

Abstract
The emergence of disease and dearth of effective pharmacological agents on most therapeutic fronts, constitutes a major threat to global public health and man’s existence. Consequently, this has created an exigency in the search for new drugs with improved clinical utility or means of potentiating available ones. To this end, accumulating empirical evidence supports molecular target therapy as a plausible egress and, β-glucuronidase (βGLU) – a lysosomal acid hydrolase responsible for the catalytic deconjugation of β-d-glucuronides has emerged as a viable molecular target for several therapeutic applications. The enzyme’s activity level in body fluids is also deemed a potential biomarker for the diagnosis of some pathological conditions. Moreover, due to its role in colon carcinogenesis and certain drug-induced dose-limiting toxicities, the development of potent inhibitors of βGLU in human intestinal microbiota has aroused increased attention over the years. Nevertheless, although our literature survey revealed both natural products and synthetic scaffolds as potential inhibitors of the enzyme, only few of these have found clinical utility, albeit with moderate to poor pharmacokinetic profile. Hence, in this review we present a compendium of exploits in the present millennium directed towards the inhibition of βGLU. The aim is to proffer a platform on which new scaffolds can be modelled for improved βGLU inhibitory potency and the development of new therapeutic agents in consequential.
Keywords: β-glucuronidase, Biomarker, Enzyme inhibition, Molecular target, Structure activity relationship
Graphical abstract

1. Introduction
The world today is embattled with an increasing paucity of effective therapeutic agents or regimen for many pathological conditions, as well as the menace of drug resistance and adverse effects of available drugs [1]. As a result, smooth and efficient clinical practice is rigidly stymied, while global public health, social security and man’s life expectancy are seriously threatened and trickles to a disquieting edge [2]. Likewise, the burdens of developing new therapeutic agents to ameliorate the status quo has become heavier on all stakeholders in drug research.
In this regard, molecular target therapy is fast becoming a spearhead in the search for new drugs with improved therapeutic effects. Amongst many targets explored, glycosyl hydrolases (GHs) are notable due to their role in many important biological processes. Their principal function is to catalytically cleave the glycosidic bond of glycans thereby eliciting different physiological responses. Therefore, inhibitors of this class of enzymes have enjoyed intense research and development owing to their potentials as antiviral, anticancer and antidiabetic agents as well as therapeutic agents for some genetic disorders [[3], [4], [5]].
GHs have been classified using different indices [6]. For example, based on substrate specificity, those cleaving O- or S-glycosides are grouped into EC 3.2.1 class, while hydrolases of N-glycosides belong to EC 3.2.2 class. Advancements in genomic science have also enabled classification into GH families based on their amino acid sequence similarities [7]. This system further groups GH families into clans, given the improved conservation of protein fold than the sequence [8]. Accordingly, the reviewed enzyme, β-glucuronidase (EC 3.2.1.31) is classified into GH family 1, 2, 30, 79, 154 and GH-A clan. β-glucuronidase (βGLU) is mainly a lysosomal hydrolase widely distributed in mammalian tissues, body fluids and microbiota; but significantly retained in the endoplasmic reticulum [9]. The enzyme is also found in plants, fishes, insects and molluscs.
Specifically, human βGLU belongs to GH family 2. It is a 332 kDa ellipsoidal and homotetrameric glycoprotein with each 75–78 kDa monomer containing 651 amino acid residues (Fig. 1 a). The monomer precursor is synthesized initially on membrane-bound ribosomes and suffers C-terminal proteolytic processing of 18 amino acid propeptide en route or after their transport to the lysosomes [[10], [11], [12], [13]]. X-ray crystallography of the protein structure reveals a dihedral symmetry for the tetramer with two identical monomers in the asymmetric unit arising from disulphide-linked dimers. Each monomer contains three structural domains (Fig. 1b). The first domain has a barrel-like structure with a jelly roll motif; the second domain exhibits a geometry identical to immunoglobulin constant domains; while the third C-terminal domain forms a TIM barrel motif (β/α)8 [14]. The active sites of human βGLU (Fig. 1c) viz. catalytic acid Glu451 (proton donor), catalytic nucleophile Glu540 (carbonium ion stabilizer), Asp207 (plausible role as Glu540) and Tyr504 (unclear catalytic role), are all housed in the third domain and in each of the four catalytic centres of the tetramer [14,15]. Moreover, the enzyme has an optimal activity at acidic pH ∼4.5, corresponding to its lysosomal environment and thermally stable up to 70 °C [10]; although hyperthermophilic variants exists in other media [16]. βGLU is encoded by the GUS gene. A deficiency arising from mutations in this encoding gene is associated with atherosclerosis [17] and lysosomal storage disease – Sly syndrome or mucopolysaccharidosis type VII [18].
Fig. 1.

3D ribbon diagram of human βGLU (A) Homotetramer (PDB ID: 3HN3) (B) Monomer structure showing the three structural domains and active site cavity (brown ring). (B) Expanded view of active site cavity. Protein structure was processed using Maestro 12.0. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
On the other hand, bacterial βGLU, which is expressed in human gut microbiota and most strains of Escherichia coli shows 45% sequence similarity with human βGLU. Also, it has a bacterial loop containing 17-amino acid residues not found in human βGLU, an optimal activity at neutral pH and active site catalytic residues as Glu413 (catalytic acid) and Glu504 (catalytic nucleophile) [19].
Consistent with the activities of lysosomal GHs, βGLU deconjugates β-d-glucuronides to their corresponding aglycone and β-d-glucuronic acid via an SN2 reaction and “configuration retaining” mechanism (Fig. 2 ). The catalytic mechanism is conceived to proceed as follows; catalytic glutamic acid residue Glu451 (or Glu413 in bacterial ortholog) protonates exocyclic glycosidic oxygen of glucuronide (1) hence releasing the aglycone via a putative oxocarbenium ion-like transition state (2). ‘Back-side’ nucleophilic attack by glutamate ion Glu540 (or Glu504 in bacterial ortholog) – the catalytic nucleophile, stabilizes the transition state and results in glucuronyl ester intermediate (3) with an inverted configuration. Finally, hydrolysis through an inverting attack of water molecule on the anomeric centre releases Glu540 to form β-d-glucuronic acid (4) and a concurrent overall retention of substrate configuration [14,15,[19], [20], [21]].
