic competency of the SARS 3C-like protease, is more dynamically stable in STI/A than that in WT. Furthermore, in our previous study (Shi et al., 2011), residues Ser139-Phe140-Leu141 of the WT maintained an extended active conformation during simulations, while those of N214A frequently jumped to sample the 310-helix conformation characteristic of the collapsed oxyanion hole as observed in R298A (Shi et al., 2008). However, in the simulations of STI/A, STIF/A and WT up to 100 ns, all three residues have similar dynamical behaviors in their backbone conformations and showed no sampling of the collapsed oxyanion hole. Therefore, the enhanced dynamical stability of these residues appears to contribute to the increased activity of STI/A and STIF/A.
Amazingly, the residues identified by MD simulations to be involved in the dynamic transmission in STI/A and STIF/A have been previously characterized to be also critical for maintaining dimerization (Fig. 5G). Therefore, our MD simulation results strongly support our previous proposal about the existence of a global network of correlated interactions constituted by the interactions of residues of both catalytic and extra domains of the SARS 3C-like protease, which is capable of coupling the dimerization and catalysis of the SARS 3C-like protease (Shi et al., 2008).
2.5. Existence of a global network of correlated motions that allosterically mediates the catalytic machinery
To gain further insights into the interaction network, we analyzed the MD simulation data of WT, N214A, STI/A and STIF/A by an approach called MutInf (McClendon et al., 2009), which is an entropy-based approach to analyze ensembles of protein conformers from molecular dynamics simulations. Briefly, this approach utilizes second-order terms from the configurational entropy expansion, called the mutual information, to identify pairs of residues with correlated conformations, or correlated motions, in an equilibrium ensemble.
As seen in the mutual information profiles of WT (Fig. 6 A), in the protomer 1 fragments of both catalytic and extra domains have highly correlated motions, which include the N-finger, helix A, Thr25, Asn28, Cys44 and catalytic dyad residue His41; Leu115-Cys156 containing CII-BII residues, oxyanion loop Ser139-Leu141, and catalytic dyad residue Cys145; Val186-Thr198 within the loop connecting the catalytic and extra domain; Asn214 Asn238 containing Asn214; and Ile281-Phe305 containing S284-T285-I286. Amazingly, these fragments with highly correlated motions contain all residues which have been previously identified to be critical for dimerization and catalysis by both experimental and simulation approaches, which together constitute a global network of correlated motions for the residues over the whole protease (Fig. 6). Strikingly, the loop residues Val186-Thr198 were also shown to be a key component of this network. So far their role in dimerization and catalysis remains unexplored and thus it is worthwhile to experimentally characterize in the further. On the other hand, although the correlation pattern in the protomer 2 of WT remains largely similar to that of the protomer 1, the significantly correlated residues slightly changed, thus resulting in the less correlation of some catalytic domain residues (Fig. 6A).
Fig. 6.

The existence of a global network of correlated motions in the SARS 3C-like protease. Mutual information matrixes calculated from the MD simulation data of WT (A), N214A (B), STI/A (C) and STIF/A (D) by MutInf program. Yellow boxes are used to highlight the regions with highly correlated motions.
Most distinguishably, although the N214A mutation has been shown to significantly provoke the dynamics of the whole protease (Shi et al., 2011), the mutual information profiles reveal that the N214A mutation globally decouples the correlation of the paired residue motions (Fig. 6B). In particular, the first half of the catalytic fold which hosts His41, one of the catalytic dyad His41-Cys145, loses the significant correlation to the rest of the protease in both protomers. The results obtained from the correlation analysis of both WT and N214A strongly suggest that in the dimeric form of the 3C-like SARS protease, there exists a global network to correlate the motions of key residues involved in dimerization and catalysis distributed over the whole protease. Most strikingly, this global network appears to be essential for implementing its catalytic action, and consequently the dynamic perturbation onto a key component even without detectable structural change is sufficient to dramatically inactivate the catalytic machinery by decoupling the global network of the correlated motions.
On the other hand, for STI/A (Fig. 6C) and STIF/A (Fig. 6D), the interpretation of the differences of their mutual information profiles from that of WT appears to be less straightforward because the changes are relatively less significant. For example, although in the protomer 1 of STI/A (Fig. 6C), the correlated motions of some pairs of residues become less significant as compare to those in WT, the correlated motions become significantly increased for residues Gly2-Arg4 with many other residues distributed over the whole protease in the second protomer of STI/A. Furthermore, in the second protomer of STI/A, the correlated motions over the majority of the residue pairs become enhanced or altered as compared to those of WT. The same situation occurs for STIF/A (Fig. 6D). As such, it is not straightforward to exactly attribute these changes to the activity enhancement in STI/A and STIF/A. Nevertheless, it appears that unlike the N214A mutation which is able to significantly decouple the global network of correlated motions, thus leading to inactivation of the catalytic machinery, the STI/A or STIF/A mutations ultimately enhance the catalytic activity by relaying the perturbation via the specific allosteric pathways, which manifests as the altered patterns of correlated motions.
In conclusion, our crystallographic and simulation studies with the WT, R298A, N214A, STI/I and STIF/A mutants established that besides the classic structurally-driven allostery, the dynamically-driven allostery also operates in the SARS 3C-like protease. Most amazingly, despite no detectable changes of the average crystal structures, the dynamically-driven allostery is sufficient to relay the perturbation of the mutations on the extra domain onto the catalytic machinery to manifest opposite effects on catalytic activity: inactivation for N214A but enhancement for STI/A and STIF/A. These results thus offer a promising avenue to design dynamically-based allosteric inhibitors for the SARS 3C-like protease by decoupling its global network of correlated motions. Recently, it has been shown that for MERS coronavirus, its 3C-like protease exists as the inactive and monomeric form in the free state but becomes dimerized upon binding to ligands (Tomar et al., 2015). It is of both fundamental and therapeutic interest to explore how the structurally- and dynamically-driven allostery operates in the MERS 3C-like protease.
