

Graphical abstract
Keywords: SARS 3CL protease, Inhibitor, Decahydroisoquinolin, Hydrophobic interaction
Abstract
The design and evaluation of a novel decahydroisoquinolin scaffold as an inhibitor for severe acute respiratory syndrome (SARS) chymotrypsin-like protease (3CLpro) are described. Focusing on hydrophobic interactions at the S2 site, the decahydroisoquinolin scaffold was designed by connecting the P2 site cyclohexyl group of the substrate-based inhibitor to the main-chain at the α-nitrogen atom of the P2 position via a methylene linker. Starting from a cyclohexene enantiomer obtained by salt resolution, trans-decahydroisoquinolin derivatives were synthesized. All decahydroisoquinolin inhibitors synthesized showed moderate but clear inhibitory activities for SARS 3CLpro, which confirmed the fused ring structure of the decahydroisoquinolin functions as a novel scaffold for SARS 3CLpro inhibitor. X-ray crystallographic analyses of the SARS 3CLpro in a complex with the decahydroisoquinolin inhibitor revealed the expected interactions at the S1 and S2 sites, as well as additional interactions at the N-substituent of the inhibitor.
1. Introduction
Although the primary epidemic of SARS (Severe Acute Respiratory Syndrome)1, 2, 3 affecting about 8500 patients and 800 dead was eventually brought under control, the recent identification of a SARS CoV (coronavirus)-like virus in Chinese bats4, 5 and of a novel coronavirus MERS-CoV (Middle East Respiratory Syndrome Corona Virus, previously known as human CoV-EMC) raise the possibility of a reemergence of SARS or related diseases.6, 7 Since no effective therapy exists for these viral infections, developing anti-SARS agents against future outbreaks remains a formidable challenge.
SARS is a positive-sense, single-stranded RNA virus featuring the largest known viral RNA which produces two large proteins with overlapping sequences, polyproteins 1a (∼450 kDa) and 1ab (∼750 kDa).8, 9, 10 SARS 3CL (chymotrypsin like) protease (3CLpro) is a key enzyme to cleave the polyproteins to yield functional polypeptides.11, 12 The 3CLpro is a cysteine protease containing a Cys-His catalytic dyad and it exists as a homodimer; each monomer contains the catalytic dyad at each active site. Due to its functional importance in the viral life cycle, 3CLpro is considered an attractive target for the structure-based design of drugs against SARS. Thus, numerous inhibitors of 3CLpro have been reported including peptide-mimics13, 14, 15, 16, 17 and small molecules derived from natural products,18, 19, 20 anti-viral agents,21, 22 anti-malaria agents,23 or high throughput screening.24, 25, 26, 27
In the course of our own studies on the SARS 3CLpro and its inhibitors,28 we found that the addition of an extra sequence to the N- or C-terminus of the mature SARS 3CLpro lowered the catalytic activity and that the mature SARS 3CLpro is sensitive to degradation at the 188Arg/189Gln site, which causes a loss of catalytic activity. The stability of 3CLpro is dramatically increased by mutating the Arg at the 188 position to Ile. The enzymatic efficiency of the R188I mutant was increased by a factor of more than 1 × 106. The potency of the mutant protease makes it possible to quantitatively evaluate substrate-based peptide-mimetic inhibitors easily by conventional HPLC using a substrate peptide containing no fluorescence derivatives. The evaluations revealed that a peptide aldehyde covering the P-site sequence of substrate, Ac-Ser-Ala-Val-Leu-NHCH(CH2CH2CON(CH3)2)–CHO, inhibits the SARS 3CLpro with an IC50 value of 37 μM. Systematic modification guided by the X-ray crystal structure of a series of peptide-mimics in a complex with R188I SARS 3CLpro resulted in 1 with an IC50 value of 98 nM (Fig. 1 ).13 All of the side-chain structures of 1 differed from the substrate sequence except at the P3 site, where the side-chain was directed outward. Kinetic inhibition data for 1 obtained from Lineweaver–Burk plots suggested that inhibitors containing an aldehyde at the C-terminus can be expected to function as competitive inhibitors.
Figure 1.
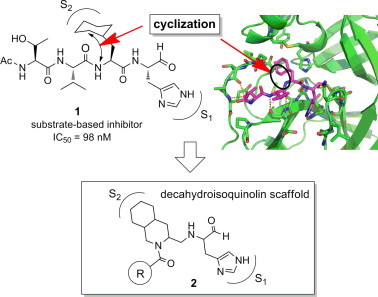
Design of a decahydroisoquinolin scaffold.
In the present study, we designed a novel non-peptide inhibitor focusing on the interactions at the S1 and S2 sites of the 3CLpro Confirmed to be critical to make the 1 potent competitive inhibitor. Among the key interactions clarified by X-ray crystallographic study, we focused on hydrophobic interactions at the cyclohexyl side-chain to design a novel inhibitor scaffold. Thus, the cyclohexyl ring is connected to the main-chain at an α-nitrogen atom of the P2 position Cha (cyclohexylalanine) via a methylene linker to yield compound 2 (Fig. 1). The resulting decahydroisoquinolin scaffold of 2 is expected to keep the hydrophobic interactions at the cyclohexyl ring of the substrate-based inhibitor at the S2 pocket. In addition, the resulting decahydroisoquinolin scaffold arranges the P1 site imidazole and active site functional aldehyde at each required position, giving the fused-ring structure of decahydroisoquinolin as a scaffold for a novel inhibitor. The acyl substituent on the nitrogen in the decahydroisoquinolin scaffold may add an extra position for the interactions with the 3CLpro.
2. Results and discussion
2.1. Chemistry
The retro synthetic route for the desired decahydroisoquinolin derivative 2 is shown in Scheme 1 . The P1 site His derivative could be introduced by a reductive amination reaction using an aldehyde derivative prepared by oxidative cleavage of the olefin bond of 3. The trans-decahydroisoquinolin scaffold of 3 could be constructed via Pd-mediated stereoselective intra-molecular cyclization29 by nucleophilic attack of a nitrogen atom to the Pd-activated olefin moiety of an allyl alcohol of 4. The olefin structure of 4 could be constructed by a Horner–Emmons reaction utilizing an aldehyde of precursor 5, and the amino group of 4 could be introduced by a Mitsunobu reaction to the alcohol of 5. The six-membered ring structure of 5 could be constructed by a Diels–Alder reaction of known ester 6 30 with butadiene.
Scheme 1.
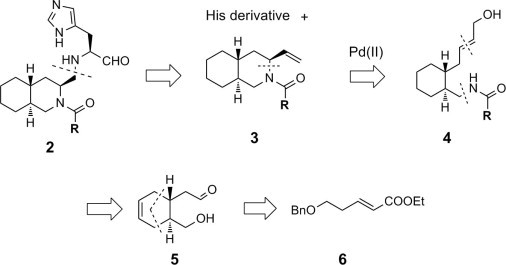
Retro synthetic route for the decahydroisoquinolin derivative.
Thus, the key intermediates 12 and 13, a precursor of the Pd-mediated cyclization, were prepared according to the route shown in Scheme 2 . The known ester 6 was first reacted with butadiene to construct the six-membered ring structure to yield 7 as an enantiomer mixture of 1,6-trans-substituted cyclohexene. The product was reduced with LAH and the resulting alcohol was then protected as tert-butyldiphenylsilyl ether to give 8. The benzyl group was removed by catalytic hydrogenation, which reduced the cyclohexene to cyclohexane at the same time. The resulting hydroxyl group was then oxidized with PCC and the resulting aldehyde was then reacted with (EtO)2P(O)CH2COOEt to yield 9. The ethyl ester of 9 was reduced with DIBALH and the resulting alcohol was protected as acetyl ester to give 10. After treatment with TBAF, the resulting alcohol was converted to the azide derivative 11 by a Mitsunobu reaction. Since the product 11 was rather unstable, 11 was immediately reduced to the corresponding amine. Without further purification, the amine derivative was coupled with p-phenylbenzoic acid using HBTU to yield 12 as an enantiomer mixture. Coupling with p-bromobenzoic acid was similarly conducted to yield a related derivative 13.
Scheme 2.
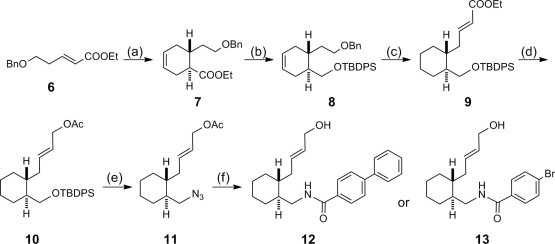
Synthesis of intermediate 12 or 13. Configurations in the racemic compounds 7–13 indicate the relative 1,6-trans configurations. Reagents: (a) 1,3-butadiene; (b) (1) LAH, (2) TBDPS-Cl/imidazole; (c) (1) H2/Pd(OH)2-C (2) PCC (3) (EtO)2P(O)CH2COOEt/NaH; (d) (1) DIBALH (2) Ac2O/pyridine/DMAP; (e) (1) TBAF, (2) (EtO)2P(O)N3/DIAD/PPh3; (f) (1) LAH, (2) 4-phenylbenzoic acid or 4-bromobenzoic acid/HBTU/DIPEA.
Construction of the decahydroisoquinolin scaffold was achieved as shown in Scheme 3 . (CH3CN)2PdCl2-mediated cyclization of 12/13 gave the desired trans-decahydroisoquinolin derivative 14/15 as a major product. The product was an enantiomer mixture which was thought to have the relative configuration of 14/15 due to the cyclization through a less hindered Pd-chelated intermediate. Thus, the vinyl substituent of the product 14/15 was thought to be axial, which was clearly confirmed by X-ray crystallographic studies of the inhibitor in a complex with the R188I mutant SARS 3CLpro as discussed below. The olefin bond of 14/15 was oxidatively cleaved by the treatment with K2OsO2(OH)4 followed by NaIO4 to yield aldehyde 16/17. Reductive amination by H-His(Trt)-N(OCH3)CH3 gave the coupling products 18 and 20 or 19 and 21 as a 1:1 diastereomer mixture which was separable on a reversed-phase column (YMC Pack ODS) by analytical HPLC (Fig. S1). The diastereomers could also be separated by conventional silica-gel column chromatography to yield diastereomers 18 and 20 or 19 and 21, each having single peak on the above reversed-phase column. Each separated diastereomer was then treated with TFA to cleave the Trt group at the imidazole ring, and the product was reduced with DIBALH to yield the desired aldehyde 22/23 or 24/25. Although the absolute configuration of each product was not determined at this stage, the purity of each product was confirmed by analytical HPLC. Since moderate but clear inhibitory activities were observed in a preliminary evaluation on the inhibitory potency of 22 and 24, the identification of the stereo-structure was then conducted.
Scheme 3.
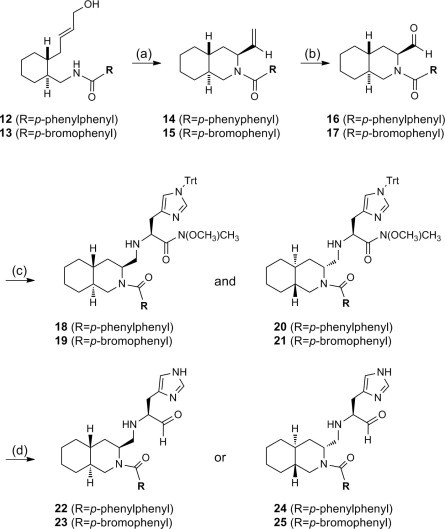
Construction of the decahydroisoquinolin scaffold. Reagents: (a) (CH3CN)2PdCl2; (b) K2OsO2(OH)4/NaIO4; (c) (1) H-His(Trt)-N(OCH3)CH3/NaBH3CN; (d) (1) TFA, (2) DIBALH.
To separately prepare the above diastereomers and estimate the absolute configurations, cyclohexene carboxylic acid obtained by a Diels–Alder reaction was converted to a salt with (R)- or (S)-α-methylbenzylamine and resolved according to the literature procedure for (1R/6S,1S/6R)-6-(2-bromophenyl)cyclohex-3-ene-1-carboxylic acid 26 31 (Scheme 4 ). Resolution of a carboxylic acid derived from compound 7 and compound 29 having the corresponding p-bromobenzyl group gave compounds showing the same polarimetric characters as the literature compounds.31 (−) Carboxylic acid 27 or 30 was obtained by salt formation with (R)-α-methylbenzylamine and following salt-liberation with HCl, whereas the salt with (S)-α-methylbenzylamine gave (+) carboxylic acid 28 or 31. Compared with the literature values, these results strongly suggest that 27 and 30 would have (1R,6S) and 28 and 31 would have (1S,6R) absolute configurations. Optical purity of each enantiomer was further confirmed using a chiral column (YMC CHIRAL Amylose-C) by HPLC (Fig. S2). Since the chemical yield from the p-bromobenzyl derivative 29 was superior to the benzyl derivative 7, enantiomer 30 or 31 was used as the starting compound for the separate synthesis of decahydroisoquinolin diastereomers.
Scheme 4.
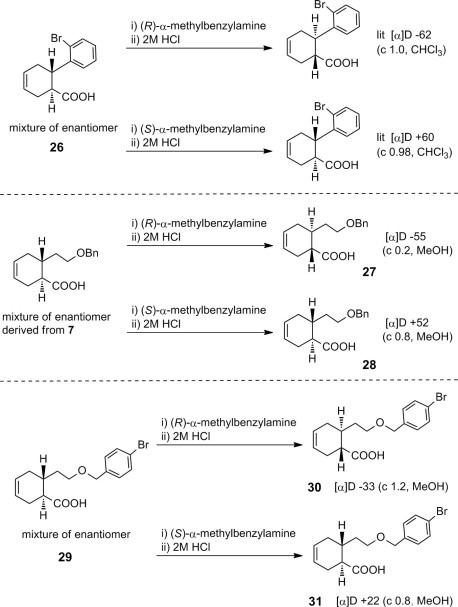
Resolution by salt formation.