Fig. 2.

Configuration retaining mechanism of βGLU catalysed hydrolysis.
Due to the increased expression of βGLU in necrotic areas and other body fluids of patients with different forms of cancer such as breast [22], cervical [23], colon [24], lung [25], renal carcinoma and leukaemia [26], compared to healthy controls, the enzyme is proffered as a reliable biomarker for tumour diagnosis and clinical therapy assessment [27]. This overexpression is also a potential diagnostic tool for other disease states such as urinary tract infection [28], HIV [29], diabetes [30], neuropathy [31] and rheumatoid arthritis [32]. In this vein, empirical data update on clinical applications of βGLU for these and other disorders is provided on BRENDA database [33].
βGLU activity is also harnessed in prodrug monotherapy. In normal body systems, drugs and other xenobiotics are detoxified via glucuronidation, an SN2 conjugation reaction and important pathway in phase II metabolism, catalysed by UDP-glucuronosyltransferases (UGTs). The resulting usually less active glucuronide metabolite is readily excreted by renal clearance due to increased polarity or sometimes via biliary clearance [34]. However, elevated levels of βGLU activity reverts this process through deglucuronidation, which hydrolyses the phase II metabolites to their active forms (Fig. 2). Hence, glycosidation of a drug to give its glucuronide enhances selective release of the active form at necrotic sites via βGLU-mediated deglucuronidation thus improving the drug’s therapeutic potential [35].
βGLU’s postulated ability to increase T Regulator cells (TReg) is also applied in low-dose immunotherapy (LDI) for managing allergic diseases [36,37], Lyme disease [38] and other chronic conditions. While it’s hydrolytic activity on glucuronide conjugates is harnessed in forensic analysis [39] and assessment of microbial water quality [40].
Nonetheless, enterobacterial βGLU deconjugation of drug and xenobiotic glucuronides in the gastrointestinal (GI) tract has been implicated in colonic genotoxicity [41] and certain drug-induced-dose-limiting toxicities. For example, the GI toxicity of anticancer drug Irinotecan (CPT-11) [42], enteropathy of non-steroidal anti-inflammatory drug (NSAID) Diclofenac [43], tissue inflammation and hepatoxicity.
Furthermore, βGLU is deemed a potential molecular target for; (1) anticancer chemotherapy considering its role in tumour growth and metastasis [44,45]. (2) Neonatal jaundice treatment due to its high expression in breast milk and role in enterohepatic bilirubin circulation (hyperbilirubinemia) [46,47]. (3) Diabetes mellitus management consequent to the positive correlations between the disease state and enzyme activity level as well as associated periodontitis [48,49]. (4) Anti-inflammatory agents development owing to its pro-inflammatory role following significant release from degranulated mast cells and neutrophils [50,51]. Expectedly, inhibition of βGLU markedly alleviated these pathological conditions and their adverse effects hence improving regimens’ efficacy.
Based on the foregoing, we extrapolate that the development of potent, specific and non-cytotoxic inhibitors of βGLU is imperative to improving the clinical efficacy of therapeutic agents and effective disease management while bearing in mind the physiological significance of both human and bacterial orthologs of the glycosyl hydrolase. However, the fate of these inhibitors rests on their inhibition constants (K i), since GHs are generally characterized by high rate enhancement (kcat/kuncat > 1017-fold). Also, accumulating evidence suggests the dependence of inhibitory potency on the ability to mimic the highly enzyme-stabilized transition state of an enzymatic reaction (K i ≈ 10−20 M) en route to catalytic product [21,52,53].
Considering the proven and encouraging potentials of enzyme inhibition and molecular target therapy in drug development, and in continuation of our exploits and expositions thereon [[54], [55], [56], [57], [58]], herein we present a comprehensive review of research undertakings in the present millennium (2000–2019) directed towards the development of potent inhibitors of βGLU that are either natural products or synthetic scaffolds. Apropos, before discussing the different inhibitors, this article will first highlight the potentials of βGLU activity as a diagnostic tool within the defined period. However, therapeutic application in prodrug monotherapy and enzyme replacement therapy (ERT) will not be covered as these have been excellently treated in other reviews [[59], [60], [61]].
Hitherto, our search of extant literature revealed that, although there exists a plethora of scholarly research on potential inhibitors of βGLU activity, no review article is exclusively devoted to the subject matter. The aim of this review is therefore to bring to light those bioactive frameworks bestowed with promising βGLU inhibitory potency. Our principal goal is to intimate the reader on key structural features of reviewed molecules crucial to their inhibitory activities and toxicity profiles, while establishing the comprehensive relationships existing between reported molecules.
2. β-Glucuronidase activity as a reliable biomarker in diagnostic science
The availability of safe, easy to use, consistent, less-invasive and cost-effective tool for early diagnosis of diseases or appraisal of therapeutic interventions is of uttermost importance in clinical medicine. Since most disease states are accompanied by elevated levels of specific enzymes in the diseased milieu (tissues, plasma and other body fluids), quantification of enzymes’ activity levels is seen as a reliable biomarker of either disease status, severity, effects, susceptibility or exposure [[62], [63], [64]]. Moreover, the substrate specificity and selective quantification of enzymes in the presence of other biomolecules makes them a tool of choice thereto [65]. A review of βGLU activity as a biomarker of some physiologically important conditions is hereby presented together with a concise summary in Table 1 .
Table 1.