3. The NS2B-NS3 two-component proteases of Dengue and Zika viruses
3.1. Dengue NS3 protease domain lacks the intrinsic capacity to fold without the NS2B co-factor
Now many crystal and NMR structures have been determined for the flaviviral NS2B-NS3 proteases, revealing that their NS3 protease domains all fold into the chymotrypsin fold as exemplified by those of Dengue and Zika viruses (Fig. 1B). Amazingly, however, the flaviviral proteases appear to adopt two distinctive conformations: the open and the closed states (Erbel et al., 2006, Noble and Shi, 2012, Noble et al., 2012, Lei et al., 2016, Luo et al., 2015, Chen et al., 2014, de la Cruz et al., 2011, Kim et al., 2013, Gibbs et al., 2018, Mahawaththa et al., 2017, Gupta et al., 2015, Pan et al., 2017, Roy et al., 2017). In both states the NS3 protease domains adopting the chymotrypsin fold remain highly similar while the NS2B co-factors assumes diverse conformations (Fig. 1B).
In 2005 when we cloned and expressed differentially-dissected fragments of the Dengue NS3 protease domain alone based on previous reports (Murthy et al., 1999, Murthy et al., 2000), all of them were found to be not refoldable and completely insoluble under a variety of solution conditions. Amazingly, in 2006 it was demonstrated that only upon being covalently linked to a core region of NS2B, the Dengue and West Nile NS3 protease domains became correctly folded and their crystal structures have been successfully determined (Erbel et al., 2006). Nevertheless, it is of fundamental interest to understand why the additional NS2B fragment is required for the NS3 protease domain to fold into the chymotrypsin fold, which is usually an independent folding unit as evidenced by the chymotrypsin fold of the SARS 3C-like proteases (Shi et al., 2004). As facilitated by our discovery in 2005 that “insoluble proteins”, even the most hydrophobic integral membrane protein fragment in nature, could in fact soluble in salt-minimized water (Li et al., 2006, Song, 2009, Song, 2013, Song, 2017, Song, 2018), we decided to address this question by high-resolution NMR characterization (Gupta et al., 2015).
Remarkably, the isolated Dengue NS3 protease domain was highly soluble in salt-minimized water but appeared to be highly disordered as evidenced by its far-UV CD and NMR HSQC spectra (Gupta et al., 2015), which is narrowly dispersed in both 1H and 15N dimensions (Fig. 7 A). Nevertheless, upon being complexed with NS2B, the NS3 protease domain suddenly became active as well as well-folded, as reflected by the well-dispersed NMR HSQC spectra (Fig. 7B), with many HSQC peaks superimposable to those of the co-expressed Dengue NS2B-NS3 protease complex (Kim et al., 2013). This observation revealed that the isolated Dengue NS3 protease domain lacks the intrinsic ability to independently fold into the chymotrypsin-like fold and the NS2B cofactor is absolutely required to co-fold with the NS3 protease domain.
Fig. 7.

The isolated Dengue NS3 protease domain lacks the intrinsic capacity to fold into a chymotrypsin fold. Two-dimensional 1H-15N NMR HSQC spectra of the Dengue NS3 protease domain in the isolated state (A), and in complex with the NS2B cofactor (B). (C) Residue specific (ΔCα-ΔCβ) values of the NS3 protease domain in the isolated state obtained in our study (red), and in complex with NS2B (blue) previously published. (D) Residue-specific {1H}–15N heteronuclear steady-state NOE (hNOE) of the NS3 protease domain in the isolated state.
Despite its narrow NMR spectral dispersions, we successfully achieved the NMR assignment of almost all non-Pro residues of the 172-residue NS3pro (14–185). As reported by (ΔCα-ΔCβ) chemical shifts (Fig. 7C), the isolated NS3 protease domain has dramatically decreased (ΔCα-ΔCβ) over the whole sequence as compared to those of the NS3 protease in complex with NS2B previously reported (Kim et al., 2013). For example, many NS3 residues in complex with NS2B have the absolute values of (ΔCα-ΔCβ) > 4 ppm, but all isolated NS3 residues have the absolute values of (ΔCα-ΔCβ) < 2 ppm. Intriguingly, however, many residues of the isolated NS3 protease domain still have the (ΔCα-ΔCβ) patterns similar to those in the complex, implying that despite lacking the well-formed secondary structures found in the complex (Fig. 7C), similar secondary structures might be weakly populated over these residues of the isolated NS3 protease domain.
Furthermore, to pinpoint the backbone dynamics on ps-ns time scale, we measured the {1H}-15N heteronuclear steady-state NOE (hNOE) of the isolated NS3 protease domain. All residues of the isolated NS3 protease domain have small or even negative hNOEs (Fig, 7D), strongly suggesting that unlike the NS3 protease domain in complex with NS2B, the isolated NS3 protease domain has largely unrestricted backbone motions on the ps-ns time scale. Nevertheless, residues over several segments such as Ile65-Leu100 have relatively large hNOE, implying that this region might have transit tertiary packing to a certain degree.
Paramagnetic relaxation enhancement (PRE) is a powerful tool in detecting transiently existing contacts in highly disordered proteins with distances even up to ∼25 Å. Therefore, by site-directed mutagenesis, we introduced Cys residue into the NS3 protease domain one by one at three locations: Gln27, Glu86 and Ser158 (Fig. 8 A), which were subsequently labelled with the nitroxide free radical probe, MTSSL for measurement of PRE by HSQC spectra. Fig. 8A shows the intensity ratios of HSQC peaks from the oxidized (paramagnetic) and reduced (diamagnetic) spectra of three mutants. For Q27C, the residues which were significant affected are all located in the first β-barrel of the chymotrypsin fold in the native NS2B-NS3pro complex. This suggests that these affected residues have long-range contacts with Cys27 with distances <25 Å. Interestingly, although residues Thr48-Glu66 assuming a short helix and β-sheet have very close contacts with Cys27 in the native structure, they were not significantly perturbed by the spin-label at Cys27. This strongly implies that the tertiary packing is non-native in the isolated NS3 protease domain. The spin-label at the position 86 significantly affected the residues on both β-barrels, suggesting that these residues have long-range contacts to Cys86 with distances <25 Å. Again, many residues have very close contacts with Cys86 in the native fold but were not significantly perturbed by the spin-label at Cys88. The residues affected by the spin-label at the position 158 are mostly located on second β-barrel, the loop connecting two β-barrels, and the last β-sheet over residues Ser68-Gly71. Taken together, the PRE measurements revealed that despite lacking of stable secondary and tight tertiary structures, the isolated NS3 protease domain is not completely extended, but instead has a loose tertiary packing. However, this tertiary packing is highly dynamic and non-native, in which the residues over the middle region of the NS3pro sequence are slightly less dynamic than the N- and C-terminal ones.