The separated (1S,6R) enantiomer 31 was then used to synthesize the corresponding decahydroisoquinolin diastereomer 40 or 41 using basically the same route as above (Scheme 5 i). (1R,6S) Enantiomer 30 was also employed for the syntheses of diastereomer 44 or 45 (Scheme 5ii). The protected intermediate 38 (R = p-phenylphenyl) from 31 and the diastereomer 42 (R = p-phenylphenyl) from (1R,6S) enantiomer 30 were co-eluted with a previously synthesized diastereomixture of 18 and 20 on a reversed-phase column (YMC Pack ODS). Intermediate 38 had the same retention time as 18, whereas intermediate 42 had the same retention time as 20 (Fig. S3). The comparison was also conducted on 39 and 43 having a p-bromophenyl N-substituent with the corresponding diastereomers 19 and 21, and the same results as above were obtained (Fig. S4). These results clearly demonstrated that the two diastereomers 18 and 20 were derived from the trans-decahydroisoquinolin structure constructed from enantiomer 7. Each protected diastereomer 38/39 and 42/43 thus synthesized was converted to the desired derivatives 40/41 and 44/45 without difficulty. Several analogs shown in Table 1 containing different N-acyl substituents of the decahydroisoquinolin scaffold were also prepared using the same synthetic route (Fig. S5).
Scheme 5.
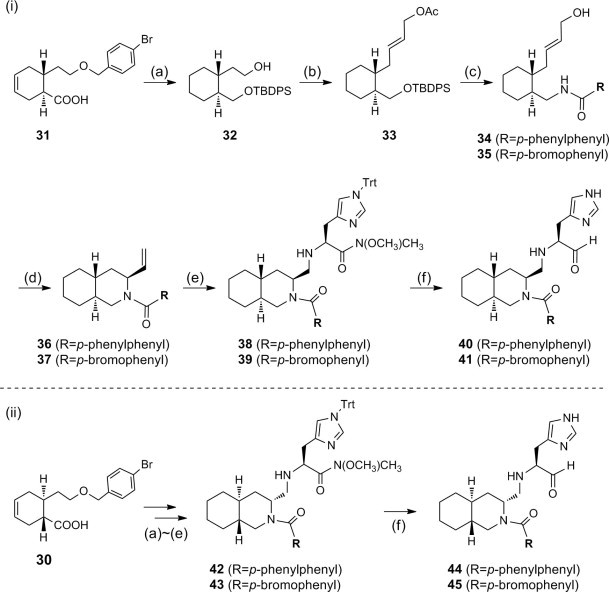
Construction of the decahydroisoquinolin scaffold starting from the separated enantiomer. Reagents: (a) (1) IBCF/NaBH4, (2) TBDPS-Cl/imidazole, (3) H2/Pd-C/sat. NaHCO3 aq.; (b) (1) PCC, (2) (EtO)2P(O)CH2COOEt/NaH, (3) DIBALH, (4) Ac2O/pyridine/DMAP; (c) (1) TBAF, (2) (EtO)2P(O)N3/DIAD/PPh3, (3) LAH, (4) 4-phenylbenzoic acid or 4-bromobenzoic acid/HBTU/DIPEA; (d) (CH3CN)2PdCl2; (e) (1) K2OsO2(OH)4/NaIO4, (2) H-His(Trt)-N(OCH3)CH3/NaBH3CN; (f) (1) TFA, (2) DIBALH.
Table 1.
| R | IC50 |
|
|---|---|---|
| (3S,4aR,8aS) | (3R,4aS,8aR) | |
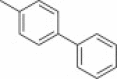 |
40 | 44 |
| 108 μM | 240 μM | |
| 46 | ||
| 135 μM | ||
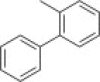 |
47 | |
| 135 μM | ||
| 48 | ||
| 68 μM | ||
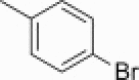 |
41 | 45 |
| 63 μM | 175 μM | |
| 49 | ||
| 57 μM | ||
2.2. Inhibitory activity
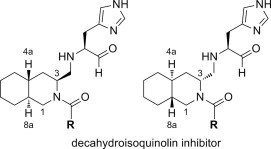
Digestion of the substrate peptide with R188I SARS 3CLpro in the presence of decahydroisoquinolin derivatives of different concentrations was conducted according to the published procedure.13 The inhibitory activities were evaluated based on IC50 values calculated from the decrease in the substrate digested by R188I SARS 3CLpro; a typical sigmoidal curve used for estimation of the IC50 value is shown in Figure S6. As summarized in Table 1, synthesized decahydroisoquinolin derivatives all showed inhibitory activities for the mutant 3CLpro. The results strongly suggest that the decahydroisoquinolin fused-ring can function as an inhibitor scaffold. Comparison of IC50 values of trans-decahydroisoquinolin diastereomers in N-4-phenylbenzoyl derivatives (40 vs 44) or N-4-bromobenzoyl derivative (41 vs 45) clearly showed that the (4aR,8aS) isomer is more potent than (4aS,8aR) isomer. The results suggest the importance of the interaction at the S2 pocket of the mutant 3CLpro. It was also demonstrated that a series of the N-benzoyl derivative was more potent than N-4-phenylbenzoyl derivatives. Substitution at the 4-position of the benzoyl substituent in 48 with halogen showed no significant effect on the inhibitory activity (41 and 49), whereas substitution at the 4-position of the phenyl group in the N-biphenylacyl derivative 40 gave a slightly more potent inhibitor than 2- or 3-substituted biphenyl derivatives (46 and 47). The results suggest that the substituent on the nitrogen atom of the decahydroisoquinolin scaffold may have some interactions with R188I SARS 3CLpro.
2.3. Evaluation of the interactions
To clarify the interactions of a newly synthesized decahydroisoquinolin inhibitor with R188I SARS 3CLpro, the structure of the protease in a complex with the inhibitor was revealed by X-ray crystallography. Subsequently, a co-crystal of the inhibitor with 3CLpro was prepared and analyzed. Structures of the 3CLpro in a complex with inhibitors 40, 41, and 44 were refined to resolutions of 1.60 Å, 2.42 Å, and 1.89 Å, respectively (PDB code 4TWY, 4TWW, and 4WY3). The data obtained are summarized in Table 2 .
Table 2.
Data collection and refinement statistics for the R188I SARS 3CL protease in complexes with compounds 40, 41, and 44
| PDB ID | 4TWY | 4TWW | 4WY3 |
|---|---|---|---|
| In complex with 40 | In complex with 41 | In complex with 44 | |
| Space group | C121 | P1 | C121 |
| Unit cell parameters | |||
| Length a | 107.83 | 54.89 | 108.11 |
| Length b | 82.128 | 59.52 | 81.82 |
| Length c | 53.271 | 68.40 | 53.24 |
| Angle α | 90 | 93.11 | 90 |
| Angle β | 104.98 | 102.82 | 104.69 |
| Angle γ | 90 | 107.30 | 90 |
| Resolution | 1.60 | 2.42 | 1.89 |
| Observations | |||
| Unique observations | 57,490 | 31,213 | 49,270 |
| Redundancy | 4.2 | 1.75 | 4.1 |
| Completeness | 88.6 | 94.3 | 93.2 |
| Mean I/σ (I) | 2.18 (at 1.60 Å) | 9.96 (at 2.42 Å) | 2.49 (at 1.89 Å) |
| R merge | 0.08 | 0.05 | 0.07 |
| Refinement | |||
| Resolution range | 25.3–1.60 | 66.1–2.42 | 30.6–1.89 |
| Rcryst | 0.29 | 0.23 | 0.27 |
| Rfree | 0.32 | 0.26 | 0.30 |
| RMSZ from ideal | |||
| Bond length (Å) | 0.93 | 0.73 | 0.86 |
| Bond angle (°) | 0.96 | 0.86 | 0.90 |
The overall structure of the 3CLpro in complex with inhibitor 41 (IC50 = 63 μM) was first compared with the substrate-based inhibitor 1 (PDB code 3ATW) (Fig. 2 ). Basically, the decahydroisoquinolin inhibitor 41 was at the active site cleft of the 3CLpro as observed in the highly potent inhibitor 1. The aldehyde group and imidazole ring of His-al, as well as the decahydroisoquinolin structure of 41, had an almost identical conformation with 1 and similarly interacted with 3CLpro. In contrast, the direction of the p-bromobenzoyl group was outward from 3CLpro and opposite to the P3 to P4 sites of 1. The N-p-bromobenzoyl group, however, was at the surface of 3CLpro, where additional hydrophobic interaction with Met of the 3CLpro may be possible (Fig. S7).
Figure 2.
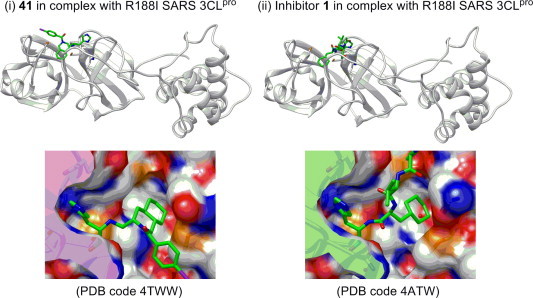
(i) X-ray structure of the inhibitor 41 in complex with R188I SARS 3CLpro (PDB code 4TWW) and molecular graphics image around the P1 and P2 sites. (ii) X-ray structure of the inhibitor 1 in complex with R188I SARS 3CLpro (Ref. 13; PDB code 4ATW) and molecular graphics image around the P1 and P2 sites.
The carbonyl carbon of the aldehyde group in 41 was detected at a distance of 2.43 Å from the active center thiol of Cys-145, and its electron density could be fitted to an sp2 carbonyl carbon as in 1 (Fig. 3 i). The results suggest that the decahydroisoquinolin inhibitor would function as a competitive inhibitor as do the peptide-aldehyde inhibitor 1.13 It was clearly confirmed that the decahydroisoquinolin scaffold of 41 took a trans-fused (4aR,8aS) configuration, as expected from the salt-resolution of enantiomixture 29. It was also confirmed that the P1 His-al substituent on the decahydroisoquinolin scaffold took an axial-configuration, as expected from the Pd(II)-mediated cyclization. The decahydroisoquinolin scaffold of 41 was inserted into a large S2 pocket created by His-41, Met-49, Met-165, and Asp-187, as in the case of a parent peptide aldehyde inhibitor, and most of the S2 pocket was occupied by the fused-ring structure of decahydroisoquinolin (Fig. 3i). The nitrogen atom of the P1 site imidazole of 41 formed a hydrogen bond with the imidazole nitrogen of His-163, resulting in close fitting at the other side of the S1 pocket formed from the Phe-140, Leu-141, and Glu-166 side chains of the protease (Fig. 3ii). These interactions, especially of the decahydroisoquinolin scaffold in the S2 pocket, function to hold the P1 site imidazole and terminal aldehyde tightly inside the active site cleft, which resulted in the compact fitting of the novel scaffold to the 3CLpro.
Figure 3.
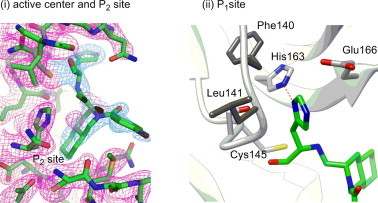
(i) Interactions of the inhibitor 41 with R188I SARS 3CLpro at the active center and P2 site. (ii) Interactions at the P1site.
To evaluate the effects of absolute configuration of the decahydroisoquinolin scaffold, structures of the 3CLpro in complex with (4aR,8aS)-N-4-phenylbenzoyl decahydroisoquinolin inhibitor 40 and (4aS,8aR)-N-4-phenylbenzoyl decahydroisoquinolin inhibitor 44 were compared (Fig. 4 i). In both inhibitors, the P1 site imidazole ring and the terminal aldehyde group had nearly the same interactions as in the (4aR,8aS)-N-bromobenzoyl decahydroisoquinolin inhibitor 41 described above. Due to the configuration change at the decahydroisoquinolin moiety, however, the (4aS,8aR) decahydroisoquinolin scaffold was clearly twisted compared to the (4aR,8aS) decahydroisoquinolin in the S2 pocket (Fig. 4ii). This conformation change of the decahydroisoquinolin scaffold transferred to the direction of the N-substituent. Thus, the substituent of (4aR,8aS) decahydroisoquinolin 40 took nearly the same conformation as the N-p-bromobenzoyl inhibitor 41 located on the surface of the 3CLpro, whereas the substituent of (4aS,8aR) decahydroisoquinolin directed outside from the protease surface. These conformational differences at the N-substituent, as well as the interactions at the S2 pocket, explain the discrepancy in the inhibitory activity between (4aR,8aS) and (4aS,8aR) decahydroisoquinolin inhibitors (41 vs 44).
Figure 4.
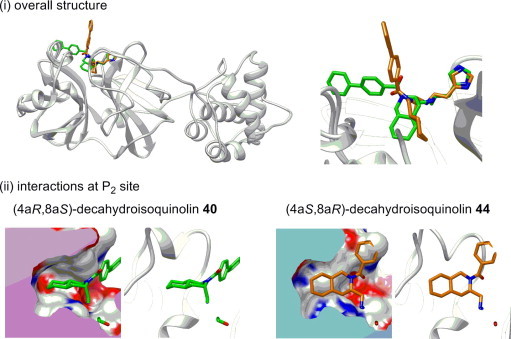
(i) X-ray structures of R188I SARS 3CLpro in complex with (4aR/8aS)-N-4-phenylbenzoyl decahydroisoquinolin inhibitor 40 (PDB code 4TWY) and (4aS/8aR)-N-4-phenylbenzoyl decahydroisoquinolin inhibitor 44 (PDB code 4WY3). (ii) Interactions of 40 and 44 at the P2 site.