Reported potential applications of βGLU activity as a biomarker.
| Pathological Condition | Significance of βGLU activity | Media | Study size | Parameter with significant correlation to βGLU activity | βGLU activity in diseased subjects compared to controls | Required βGLU activity | Ref |
|---|---|---|---|---|---|---|---|
| Periodontal Disease | Biomarker of
|
Saliva | 380 |
|
|
≥100 units | [69] |
| Biomarker of disease susceptibility | 70 |
|
˃ 100 units | [70] | |||
| 200 | NS | NS | [71] | ||||
| Biomarker of disease status | GCF | 14 |
|
[72] | |||
| Biomarker of disease severity and assessment of non-surgical therapy | Saliva | 31 | NS | [73] | |||
| Diabetes and Periodontitis | Biomarker of disease risk | 80 |
|
|
[86] | ||
| Biomarker of disease status and severity | Serum | 350 |
|
[87] | |||
| Peripheral venous blood neutrophilic leukocytes | 165 |
|
[88] | ||||
| Saliva | 192 | NS |
|
[89] | |||
| GCF | 45 |
|
[90] | ||||
| Colon Cancer | Tumour biomarker | Serum | 38 |
|
|
>208.10 pKat/mL | [96] |
| Ovarian and Endometrial Cancer | Biomarker of tumour status and severity | Peritoneal fluid | 35 |
|
|
NS | [97] |
| Pelvic inflammatory disease | Biomarker of disease status | NS |
|
||||
| Bacterial peritonitis | Biomarker for early disease diagnosis and assessment of therapeutic intervention | 71 |
|
[98] | |||
| Bacterial meningitis | CSF | 140 |
|
[99] | |||
| Sterile CSF pleocytosis due to UTI or meningitis | Biomarker for differential diagnosis | 92 |
|
[100] | |||
| Bacterial lung infection | Biomarker for disease diagnosis, prognosis and differential BALF bacterial culture screening | BALF | 92 | BALF levels of
|
|
*43 nmol 4MU/ml/h | [101] |
| Organophosphorus pesticide poisoning | Biomarker of poisoning severity | Plasma | 74 | NS |
|
NS | [108] |
| 108 |
|
[109] | |||||
| Serum | 21 | NS | [110] | ||||
| 40 |
|
|
[111] | ||||
| Plasma | 220 |
|
[112] | ||||
| 284** | AChE activity
|
|
[113] |
NS: Not specified; W: weak correlation; N: No correlation; PI: Russell periodontal index; *threshold to distinguish culture positive from culture negative BALF; ** acute exposure (5), chronic exposure (230).
2.1. Periodontal disease
Periodontal disease is a group of inflammatory disorders triggered by host’s immune response to the actions of virulent subgingival plaque bacteria biofilms which activates the release of polymorphonuclear leukocytes and macrophages into the gingival crevice. This leads to gingivitis – an inflammation of periodontal tissues and distortion of periodontal histology that is reversible with improved oral hygiene; or, subsequent tissue destruction, alveolar bone resorption and tooth loss if left unattended i.e. periodontitis [66,67]. Therefore, a reliable tool to ascertain disease status, severity, risk or efficacy of administered therapy is highly desirous to clinicians.
However, conventional diagnosis involving the measurement of periodontal clinical parameters such as probing depth (PD), clinical attachment level (CAL), gingival index (Gg-I), bleeding on probing (BOP) and alveolar bone loss (ABL), suffers from intrinsic limitations. They only define the status of patient’s periodontium at the time of examination and not periodontal disease susceptibility or risk [68]. Thus, since periodontitis is characterized by an influx of inflammatory mediators and corresponding enzymes into the gingival sulcus, the quantification of neutrophil-derived βGLU activity in gingival crevicular fluid (GCF) or GCF’s outflow into the oral cavity and subsequent less invasive estimation of βGLU activity in saliva is considered a reliable biomarker for periodontal disease diagnosis.
To this effect, the relationship between salivary βGLU activity and periodontal clinical parameters (PD, CAL and Gg-I) was investigated in subjects with different stages of periodontal disease [69]. The mean PD and Gg-I, number of sites with PD ≥ 5 mm and total number of white blood cells, blood neutrophils and monocytes all showed highly significant correlations with enzyme’s activity, while CAL had a weaker correlation. Using logistic regression modelling and the presence of at least 1 or 4 sites with PD ≥ 5 mm as disease criterion, βGLU activity showed promising potentials as a tool for periodontal disease screening or assessment of therapeutic intervention. The study also observed smoking status to be insignificant on enzyme activity. However, in a similar study, only PD, CAL and lymphocyte count exhibited positive correlation with salivary enzyme activity while no significant relationship was observed for Gg-I [70]. Recently, subjects with chronic generalized periodontitis were also found to have significant increase in βGLU activity (8-fold) compared to normal ones. Although, a reduction in enzyme activity persisted in smokers regardless of periodontal status [71].
The efficacy of therapeutic intervention using amoxicillin and metronidazole to downregulate amplified neutrophil activity was studied in 14 patients with aggressive periodontitis [72]. Treatment involved seven consecutive days of antibiotic administration with concurrent scaling, root planning and surgical therapy and a total of 36 months posttreatment evaluation period. Subsequently, a markedly downregulated neutrophil activity with approximately 50% inhibition of βGLU activity in GCF was observed. Periodontal health was also restored and maintained during posttreatment evaluations. In another study, βGLU activity was posited as a better biomarker compared to alkaline phosphatase for evaluating the response to non-surgical periodontal therapy in patients with different stages of periodontal disease [73]. Taken together, these results articulate the potentials of βGLU activity as an indication of PD, tissue inflammation or destruction as well as a biomarker of neutrophil influx, disease risk, susceptibility, status, or severity for timely diagnosis of the inflammatory disorder. However, administration of doxycycline hyclate in 16 subjects with aggressive periodontitis was inefficient on salivary βGLU activity [74]. Surprisingly, an increase in enzyme activity was found even after 2 months of treatment in 12 patients and a decrease in 4. Although, the authors concluded βGLU concentration only facilitated the detection of periodontal inflammation and not worthy as biomarker of susceptibility, their contrasting result is linkable to periodontal pre-treatment of subjects prior to examination and short treatment time using doxycycline.
Empirical evidence affirms the role of inflammatory mediators and signalling pathways in the pathogenesis of insulin resistance and β-cell dysfunction in diabetes mellitus [[75], [76], [77]]. In parallel, these inflammatory mediators e.g. cytokines and MMPs are also produced in periodontal tissues [[78], [79], [80], [81], [82]]; hence, leading to compromised glycaemic control after accessing systemic circulation. The susceptibility to periodontitis is therefore heightened in persons with diabetes or a history of the hormonal imbalance and vice versa. Putatively, an effective therapy for one affords an improved management of the other [[83], [84], [85]].