Fig. 8.

The disordered Dengue NS3 protease domain has non-native long-range contacts. Intensity ratios of HSQC peaks and structures for the spin-labeled Q27C (A), E86C (B) and S158C (C) in the paramagnetic state of the MTSL probe and diamagnetic state after the MTSL probe was reduced. NS3 protease domains with the residue colored in green if its intensity ratio <0.85. (D) A proposed model for the co-folding of the NS2B-NS3 two-component protease.
In summary, as illustrated by Fig. 8D, our study decodes that in the absence of the NS2B co-factor, the isolated Dengue NS3 protease domain lacks the intrinsic ability to fold into the chymotrypsin fold and thus exists in a highly disordered state without any stable secondary and tertiary structures, as well as enzymatic activity, but however, owns some non-native long-range contacts. However, upon being complexed with the NS2B cofactor, they will co-fold into a well-defined and active complex. As such, discovery/design of molecules that can block the interaction between NS2B and NS3 may represent a promising approach to inhibit the folding of the Dengue NS2B-NS3 protease for therapeutic application.
3.2. Zika NS3 protease domain also lacks the intrinsic capacity to fold
Immediately after the Zika outbreak in 2016, we started to clone and express the Zika NS2B and NS3 protease in both separated and covalently-linked forms (Roy et al., 2017). Interestingly, the isolated Zika NS3 protease domain is partially soluble in buffer but nevertheless, like the Dengue NS3 protease domain, it is also highly disordered as judged by its far-UV CD and narrowly-dispersed HSQC spectra (Fig. 9 A). Nevertheless, upon being complexed with the Zika NS2B, it folded into an active and well-defined structure with a well-dispersed HSQC spectrum (Fig. 9B). This reveals that like the dengue one, the Zika NS3 protease domain is also lacking of the intrinsic capacity to independently fold and thus absolutely needs the NS2B cofactor to co-fold into the chymotrypsin fold to harbour the active catalytic machinery.
Fig. 9.

The unique properties of the Zika NS2B-NS3 protease. Two-dimensional 1H-15N NMR HSQC spectra of the Zika NS3 protease domain in the isolated state (A), and in complex with the NS2B cofactor (B). (C) Superimposition of HSQC spectra of the 15N-labeled Zika NS2B in the isolated state (blue) and in complex with the unlabeled NS3 protease domain (red). (D) Superimposition of HSQC spectra of the 15N-labeled Zika NS2B in the isolated state (blue), and in complex with unlabeled NS3pro in the presence of unlabelled bovine pancreatic trypsin inhibitor (BPTI) at a molar ratio of 1:2 (red). (E) The crystal structure of the open state of the Zika NS2B-NS3 protease complex (PDB ID of 5T1V). (F) A proposed diagram showing the conformational transformation of NS2B from the open to closed states as triggered by complexing with BPTI. Blue arrows are used for indicating β-strands formed over NS2B, while purple dashed lines are for flexible regions of NS2B.
We subsequently achieved NMR assignment of the Zika NS2B (48–100) in the free state and in complex with the NS3 protease domain (Fig. 9C). The small absolute values of (ΔCα-ΔCβ) chemical shifts clearly suggested that the isolated Zika NS2B lacks any stable secondary and tertiary structure (Roy et al., 2017). Most unexpectedly, our NMR results indicated that about a half of the ZIKV NS2B residues (Arg73-Lys100) remained similarly disordered both in the free state and in the complexed form. This is significantly different from what was observed on the Dengue NS2B-NS3 protease complex in which both N- and C-terminal residues of NS2B have packing contacts with the NS3 protease domain. Furthermore, upon being complexed with an inhibitor BPTI as monitored by NMR HSQC spectra (Fig. 9D), more NS2B residues appeared to established contacts with the NS3 protease domain but residues of Leu86-Lys100 still remain similarly disordered as in the isolated form. This unique feature of the Zika NS2B-NS3 protease complex was also supported by the crystal structure of the apo-form (Chen et al., 2016), in which the C-half of the Zika NS2B is completely invisible (Fig. 9E). Furthermore, we also generate a truncated Zika NS2B with the C-terminal residues 75–100 deleted. Interestingly, this shorter NS2B could still form a partially-folded intermediate with the NS3 protease domain which has partially-formed secondary structures and tertiary packing, as well as significant μs-ms conformational dynamics. Most strikingly, this complex is catalytically inactive, thus suggesting that despite absence of contacts with the NS3 protease domain in the apo-form, the C-half of the Zika NS2B is still required to form the closed active state upon binding to substrates or inhibitors such as BPTI (Fig. 9F).