3. Conclusion
A novel non-peptide inhibitor based on the interactions at the S1 and S2 sites of SARS 3CLpro was designed and synthesized. Focusing on cleavage site interaction at the S1 site and hydrophobic interaction at the S2 site, a decahydroisoquinolin scaffold was designed. Using a cyclohexene enantiomer obtained by salt resolution using chiral amine, the trans-decahydroisoquinolin derivative was synthesized as an enantiomer. Several analogs containing different N-substituents were also prepared similarly. All decahydroisoquinolin inhibitors showed moderate but clear inhibitory activities for SARS 3CLpro, which confirmed that the fused ring structure of the decahydroisoquinolin scaffold functions as an inhibitor for SARS 3CLpro. By X-ray crystallographic studies, it was confirmed that the decahydroisoquinolin inhibitors were at the active site cleft of 3CLpro, as observed in the highly potent peptide–aldehyde inhibitor. The decahydroisoquinolin scaffold was inserted into a large S2 pocket and occupied most of the pocket. The P1 site imidazole was inserted into the S1 pocket as expected. These interactions were effective to hold the terminal aldehyde tightly inside the active site cleft, which resulted in the compact fitting of the novel scaffold to 3CLpro. The acyl substituent on the nitrogen in the decahydroisoquinolin scaffold was at the surface of the 3CLpro, where additional interactions with the 3CLpro may be possible. Evaluations on the analogs focusing on the interactions at the N-substituent are now underway.
4. Experimental
4.1. General
All solvents were of reagent grade. THF was distilled from sodium and benzophenone ketyl. CH2Cl2 was distilled from CaH2. All commercial reagents were of the highest purity available. Analytical TLC was performed on silica gel (60 F-254, 0.25 mm Plates). Column chromatography was carried out on Wakogel C-200E (particle size, 75–150 μm) or Wakogel FC-40 (particle size, 20–40 μm). 1H NMR spectra were recorded in CDCl3 (unless otherwise stated) on agilent UNITY INOVA 400 NB, JEOL JNM-ECS 400, Bruker AM-300, or JEOL JNM-LA 500 spectrometers. Chemical shifts are expressed in ppm relative to tetramethylsilane (0 ppm) or CHCl3 (7.28 ppm). The coupling constants are given in Hz. 13C NMR spectra were recorded on the same spectrometers at 100 or 125 MHz, using the central resonance of CDCl3 (δ C 77.0 ppm) as the internal reference unless otherwise stated. High-resolution mass spectra (HRMS) were obtained on a JMS-HX-110A (FAB), and Shimadzu LCMS-IT-TOF (ESI). Low-resolution mass spectra (LRMS) were obtained on a Shimadzu LCMS-2010EV (ESI). Optical rotations were determined with a HORIBA SEPA-300 polarimeter. Preparative HPLC was performed using a COSMOSIL 5C18-ARII column (20 × 250 mm) with a linear gradient of CH3CN in 0.1% aqueous TFA at a flow rate of 5.0 mL/min on a HITACHI LaChrom system (OD, 254 nm). For analytical HPLC, unless otherwise noted, a COSMOSIL 5C18-ARII column (4.6 × 150 mm) was employed with a linear gradient of CH3CN in 0.1% aqueous TFA at a flow rate of 0.9 mL/min on a HITACHI LaChrom system (OD, 254 nm). The purity of the test compounds was determined by analytical HPLC. All test compounds showed ⩾95% purity.
4.1.1. (1S/R,6R/S)-Ethyl 6-[2-(benzyloxy)ethyl]cyclohex-3-enecarboxylate 7
To a solution of 1,3-butadiene (20 wt% solution in hexane, 17 mL, 40 mmol) was added ester 6 (2.34 g, 10.0 mmol), heated at 250 °C for 60 h. After the reaction mixture was cooled to room temperature, water was added and the whole was extracted with AcOEt. The organic layer was washed with 1 M HCl and brine, dried over MgSO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 30:1) to give 7 (1.87 g, 65%) as a yellow pale oil. 1H NMR (400 MHz): δ = 7.36–7.31 (m, 4H), 7.29–7.26 (m, 1H), 5.64 (m, 2H), 4.51 (d, J = 11.6 Hz, 1H), 4.46 (d, J = 12.0 Hz, 1H), 4.14 (q, J = 7.2 Hz, 2H), 3.54–3.50 (m, 2H), 2.41–2.35 (m, 1H), 2.31–2.20 (m, 3H), 2.09–2.04 (m, 1H), 1.84–1.73 (m, 2H), 1.54–1.45 (m, 1H), 1.25 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz): δ = 175.8, 138.5, 128.3, 127.6, 127.5, 125.7, 124.7, 72.8, 67.9, 60.2, 45.3, 33.7, 32.4, 29.9, 28.0, 14.2; HRMS (EI) calcd for C18H24O3 [M]+: 288.1725. Found: 288.1722.
4.1.2. {(1S/R,6R/S)-6-[2-(Benzyloxy)ethyl]cyclohex-3-en-1-yl}methanol
To a suspension of LiAlH4 (387 mg, 10.2 mmol) in ether (30 mL) was added 7 (1.47 g, 5.12 mmol) at 0 °C. After being stirred for 15 min at 0 °C, the reaction was quenched with H2O. The mixture was warmed to room temperature and filtered through Celite and a silica gel layer, and the filtrate was concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 1:1) to give a title alcohol (1.25 g, quant.) as a colorless oil. 1H NMR (400 MHz): δ = 7.37–7.32 (m, 4H), 7.30–7.26 (m, 1H), 5.65–5.57 (m, 2H), 4.51 (s, 2H), 3.66 (dd, J = 10.8, 6.0 Hz, 1H), 3.62–3.48 (m, 3H), 2.14–2.09 (m, 2H), 2.01–1.75 (m, 5H), 1.66–1.59 (m, 1H), 1.55–1.48 (m, 1H); 13C NMR (100 MHz): δ = 138.3, 128.4, 127.7, 127.6, 125.8, 125.5, 73.1, 68.5, 65.0, 39.7, 32.9, 31.0, 29.5, 26.7; HRMS (EI) calcd for C16H22O2 [M]+: 246.1620. Found: 246.1618.
4.1.3. ({(1S/R,6R/S)-6-[2-(Benzyloxy)ethyl]cyclohex-3-en-1-yl}methoxy)(tert-butyl)diphenylsilane 8
TBDPS-Cl (3.6 mL, 13.1 mmol) was added to a solution of the above alcohol (2.92 g, 11.9 mmol) and imidazole (1.21 g, 17.8 mmol) in CH2Cl2 (30 mL) and the mixture was stirred for 16 h. The reaction was quenched with saturated aqueous NH4Cl, and the whole was extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 20:1) to give 8 (5.76 g, quant.) as a colorless oil. 1H NMR (400 MHz): δ = 7.67–7.65 (m, 4H), 7.43–7.30 (m, 10H), 7.28 (m, 1H), 5.63–5.54 (m, 2H), 4.49 (d, J = 12.0 Hz, 1H), 4.45 (d, J = 12.0 Hz, 1H), 3.68 (dd, J = 10.0, 5.2 Hz, 1H), 3.62 (dd, J = 9.8, 7.0 Hz, 1H,), 3.54–3.45 (m, 2H), 2.17–2.06 (m, 2H), 2.02–1.95, (m, 1H), 1.87–1.80 (m, 2H), 1.73–1.67 (m, 2H), 1.51–1.42 (m, 1H), 1.05 (s, 9H); 13C NMR (100 MHz): δ = 138.6, 135.62, 135.61, 133.98, 133.95, 129.5, 128.3, 127.58, 127.56, 127.4, 125.8, 125.4, 72.9, 68.6, 65.9, 39.6, 32.9, 30.9, 29.1, 26.9, 26.7, 19.3; HRMS (FAB) calcd for C32H41O2Si [M+H]+: 485.2876. Found: 485.2870.
4.1.4. 2-[(1R/S,6S/R)-6-{[(tert-Butyldiphenylsilyl)oxy]methyl}cyclohexyl]ethanol
To a solution of 8 (3.40 g, 7.01 mmol) in CH3OH/AcOEt/CH2Cl2 (10:10:1, 21 mL) Pd(OH)2–C (610 mg) was added and stirred under a hydrogen gas atmosphere at room temperature for 12 h. The mixture was filtered through Celite and a silica gel layer, and the filtrate was dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 3:1) to give a title alcohol (2.78 g, quant.) as a colorless oil. 1H NMR (400 MHz): δ = 7.68–7.65 (m, 4H), 7.45–7.36 (m, 6H), 3.68–3.54 (m, 4H), 1.78–1.66 (m, 5H), 1.37–1.18 (m, 7H), 1.06 (s, 9H), 1.01–0.96 (m, 1H); 13C NMR (100 MHz): δ = 135.69, 135.66, 133.92, 133.90, 129.55, 129.54, 127.60, 127.57, 66.6, 61.1, 44.5, 36.5, 35.5, 31.9, 30.0, 26.9, 26.1, 26.0, 19.3; HRMS (FAB) calcd for C25H37O2Si [M+H]+: 397.2563. Found: 397.2569.
4.1.5. (E)-Ethyl 4-[(1R/S,2S/R)-2-{[(tert-butyldiphenylsilyl)oxy]methyl}cyclohex-1-yl]but-2-enoate 9
To a solution of PCC (3.45 g, 16.0 mmol) and Celite (3.5 g) in CH2Cl2 (20 mL), above alcohol (2.50 g, 6.30 mmol) was added at 0 °C. The temperature was gradually raised to room temperature. After being stirred for 6 h, the reaction mixture was filtered through a silica gel layer and the filtrate was concentrated. This compound was immediately used for the next step without purification. Triethylphosphonoacetate (1.5 mL, 7.7 mmol) was added to a suspension of NaH [60% in mineral oil (308 mg, 7.70 mmol)] in THF (10 mL) at −20 °C under an argon gas atmosphere and the mixture was stirred for 0.5 h. The oxidized product was added drop-wise to the reaction mixture and stirred for 1.5 h at −20 °C. The reaction was quenched with saturated aqueous NH4Cl, and the whole was extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 20:1) to give 9 (2.70 g, 92%, 2 steps) as a colorless oil. 1H NMR (400 MHz): δ = 7.67–7.64 (m, 4H) 7.45–7.36 (m, 6H), 6.91 (ddd, J = 15.4, 8.8, 6.4 Hz, 1H), 5.72 (d, J = 15.6 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 3.63–3.57 (m, 2H), 2.38–2.32 (m, 1H), 1.97 (td, J = 14.8, 8.1 Hz, 1H), 1.79–1.76 (m, 1H), 1.71–1.69 (m, 4H), 1.54–1.49 (m, 1H), 1.32–1.18 (m, 4H), 1.29 (t, J = 7.2 Hz, 3H), 1.05 (s, 9H), 1.03–0.97 (m, 1H); 13C NMR (100 MHz): δ = 166.6, 148.2, 135.62, 135.61, 133.82, 133.80, 129.59, 129.55, 127.62, 127.59, 122.4, 66.2, 60.1, 43.9, 37.8, 36.4, 31.9, 30.0, 26.9, 26.1, 26.0, 19.3, 14.3; HRMS (FAB) calcd for C29H40NaO3Si [M+Na]+: 487.2644. Found: 487.2651.
4.1.6. (E)-4-[(1R/S,2S/R)-2-{[(tert-Butyldiphenylsilyl)oxy]methyl}cyclohexyl]but-2-en-1-ol
To a solution of 9 (1.92 g, 4.13 mmol) in CH2Cl2 (20 mL), DIBALH (1.0 mol/L solution in hexane, 12.4 mL, 12.4 mmol) was added at −78 °C. After being stirred for 15 min at the same temperature, the reaction was quenched with CH3OH (5.0 mL). The mixture was warmed to room temperature, and filtered through Celite and a silica gel layer. The filtrate was dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 1:1) to give a title alcohol (1.74 g, quant.) as a colorless oil. 1H NMR (400 MHz): δ = 7.68–7.65 (m, 4H), 7.44–7.36 (m, 6H), 5.64–5.48 (m, 2H), 4.04 (d, J = 6.0 Hz, 2H), 3.66 (dd, J = 10.0, 2.8 Hz, 1H), 3.58 (dd, J = 9.8, 5.4 Hz, 1H), 2.23–2.17 (m, 1H), 1.87–1.79 (m, 2H), 1.72–1.69 (m, 3H), 1.43–1.32 (m, 1H), 1.30–1.18 (m, 4H), 1.05 (s, 9H), 1.01–0.94 (m, 1H); 13C NMR (100 MHz): δ = 135.64, 135.63, 134.0, 131.6, 130.2, 129.52, 129.50, 127.58, 127.55, 66.3, 63.8, 43.9, 38.1, 36.2, 31.7, 30.0, 26.9, 26.2, 26.1, 19.4; HRMS (FAB) calcd for C27h38nao2si [M+Na]+: 445.2539. Found: 445.2541.