Accordingly, βGLU activity was found to be significantly elevated in the saliva of patients with chronic periodontitis and diabetes compared to nondiabetic ones [86]. A significant correlation to βGLU activity was observed for PD and CAL and not Gg-I in nondiabetic subjects with periodontitis; whereas, these periodontal parameters were similar in diabetics. The increased disease burden was also established when βGLU activity was measured in the sera of patients with both diabetes and periodontitis [87]. Compared to controls, enzyme activity was 9-fold higher in diabetic subjects with periodontitis and only 2-fold higher in diabetic subjects without periodontitis. This difference was attributed to damaged lysosomal membrane and consequent enzyme leakage into the cytosol.
The quantification of βGLU activity in neutrophil leukocytes exposed to bacteria stimuli has shown that diabetic patients with chronic periodontitis have strikingly higher enzyme activity compared to nondiabetics burdened with periodontitis and healthy subjects [88]. Using discriminant analysis, the study established that βGLU activity has a diagnostic potential with great accuracy in distinguishing healthy subjects from diseased ones. βGLU activity stimulated by nonopsonized Staphylococcus aureus showed strongest correlation with the intensity of periodontal parameters compared to opsonized zymosan and prodigiosan, while the highest enzyme activity was stimulated by opsonized prodigiosan.
The strength of association between salivary βGLU activity, periodontitis and type 2 diabetes mellitus has been examined in dentate patients with different stages of periodontal disease, diabetic patients and edentulous patients [89]. In all subjects, diabetic status contributed significantly to βGLU activity, while periodontal status had greater influence on enzyme activity. Higher enzyme activity was also found in nondiabetic dentate patients with periodontitis compared to edentulous controls. Overall, compared to IL-1β, βGLU activity level was more reliable as biomarker of disease severity for periodontitis than it was for the presence of diabetes. In a predating study [90], GCF βGLU activity also correlated strongly with PD, CAL and BOP regardless of diabetic status. Lower enzyme activity was seen in diabetic subjects compared to those with periodontitis. The results suggested a lower release of βGLU in response to systemic inflammation (diabetes) due to reduced deficiency in neutrophil activity, in contrast to amplified activity in response to local inflammation (periodontitis).
2.2. Cancer
Despite landmark developments in oncology, the high rates of morbidity and mortality and increased medical costs associated with all forms of cancer coupled with patients’ psychological trauma on disease diagnosis has remained a major threat to global public health. The successful disease management and sustained wellbeing of affected individuals is however subject to early disease diagnosis and constant evaluation of administered therapy. This in turn relies on efficient tumour markers to ascertain disease risk or status i.e. early stage or metastatic cancer [91]. Although a vast number of biomarkers have been identified for cancer diagnosis, only few have gained clinical approval due to inconsistencies and false positives in their utility [92]. A successful biomarker is that which will not only be specific and selective but will also predict treatment response, while differentiating lethargic and aggressive tumours.
The aetiology of cancer is known to be closely associated with inflammatory pathways and oxidative stress, which jointly create microenvironments favouring neoplasia [93,94]. Hence, in the tumour milieu, an increase in extracellular activity of lysosomal exoglycosidases responsible for the catalytic degradation of glycoconjugates occur, due to malignancy-mediated cell-death and/or lysosomal damage. In this vein, available practical data supports the overexpression of βGLU in extracellular fluids and tissues around tumour sites as a prime factor in cancer aetiology [24]; thus, suggesting the enzyme’s viability as cancer biomarker [95].
For example, βGLU activity was 2-folds higher in the blood samples of 21 patients with colorectal adenocarcinoma compared to healthy controls [96]. Based on cell maturity and clinical grading, enzyme activity was highest in subjects with low or moderately differentiated cells and in subjects with tumours infiltrating surrounding tissues and organs or visceral peritoneum respectively. Estimation of serum βGLU activity in this case proved to be 80% sensitive and 82% specific in distinguishing diseased and healthy subjects. Likewise, enzyme activity was elevated by 6-fold in the peritoneal fluids of patients with ovarian or endometrial cancer compared to women with infertility used as controls [97]. The stronger correlation between cancer stage and βGLU activity, compared to the correlations of other lysosomal exoglycosidases i.e. β-galactosidase and α-mannosidase, reiterated the improved clinical viability of βGLU activity as biomarker of gynaecologic tumour status and severity. However, βGLU activity was equally elevated in the peritoneal fluid of patients with pelvic inflammation hence compromising its application for differential diagnosis of gynaecologic cancer and pelvic inflammatory disease.
2.3. Bacterial inflammation
Bacterial peritonitis is a life-threatening inflammation of the peritoneum – the tissues lining of the inner abdominal walls. As with other inflammatory conditions, cellular damage and polymorphonuclear leukocytes activity increases the level of lysosomal enzymes in the extracellular space via enzyme leakage through the cellular membrane. Thus, quantification of these enzymes provides a potential diagnostic platform.
In fact, βGLU activity in the peritoneal fluid of patients with culture positive bacterial peritonitis was 9 and 33-fold greater compared to that of patients with acute mesenteric lymphadenitis and controls respectively [98]. Peritoneal fluid-βGLU activity measurement in this case holds greater clinical potential compared to β-galactosidase and α-mannosidase for early disease diagnosis and evaluating patient’s response to treatment.
Similarly, considering the current global prevalence of antimicrobial resistance, timely diagnosis of bacteria-mediated meningeal inflammation (bacterial meningitis) is crucial to the outcome of therapeutic intervention. It has been shown that βGLU activity is elevated in cell-free cerebrospinal fluid (CSF) of bacterial meningitis patients early in the disease pathogenesis, even when traditional laboratory parameters such as number of CSF cells, CSF-blood glucose ratio and protein concentration indicated normal status [99]. CSF-βGLU as biomarker was superior to these traditional parameters for early and sensitive prediction of patient’s response to antibiotic treatment.