3.3. NS2B co-factors coordinate functional dynamics of Dengue and Zika proteases
To understand the role of Dengue and Zika NS2B co-factors in functional dynamics, we have also conducted MD simulations of the NS3 proteases in the isolated state and in complex with the NS2B cofactors for both Dengue and Zika proteases, followed by analysis with MutInf to obtain the profiles of correlated motions. Interestingly, like what we previously observed on the SARS 3C-like protease, global networks of correlated motions do exist in both Dengue and Zika NS2B-NS3 protease complexes (Fig. 10 ). Amazingly, in both networks, the majority of the significant correlated motions are established between the NS2B cofactor and NS3 protease residues. Most distinguishably, the global networks of correlated motions are largely eliminated in the isolated Dengue and Zika NS3 protease domains. The results indicate that for the NS2B-NS3 two component proteases, their global networks of correlated motions are coordinated by the NS2B co-factors. Therefore, for flaviviral NS2B-NS3 proteases, the NS2B co-factors are not only absolutely required for the correct folding of the NS3 protease domains into the enzymatically active complex, but also essential for coordinating the functional dynamics needed for implementing catalytic actions in the folded complexes.
Fig. 10.

The existence of a global network of correlated motions in the Dengue and Zika proteases coordinated by the NS2B cofactors. Mutual information matrixes calculated from the MD simulation data of the Dengue and Zika proteases by MutInf program. Yellow boxes are used for indicating the NS2B cofactor residues while pink boxes for the NS3 protease residues which have significant correlated motions.
3.4. Dengue and Zika proteases are susceptible to allosteric inhibition by natural products
Previous efforts for drug development targeting the flaviviral proteases revealed that one major challenge in rational design of their active site inhibitors results from the fact that the NS2B-NS3 protease active site is flat. Furthermore, another difficulty is that they appear to prefer positively charged amino acids in peptides and peptidomimetics, which thus limits further optimization of them into effective inhibitors in vivo (Li et al., 2005, Erbel et al., 2006, Noble and Shi, 2012, Noble et al., 2012, Lei et al., 2016, Luo et al., 2015, Gibbs et al., 2018).
Therefore, also to respond to the urgency to fight ZIKV infection, we decided instead to screen out small molecule inhibitors from a list of selected natural products isolated from edible plants on the unlinked Zika NS2B-NS3pro complex which better mimics the situation in vivo (Roy et al., 2017). Indeed, we have successfully identified six compounds with significant inhibitory effects, which belong to flavonoid and natural phenol (Fig. 11 A). Intriguingly, despite sharing the same chemical scaffold, five flavonoids have very diverse inhibitory effects, with Ki values ranging from 0.77 μM for Myricetin to 34.02 μM for Apigenin. In paricular, a significant inhibitory effect (IC50 of 3.45 μM and Ki of 2.61 μM) was detected for Curcumin, a natural phenol isolated from a very popular spice yellow ginger. Most strikingly, the six compounds inhibit Zika NS2B-NS3 protease by changing Vmax but not Km (Roy et al., 2017), thus indicating that these compounds inhibit Zika NS2B-NS3 protease in a non-competitive mode. In other words, six compounds act as allosteric inhibitors for the Zika NS2B-NS3 protease with their binding sites having no significant overlap with that for the substrate.
Fig. 11.

The Zika NS2B-NS3 protease is susceptible to allosteric inhibition by natural products. (A)Chemical structures of six natural products identified to allosterically inhibit the Zika NS2B-NS3 protease. (B) The crystal structure (PDB code of 5LC0) of the Zika NS2B-NS3 protease determined with an active site inhibitor cn-716 (in spheres); onto which six natural products (in sticks) were docked. (C) The Zika NS2B-NS3 protease in complex with cn-716 and six natural products in which NS2B is displayed in ribbon and NS3 protease domain in the electrostatic potential surface. (D) Expanded binding pockets of the Zika NS2B-NS3 protease in complex with six natural products.
To facilitate the visualization of the NMR experimental results and to elucidate structure-activity relationship of the six compounds, we docked six small molecules to the crystal structure (5LC0) of ZIKV NS2B-NS3 protease with the substrate-derived inhibitor cn-716 removed. Indeed, all six compounds bind to the pockets on the back of the active site (Fig. 11B). Noticeably in the complexes, the short β-sheet formed by NS2B residue Leu74-Leu78 and Asp83-Leu86 has direct contacts with the active site inhibitor cn-716 on one side, and with the six compounds on another side (Fig. 11C). Indeed, the binding pocket for six compounds is constituted by the surfaces provided by both Zika NS2B and NS3 protease domain (Fig. 11C), with the inner surfaces relatively polar and negatively charged. Interestingly five flavonoids are highly superimposable in the pocket. On the other hand, while one phenyl ring of Curcumin also occupies the same pocket like flavonoids, another phenyl ring has established an addition binding to a new pocket on the NS3 protease domain, which is not observed for the five flavonoids (Fig. 11D). Further analysis revealed that the binding is largely mediated by the formation of the hydrogen bond networks between the Zika NS2B-NS3 protease and six compounds. In fact, the inhibitory activity of these compounds appears to be largely dependent on the number of hydrogen bonds formed between the compound and the Zika NS2B-NS3 protease (Roy et al., 2017).
4. Concluding remarks
Enzymes are characterized by their dynamics over a large range of time scales and allosteric regulation as compared to man-made catalysts. Nevertheless, the role of dynamics in catalysis is hotly debated, and the relationship between dynamics and allosteric regulation still remains poorly understood (Tousignant and Pelletier, 2004, Laskowski et al., 2009, Guarnera and Berezovsky, 2016, Motlagh et al., 2014, Lisi and Loria, 2017, Nussinov and Tsai, 2013). The viral 3C-like proteases of Coronavirus such as the SARS-CoV and NS2B-NS3 proteases of Flavivirus including Dengue and Zika viruses utilize the chymotrypsin fold to harbour their catalytic machineries, which represent one of the largest and most comprehensively studied of all enzyme families (Perona and Craik, 1997, Khosla and Harbury, 2001). Amazingly, however, in addition to the chymotrypsin fold, all three viral proteases evolutionarily acquired extra domains/co-factors: while the Coronavirus 3C-like proteases gain an extra helical domain of >100 residues, the Flavivirus proteases need a NS2B co-factor of ∼40 residues for their activity. Therefore, the three proteases not only represent validated targets for development of antiviral drugs, but can also serve as amazing models for delineating the mechanisms for protein dynamics and allostery to regulate enzymatic catalysis by characterizing the roles of the extra domains/co-factors in folding, structures and dynamics of the enzymes.