4.1.7. (E)-4-[(1R/S,2S/R)-2-{[(tert-Butyldiphenylsilyl)oxy]methyl}cyclohexyl]but-2-en-1-yl acetate 10
To a solution of above alcohol (1.74 g, 4.11 mmol) in CH2Cl2 (20 mL), pyridine (0.50 mL, 6.2 mmol), acetic anhydride (0.59 mL, 6.19 mmol), and DMAP (50 mg, 0.41 mmol) were added at 0 °C. The mixture was stirred at room temperature for 1 h. The reaction was quenched with saturated aqueous NH4Cl. The mixture was extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 30:1) to give 10 (1.81 g, 95%) as a colorless oil. 1H NMR (400 MHz): δ = 7.67–7.64 (m, 4H), 7.44–7.36 (m, 6H), 5.71–5.64 (m, 1H), 5.49–5.42 (m, 1H), 4.47 (d, J = 6.4 Hz, 2H), 3.65 (dd, J = 9.8, 3.0 Hz, 1H), 3.57 (dd, J = 10.0, 4.8 Hz, 1H), 2.23–2.18 (m, 1H), 2.05 (s, 3H), 1.87–1.79 (m, 2H), 1.71–1.68 (m, 3H), 1.43–1.35 (m, 1H), 1.30–1.18 (m, 4H), 1.05 (s, 9H), 1.00–0.94 (m, 1H); 13C NMR (100 MHz): δ = 170.9, 135.6, 134.8, 133.94, 133.93, 129.5, 127.6, 125.0, 66.3, 65.3, 43.8, 38.0, 36.3, 31.7, 30.0, 26.9, 26.2, 26.1, 21.0, 19.3; HRMS (FAB) calcd for C29H40NaO3Si [M+Na]+: 487.2644. Found: 487.2642.
4.1.8. (E)-4-[(1R/S,2S/R)-2-(Hydroxymethyl)cyclohexyl]but-2-en-1-yl acetate
To a solution of 10 (1.81 g, 3.89 mmol) in THF (20 mL), TBAF [1.0 M solution in THF (7.8 mL, 7.8 mmol)] was added at room temperature. After the mixture was stirred for 12 h, the reaction was quenched with saturated aqueous NH4Cl and the whole was extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 6:1) to give a title alcohol (1.03 g, quant.) as a colorless oil. 1H NMR (400 MHz): δ = 5.80–5.72 (m, 1H), 5.60–5.53 (m, 1H), 4.51 (d, J = 6.4 Hz, 2H), 3.69 (dd, J = 10.8, 3.2 Hz, 1H), 3.59 (dd, J = 10.8, 5.6 Hz, 1H), 2.33–2.27 (m, 1H), 2.06 (s, 3H), 2.02–1.90 (m, 1H), 1.81–1.79 (m, 1H), 1.74–1.67 (m, 3H), 1.37–1.11 (m, 5H), 1.05–0.95 (m, 1H); 13C NMR (100 MHz): δ = 170.9, 134.5, 125.3, 65.7, 65.2, 43.8, 38.0, 36.4, 31.7, 29.5, 26.0, 25.8, 21.0; HRMS (FAB) calcd for C13H22NaO3 [M+Na]+: 249.1467. Found: 249.1460.
4.1.9. N-({(1S/R,2R/S)-2-[(E)-4-Hydroxybut-2-en-1-yl]cyclohexyl}methyl)-[1,1′-biphenyl]-4-carboxamide 12
DPPA (2.4 mL, 11 mmol) was added drop-wise to a solution of above alcohol (1.03 g, 4.56 mmol), triphenylphosphine (2.80 g, 10.8 mmol), and DEAD (40% solution in toluene, 4.2 mL, 10.8 mmol) in THF (10 mL) at 0 °C. The mixture was stirred for 16 h at the same temperature, and then the reaction mixture was concentrated. The residue was roughly purified by silica gel column chromatography (hexane/AcOEt = 30:1) to give 11. 1H NMR (400 MHz): δ = 5.77–5.69 (m, 1H), 5.62–5.54 (m, 1H), 4.52 (d, J = 6.0 Hz, 2H), 3.40 (dd, J = 12.0, 3.2 Hz, 1H), 3.25 (dd, J = 12.2, 6.2 Hz, 1H), 2.29–2.24 (m, 1H), 2.06 (s, 3H), 2.00–1.91 (m, 1H), 1.80–1.65 (m, 4H), 1.34–1.29 (m, 2H), 1.27–1.11 (m, 3H), 1.05–0.95 (m, 1H).
The crude 11 was dissolved in ether (10 mL) and added to a suspension of LiAlH4 (1.04 g, 27.4 mmol) in ether (10 mL) at 0 °C. The reaction was quenched with CH3OH and concentrated. The mixture was stirred for 6 h under reflux. The reaction mixture cooled to room temperature and then quenched with CH3OH and concentrated to give a corresponding amine derivative. 1H NMR (400 MHz): δ = 5.63–5.48 (m, 2H), 3.94–3.92 (m, 2H), 2.81 (dd, J = 12.6, 3.0 Hz, 1H), 2.43 (dd, J = 12.8, 7.6 Hz, 1H), 2.20–2.15 (m, 1H), 1.97–1.86 (m, 1H), 1.79–1.75 (m, 1H), 1.69–1.60 (m, 3H), 1.24–1.09 (m, 5H), 1.05–0.96 (m, 1H).
The residue was used in the next step without purification. The crude product in CH2Cl2 (10 mL) was added to a solution of HBTU (4.32 g, 11.4 mmol), DIPEA (2.4 mL, 14 mmol), and 4-biphenyl carboxylic acid (903 mg, 4.56 mmol) in CH2Cl2 (10 mL) at 0 °C. The mixture was stirred for 3 h at room temperature. The reaction was quenched with saturated aqueous NH4Cl and extracted with CH2Cl2. The organic layer was washed with brine and dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 1:1) to afford 12 (1.04 g, 63%, 3 steps) as a colorless oil. 1H NMR (400 MHz): δ = 7.84–7.82 (m, 2H), 7.67–7.59 (m, 4H), 7.48–7.44 (m, 2H), 7.41–7.37 (m, 1H), 6.28 (br s, 1H), 5.79–5.67 (m, 2H), 4.10 (d, J = 4.4 Hz, 2H), 3.78 (ddd, J = 13.6, 6.0, 3.6 Hz, 1H), 3.20 (ddd, J = 13.7, 8.1, 5.9 Hz, 1H), 2.32–2.27 (m, 1H), 2.21–2.12 (m, 1H), 1.87–1.84 (m, 1H), 1.74–1.72 (m, 3H), 1.52–1.41 (m, 1H), 1.32–1.04 (m, 5H); 13C NMR (100 MHz): δ = 167.2, 144.2, 140.0, 133.3, 131.2, 130.5, 128.9, 128.0, 127.3, 127.23, 127.17, 63.8, 43.3, 41.1, 39.6, 36.5, 31.9, 30.6, 26.0, 25.7; HRMS (EI) calcd for C24H29NO2 [M]+:363.2198. Found: 363.2207.
4.1.10. 4-Bromo-N-({(1S/R,2R/S)-2-[(E)-4-hydroxybut-2-en-1-yl]cyclohexyl}methyl)benzamide 13
A title compound was similarly prepared from 10 as above. Colorless oil; yield 50% (3 steps): 1H NMR (400 MHz): δ = 7.64–7.61 (m, 2H), 7.58–7.56 (m, 2H), 6.16 (m, 1H), 5.78–5.66 (m, 2H), 4.10 (d, J = 4.8 Hz, 2H), 3.76 (ddd, J = 13.4, 5.8, 3.8 Hz, 1H), 3.16 (ddd, J = 13.7, 8.1, 5.9 Hz, 1H), 2.29–2.25 (m, 1H), 2.18–2.11 (m, 1H), 1.84–1.80 (m, 1H), 1.73–1.71 (m, 3H), 1.50–1.41 (m, 1H), 1.28–0.96 (m, 5H); 13C NMR (100 MHz): δ = 166.5, 133.5, 131.8, 131.2, 130.4, 128.5, 126.0, 63.7, 43.3, 41.0, 39.6, 36.4, 31.9, 30.5, 26.0, 25.7; HRMS (EI) calcd for C18H24BrNO2 [M]+ 365.0990. Found 365.0996.
4.1.11. (1,1′-Biphenyl)-4-yl{(3S/R,4aR/S,8aS/R)-3-vinyloctahydroisoquinolin-2(1H)-yl}methanone 14
To a solution of 12 (120 mg, 0.331 mmol) in dry CH2Cl2 (1 mL), (CH3CN)2PdCl2 (15 mg, 0.056 mmol) was added at 0 °C under an argon gas atmosphere, and the mixture was stirred at the same temperature for 4 h. The reaction mixture was filtered and concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 10:1) to give 14 (100 mg, 88%) as a colorless oil. 1H NMR (400 MHz): δ = 7.64–7.58 (m, 4H), 7.49–7.43 (m, 4H), 7.38–7.35 (m, 1H), 5.87 (ddd, J = 17.5, 10.7, 3.7 Hz, 0.4H), 5.78 (ddd, J = 17.5, 10.7, 3.5 Hz, 0.6H), 5.55 (br s, 0.4H), 5.31–5.28 (m, 1H), 5.23–5.16 (m, 1H), 4.54 (br s, 0.6H), 4.49 (dd, J = 13.2, 4.0 Hz, 0.6H), 3.49 (dd, J = 13.0, 3.8 Hz, 0.4H), 2.86 (dd, J = 13.2, 11.6 Hz, 0.4H), 2.61 (dd, J = 12.8, 11.6 Hz, 0.6H), 1.84–1.52 (m, 5H), 1.47–1.18 (m, 5H), 1.15–1.13 (m, 0.4H), 1.03–0.98 (m, 1H), 0.90–0.84 (m, 0.6H); 13C NMR (100 MHz): δ = 171.1, 170.4, 142.3, 142.2, 140.3, 137.1, 136.7, 135.4, 128.8, 127.69, 127.66, 127.4, 127.1, 126.8, 116.6, 116.1, 57.2, 50.8, 49.7, 43.5, 42.8, 41.9, 37.5, 36.8, 35.9, 32.9, 29.9, 29.7, 26.2, 26.1, 25.8, 25.7; HRMS (EI) calcd for C24H27NO [M]+: 345.2093. Found: 345.2090.
4.1.12. (4-Bromophenyl)((3S,4aR/S,8aS/R)-3-vinyloctahydroisoquinolin-2(1H)-yl)methanone 15
A title compound was similarly prepared as above. Colorless oil; yield 57%: 1H NMR (400 MHz): δ = 7.56–7.50 (m, 2H), 7.29–7.27 (m, 2H), 5.84 (ddd, J = 17.4, 10.6, 3.8 Hz, 0.4H), 5.74 (ddd, J = 17.5, 10.7, 3.5 Hz, 0.6H), 5.49 (br s, 0.4H), 5.29–5.26 (m, 1H), 5.19–5.10 (m, 1H), 4.44 (dd, J = 13.4, 3.8 Hz, 0.6H), 4.39 (s, 0.6H), 3.33 (dd, J = 13.2, 3.6 Hz, 0.4H), 2.82 (dd, J = 13.0, 11.8 Hz, 0.4H), 2.57 (dd, J = 13.0, 11.4 Hz, 0.6H), 1.83–1.49 (m, 5H), 1.43–1.19 (m, 5H), 1.13–1.04 (m, 0.4H), 0.99–0.96 (m, 1H), 0.88–0.83 (m, 0.6H); 13C NMR (100 MHz): δ = 170.2, 169.6, 136.9, 136.5, 135.4, 131.7, 131.6, 128.6, 128.0, 123.7, 123.6, 116.7, 116.2, 57.2, 50.8, 49.6, 43.5, 42.8, 41.8, 37.5, 36.7, 35.9, 32.8, 29.9, 29.6, 26.1, 26.0, 25.7, 25.6; HRMS (EI) Calcd for C18H22BrNO [M]+: 347.0885. Found: 347.0879.
4.1.13. (S)-2-({[(3S,4aR,8aS)-2-[(1,1′-Biphenyl)-4-carbonyl]decahydroisoquinolin-3-yl]methyl}amino)-N-methoxy-N-methyl-3-(1-trityl-1H-imidazol-4-yl)propanamide 18
To a solution of K2OsO2(OH)4 (3.1 mg, 0.0083 mmol) and N-methylmorpholine N-oxide (389 mg, 3.32 mmol), 14 (286 mg, 0.829 mmol) was added in THF/H2O (3:1, 10 mL). After being stirred for 12 h, NaIO4 (710 mg, 3.32 mmol) was added to the mixture. The resultant mixture was stirred for 30 min. The reaction was quenched with H2O, and the whole was extracted with AcOEt. The organic layer was washed with brine, dried over MgSO4, filtered, and concentrated. The residue was roughly purified by silica gel column chromatography (hexane/AcOEt = 3:1) to give 16. 1H NMR (400 MHz): δ = 9.69 (s, 0.75H), 9.65 (s, 0.25H), 7.69–7.54 (m, 5H), 7.49–7.37 (m, 4H), 5.50 (d, J = 6.4 Hz, 0.75H), 4.62–4.59 (m, 0.25H), 4.44 (d, J = 5.6 Hz, 0.25H), 3.69–3.65 (m, 0.75H), 2.81 (dd, J = 13.2, 11.6 Hz, 0.75H), 2.40 (t, J = 12.6 Hz, 0.25H), 2.33 (d, J = 13.6 Hz, 0.75H), 2.15 (dd, J = 13.6 Hz, 0.25H), 1.74–1.69 (m, 3H), 1.59–1.50 (m, 1H), 1.44–1.41 (m, 1H), 1.25–1.11 (m, 3H), 1.08–0.96 (m, 2H), 0.92–0.76 (m, 1H).
The product was used without further purification. To a solution of 16 and H-His(Trt)-N(OCH3)CH3 (410 mg, 0.930 mmol) in CH2Cl2 (1 mL), AcOH (0.05 mL, 0.8 mmol) was added. The mixture was stirred at room temperature for 2 h and then NaBH3CN (181 mg, 2.88 mmol) was added. The resultant mixture was stirred for 30 min. The reaction was quenched with 1 M HCl and the whole was extracted with AcOEt. The organic layer was washed with saturated aqueous NaHCO3 and brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography (CHCl3/CH3OH = 25:1) to give 18 and 20.