CSF-βGLU activity in neonates and infants has also been studied as biomarker for differential diagnosis of sterile CSF pleocytosis due to urinary tract infection (UTI) or meningitis [100]. Median βGLU activity in CSF of patients showed significant difference without overlapping in each disease state i.e., bacterial meningitis (168), viral meningitis (26.5) and UTI with sterile CSF pleocytosis (44.1); median enzyme activity was lowest (19.1) in febrile subjects without CSF pleocytosis used as controls. This proffers an unambiguous diagnosis of each condition with 100% sensitivity and specificity. In contrast, broad overlapping was found with classic CSF laboratory parameters viz., CSF cell number, neutrophil number, protein concentration and CSF-blood glucose ratio.
Recently, a case-control study concluded that βGLU activity in bronchoalveolar lavage fluid (BALF) is clinically useful as biomarker of bacterial lung infection [101]. In BALF samples from 92 children, enzyme activity was significantly higher in patients with positive BALF bacterial culture (C+) compared to those with culture negative BALF (C–). 43 nmol 4-methylumbelliferone (4MU)/ml/h was identified as optimum activity value, which allowed differential sample screening (i.e. C+ from C–) with 84.8% sensitivity and 78.3% specificity. Moreover, receiver operating characteristics (ROC) curve analysis established the superiority and higher prognostic value of βGLU activity for bacterial lung infection compared to % polymorphonuclear cell count, human leukocyte elastase, IL-8 and TNF-α.
2.4. Organophosphorus pesticide poisoning
Repeated exposure to organophosphorus compounds (OP) in pesticides is responsible for the lethal poisonings seen in agricultural and veterinary workers; particularly, those in developing countries. OP exerts their fatal effects by inhibiting acetylcholinesterase (AChE) in nervous tissues, leading to muscarinic and nicotinic effects with central nervous disturbance [102]. On the other hand, βGLU is retained in liver microsomal endoplasmic reticulum by forming non-covalent binding complexes with its accessory 64 kda glycoprotein, egasyn, a carboxylesterase isozyme [103,104]. Liver intake of OP however cleaves microsomal βGLU-egasyn complex thus elevating the level of βGLU in plasma to consequently making it an alternative biomarker for OP pesticide poisoning diagnosis [[105], [106], [107]].
A cross-sectional study of pest control workers [108] and plastic greenhouse workers [109], established that plasma βGLU activity was more sensitive as biomarker of OP poisoning compared to butyrylcholinesterase (BuChE) and acid phosphatase (AcP). The enzyme’s activity was higher in subjects with increased exposure than those with low exposure and controls. βGLU activity correlated significantly with BuChE and AcP activities. Likewise, in a case-control study, serum βGLU activity was significantly increased in patients with severe poisoning compared to mildly affected patients and controls [110]. The difference in enzyme activity between the latter groups was insignificant.
Nonetheless, mildly exposed subjects have been reported to show elevated βGLU activity than severely exposed and control subjects, which suggests the enzyme’s utility for diagnosing low-levels of OP exposure. For instance, a study [111], observed the order of serum βGLU activity based on poisoning severity as mild > severe > moderate poisoning, while the order after 12 and 24 h of admission was mild > severe ≈ moderate poisoning; strong correlation also persisted between serum βGLU and BuChE activities. Similar results were also obtained in a recent cross-sectional study [112]. Therein, moderately exposed subjects showed higher enzyme activity than the highly exposed, while non-significant statistical difference persisted between control and highly exposed groups. Notably, βGLU activity correlated well with diabetes propensity, lipid profile, liver function and BuChE but not AChE.
In another cross-sectional study, significant difference in plasma βGLU activity only exists between controls and subjects with chronic exposure (1–45 years) to OP [113]. Activity level was similar in controls and patients with acute poisoning; although 3 out of the 5 examined acutely poisoned patients showed increased level of plasma βGLU activity. However, a case report of an acute OP self-poisoned patient reached contrasting conclusion [114]. The opposing result can be linked to sample size and limited data on patient’s medical history. Moreover, the reduced susceptibility of βGLU-egasyn complex to OP in humans is another primal factor [115]. In contrast to murine βGLU-egasyn interaction, binding in humans is independent of the C-terminal 18 amino acids propeptide in βGLU and esterase active site of egasyn. Rather, it involves the 51 amino residues in βGLU internal segment i.e. residues 228 to 279.
3. Inhibitors of β-glucuronidase activity
3.1. Natural products derived βGLU inhibitors
The progress and momentous achievements in drug discovery cannot be isolated from the wealth of chemical entities i.e. natural products, gifted to man by nature. Many successful molecular candidates of pharmaceutical drug discovery programmes are indebted to the presence of natural product-derived/inspired fragments in their scaffolds [116]. Therefore, the chemical space of natural products (both plants and microbes) has continued to be a much-researched depository for therapeutically significant molecules. In this section, we review some application of natural products including isolated compounds, whole plant extracts and natural product-inspired molecules as potent inhibitors of βGLU. The structures of selected inhibitors (IC50 ≤ 5 μM) are presented in Table 2 at the end of the section.
Table 2.
Most active natural product-derived βGLU inhibitors with IC50 ≤ 5 μM.
| Class | Compound | Structure | IC50 μM | Ref. |
|---|---|---|---|---|
| Flavonoids | 20 |  |
EcoGUS = 0.40 SpasGUS = 0.33 |
[136] |
| 21 | 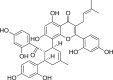 |
EcoGUS = 1.60 SpasGUS = 0.98 |
||
| Thiosulfinate | 52 |  |
3.60 | [165] |
| Iminosugars | 60 | 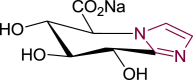 |
12 nMa | [175] |
| 61 | 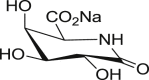 |
31 nMa | [174] | |
| 62 | 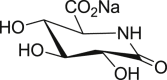 |
32 nMa | ||
| 63 |  |
25 nMa | ||
| 77 | 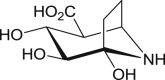 |
60 nMa,b | [179] | |
| 87 |  |
3.30c | [182] |
Inhibition constant Ki.
Potency against E. coli βGLU.
Absolute selectivity for E. coli βGLU.