First of all, our results indicate that the chymotrypsin folds of the Coronavirus 3C-like proteases and Flavivirus proteases have fundamentally distinctive capacity in folding. While the chymotrypsin domain of the SARS 3C-like protease can independently fold without needing for the extra domain, those of the Dengue and Zika proteases lack the intrinsic capacity to correctly fold without the NS2B co-factors. As such, the isolated Dengue NS3 chymotrypsin domain exists in an inactive and highly disordered state without any stable secondary and tight tertiary packing, as well as restricted ps-ns backbone motions. Nevertheless, it is not completely extended but instead has dynamic and non-native long-range tertiary packing. As such, discovery/design of molecules that can specifically disrupt the interaction between the NS3 chymotrypsin domains and NS2B cofactors of Flavivirus proteases are expected to block the correct folding of the enzymes, thus representing a promising strategy for developing antiviral drugs.
Secondly, even the chymotrypsin domain of the SARS 3C-like protease can independently fold, without the extra domain it is enzymatically inactive due to the elimination of dimerization. Determination of high-resolution structures of its monomeric mutants deciphered that in the monomeric enzyme, the catalytic machinery is frozen in the inactive collapsed state by the structurally-driven allostery. The inactive collapsed state is structurally characteristic of the chameleon formation of a short 310 helix from a dynamic loop in the active state. The coupling of two interaction networks, one for dimerization and another for catalysis, appears to provide the basis for the structurally-driven allostery of the SARS 3C-like protease. Therefore, the molecules that can block the enzymatic dimerization may also have the potential to be developed into antiviral drugs.
Thirdly, for the SARS 3C-like protease, the mutation perturbations onto the extra domain without changing the average structures can be relayed to the catalytic machinery to achieve either inactivating or enhancing effects. MD simulations decode the existence of a global network of correlated motions in the SARS 3C-like protease which appears to be essential for implementing the catalytic actions as well as for underlying the dynamically-driven allostery. The N214A mutation inactivates the catalytic machinery by globally decoupling this network, while STI/A and STIF/A enhance the catalytic machinery by globally altering the patterns of this network. Strikingly, despite having capacity in folding fundamentally different from that of the SARS 3C-like protease, the Dengue and Zika NS2B-NS3 proteases also have global networks of correlated motions, which are amazingly coordinated by the NS2B co-factors. This implies that the NS2B co-factors not only participate in co-folding with the NS3 chymotrypsin domain, but are also essential for coordinating the functional dynamics of the active enzyme complexes.
Finally, results together reveal that protein dynamics also play a central role in enzymatic catalysis, and allosteric actions operate in all three proteases, as we previously characterized on the dynamics of the transmembrane Eph receptor on different time scales (Qin et al., 2012, Qin et al., 2015, Huan et al., 2013). Therefore, it is promising and feasible to discover/design molecules that allosterically inhibit three enzymes. Indeed, two categories of natural products extensively existing in edible plants have been successfully identified to allosterically inhibit the Zika and Dengue protease by binding to a pocket on the back of the active site. This result may bear significant implications in further design of therapeutic inhibitors particularly for the Flavivirus proteases because they have relatively flat active sites and general preference for positively charged amino acids in peptides and peptidomimetics.
In conclusion, by introducing extra domains/cofactors to the chymotrypsin folds which harbour the entire catalytic machinery, nature achieves regulation of enzymatic catalysis of the viral proteases of Coronavirus and Flavivirus with extremely diverse strategies, which range from participation in folding to mediation of structurally- and dynamically-driven allostery. Remarkably, discovery/design of molecules to interfere in all these processes may offer promising avenues for further developments of specific inhibitors to these enzymes for therapeutic applications.
Funding
The studies were supported by Ministry of Education - Singapore Tier 3 Grant MOE2012-T3-1-008 (R-154-002-580-112), and also partly by Tier 2 Grant MOE 2015-T2-1-111 to Jianxing Song.
References
- Allaire M., Chernaia M.M., Malcolm B.A., James M.N. Picornaviral 3C cysteine proteinases have a fold similar to chymotrypsin-like serine proteinases. Nature. 1994;369:72–76. doi: 10.1038/369072a0. [DOI] [PubMed] [Google Scholar]
- Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baronti C., Piorkowski G., Charrel R.N., Boubis L., Leparc-Goffart I., de Lamballerie X. Complete coding sequence of Zika virus from a French Polynesia outbreak in 2013. Genome Announc. 2014;2:e00500.14. doi: 10.1128/genomeA.00500-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrila J., Bacha U., Freire E. Long-range cooperative interactions modulate dimerization in SARS 3CLpro. Biochemistry. 2006;45:14908–14916. doi: 10.1021/bi0616302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrila J., Gabelli S.B., Bacha U., Amzel L.M., Freire E. Mutation of Asn28 disrupts the dimerization and enzymatic activity of SARS 3CL(pro) Biochemistry. 2010;49:4308–4317. doi: 10.1021/bi1002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S., Gething P.W., Brady O.J., Messina J.P., Farlow A.W., et al. The global distribution and burden of dengue. Nature. 2013;496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoch, Brady O.J., Kraemer M.U., German M., Creatore M.I., et al. Potential for Zika virus introduction and transmission in resource-limited countries in Africa and the Asia-Pacific region: a modelling study. Lancet Infect. Dis. 2016;16:1237–1245. doi: 10.1016/S1473-3099(16)30270-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers T.J., Hahn C.S., Galler R., Rice C.M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990;44:649–688. doi: 10.1146/annurev.mi.44.100190.003245. [DOI] [PubMed] [Google Scholar]