Compound 18: [80 mg, 13% (50% max.), 3 steps] as a colorless oil. [α]28 D −20 (c 0.48, CHCl3); 1H NMR (400 MHz): δ = 7.59–7.53 (m, 4H), 7.47–7.41 (m, 4H), 7.37–7.29 (m, 11H), 7.13–7.09 (m, 6H), 6.62 (m, 0.6H), 6.56 (m, 0.4H), 4.94 (br s, 0.6H), 4.41 (dd, J = 13.0, 3.0 Hz, 0.4H), 4.12–4.11 (m, 0.4H), 3.93 (m, 1H), 3.69 (s, 1.8H), 3.50 (s, 1.2H), 3.44–3.41 (m, 0.6H), 3.14 (s, 1.8H), 3.08 (s, 1.2H), 2.93–2.84 (m, 2.4H), 2.76–2.66 (m, 2H), 2.46 (t, J = 12.2 Hz, 0.6H), 1.80–1.70 (m, 3H), 1.61–1.54 (m, 1H), 1.43–1.17 (m, 6H), 1.08–0.85 (m, 2H); 13C NMR (100 MHz): δ = 175.4, 175.2, 171.3, 170.5, 142.44, 142.38, 141.88, 141.86, 140.42, 140.35, 138.2, 138.1, 137.6, 137.2, 135.9, 135.7, 129.72, 129.66, 129.3, 128.8, 128.7, 127.91, 127.87, 127.54, 127.46, 127.4, 127.2, 127.1, 127.04, 126.99, 119.3, 115.6, 77.2, 75.03, 75.02, 61.6, 61.5, 57.8, 57.4, 55.5, 49.5, 48.3, 47.1, 46.6, 43.1, 42.6, 42.1, 36.4, 36.2, 34.4, 33.0, 32.9, 32.6, 32.2, 32.0, 29.9, 29.7, 26.14, 26.05, 25.8, 25.7; HRMS (EI) calcd for C50H53N5O3 [M]+: 771.4148. Found: 771.4141.
Compound 20: [75 mg, 12% (50% max.), 3 steps] as a colorless oil. [α]D 28 +32 (c 2.3, CHCl3); 1H NMR (400 MHz): δ = 7.58–7.24 (m, 19H), 7.13–7.07 (m, 6H), 6.58 (m, 0.4H), 6.55 (m, 0.6H), 5.02–4.97 (m, 0.4H), 4.46 (dd, J = 13.2, 3.6 Hz, 0.6H), 4.13 (br s, 0.4H), 3.95 (m, 1H), 3.65 (s, 1.2H), 3.62–3.58 (m, 0.6H), 3.50 (s, 1.8H), 3.44 (dd, J = 13.4, 3.4 Hz, 0.4H), 3.14 (s, 1.2H), 3.11 (s, 1.8H), 3.01–2.94 (m, 1H), 2.89–2.81 (m, 2H), 2.65 (dd, J = 11.8, 6.6 Hz, 0.4H), 2.52 (dd, J = 12.0, 6.8 Hz, 0.6H), 2.50–2.44 (m, 0.6H), 2.26–2.24 (br s, 1H), 1.71–1.69 (m, 3H), 1.60–1.52 (m, 2H), 1.45–1.16 (m, 5H), 1.07–0.83 (m, 2H); 13C NMR (100 MHz): δ = 175.6, 175.3, 171.1, 170.8, 142.44, 142.37, 141.9, 141.8, 140.41, 140.39, 138.12, 138.08, 137.5, 137.3, 135.7, 129.72, 129.66, 128.73, 128.68, 127.9, 127.5, 127.4, 127.1, 127.05, 127.03, 126.95, 119.5, 119.3, 77.2, 75.0, 61.6, 61.5, 57.7, 57.5, 55.4, 49.3, 48.4, 47.4, 47.2, 43.0, 42.8, 42.0, 36.7, 36.5, 34.6, 33.5, 33.00, 32.96, 32.3, 32.1, 29.9, 29.7, 29.6, 26.2, 26.0, 25.8, 25.7; HRMS (EI) calcd for C50H53N5O3 [M]+: 771.4148. Found: 771.4154.
4.1.14. (S)-2-({[(3S,4aR,8aS)-2-(4-Bromobenzoyl)decahydroisoquinolin-3-yl]methyl}amino)-N-methoxy-N-methyl-3-(1-trityl-1H-imidazol-4-yl)propanamide 19
Compound 19 was similarly synthesized as 18. Colorless oil; yield 11% (50% max., 3 steps): [α]D 28 −31 (c 0.83, CHCl3); 1H NMR (400 MHz): δ = 7.47 (d, J = 8.4 Hz, 1.2H), 7.44 (d, J = 8.4 Hz, 0.8H), 7.34–7.31 (m, 10.8H), 7.22 (d, J = 8.4 Hz, 1.2H), 7.12–7.11 (m, 6H), 6.60 (br s, 0.6H), 6.55 (br s, 0.4H), 4.87 (m, 0.6H), 4.37 (dd, J = 13.2, 3.6 Hz, 0.4H), 4.10 (br s, 0.6H), 3.89 (br s, 0.4H), 3.78 (m, 0.6H), 3.64 (s, 1.8H), 3.51 (s, 1.2H), 3.24 (dd, J = 13.2, 3.6 Hz, 0.6H), 3.13 (s, 1.8H), 3.11 (s, 1.2H), 2.91–2.80 (m, 2.4H), 2.73–2.62 (m, 2H), 2.47–2.41 (m, 0.4H), 1.76–1.65 (m, 3.4H), 1.60–1.54 (m, 1.6H), 1.36–1.25 (m, 5H), 1.00–0.82 (m, 2H); 13C NMR (100 MHz): δ = 175.5, 175.1, 170.4, 169.7, 142.5, 142.4, 138.3, 138.1, 137.7, 137.2, 135.9, 135.8, 131.52, 131.49, 129.75, 129.72, 128.7, 128.4, 127.94, 127.91, 123.24, 123.21, 119.26, 119.25, 77.2, 75.1, 61.6, 61.5, 57.8, 57.5, 55.6, 49.3, 48.4, 47.1, 46.6, 43.1, 42.6, 42.0, 36.4, 36.2, 34.5, 33.0, 32.9, 32.7, 32.3, 32.0, 29.9, 29.7, 26.1, 26.0, 25.8, 25.7; HRMS (EI) calcd for C44H48BrN5O3 [M]+: 773.2941. Found: 773.2948.
4.1.15. (S)-2-({[(3R,4aS,8aR)-2-(4-Bromobenzoyl)decahydroisoquinolin-3-yl]methyl}amino)-N-methoxy-N-methyl-3-(1-trityl-1H-imidazol-4-yl)propanamide 21
Compound 21 was similarly synthesized as 20. Colorless oil; yield 11% (50% max., 3 steps): [α]D 28 +4.5 (c 0.42, CHCl3); 1H NMR (400 MHz): δ = 7.47–7.43 (m, 2H), 7.37–7.29 (m, 12H), 7.12–7.10 (m, 6H), 6.55 (m, 1H), 4.98–4.95 (m, 0.4H), 4.40 (dd, J = 13.2, 3.6 Hz, 0.6H), 4.10 (br s, 0.4H), 3.90 (br s, 0.6H), 3.84–3.81 (m, 0.6H), 3.64–3.58 (m, 0.4H), 3.63 (s, 1.8H), 3.54 (s, 1.2H), 3.28 (dd, J = 13.2, 3.6 Hz, 0.4H), 3.13 (s, 1.8H), 3.11 (s, 1.2H), 2.98–2.91 (m, 1H), 2.86–2.74 (m, 2.6H), 2.60 (dd, J = 11.6, 6.0 Hz, 0.6H), 2.47–2.41 (m, 1.4H), 1.72–1.65 (m, 3H), 1.59–1.47 (m, 2H), 1.43–1.12 (m, 5H), 1.04–0.79 (m, 2H); 13C NMR (100 MHz): δ = 175.6, 175.2, 170.3, 170.0, 142.42, 142.37, 138.2, 138.1, 137.35, 137.25, 135.70, 135.66, 131.47, 131.45, 129.73, 129.70, 128.9, 128.7, 127.93, 127.91, 123.32, 123.26, 119.5, 119.3, 77.2, 75.1, 75.0, 61.5, 57.5, 57.4, 55.5, 49.2, 48.4, 47.4, 47.2, 42.9, 42.8, 42.0, 36.6, 36.5, 34.7, 33.6, 33.0, 32.9, 32.3, 32.0, 29.8, 29.6, 26.1, 26.0, 25.8, 25.6; HRMS (EI) calcd for C44H48BrN5O3 [M]+: 773.2941. Found: 773.2944.
4.1.16. (S)-2-[({(3S,4aR,8aS)-2-[(1,1′-Biphenyl)-4-carbonyl]decahydroisoquinolin-3-yl}methyl)amino]-3-(1H-imidazol-4-yl)-N-methoxy-N-methylpropanamide
TFA/CH2Cl2/TIS/H2O (10:10:1.0:1.0, 5.5 mL) was added to 18 (40 mg, 0.052 mmol). The mixture was stirred at room temperature for 4 h. The mixture was concentrated under reduced pressure. The residue was diluted with AcOEt and basified by saturated aqueous NaHCO3. The whole was extracted with AcOEt and the organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (CHCl3/CH3OH = 10:1) to give the de-tritylated product (25 mg, 90%) as a yellowish oil. [α]D 28 −33 (c 0.51, CHCl3); 1H NMR (400 MHz): δ = 7.68–7.36 (m, 10H), 6.84 (s, 0.6H), 6.82 (s, 0.4H), 5.03–5.01 (m, 0.4H), 4.31–4.27 (m, 0.6H), 4.15 (br s, 0.6H), 3.86 (br s, 0.4H), 3.73 (s, 1.2H), 3.66 (s, 1.8H), 3.54–3.51 (m, 1H), 3.25 (s, 1.2H), 3.19 (s, 1.8H), 3.00–2.86 (m, 1H), 2.75–2.62 (m, 2H), 2.52–2.44 (m, 2H), 1.77–1.68 (m, 3.4H), 1.62–1.59 (m, 1.6H), 1.49–1.23 (m, 5H), 1.17–1.11 (m, 0.4H), 1.07–0.99 (m, 1H), 0.89–0.85 (m, 0.6H); 13C NMR (100 MHz): δ = 174.4, 171.0, 143.1, 142.4, 140.2, 140.1, 135.8, 135.3, 135.2, 134.8, 128.84, 128.83, 128.2, 127.8, 127.7, 127.5, 127.20, 127.18, 127.14, 127.08, 77.2, 61.7, 59.8, 58.4, 55.7, 49.5, 49.4, 48.6, 48.1, 43.5, 42.6, 42.0, 36.7, 34.3, 34.1, 33.0, 32.9, 32.2, 30.0, 29.6, 26.2, 26.0, 25.8, 25.6; HRMS (EI) calcd for C31H39N5O3 [M]+: 529.3053. Found: 529.3057.
Compounds 19, 20, and 21 were similarly treated with TFA/CH2Cl2/TIS/H2O (10:10:1.0:1.0, 5.5 mL) as above to yield the corresponding de-tritylated products.
4.1.17. From 19: (S)-2-({[(3S,4aR,8aS)-2-(4-bromobenzoyl)decahydroisoquinolin-3-yl]methyl}amino)-3-(1H-imidazol-4-yl)-N-methoxy-N-methylpropanamide
Yellowish oil; yield, 70%: [α]D 28 −33.9 (c 0.415, CHCl3); 1H NMR (400 MHz): δ = 7.66 (s, 0.6H), 7.56–7.53 (m, 2H), 7.38 (s, 0.4H), 7.32 (d, J = 8.4 Hz, 1.2H), 7.19 (d, J = 8.4 Hz, 0.8H), 6.83 (s, 0.6H), 6.81 (s, 0.4H), 4.97–4.95 (m, 0.4H), 4.26–4.22 (m, 0.6H), 4.00–3.98 (m, 0.6H), 3.85–3.84 (m, 0.4H), 3.72 (s, 1.2H), 3.66 (s, 1.8H), 3.56–3.53 (m, 0.6H), 3.35 (dd, J = 13.4, 3.8 Hz, 0.4H), 3.24 (s, 1.2H), 3.20 (s, 1.8H), 2.99–2.83 (m, 2H), 2.71–2.60 (m, 2H), 2.54–2.41 (m, 2H), 1.77–1.58 (m, 4H), 1.51–1.33 (m, 1H), 1.30–1.17 (m, 5H), 1.05–0.80 (m, 2H); 13C NMR (100 MHz): δ = 174.3, 173.1, 170.2, 135.8, 135.4, 135.3, 134.7, 131.9, 131.7, 129.3, 128.3, 124.2, 123.7, 77.2, 61.7, 59.7, 58.4, 55.8, 49.6, 49.4, 48.5, 48.0, 43.5, 42.6, 42.0, 36.6, 34.2, 34.0, 33.0, 32.8, 32.6, 29.9, 29.6, 26.1, 26.0, 25.8, 25.6; HRMS (EI) calcd for C25H34BrN5O3 [M]+: 531.1845. Found: 531.1839.