3.1.1. Natural acids and lactones
The clarification [117,118] that d-saccharic acid-1,4-lactone (5, Fig. 3 ) (saccharolactone or d-glucaric acid-1,4-lactone) is the active and non-toxic βGLU inhibitor responsible for the strong inhibitory potency found with saccharate solutions has precipitated an increased interest in the compound. Despite the poor stability at physiological pH, d-saccharic acid-1,4-lactone (d-SAL) has been explored extensively for its therapeutic significance. Although an IC50 value of 3.6 μM was reported in the pioneering work [117], a value ca. 40 μM is recurrent in literature. Nevertheless, at a concentration of 1 mM, d-SAL completely inhibited the hydrolytic action of human liver-derived βGLU on quercetin glucuronides [119], while over 90% inhibition of βGLU in breast milk was recorded at 10 μM [120]. Also, using urine samples of male Sprague-Dawley rats, d-SAL was identified via metabolomics strategy as one of the therapeutic constituents in LiuWeiDiHuang pills, a famous traditional Chinese prescription for cancer treatment and prevention [121]. Whereas, in a 6-days cumulative study, intraperitoneal pre or cotreatment of female Wistar rats with 3 mg/ml, 10 mg/ml or 10 mg/0.5 ml d-SAL reduced the severity of CPT-11 induced small-intestine mucosal damage assessed by the number of apoptotic cells or mitotic figures, compared to CPT-11 treated controls [122]. Damage reduction was independent of treatment schedule.
Fig. 3.

Lactones and l-aspartic acid based βGLU inhibitors.
The synthetic and natural precursors of d-SAL i.e., 2,5-di-O-acetyl-d-glucaro-1,4:6,3-dilactone (d-SDL, 6, Fig. 3) and d-glucurono-γ-lactone (d-GL, 7) respectively, have also potently inhibited βGLU activity in male Fischer rats thereby providing chemopreventive effects against azoxymethane-induced colon carcinogenesis [123]. Diet supplementation with 0.5 or 2% d-SDL for 5 weeks significantly reduced aberrant crypt foci formation (i.e. pre-neoplastic lesions) by over 48.6 and 55.3% respectively, compared to azoxymethane-controls. d-GL did not afford any significant reduction in colonic tumour incidence during this initiation phase. In addition, 32 weeks treatment with d-SDL or high dose (2%) of d-GL, after 3-weeks subcutaneous injections of 15 mg/kg azoxymethane, provided over 70% inhibition of colon carcinogenesis during the post-initiation phase. It was suggested that d-SDL is a blocking agent which may inhibit pre-neoplasia during the initiation phase, while d-GL is only active during post-initiation of colon carcinogenesis. The increased hydrolytic stability of d-SDL to d-SAL in vivo might also be responsible for these observed phenomena [124].
In another study, d-SAL was more therapeutically efficient compared to its natural precursor (d-GL), for reducing epidermal hyperplasia (lethargic tumour promoter) and inflammation in 7,12-dimethylbenz(α)anthracene (DMBA)-induced complete skin carcinogenesis of SENCAR mice [125]. Pre and cotreatment of murine models with d-SAL for 4-weeks (twice weekly) by topical administration (0.5–4 mg) or dietary treatment (0.5 and 1%), both significantly reduced epidermal hyperplasia and inflammation by up to 57% of DMBA-controls in a dose-dependent manner. d-SAL also inhibited the initiation of carcinogenesis by reducing DMBA-induced oxidative DNA damage (C-8 hydroxylation of guanine) and mutations in codon 61 of Ha-ras gene, by up to 78%. In contrast, d-GL only inhibited epidermal hyperplasia with topical treatment and inflammation by dietary treatment (5% in diet); albeit with inferior potency compared to d-SAL.
Interestingly, another lactone-based βGLU inhibitor (8, Fig. 3), exhibited 8-fold superior potency compared to d-SAL [126]. Isolated from the ethyl acetate extracts of Aspergillus terreus, an endophytic fungus initially isolated from marine alga Laurencia ceylanica, butyrolactone (8) with IC50 = 6.2 μM possesses 16-fold stronger potency than its prenylated isomer (9).
βGLU in breast milk is believed to be involved in neonatal jaundice and hyperbilirubinemia, by increasing serum bilirubin levels via enterohepatic bilirubin circulation in breastfed newborns, in contrast to those fed with infant formulas [47]. Therefore, the suppression of βGLU-mediated deglucuronidation has been proffered as a practicable regimen. l-aspartic acid (10, Fig. 3) in casein hydrolysate formulas was identified as the active βGLU inhibitor responsible for the lower levels of neonatal jaundice observed in newborns receiving such formulae [127]. At 100 μM, the natural amino acid showed ∼86% inhibition of βGLU that is, 100-fold more potent than d-isomer. Moreover, in a randomized and double-blind clinical trial involving 64-newborns, supplementing breastfeeding in the first week of life with 6 doses of l-aspartic acid (180 mg/5 ml of water/day) was more potent against βGLU activity than higher concentrations of enzymatically hydrolysed casein (infant formula containing the inhibitor) or whey/casein (routine formula lacking the inhibitor) [128]. l-aspartic acid supplementation at minimal aliquot concentration significantly lowered transcutaneous bilirubin levels (25% lower than control), leading to higher faecal bilirubin excretion and reducing neonatal jaundice, with no adverse effects.
3.1.2. Flavonoids
Flavonoids are evidently an indispensable class of natural products due to their ubiquity in the vegetal domain and their therapeutic significance. Found in a variety of plant parts (leaves, flowers, stems, nuts, seeds etc.), they perform important functions especially plant’s growth and protection against pathogenic invasion. This intrinsic property encourages their utility as a major constituent of different local medicinal formulations and diets, while their acceptable toxicity profile and physiologic tolerance further presents them as druggable subjects. Flavonoids are typified by the C6–C3–C6 ring system that is, their basic structural skeleton consists of two benzene rings (A and B) linked by heterocyclic pyran ring C (Fig. 4 ). Variations on the chromane core (ring A and C) and attachment position of ring B due to biosynthetic origins lead to classification into flavones, flavanols, flavanones, flavonols, isoflavones, neoflavonoids, anthocyanins and chalcones [129]. This ring system and the presence of hydroxyl units has availed flavonoids with different pharmacological properties such as antidiabetic, antioxidant, antimicrobial, antiplasmodial, antiproliferative and particularly enzyme inhibition [130,131].