- Chan J.F.-W., Zhang A.J., Chan C.C.-S., Yip C.C.-Y., Mak W.W.-N., et al. Zika virus infection in dexamethasone-immunosuppressed mice demonstrating disseminated infection with multi-organ involvement including orchitis effectively treated by recombinant type I interferons. Biomedicine. 2016;14:112–122. doi: 10.1016/j.ebiom.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F., Choi G.K., Yip C.C., Cheng V.C., Yuen K.Y. Zika fever and congenital Zika syndrome: an unexpected emerging arboviral disease. J. Infect. 2016;72:507–524. doi: 10.1016/j.jinf.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Wei P., Huang C., Tan L., Liu Y., et al. Only one protomer is active in the dimer of SARS 3C-like proteinase. J. Biol. Chem. 2006;281:13894–13898. doi: 10.1074/jbc.M510745200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Hu T., Zhang J., Chen J., Chen K., et al. Mutation of Gly-11 on the dimer interface results in the complete crystallographic dimer dissociation of severe acute respiratory syndrome coronavirus 3C-like protease: crystal structure with molecular dynamics simulations. J. Biol. Chem. 2008;283:554–564. doi: 10.1074/jbc.M705240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.-N., Loscha K.V., Nitsche C., Graham B., Otting G. The dengue virus NS2B-NS3 protease retains the closed conformation in the complex with BPTI. FEBS Lett. 2014;588:2206–2211. doi: 10.1016/j.febslet.2014.05.018. [DOI] [PubMed] [Google Scholar]
- Chen X., Yang K., Wu C., Chen C., Hu C., et al. Mechanisms of activation and inhibition of Zika virus NS2B-NS3 protease. Cell Res. 2016;26:1260–1263. doi: 10.1038/cr.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cugola F.R., Fernandes I.R., Russo F.B., Freitas B.C., Dias J.L., et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature. 2016;534:267–271. doi: 10.1038/nature18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz L., Nguyen T.H., Ozawa K., Shin J., Graham B., Huber T., Otting G. Binding of low molecular weight inhibitors promotes large conformational changes in the dengue virus NS2B-NS3 protease: fold analysis by pseudocontact shifts. J. Am. Chem. Soc. 2011;133:19205–19215. doi: 10.1021/ja208435s. [DOI] [PubMed] [Google Scholar]
- Dick G., Kitchen S., Haddow A. Zika virus (I). Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952;46:509–520. doi: 10.1016/0035-9203(52)90042-4. [DOI] [PubMed] [Google Scholar]
- Dodson G.G., Lane D.P., Verma C.S. Molecular simulations of protein dynamics: new windows on mechanisms in biology. EMBO Rep. 2008;9:144–150. doi: 10.1038/sj.embor.7401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ortenzio E., Matheron S., de Lamballerie X., Hubert B., Piorkowski G., et al. Evidence of sexual transmission of Zika virus. N. Engl. J. Med. 2016;374:2195–2198. doi: 10.1056/NEJMc1604449. [DOI] [PubMed] [Google Scholar]
- Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Erbel P., Schiering N., D'Arcy A., Renatus M., Kroemer M., et al. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 2006;13:372–373. doi: 10.1038/nsmb1073. [DOI] [PubMed] [Google Scholar]
- Gibbs A.C., Steele R., Liu G., Tounge B.A., Montelione G.T. Inhibitor Bound Dengue NS2B-NS3pro reveals multiple dynamic binding modes. Biochemistry. 2018;57:1591–1602. doi: 10.1021/acs.biochem.7b01127. [DOI] [PubMed] [Google Scholar]
- Guarnera E., Berezovsky I.N. Allosteric sites: remote control in regulation of protein activity. Curr. Opin. Struct. Biol. 2016;37:1–8. doi: 10.1016/j.sbi.2015.10.004. [DOI] [PubMed] [Google Scholar]
- Gupta G., Lim L., Song J. NMR and MD studies reveal that the isolated dengue NS3 protease is an intrinsically disordered chymotrypsin fold which absolutely requests NS2B for correct folding and functional. PLoS One. 2015;10:e0134823. doi: 10.1371/journal.pone.0134823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman M.G., Halstead S.B., Artsob H., Buchy P., Farrar J., et al. Dengue: a continuing global threat. Nat. Rev. Microbiol. 2010;8:S7–S16. doi: 10.1038/nrmicro2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman M.G., Harris E. Dengue. Lancet. 2015;385:453–465. doi: 10.1016/S0140-6736(14)60572-9. [DOI] [PubMed] [Google Scholar]
- Hammes-Schiffer S., Benkovic S.J. Relating protein motion to catalysis. Annu. Rev. Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281:4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T., Zhang Y., Li L., Wang K., Chen S., et al. Two adjacent mutations on the dimer interface of SARS coronavirus 3C-like protease cause different conformational changes in crystal structure. Virology. 2009;388:324–334. doi: 10.1016/j.virol.2009.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan X., Shi J., Lim L., Mitra S., Zhu W., et al. Unique structure and dynamics of the EphA5 ligand binding domain mediate its binding specificity as revealed by X-ray crystallography, NMR and MD simulations. PLoS One. 2013;8:e74040. doi: 10.1371/journal.pone.0074040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito G., Rezza G. Preface - emerging viruses: from early detection to intervention. Adv. Exp. Med. Biol. 2017;972:1–5. doi: 10.1007/5584_2017_33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus M., McCammon J.A. Dynamics of proteins: elements and function. Annu. Rev. Biochem. 1983;52:263–300. doi: 10.1146/annurev.bi.52.070183.001403. [DOI] [PubMed] [Google Scholar]
- Khosla C., Harbury P.B. Modular enzymes. Nature. 2001;409:247–252. doi: 10.1038/35051723. [DOI] [PubMed] [Google Scholar]