4.1.18. From 20: (S)-2-[({(3R,4aS,8aR)-2-[(1,1′-biphenyl)-4-carbonyl]decahydroisoquinolin-3-yl}methyl)amino]-N-methoxy-N-methyl-3-(1H-imidazol-4-yl)propanamide
Yellowish oil; yield, quantitative: [α]D 28 −41 (c 0.45, CHCl3); 1H NMR (400 MHz): δ = 7.65–7.59 (m, 4H), 7.54 (s, 1H), 7.49–7.44 (m, 4H), 7.39–7.36 (m, 1H), 6.78 (m, 1H), 5.21–5.20 (m, 0.75H), 4.52–4.49 (m, 0.25H), 4.12 (m, 0.25H), 3.90–3.88 (m, 0.75H), 3.67 (s, 2.25H), 3.67–3.65 (m, 0.75H), 3.56 (s, 0.75H), 3.56–3.49 (m, 0.75H), 3.25 (s, 2.25H), 3.25–3.21 (m, 0.25H), 3.21 (s, 0.75H), 3.11–3.05 (m, 0.25H), 2.98–2.95 (m, 0.75H), 2.89–2.83 (m, 0.75H), 2.63–2.52 (m, 1.5H), 2.37 (dd, J = 12.0, 4.4 Hz, 1H), 2.29 (m, 1H), 1.72 (br s, 2H), 1.62–1.41 (m, 4H), 1.30–1.22 (m, 4H), 1.19–1.06 (m, 0.75H), 1.00–0.85 (m, 1.25H); 13C NMR (100 MHz): δ = 174.9, 171.6, 171.0, 142.4, 140.2, 135.6, 135.4, 135.2, 134.4, 128.9, 128.8, 127.7, 127.4, 127.23, 127.16, 127.1, 127.0, 77.2, 61.7, 58.5, 55.5, 49.4, 49.1, 47.5, 42.8, 42.3, 36.9, 36.8, 35.2, 34.3, 33.1, 32.9, 32.3, 29.9, 29.65, 29.56, 29.2, 26.2, 26.0, 25.8, 25.6; HRMS (EI) calcd for C31H39N5O3 [M]+: 529.3053. Found: 529.3060.
4.1.19. From 21: (S)-2-({[(3R,4aS,8aR)-2-(4-bromobenzoyl)decahydroisoquinolin-3-yl]methyl}amino)-N-methoxy-N-methyl-3-(1H-imidazol-4-yl)propanamide
Yellowish oil; yield, 65%: [α]D 28 −27.7 (c 0.96, CHCl3); 1H NMR (400 MHz): δ = 7.57–7.47 (m, 3H), 7.30–7.27 (m, 2H), 6.79 (s, 0.25H), 6.78 (s, 0.75H), 5.17–5.14 (m, 0.75H), 4.45 (dd, J = 13.4, 3.4 Hz, 0.25H), 3.94 (br s, 0.25H), 3.87–3.86 (m, 0.75H), 3.67 (s, 2.25H), 3.59 (s, 0.75H), 3.39 (dd, J = 13.6, 3.2 Hz, 0.75H), 3.25 (s, 2.25H), 3.21 (s, 0.75H), 3.19–3.16 (m, 0.75H), 3.07–3.01 (m, 0.25H), 2.98–2.89 (m, 1H), 2.82 (dd, J = 13.4, 11.8 Hz, 0.75H), 2.70–2.48 (m, 1.5H), 2.36 (dd, J = 12.2, 4.6 Hz, 0.75H), 2.30 (dd, J = 11.8, 5.8 Hz, 0.25H), 1.82–1.61 (m, 5H), 1.48–1.28 (m, 5H), 1.09–1.04 (m, 0.75H), 0.98–0.87 (m, 1.25H); 13C NMR (100 MHz): δ = 174.9, 170.7, 170.2, 135.6, 135.31, 135.26, 134.5, 131.8, 131.6, 128.7, 128.4, 123.8, 123.5, 77.2, 61.7, 58.4, 58.0, 55.4, 49.5, 49.1, 47.5, 47.4, 42.8, 42.7, 42.2, 36.8, 36.7, 35.1, 34.2, 33.0, 32.9, 32.3, 29.8, 29.5, 29.2, 26.1, 26.0, 25.8, 25.6; HRMS (EI) Calcd. For C25H34BrN5O3 [M]+: 531.1845. Found: 531.1839.
4.1.20. (S)-2-[({(3S,4aR,8aS)-2-[(1,1′-Biphenyl)-4-carbonyl]decahydroisoquinolin-3-yl}methyl)amino]-3-(1H-imidazol-4-yl)propanal 22
To a solution of above de-tritylated product of 18 (33 mg, 0.61 mmol) in CH2Cl2 (1 mL), DIBALH (1.0 mol/L solution in hexane, 1.2 mL, 1.2 mmol) was added drop-wise at −78 °C. The reaction mixture was stirred for 5 min. The reaction was quenched with CH3OH and concentrated. The residue was dissolved in CH3OH and filtered through a silica gel layer. The filtrate was concentrated. The residue was purified by HPLC to give 22 (10.5 mg, 28%) as a colorless oil. [α]D 28 −3.2 (c 0.48, CH3OH); 1H NMR (500 MHz, CD3OD, referenced to residual CH3OH): δ = 8.80 (br s, 1H), 7.75–7.73 (m, 2H), 7.67–7.65 (m, 2H), 7.58 (d, J = 8.4 Hz, 2H), 7.50 (br s, 1H), 7.48–7.45 (m, 2.5H), 7.40–7.36 (m, 1.5H), 5.13 (m, 1H), 4.82 (dd, J = 8.4, 3.2 Hz, 1H), 3.88–3.79 (m, 2H), 3.63–3.59 (m, 1H), 3.43–3.40 (m, 1H), 3.34 (s, 1H), 2.97 (t, J = 12.6 Hz, 1H), 1.77–1.68 (m, 5H), 1.45–1.34 (m, 5H), 1.06–0.98 (m, 2H); 13C NMR (125 MHz, CD3OD, referenced to CD3OD): δ = 175.1, 175.0, 163.0, 162.7, 144.70, 144.66, 141.1, 141.04, 135.5, 134.86, 134.80, 129.87, 129.86, 129.00, 128.97, 128.92, 128.0, 127.9, 118.6, 95.0, 94.9, 61.3, 61.0, 50.7, 50.5, 49.6, 47.2, 47.1, 43.2, 43.1, 37.59, 37.56, 35.2, 33.5, 30.2, 26.9, 26.5, 23.1, 22.9; HRMS (ESI) calcd for C29H35N4O2 [M+H]+: 471.2760. Found: 471.2760.
4.1.21. (S)-2-((((3S,4aR,8aS)-2-(4-Bromobenzoyl)decahydroisoquinolin-3-yl)methyl)amino)-3-(1H-imidazol-4-yl)propanal 23
A title compound 23 was synthesized from the de-tritylated product of 19 as above. Colorless oil; yield, 36%: [α]D 28 −1.1 (c 0.40, CH3OH); 1H NMR (400 MHz, CD3OD, referenced to residual CH3OH): δ = 8.72 (br s, 1H), 7.66–7.64 (m, 2H), 7.46 (br s, 1H), 7.41 (d, J = 8.4 Hz, 2H), 5.11–5.03 (m, 1H), 4.78 (dd, J = 11.0, 3.0 Hz, 1H), 3.85–3.75 (m, 2H), 3.47–3.39 (m, 2H), 3.26–3.24 (m, 1H), 2.96–2.84 (m, 1H), 1.78–1.54 (m, 5H), 1.43–1.22 (m, 5H), 1.07–0.93 (m, 2H); 13C NMR (100 MHz, CD3OD, referenced to CD3OD): δ = δ = 174.1, 173.5, 163.1, 162.6, 135.7, 135.45, 135.39, 133.0, 130.39, 130.35, 130.2, 125.84, 125.78, 118.8, 118.7, 95.1, 94.9, 61.3, 61.0, 50.64, 50.58, 43.3, 43.2, 37.71, 37.70, 37.68, 35.25, 35.22, 33.7, 30.4, 30.3, 27.0, 26.6, 23.2, 23.0; HRMS (ESI) calcd for C23H30BrN4O2 [M+H]+: 473.1552. Found: 473.1543.
4.1.22. (S)-2-[({(3R,4aS,8aR)-2-[(1,1′-Biphenyl)-4-carbonyl]decahydroisoquinolin-3-yl}methyl)amino]-3-(1H-imidazol-4-yl)propanal 24
A title compound 24 was synthesized from the de-tritylated product of 20 as above. Colorless oil; yield, 30%: [α]D 29 −2.3 (c 0.61, CH3OH); 1H NMR (500 MHz, CD3OD, referenced to residual CH3OH): δ = 8.76 (s, 1H), 7.74 (d, J = 6.4 Hz, 2H), 7.66 (d, J = 6.0 Hz, 2H), 7.56 (d, J = 6.4 Hz, 2H), 7.48–7.45 (m, 3.5H), 7.40–7.37 (m, 1.5H), 5.09 (br s, 1H), 3.86–3.75 (m, 2H), 3.64–3.59 (m, 1H), 3.55–3.48 (m, 1H), 3.35–3.32 (m, 1H), 3.28–3.26 (m, 1H), 2.93–2.91 (m, 1H), 1.79–1.66 (m, 5H), 1.47–1.28 (m. 5H), 1.07–0.97 (m, 2H); 13C NMR (125 MHz, CD3OD, referenced to CD3OD): δ = 175.5, 175.4, 163.1, 162.8, 145.0, 141.2, 135.7, 135.6, 134.9, 130.1, 129.2, 129.1, 128.2, 128.1, 119.0, 95.4, 95.1, 62.1, 61.6, 50.8, 43.2, 43.1, 37.7, 35.5, 35.4, 33.8, 33.7, 30.4, 27.0, 26.6, 24.5, 24.1; LRMS (ESI) calcd for C29H35N4O2 [M+H]+: 471.28. Found: 471.30.
4.1.23. (S)-2-({[(3R,4aS,8aR)-2-(4-Bromobenzoyl)decahydroisoquinolin-3-yl]methyl}amino)-3-(1H-imidazol-4-yl)propanal 25
A title compound 25 was synthesized from the de-tritylated product of 21 as above. Colorless oil; yield, 28%: [α]D 29 −7.8 (c 0.36, CH3OH); 1H NMR (500 MHz, CD3OD, referenced to residual CH3OH): δ = 8.72 (br s, 1H), 7.66–7.62 (m, 2H), 7.45 (s, 1H), 7.43–7.36 (m, 2H), 5.06 (m, 1H), 3.83–3.75 (m, 2H), 3.49–3.46 (m, 2H), 3.34–3.33 (m, 1H), 3.28–3.23 (m, 1H), 2.92–2.85 (m, 1H), 1.76–1.58 (m, 5H), 1.44–1.26 (m, 5H), 1.06–0.93 (m, 2H); 13C NMR (125 MHz, CD3OD, referenced to CD3OD): δ = 174.43, 174.35, 163.1, 162.8, 135.7, 135.6, 135.3, 133.0, 130.3, 125.9, 118.8, 95.4, 95.1, 62.1, 61.6, 50.7, 43.1, 43.0, 37.7, 37.6, 35.4, 35.3, 33.7, 30.3, 27.0, 26.6, 24.6, 24.1; LRMS (ESI) calcd for C23H30BrN4O2 [M+H]+: 473.16. Found: 473.25.
4.1.24. (1S,6R)-6-{2-[(4-Bromobenzyl)oxy]ethyl}cyclohex-3-enecarboxylic acid 31
To a solution of 1,3-butadiene (20 wt% solution in toluene, 108 mL, 255 mmol) was added (E)-ethyl 5-[(4-bromobenzyl)oxy]pent-2-enoate32 (20.0 g, 63.9 mmol), and the mixture was heated at 225 °C for 60 h. After the reaction mixture was cooled to room temperature, water was added and the whole was extracted with AcOEt. The organic layer was washed with 1 M HCl and brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 35:1) to give an ethyl ester of 29, (1S/R, 6R/S)-ethyl 6-{2-[(4-bromobenzyl)oxy]ethyl)}cyclohex-3-enecarboxylate, (11.7 g, 50%) as a yellow pale oil. 1H NMR (400 MHz): δ = 7.47–7.45 (m, 2H), 7.22 (d, J = 8.4 Hz, 2H), 5.65 (m, 2H), 4.43 (dd, J = 18.8, 12.0 Hz, 2H) 4.14 (q, J = 7.2 Hz, 2H), 3.52–3.49 (m, 2H), 2.41–2.20 (m, 4H), 2.08–2.03 (m, 1H), 1.81–1.72 (m, 2H), 1.53–1.46 (m, 1H), 1.26 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz): δ = 175.8, 137.5, 131.4, 129.2, 125.7, 124.8, 121.3, 72.1, 68.1, 60.3, 45.3, 33.7, 32.4, 29.9, 28.1, 14.3; HRMS (EI) Calcd for C18H23BrO3 [M]+: 366.0831. Found: 366.0826.
The above ester (31.8 g, 86.6 mmol) was dissolved in 2 M NaOH/THF (1:1, 100 mL). After being stirred for 15 h under reflux, the reaction mixture was cooled to room temperature. The mixture was acidified with 2 M HCl, and the whole was extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue purified by silica gel column chromatography (hexane/AcOEt = 3:1). The product was dissolved in AcOEt (300 mL) and then (S)-(−)-phenylethylamine (11 mL, 87 mmol) was added. After 12 h, the solid was collected by suction filtration. The free acid was liberated from the salt by treatment with 2 M HCl and extraction with AcOEt. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 3:1) to give 31 [7.63 g, 26% (50% max.)] as a colorless oil. [α]D 28 +22 (c 0.78, CHCl3); 1H NMR (400 MHz): δ = 7.47–7.45 (m, 2H), 7.20 (d, J = 8.0 Hz, 2H), 5.69–5.66 (m, 2H), 4.44 (dd, J = 17.0, 12.2 Hz, 2H) 3.56–3.49 (m, 2H), 2.47–2.20 (m, 4H), 2.12–2.07 (m, 1H), 1.91–1.75 (m, 2H), 1.60–1.51 (m, 1H); 13C NMR (100 MHz): δ = 181.1, 137.3, 131.5, 129.3, 125.7, 124.5, 121.4, 72.2, 68.0, 44.9, 33.6, 32.1, 29.5, 27.7; HRMS (EI) calcd for C16H19BrO3 [M]+: 338.0518. Found: 338.0520.