Fig. 4.

Natural flavonoids (most active in blue box) and SAR analysis for E. coli βGLU inhibition. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Luteolin (11, Fig. 4), a dietary 3′, 4’, 5, 7-tetrahydroxyflavone has been reported as a viable chemopreventive and anticarcinogenic agent plausibly by inhibiting bacterial βGLU-mediated enterohepatic circulation of colonic carcinogens [132]. The 30-weeks cumulative study using male Wister rats showed that pre or co-treatment with luteolin by intragastric gavage per os (p.o.) at 0.1, 0.2 or 0.3 mg/kg body weight/day, significantly reduced bacterial βGLU activity thereby suppressing 1,2-dimethylhydrazine (DMH)-induced colon adenocarcinomas in a dose-dependent manner compared to DMH-controls. Although 0.2 and 0.3 mg kg−1 day−1 produced similar result. Luteolin supplementation also reduced tumour size from 2 cm to 0.25 and 0.50 cm during the initiation and post-initiation stages respectively, as well up to 90% reduction in tumour incidence. In addition, luteolin (11), its 7-O-glycoside (12), apigenin-7-O-glycoside (13) and catechin (14) were identified as key constituents in the leaf extracts of Pistacia terebinthus responsible for the high E. coli βGLU inhibitory potency [133]. At the highest test concentration (8.2 μg/ml), the leaf extracts exhibited 97.2% inhibition of βGLU activity, corresponding to an IC50 value of 2.11 μg/ml.
In another exploit, 32 natural flavonoids were evaluated for their inhibitory strengths against E. coli βGLU [134]. It was established that luteolin (11), similar flavones – baicalein (15), scutellarein (16) and its glucuronidated analogue scutellarin (17), as well as dietary and ubiquitously occurring flavonol – quercetin (18), are superior inhibitors (IC50 = 5.76–29.64 μM) compared to reference inhibitor d-SAL (IC50 = 36.07 μM); isoflavones and dihydroflavones displayed weaker inhibition. Overall, luteolin (11) and scutellarein (16) emerged the most potent flavone-based E. coli βGLU inhibitors with IC50 = 8.68 and 5.76 μM respectively. SAR analysis (Fig. 4) revealed the importance of 5,6,7-trihydroxy (pyrogallol) unit to bacterial βGLU inhibition, as unsubstitution or replacement of a hydroxy unit with methoxy or glycosyl unit led to significant loss of inhibitory activity. O-methylation at positions-6, 7 and 4′ and installing OH unit at position-3 was also detrimental to potency, whereas the presence of hydroxy unit at C-4’ favoured potency. In addition, molecular docking studies of luteolin and scutellarein in the active site of E. coli βGLU showed hydrogen bonding (H-b) interactions of phenolic OH units at C-5 and C-7 with catalytic acid Glu413 and Arg562 as well as hydrophobic contacts with Ser360 and Leu361 in the bacterial loop.
Furthermore, subjecting the methanolic extracts of edible flower and pedicel of aquatic rhizomatous herb – Nymphaea pubescens (water lily) to bioactivity-guided fractionation established the superior (3-fold) bacterial βGLU inhibitory potency of crude flower extracts compared to pedicel extracts and silymarin – a marketed natural βGLU inhibitor [135]. Kaempferol (19) was subsequently identified as one of the active metabolites with promising activity; IC50 = 36.47 μM and 76-fold superior to silymarin. Interestingly, the lower activity of kaempferol (19) compared to flavonoids (11, 15–18) affirmed the SAR results described earlier.
Amidst 21 constituents found in commonly used Chinese herbal medicines, two prenylflavonoids, Sanggenon C (20, Fig. 5 ) and Kuwanon G (21) emerged as the most potent broad-spectrum inhibitors (>70% inhibition; IC50 = 12.5 and 7.4 μM respectively) of whole human gut bacterial βGLU (consisting of eight bacterial isolates), compared to reference compound amoxapine (7.3% inhibition) [136]. Compound 20 exhibited improved potency against recombinant βGLU from E. coli (EcoGUS) and S. pasteuri (SpasGUS), IC50 = 0.40 and 0.33 μM respectively, compared to compound 21 (IC50 = 1.6 and 0.98 μM respectively). However, compound 21 was a stronger inhibitor of βGLU from three representative bacterial isolates (E. coli, E. fergusonni and S. pasteuri) compared to compound 20. Additionally, molecular docking studies of 20 and 21 in the binding pockets of EcoGUS and SpasGUS revealed both flavonoids bind (via their hydroxy groups) in the allosteric site and not the active site of the recombinant enzymes. The higher number of H-b interactions formed by compound 21 supported the overall increased potency compared to compound 20.
Fig. 5.

Potent prenylflavonoids for bacterial βGLU inhibition.
The realization that pro-inflammatory mediators such as βGLU, released from degranulated mast cells or neutrophils, play significant roles in inflammatory disorders has sparked increased interest in the search for potent inhibitors of such processes as anti-inflammatory drug candidates. In this regard, dihydroxychalcones 22 and 23 (Fig. 6 ) isolated from the roots of Hypericum geminiflorum holds therapeutic promise [137]. Compounds 22 and 23 potently inhibited the release of βGLU from degranulated rat neutrophils with IC50 values of 5.80 and 6.60 μM respectively, better than the reference inhibitor trifluoperazine. However, only compound 23 showed moderate activity against the enzyme’s release from degranulated rat peritoneal mast cells with IC50 = 70.0 μM. Conversely, an isomer of luteolin – norartocarpetin 24 (Fig. 6) and flavonoid-like mornigrol D 25 (Fig. 6), both isolated from the barks of Morus nigra (black mulberry), were moderately potent βGLU inhibitors [138]. At 100 μM, compounds 24 and 25 showed 67.7% and 65.9% inhibition respectively against the release of βGLU from activated rat neutrophils. However, chiral flavonoid alkaloids isolated from the ethanolic extracts of Scutellaria moniliorrhiza and subsequently separated using chiral HPLC, – scumonilines 26–29 (Fig. 6), are better drug candidates compared to compounds 24 and 25, but similar to compounds 22 and 23 [139]. Scumonilines 26–29 displayed ∼63% inhibition at 10 μM against βGLU release from activated rat neutrophils with IC50 values of 5.21, 5.85, 5.47 and 5.16 μM respectively; better than reference inhibitor ginkgolide B (IC50 = 6.63 μM). Compounds 26–29 were also more potent lead molecules compared to both glucuronate esters 30–34 (Fig. 6) isolated from similar Scutellaria regeliana [140] and chiral egenine-based alkaloids 35–37 isolated from the rhizomes of Corydalis decumbens [141]. Compounds 30–34 showed 43.7–47.1% inhibition at 10 μM, whereas compounds 35–37 were more inferior inhibitors at the same concentration with 32.4–41.3% inhibition.