- Kim Y.M., Gayen S., Kang C., Joy J., Huang Q., et al. NMR analysis of a novel enzymatically active unlinked dengue NS2B-NS3 protease complex. J. Biol. Chem. 2013;288:12891–12900. doi: 10.1074/jbc.M112.442723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Lai M.M., Cavanagh D. The molecular biology of coronaviruses. Adv. Virus Res. 1997;48:1–100. doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R.A., Gerick F., Thornton J.M. The structural basis of allosteric regulation in proteins. FEBS Lett. 2009;583:1692–1698. doi: 10.1016/j.febslet.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Lee N.D., Hui A., Wu P., Chan P., Cameron G.M., et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- Lei J., Hansen G., Nitsche C., Klein C.D., Zhang L., Hilgenfeld R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science. 2016;353:503–505. doi: 10.1126/science.aag2419. [DOI] [PubMed] [Google Scholar]
- Li C., Xu D., Ye Q., Hong S., Jiang Y., et al. Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell. 2016;19:120–126. doi: 10.1016/j.stem.2016.04.017. [DOI] [PubMed] [Google Scholar]
- Li C., Teng X., Qi Y., Tang B., Shi H., et al. Conformational flexibility of a short loop near the active site of the SARS-3CLpro is essential to maintain catalytic. Activity Sci Rep. 2016;6:20918. doi: 10.1038/srep20918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Lim S.P., Beer D., Patel V., Wen D., et al. Functional profiling of recombinant NS3 proteases from all four serotypes of dengue virus using tetrapeptide and octapeptide substrate libraries. J. Biol. Chem. 2005;280:28766–28774. doi: 10.1074/jbc.M500588200. [DOI] [PubMed] [Google Scholar]
- Li M., Liu J., Ran X., Fang M., Shi J., et al. Resurrecting abandoned proteins with pure water: CD and NMR studies of protein fragments solubilized in salt-free water. Biophys. J. 2006;91:4201–4209. doi: 10.1529/biophysj.106.093187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L., Shi J., Mu Y., Song J. Dynamically-driven enhancement of the catalytic machinery of the SARS 3C-like protease by the S284-T285-I286/A mutations on the extra domain. PLoS One. 2014;9:e101941. doi: 10.1371/journal.pone.0101941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisi G.P., Loria J.P. Allostery in enzyme catalysis. Curr. Opin. Struct. Biol. 2017;47:123–130. doi: 10.1016/j.sbi.2017.08.002. [DOI] [PubMed] [Google Scholar]
- Lu G., Gong P. A structural view of the RNA-dependent RNA polymerases from the Flavivirus genus. Virus Res. 2017;234:34–43. doi: 10.1016/j.virusres.2017.01.020. [DOI] [PubMed] [Google Scholar]
- Luo D., Vasudevan S.G., Lescar J. The flavivirus NS2B-NS3 protease±helicase as a target for antiviral drug development. Antivir. Res. 2015;118:148–158. doi: 10.1016/j.antiviral.2015.03.014. [DOI] [PubMed] [Google Scholar]
- Ma B., Nussinov R. Enzyme dynamics point to stepwise conformational selection in catalysis. Curr. Opin. Chem. Biol. 2010;14:652–659. doi: 10.1016/j.cbpa.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahawaththa M.C., Pearce B.J.G., Szabo M., Graham B., Klein C.D., et al. Solution conformations of a linked construct of the Zika virus NS2B-NS3 protease. Antivir. Res. 2017;142:141–147. doi: 10.1016/j.antiviral.2017.03.011. [DOI] [PubMed] [Google Scholar]
- McClendon C.L., Friedland G., Mobley D.L., Amirkhani H., Jacobson M.P. Quantifying correlations between allosteric sites in thermodynamic ensembles. J. Chem. Theor. Comput. 2009;5:2486–2502. doi: 10.1021/ct9001812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesters J.R., Tan J., Hilgenfeld R. Viral enzymes. Curr. Opin. Struct. Biol. 2006;16:776–786. doi: 10.1016/j.sbi.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlakar J., Korva M., Tul N., Popović M., PoljsÏak-Prijatelj M., et al. Zika virus associated with microcephaly. N. Engl. J. Med. 2016:951–958. doi: 10.1056/NEJMoa1600651. 2016. [DOI] [PubMed] [Google Scholar]
- Motlagh H.N., Wrabl J.O., Li J., Hilser V.J. The ensemble nature of allostery. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy H.M., Clum S., Padmanabhan R. Dengue virus NS3 serine protease. Crystal structure and insights into interaction of the active site with substrates by molecular modeling and structural analysis of mutational effects. J. Biol. Chem. 1999;274:5573–5580. doi: 10.1074/jbc.274.9.5573. [DOI] [PubMed] [Google Scholar]
- Murthy H.M., Judge K., DeLucas L., Padmanabhan R. Crystal structure of Dengue virus NS3 protease in complex with a Bowman-Birk inhibitor: implications for flaviviral polyprotein processing and drug design. J. Mol. Biol. 2000;301:759–767. doi: 10.1006/jmbi.2000.3924. [DOI] [PubMed] [Google Scholar]
- Nichol S.T., Arikawa J., Kawaoka Y. Emerging viral diseases. Proc. Natl. Acad. Sci. U. S. A. 2000;97:12411–12412. doi: 10.1073/pnas.210382297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble C.G., Shi P.Y. Structural biology of dengue virus enzymes: towards rational design of therapeutics. Antivir. Res. 2012;96:115–126. doi: 10.1016/j.antiviral.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Noble C.G., Seh C.C., Chao A.T., Shi P.Y. Ligand-bound structures of the dengue virus protease reveal the active conformation. J. Virol. 2012;86:438–446. doi: 10.1128/JVI.06225-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R., Tsai C.J. Allostery in disease and in drug discovery. Cell. 2013;153:293–305. doi: 10.1016/j.cell.2013.03.034. [DOI] [PubMed] [Google Scholar]
- Pan A., Saw W.G., Subramanian Manimekalai M.S., Grüber A., Joon S., et al. Structural features of NS3 of Dengue virus serotypes 2 and 4 in solution and insight into RNA binding and the inhibitory role of quercetin. Acta Crystallogr D Struct Biol. 2017;73:402–419. doi: 10.1107/S2059798317003849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Y.P. Three-dimensional model of a substrate-bound SARS chymotrypsin-like cysteine proteinase predicted by multiple molecular dynamics simulations: catalytic efficiency regulated by substrate binding. Proteins. 2004;57:747–757. doi: 10.1002/prot.20249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera R., Kuhn R.J. Structural proteomics of dengue virus. Curr. Opin. Microbiol. 2008;11:369–377. doi: 10.1016/j.mib.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perona J.J.1, Craik C.S. Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. J. Biol. Chem. 1997;272:29987–29990. doi: 10.1074/jbc.272.48.29987. [DOI] [PubMed] [Google Scholar]