4.1.25. [(1S,6R)-6-{2-[(4-Bromobenzyl)oxy]ethyl}cyclohex-3-en-1-yl]methanol
To a solution of 31 (7.70 g, 22.7 mmol) in THF (80 mL), Et3N (6.4 mL, 46 mmol) and IBCF (4.5 mL, 34 mmol) were added at −20 °C. After being stirred for 15 min at the same temperature, NaBH4 (3.47 g, 91.2 mmol) and H2O (10 drops from a pipette) was added. The mixture was warmed up to room temperature and then the reaction was quenched with saturated aqueous NH4Cl. The whole was extracted with AcOEt and the organic layer was washed with brine and dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 3:1) to give a title alcohol (5.68 g, 77%) as a colorless oil. [α]D 29 +22 (c 0.65, CHCl3); 1H NMR (400 MHz): δ = 7.48–7.46 (m, 2H), 7.20 (d, J = 8.4 Hz, 2H), 5.65–5.58 (m, 2H), 4.45 (s, 2H), 3.68 (dd, J = 10.8, 6.4 Hz, 1H), 3.62 (dd, J = 10.8, 5.2 Hz, 1H), 3.58–3.47 (m, 2H), 2.15–2.09 (m, 2H), 2.00–1.76 (m, 4H), 1.66–1.49 (m, 3H); 13C NMR (100 MHz): δ = 137.4, 131.5, 129.3, 125.8, 125.5, 121.4, 72.3, 68.7, 65.0, 39.7, 32.9, 31.1, 29.5, 26.6; HRMS (EI) calcd for C16H21BrO2 [M]+: 324.0725. Found: 324.0732.
4.1.26. {[(1S,6R)-6-{2-[(4-Bromobenzyl)oxy]ethyl}cyclohex-3-en-1-yl]methoxy}(tert-butyl)diphenylsilane
TBDPS-Cl (5.0 mL, 19 mmol) was added to a solution of above alcohol (5.66 g, 17.4 mmol) and imidazole (1.43 g, 21.0 mmol) in CH2Cl2 (50 mL), and the mixture was stirred for 8 h at room temperature. The reaction was quenched with saturated aqueous NH4Cl, and the whole was extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 30:1) to give a di-protected alcohol compound (9.81 g, quant.) as a colorless oil. [α]D 28 +18.6 (c 1.74, CHCl3); 1H NMR (400 MHz): δ = 7.67–7.64 (m, 4H), 7.44–7.34 (m, 8H), 7.17 (d, J = 8.4 Hz, 2H), 5.63–5.54 (m, 2H), 4.41 (dd, J = 15.2, 12.0 Hz, 2H), 3.68 (dd, J = 9.8, 5.4 Hz, 1H), 3.62 (dd, J = 10.0, 6.8 Hz, 1H), 3.50–3.46 (m, 2H), 2.16–1.96 (m, 3H), 1.87–1.81 (m, 2H), 1.71–1.68 (m, 2H), 1.48–1.44 (m, 1H), 1.05 (s, 9H); 13C NMR (100 MHz): δ = 137.7, 135.61, 135.60, 133.94, 133.92, 131.4, 129.5, 129.1, 127.6, 125.8, 125.3, 121.2, 72.1, 68.7, 65.9, 39.6, 32.9, 30.8, 29.0, 26.9, 26.7, 19.3; HRMS (FAB) Calcd. For C32H40BrO5 [M+H]+: 563.1981. Found: 563.1988.
4.1.27. 2-[(1R,2S)-2-{[(tert-Butyldiphenylsilyl)oxy]methyl}cyclohexyl]ethanol 32
To a solution of above di-protected alcohol (9.81 g, 17.4 mmol) in CH3OH/EtOAc/saturated aqueous NaHCO3 (5:5:1, 110 mL), Pd-C (3.8 g) was added, and the mixture was stirred under a hydrogen gas atmosphere at room temperature for 6 h. The mixture was filtered through Celite and a silica gel layer, and the filtrate was dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (hexane/AcOEt = 6:1) to give 32 (6.90 g, quant.) as a colorless oil. [α]D 28 +12 (c 0.65, CHCl3); 1H NMR (400 MHz): δ = 7.68–7.65 (m, 4H), 7.43–7.36 (m, 6H), 3.67–3.56 (m, 4H), 1.78–1.70 (m, 5H), 1.37–1.21 (m, 6H), 1.06 (s, 9H), 1.02–0.96 (m, 1H); 13C NMR (100 MHz): δ = 135.69, 135.66, 133.91, 133.89, 129.55, 129.54, 127.60, 127.57, 66.5, 61.0, 44.5, 36.5, 35.5, 31.9, 30.0, 26.9, 26.1, 26.0, 19.3; HRMS (FAB) calcd for C25H37O2Si [M+H]+: 397.2563. Found: 397.2558.
4.1.28. (S)-2-[({(3S,4aR,8aS)-2-[(1,1′-Biphenyl)-4-carbonyl]decahydroisoquinolin-3-yl}methyl)amino]-3-(1H-imidazol-4-yl)propanal 40
Title compound was prepared from 32 according to the same procedure33 employed for the synthesis of 22 starting from enantiomer mixture 7. Colorless solid; yield, 30%: [α]D 28 −4.3 (c 0.83, CH3OH); 1H NMR (500 MHz, CD3OD, referenced to residual CH3OH): δ = 8.81 (br s, 1H), 7.75–7.73 (m, 2H), 7.67–7.65 (m, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.51 (br s, 1H), 7.48–7.45 (m, 2.5H), 7.40–7.36 (m, 1.5H), 5.14–5.13 (m, 1H), 4.81 (dd, J = 9.8, 2.6 Hz, 1H), 3.89–3.80 (m, 2H), 3.63–3.59 (m, 1H), 3.44–3.39 (m, 1H), 3.34 (s, 1H), 2.97 (t, J =12.6 Hz, 1H), 1.82–1.62 (m, 5H), 1.45–1.28 (m, 5H), 1.09–0.89 (m, 2H); 13C NMR (125 MHz, CD3OD, referenced to CD3OD): δ = 175.24, 175.17, 163.2, 162.8, 144.9, 144.8, 141.21, 141.20, 135.6, 135.03, 134.97, 130.1, 129.22, 129.18, 129.12, 128.2, 128.1, 118.98, 118.95, 95.0, 94.9, 61.3, 60.9, 50.73, 50.69, 49.8, 47.1, 47.0, 43.33, 43.30, 37.8, 37.7, 35.4, 33.7, 30.4, 27.0, 26.6, 23.1, 22.9; HRMS (ESI) Calcd. For C29H35N4O2 [M+H]+: 471.2760. Found: 471.2765.
Compounds 41, 44, and 45–49 listed in Table 1 were similarly prepared as above.
4.1.29. Compound 41
4.1.29.1. (S)-2-({[(3S,4aR,8aS)-2-(4-Bromobenzoyl)decahydroisoquinolin-3-yl]methyl}amino)-3-(1H-imidazol-4-yl)propanal 41
Colorless solid; yield, 23%: [α]D 28 −0.64 (c 0.88, CH3OH); 1H NMR (400 MHz, CD3OD, referenced to residual CH3OH): δ = 8.80 (br s, 1H), 7.65–7.63 (m, 2H), 7.49 (br s, 1H), 7.42 (d, J = 8.4 Hz, 2H), 5.11 (m, 1H), 4.81–4.78 (m, 1H), 3.86–3.78 (m, 2H), 3.47–3.34 (m, 2H), 2.97–2.91 (m, 1H), 1.75–1.60 (m, 5H), 1.42–1.24 (m, 5H), 1.04–0.96 (m, 2H); 13C NMR (100 MHz, CD3OD, referenced to CD3OD): δ = 174.2, 174.1, 163.2, 162.8, 135.6, 135.44, 135.39, 132.9, 130.40, 130.36, 130.0, 125.8, 125.7, 119.0, 118.9, 95.0, 94.9, 61.2, 60.8, 50.64, 50.60, 43.24, 43.22, 37.69, 37.66, 35.25, 35.23, 33.7, 30.4, 30.3, 27.0, 26.6, 23.1, 22.9; HRMS (ESI) calcd for C23H30BrN4O2 [M+H]+: 473.1552. Found: 473.1546.
4.1.30. Compound 44
Colorless solid; yield, 31%: [α]D 28 −1.62 (c 1.23, CH3OH); 1H NMR (500 MHz, CD3OD, referenced to residual CH3OH): δ = 8.75 (s, 1H), 7.75 (d, J = 8.0 Hz, 2H), 7.66 (d, J = 7.2 Hz, 2H), 7.57 (d, J = 8.0 Hz, 2H), 7.47–7.45 (m, 3.5H), 7.40–7.37 (m, 1.5H), 5.09 (br s, 1H), 3.80 (m, 2H), 3.66–3.63 (m, 1H), 3.51 (m, 1H), 3.26 (m, 1H), 2.93–2.91 (m, 1H), 1.77–1.68 (m, 5H), 1.45–1.35 (m, 5H), 1.07–0.97 (m, 2H); 13C NMR (125 MHz, CD3OD, referenced to CD3OD): δ = 175.5, 175.4, 163.2, 162.8, 144.9, 141.2, 135.6, 135.5, 134.9, 130.1, 129.2, 129.1, 128.2, 128.1, 119.1, 95.2, 95.0, 62.0, 61.4, 50.8, 43.12, 43.10, 37.73, 37.69, 35.5, 35.4, 33.7, 30.4, 27.0, 26.6, 24.4, 23.9; HRMS (ESI) calcd for C29H35N4O2 [M+H]+: 471.2760. Found: 471.2756.
4.1.31. Compound 45
Colorless solid; yield, 30%: [α]D 28 −6.1 (c 1.0, CH3OH); 1H NMR (500 MHz, CD3OD, referenced to residual CH3OH): δ = 8.82 (br s, 1H), 7.65 (d, J = 8.4 Hz, 2H), 7.49 (s, 1H), 7.40 (d, J = 8.0 Hz, 2H), 5.06 (m, 1H), 4.83 (m, 1H), 3.86–3.76 (m, 2H), 3.51–3.43 (m, 2H), 3.27–3.25 (m, 1H), 2.93–2.90 (m, 1H), 1.76–1.66 (m, 5H), 1.44–1.30 (m, 5H), 1.04–0.96 (m, 2H); 13C NMR (125 MHz, CD3OD, referenced to CD3OD): δ = 174.43, 174.35, 163.1, 162.8, 135.6, 135.5, 135.3, 133.0, 130.4, 125.9, 119.1, 95.3, 95.0, 61.8, 61.3, 50.7, 43.03, 43.01, 37.7, 37.6, 35.4, 35.3, 33.7, 30.33, 30.31, 27.0, 26.6, 24.4, 23.9; HRMS (ESI) calcd for C23H30BrN4O2 [M+H]+: 473.1552. Found: 4731537. Found: 4731537.
4.1.32. Compound 46
Colorless solid; yield, 31%: [α]D 29 −6.7 (c 0.10, CH3OH); 1H NMR (400 MHz, CD3OD, referenced to residual CH3OH): δ = 8.66 (br s, 1H), 7.79–7.73 (m, 2H), 7.64–7.55 (m, 4H), 7.48–7.45 (m, 3.5H), 7.41–7.37 (m, 1.5H), 5.19–5.18 (m, 1H), 4.77 (dd, J = 12.6, 3.2 Hz, 1H), 3.87–3.79 (m, 2H), 3.63–3.58 (m, 1H), 3.46–3.40 (m, 1H), 3.36–3.34 (m, 0.5H), 3.25–3.23 (m, 1.5H), 2.99–2.93 (m, 1H), 1.79–1.62 (m, 5H), 1.42–1.21 (m, 5H), 1.10–0.89 (m, 2H); LRMS (ESI) calcd for C29H35N4O2 [M+H]+: 471.28. Found: 471.35.
4.1.33. Compound 47
Colorless solid; yield, 27% (obtained as the mixture of a diastereomer derived from Pd-mediated cyclization): 1H NMR (400 MHz, CD3OD, referenced to residual CH3OH): δ = 8.84 (br s, 1H), 7.59–7.38 (m, 11H), 5.03 (m, 1H), 4.81 (dd, J = 10.0, 2.8 Hz, 1H), 3.90 (m, 1H), 3.69–3.59 (m, 1H), 3.41 (m, 1H), 3.34 (s, 1H), 3.27–3.24 (m, 1H), 2.93–2.87 (m, 1H), 2.61–2.54 (m, 1H), 1.59–1.56 (m, 2H), 1.50 (d, J = 10.4 Hz, 1H), 1.42 (d, J = 12.4 Hz, 1H), 1.18–1.08 (m, 2H), 0.99–0.88 (m, 3H), 0.70–0.61 (m, 1H), 0.46–0.44 (m, 1H); LRMS (ESI) calcd for C29H35N4O2 [M+H]+: 471.28. Found: 471.35.
4.1.34. Compound 48
Colorless solid; yield, 25%: [α]D 29 −3.7 (c 0.15, CH3OH); 1H NMR (400 MHz, CD3OD, referenced to residual CH3OH): δ = 8.61 (br s, 1H), 7.52–7.46 (m, 6H), 7.45–7.41 (m, 1H), 5.13–5.11 (m, 2H), 4.77 (dd, J = 11.8, 3.4 Hz, 1H), 3.79–3.66 (m, 1H), 3.56–3.50 (m, 1H), 3.42–3.32 (m, 1H), 3.26–3.20 (m, 1H), 2.97–2.91 (m, 1H), 1.78–1.59 (m, 5H), 1.40–1.20 (m, 5H), 1.08–0.86 (m, 2H); LRMS (ESI) calcd for C23H31N4O2 [M+H]+: 395.24. Found: 395.30.
4.1.35. Compound 49
Colorless solid; yield, 18%: [α]D 29 −3.6 (c 0.18, CH3OH); 1H NMR (400 MHz, CD3OD, referenced to residual CH3OH): δ = 8.68 (br s, 1H), 7.56–7.53 (m, 2H), 7.44 (br s, 1H), 7.25–7.19 (m, 3H), 5.10 (m, 1H), 4.77 (dd, J = 11.4, 3.2 Hz, 1H), 3.84–3.75 (m, 2H), 3.52–3.47 (m, 1H), 3.40–3.34 (m, 1H), 3.25–3.23 (m, 1H), 2.96–2.89 (m, 1H), 1.78–1.65 (m, 5H), 1.43–1.23 (m, 5H), 1.05–0.93 (m, 2H); LRMS (ESI) calcd for C23H30FN4O2 [M+H]+: 413.24. Found: 413.35.
4.2. Estimation of IC50 values
Peptide substrate [H-Thr-Ser-Ala-Val-Leu-Gln-Ser-Gly-Phe-Arg-Lys-NH2]28 (111 μM) in a reaction solution (25 μL of 20 mM Tris–HCl buffer pH 7.5 containing 7 mM DTT) was incubated with the R188I SARS 3CLpro 28 (56 nM) at 37 °C for 60 min in the presence of various inhibitor concentrations at 37 °C for 60 min. The cleavage reaction was monitored by analytical HPLC [Cosmosil 5C18 column (4.6 × 150 mm), a linear gradient of CH3CN (10–20%) in an aq0.1% TFA over 30 min], and the cleavage rates were calculated from the reduction in the substrate peak area. Each IC50 value was obtained from the sigmoidal dose-response curve (see Fig. S1 for a typical sigmoidal curve). Each experiment was repeated 3 times and the results were averaged.
4.3. X-ray crystallography
The purified SARS 3CLpro in 20 mM Bis–Tris pH 5.5, 10 mM NaCl, and 1 mM DTT was concentrated to 8 mg/mL.13 Crystals of SARS 3CLpro were grown at 4 °C using a sitting-drop vapor diffusion method by mixing it with an equal volume of reservoir solution containing 100 mM MES pH 6.2, 5–10% PEG20000, and 5 mM DTT. Cubic-shaped crystals with dimensions of 0.3 mm × 0.3 mm × 0.3 mm grew within 3 days. The crystals were then soaked for 24 h with reservoir-based solution of 100 mM MES pH 6.2, 5–8% PEG20000, and 5 mM DTT containing 3 mM of 40 or 44. Crystals were then transferred into a cryobuffer of 100 mM MES pH 6.2, 10% PEG20000, 5 mM DTT, 15% ethylene glycol containing 3 mM of 40 or 44, and flash-frozen in a nitrogen stream at 100 K. X-ray diffraction data of SARS 3CLpro in complexes with inhibitor 40 or 44 were collected at the SPring-8, beamline BL44XU with a Rayonix MX300HE CCD detector at a wavelength of 0.900 Å.
Crystals of SARS 3CLpro in a complex with 41 were obtained by co-crystallization using sitting-drop vapor diffusion at 4 °C and mixing an equal volume of protein-inhibitor complex (final inhibitor concentration of 3 mM) and a reservoir solution containing 100 mM MES pH 6.0, 5–6% PEG20000, and 5 mM DTT. Cubic-shaped crystals with dimensions of 0.2 mm × 0.2 mm × 0.2 mm were obtained within 3 days. Crystals were transferred into cryobuffer with 100 mM MES pH 6.0, 6% PEG20000, 5 mM DTT, 15% ethylene glycol, and 3 mM of 41 and then flash-frozen in a nitrogen stream at 100 K. X-ray diffraction data were collected on a Rigaku RAXIS VII imaging-plate detector at a wavelength of 1.5418 Å equipped with an in-house rotating anode FR-E/Super Bright X-ray generator and Confocal VariMax (VariMax HF) optics system.
The structures of SARS 3CLpro in a complex with inhibitors were determined by molecular replacement using the Molrep34 program with a R188I SARS 3CLpro structure (PDB code 3AW113) as the search model. Rigid body refinement and subsequent restrained refinement protocols were performed with the program Refmac 535 of the CCP package.36 The Coot program37 was used for manual model rebuilding. Water molecules were added using Coot only after the refinement of protein structures had converged. Ligands generated on JLigand38 software were directly built into the corresponding difference in electron density, and the model was then subjected to an additional round of refinement. The figures for structural representation were generated on Pymol39 or chimera40 software.
5. PDB ID codes
4TWY, 4TWW, and 4WY3.
Acknowledgments
This work was supported, in part, by a Grant-in-aid for Scientific Research 25460160 to K.A. from the Japan Society for the Promotion of Science and by a grant for Adaptable and Seamless Technology Transfer Program through Target-driven R&D AS251Z01976Q to Y.H. from Japan Science and Technology Agency. We thank Shimadzu Co. for the measurements of mass spectra using LCMS-IT-TOF MS.
Footnotes
Supplementary data (the HPLC data for the evaluation of purities using a reversed-phase or chiral column, typical sigmoidal curves used to obtain IC50 values, and NMR data of synthesized compounds) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2014.12.028.
Supplementary data
The HPLC data for the evaluation of purities using a reversed-phase or chiral column, typical sigmoidal curves used to obtain IC50 values, and NMR data of synthesized compounds.
References and notes
- 1.Lee N., Hui D., Wu A., Chan P., Cameron P., Joynt F.M., Ahuja A., Yung M.Y., Leung C.B., To K.F., Leu M.D., Szeto C.C., Chung S., Sung J.J.Y. N. Engl. J. Med. 1986;2003:348. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 2.Drosten C., Günther S., Preiser W., Ven der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolensnikova L., Fouchier R.A.M., Berger A., Burguière A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J., Müller S., Rickerts V., Stürmer M., Vieth S., Klenk H.D., Osterhaus A.D.M.E., Schmitz H., Doerr H.W. N. Engl. J. Med. 1967;2003:348. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 3.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Engl. J. Med. 1953;2003:348. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H., Wang H., Crameri G., Hu Z., Zhang J., McEachern J., Field H., Daszak P., Eaton B.T., Zhang S., Wang L.F. Science. 2005;310:676. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 5.Lau S.K.P., Woo P.C.Y., Li K.S.M., Huang Y., Tsoi H.W., Wong B.H.L., Wong S.S.Y., Leung S.Y., Chan K.H., Yuen K.Y. Proc. Natl. Acad. Sci. U.S.A. 2005;102:14040. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ren Z., Yan L., Zhang N., Guo Y., Yang C., Lou Z., Rao Z. Protein Cell. 2013;4:248. doi: 10.1007/s13238-013-2841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kilianski A., Mielech A.M., Deng X., Baker S. J. Virol. 2013;87:11955. doi: 10.1128/JVI.02105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chem M.H., Tong W., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 9.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Sott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bermard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. Science. 2003;300:1399. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 10.Thiel V., Ivanov K.A., Putics Á., Hertzig T., Schelle B., Bayer S., Weiβbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. J. Gen. Viol. 2003;84:2305. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 11.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Science. 2003;300:1763. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 12.Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L., Lai B., Pei J., Liu Y., Chen J., Lai L. J. Biol. Chem. 2004;279:1637. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akaji K., Konno H., Mitsui H., Teruya K., Shimamoto Y., Hattori Y., Ozaki T., Kusunoki M., Sanjoh A. J. Med. Chem. 2011;54:7962. doi: 10.1021/jm200870n. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 14.Thanigaimalai P., Konno S., Yamamoto T., Koiwai Y., Taguchi A., Takayama K., Yakushiji F., Akaji K., Kiso Y., Kawasaki Y., Chen S.-E., Naser-Tavakolian A., Schön A., Freire E., Hayashi Y. Eur. J. Med. Chem. 2013;65:436. doi: 10.1016/j.ejmech.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chuck C.-P., Chen C., Ke Z., Wan D.C.-C., Chow H.-F., Wong K.-B. Eur. J. Med. Chem. 2013;59:1. doi: 10.1016/j.ejmech.2012.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Regnier T., Sarma D., Hidaka K., Bacha U., Freire E., Hayashi Y., Kiso Y. Bioorg. Med. Chem. Lett. 2009;19:2722. doi: 10.1016/j.bmcl.2009.03.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh A.K., Xi K., Grum-Tokars V., Xu X., Ratia K., Fu W., Houser K.V., Baker S.C., Johnson M.E., Mesecar A.D. Bioorg. Med. Chem. Lett. 2007;17:5876. doi: 10.1016/j.bmcl.2007.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park J.-Y., Kim J.H., Kwon J.M., Kwon H.-J., Jeong H.J., Kim Y.M., Kim D., Lee W.S., Ryu Y.B. Bioorg. Med. Chem. 2013;21:3730. doi: 10.1016/j.bmc.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryu Y.B., Park S.-J., Kim Y.M., Lee J.-Y., Seo W.D., Chang J.S., Park K.H., Rho M.-C., Lee W.S. Bioorg. Med. Chem. Lett. 1873;2010:20. doi: 10.1016/j.bmcl.2010.01.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wen C.-C., Kuo Y.-H., Jan J.-T., Liang P.-H., Wang S.-Y., Liu H.-G., Lee C.-K., Chang S.-T., Kuo C.-J., Lee S.-S., Hou C.-C., Hsiao P.-W., Chien S.-C., Shyur L.-F., Yang N.-S. J. Med. Chem. 2007;50:4087. doi: 10.1021/jm070295s. [DOI] [PubMed] [Google Scholar]
- 21.Ikejiri M., Saijo M., Morikawa S., Fukushi S., Mizutani T., Kurane I., Maruyama T. Bioorg. Med. Chem. Lett. 2007;17:2470. doi: 10.1016/j.bmcl.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho J.H., Bernard D.L., Sidwell R.W., Kern E.R., Chu C.K. J. Med. Chem. 2006;49:1140. doi: 10.1021/jm0509750. [DOI] [PubMed] [Google Scholar]
- 23.Biot C., Daher W., Chavain N., Fandeur T., Khalife J., Dive D., Clercq E.D. J. Med. Chem. 2006;49:2845. doi: 10.1021/jm0601856. [DOI] [PubMed] [Google Scholar]
- 24.Liu W., Zhu H.-M., Niu G.-J., Shi E.-Z., Chen J., Sun B., Chen W.-Q., Zhou H.-G., Yang C. Bioorg. Med. Chem. 2014;22:292. doi: 10.1016/j.bmc.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee H., Mittal A., Patel K., Gatuz J.L., Truong L., Torres J., Mulhearn D.C., Johnson M.E. Bioorg. Med. Chem. 2014;22:167. doi: 10.1016/j.bmc.2013.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobs J., Grum-Tokars V., Zhou Y., Turlington M., Saldanha S.A., Chase P., Eggler A., Dawson E.S., Baez-Santos Y.M., Tomar S., Mielech A.M., Baker S.C., Lindsley C.W., Hodder P., Mesecar A., Stauffer S.R. J. Med. Chem. 2013;56:534. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramajayam R., Tan K.-P., Liu H.-G., Liang P.-H. Bioorg. Med. Chem. 2010;18:7849. doi: 10.1016/j.bmc.2010.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akaji K., Konno H., Onozuka M., Makino A., Saito H., Nosaka K. Bioorg. Med. Chem. 2008;16:9400. doi: 10.1016/j.bmc.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Makabe H., Looi K.K., Hirota M. Org. Lett. 2003;5:27. doi: 10.1021/ol0201916. [DOI] [PubMed] [Google Scholar]
- 30.Azzena F., Calvani F., Crotti P., Gardelli C., Macchia F., Pineschi M. Tetrahedron. 1995;51:10601. [Google Scholar]
- 31.Crane S.N., Black W.C., Palmer J.T., Davis D.E., Setti E., Robichaud J., Paquet J., Oballa R.M., Bayly C.I., McKay D.J., Somoza J.R., Chauret N., Seto C., Scheigetz J., Wesolowski G., Massé F., Desmarais S., Ouellet M. J. Med. Chem. 2006;49:1066. doi: 10.1021/jm051059p. [DOI] [PubMed] [Google Scholar]
- 32.Synthesis of (E)-ethyl 5-[(4-bromobenzyl)oxy]pent-2-enoate is included in Supplementary data.
- 33.Procedures and data for the syntheses of 40 from 32 as well as the related compounds 41, 44, and 45–49 are included in Supplementary data.
- 34.Vagin A., Teplyakov A. Acta Crystallogr. D Biol. Crystallogr. 2010;66:22. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov G.N., Vagin A.A., Dodson E.J. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A., McNicholas S.J., Murshudov G.N., Pannu N.S., Potterton E.A., Powell H.R., Read R.J., Vagin A., Wilson K.S. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P., Cowtan K. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Lebedev A.A., Young P., Isupov M.N., Moroz O.V., Vagin A.A., Murshudov G.N. Acta Crystallogr. D Biol. Crystallogr. 2012;68:431. doi: 10.1107/S090744491200251X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger LLC.
- 40.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. J. Comput. Chem. 2004;25:1605. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The HPLC data for the evaluation of purities using a reversed-phase or chiral column, typical sigmoidal curves used to obtain IC50 values, and NMR data of synthesized compounds.