- Poutanen S.M.D.E., Low B., Henry S., Finkelstein D., Rose K., et al. Identification of severe acute respiratory syndrome in Canada. N. Engl. J. Med. 2003;348:1995–2005. doi: 10.1056/NEJMoa030634. [DOI] [PubMed] [Google Scholar]
- Qin H., Lim L., Song J. Protein dynamics at Eph receptor-ligand interfaces as revealed by crystallography, NMR and MD simulations. BMC Biophys. 2012;5:2. doi: 10.1186/2046-1682-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H., Lim L.Z., Song J. Dynamic principle for designing antagonistic/agonistic molecules for EphA4 receptor, the only known ALS modifier. ACS Chem. Biol. 2015;10:372–378. doi: 10.1021/cb500413n. [DOI] [PubMed] [Google Scholar]
- Ramos da Silva S., Gao S.-J. Zika virus: an update on epidemiology, pathology, molecular biology, and animal model. J. Med. Virol. 2016;88:1291–1296. doi: 10.1002/jmv.24563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A., Lim L., Srivastava S., Lu Y., Song J. Solution conformations of Zika NS2B-NS3pro and its inhibition by natural products from edible plants. PLoS One. 2017;12:e0180632. doi: 10.1371/journal.pone.0180632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp T.M., Muñoz-JordaÂn J., Perez-Padilla J., Bello-PagaÂn M.I., Rivera A., et al. Zika virus infection associated with severe thrombocytopenia. Clin. Infect. Dis. 2016;63(9):1198–1201. doi: 10.1093/cid/ciw476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Wei Z., Song J. Dissection study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme: defining the extra domain as a new target for design of highly specific protease inhibitors. J. Biol. Chem. 2004;279:24765–24773. doi: 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Song J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006;273:1035–1045. doi: 10.1111/j.1742-4658.2006.05130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Sivaraman J., Song J. Mechanism for controlling the dimer-monomer switch and coupling dimerization to catalysis of the severe acute respiratory syndrome coronavirus 3C-like protease. J. Virol. 2008;82:4620–4629. doi: 10.1128/JVI.02680-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Han N., Lim L., Lua S., Sivaraman J., Wang L., Mu Y., Song J. Dynamically-driven inactivation of the catalytic machinery of the SARS 3C-like protease by the N214A mutation on the extra domain. PLoS Comput. Biol. 2011;7:e1001084. doi: 10.1371/journal.pcbi.1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikka V., Chattu V.K., Popli R.K., Galwankar S.C., Kelkar D., et al. The emergence of Zika virus as a global health security threat: a review and a consensus statement of the INDUSEM Joint Working Group (JWG) J. Global Infect. Dis. 2016;8:3. doi: 10.4103/0974-777X.176140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J. Insight into "insoluble proteins" with pure water. FEBS Lett. 2009;583:953–959. doi: 10.1016/j.febslet.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Song J. Why do proteins aggregate? “Intrinsically insoluble proteins” and “dark mediators” revealed by studies on “insoluble proteins” solubilized in pure water. F1000 Res. 2013;2:94. doi: 10.12688/f1000research.2-94.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J. Transforming cytosolic proteins into “insoluble” and membrane-toxic forms triggering diseases/aging by genetic, pathological or environmental factors. Protein Pept. Lett. 2017;24:294–306. doi: 10.2174/0929866524666170209154001. [DOI] [PubMed] [Google Scholar]
- Song J. Environment-transformable sequence-structure relationship: a general mechanism for proteotoxicity. Biophys Rev. 2018;10:503–516. doi: 10.1007/s12551-017-0369-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J., Verschueren K.H., Anand K., Shen J., Yang M., et al. pH-dependent conformational flexibility of the SARS-CoV main proteinase (M(pro)) dimer: molecular dynamics simulations and multiple X-ray structure analyses. J. Mol. Biol. 2005;354:25–40. doi: 10.1016/j.jmb.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomar S., Johnston M.L., St John S.E., Osswald H.L., Nyalapatla P.R., et al. Denison MR, mesecar AD. Ligand-induced dimerization of Middle East respiratory syndrome (MERS) coronavirus nsp5 protease (3CLpro): implications for nsp5 regulation and the development of antivirals. J. Biol. Chem. 2015;290:19403–19422. doi: 10.1074/jbc.M115.651463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L. Viral proteases. Chem. Rev. 2002;102:4609–4626. doi: 10.1021/cr010184f. [DOI] [PubMed] [Google Scholar]
- Tousignant A., Pelletier J.N. Protein motions promote catalysis. Chem. Biol. 2004;11:1037–1042. doi: 10.1016/j.chembiol.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Wikan N., Smith D.R. Zika virus: history of a newly emerging arbovirus. Lancet Infect. Dis. 2016;16:e119–e126. doi: 10.1016/S1473-3099(16)30010-X. [DOI] [PubMed] [Google Scholar]
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J., Niu C., Cherney M.M., Zhang J., Huitema C., et al. A mechanistic view of enzyme inhibition and peptide hydrolysis in the active site of the SARS CoV 3C-like peptidase. J. Mol. Biol. 2007;371:1060–1074. doi: 10.1016/j.jmb.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong N., Zhang S., Zou P., Chen J., Kang X., et al. Without its N-finger, the main protease of severe acute respiratory syndrome coronavirus can form a novel dimer through its C-terminal domain. J. Virol. 2008;82:4227–4234. doi: 10.1128/JVI.02612-07. [DOI] [PMC free article] [PubMed] [Google Scholar]