Fig. 6.

Reported natural inhibitors of βGLU release from activated neutrophils and most potent chiral flavonoid alkaloid (in blue box). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.1.3. Plant extracts and ethnomedicinal preparations
Evidently due to its phycocyanin-rich content and potent reduction of zymosan-induced damage to knee joint histological architecture, edible blue-green microalgae – spirulina is deemed a therapeutically viable anti-arthritic agent [142]. An analysis of the synovial fluid of female OF1 mice knee joints after 4-days intra-articular injection of zymosan, followed by 8-days oral administration of spirulina water-suspension at 100 mg/kg and 400 mg/kg, showed 78.7% and 89.2% inhibition of βGLU activity respectively. Subsequently, arthritic parameters such as tibial articular cartilage destruction, erosion of bone structure, articular tissue inflammation, loss of general joint architecture and pannus formation, were all markedly reduced in spirulina fed mice compared to those receiving zymosan only. These anti-inflammatory and anti-arthritic effects were comparable to reference drug Triamcinolone, a glucocorticoid with 94.1% βGLU inhibition at 10 mg/kg of body weight.
Tuber extracts of Arisaema tortuosum (Whipcord Cobra Lily) has shown moderate anti-inflammatory effect via βGLU inhibition in a dose-dependent manner [143]. Conceivably, the presence of quercetin, rutin and lectin, identified through chromatographic profiling, facilitated maximum inhibition of 92.9% at 100 mg/ml, superior to reference inhibitor salicylic acid. Employing a similar approach with ginger rhizomes [144], only 6-Gingerol (38, Fig. 7 ) was identified with 85% inhibition of βGLU at 1 mM, comparable to salicylic acid (82% inhibition). It is also noteworthy that constituents profiling of essential oils extracted from the leaves of seven local varieties of Piper betle L. identified eugenol (39) with an IC50 value of 616.68 μg/ml similar to 794.62 μg/ml of silymarin; although the essential oils were only moderate βGLU inhibitors with IC50 ≥ 5.26 mg/ml [145]. However, metabolomics profiling of the leafy shoots-methanolic extracts of three Swertia species viz. S. chirayita, S. decussata and S. bimaculate, presented S. chirayita as the strongest inhibitor of βGLU and xanthones as the most active metabolites responsible for the Swertia species’ potency [146]. The C-2-glycoside of norathyriol, mangiferin (40) emerged as the most active and therapeutically promising xanthone with IC50 = 38 μM and 50-fold more potent than silymarin.
Fig. 7.

Natural βGLU inhibitors profiled from plant extracts and ethnomedicinal preparations.
Shikunshito-Kamiho (SKTK), a traditional oriental formulation and Fenugreek seeds (FGS), an Indian spice, are natural ethnotherapeutic agents with potentials identical to luteolin (11) i.e., attenuation of colon carcinogenesis via potent inhibition of bacterial βGLU activity [147,148]. 5-weeks oral administration of SPF male ICR mice’s diet supplemented with 20 mg/kg or 60 mg/kg SKTK water extracts, after 10 weeks subcutaneous injection of DMH (weekly), equipotently reduced βGLU activity during and after treatment by 14.9% and 21.3% of controls (DMH alone) respectively. Tumour incidence in colon, measured by the number of aberrant crypt foci was also significantly reduced from 21.6 (distributed in sigmoid and descending colons) in controls, to 6.9 and 6.4 (distributed in rectum) at 20 mg/kg and 60 mg/kg doses respectively. Similarly, diet supplementation with FGS significantly reduced intestinal βGLU activity, leading to a marked reduction in tumour incidence from 93.3% in male Wister rats receiving DMH alone for 15 weeks to 16.6% in rats receiving DMH + 2 g/kg FGS water extracts for 30-weeks cumulative period. Flavonoid, saponin and fibre rich content of FGS were presumed to be responsible for its anticarcinogenic activity.
The hepatoprotective activity of Chinese medicinal formulation – Reduohanxiao-tang, used in the treatment of stroke and liver diseases has also been attributed to its ability to inhibit bacterial βGLU activity in vivo [149]. Water extracts of this local formulation and two of its ingredients viz. rhizomes of Pueraria thunbergiana and Scutellaria baicalensis potently inhibited the activities of E. coli and rat liver-derived βGLU with IC50 values ranging from 0.35 to 1.42 mg/ml. Subsequently, oral administration of these extracts at 100 mg/kg alleviated CCl4-induced liver injury measured by significant reduction in serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), and lactic acid dehydrogenase (LDH) levels, compared to controls. The superior hepatoprotective effect of Pueraria thunbergiana was accredited to isoflavone daidzein (41, Fig. 7), an aglycone metabolite of main components puerarin and daidzin by intestinal bacteria. Daidzein inhibited E. coli and rat liver βGLU with IC50 = 0.41 and 0.50 mg/ml respectively whereas puerarin and daidzin were inactive.
Fascinatingly, structurally similar tectorigenin (42), an aglycone metabolite of 7-O-glycoside – tectoridin (43), isolated from the flowers of Pueraria thunbergiana, exhibited better inhibitory potency (IC50 = 0.30 mg/ml) than its congeners [150]. 50 mg/kg intraperitoneal pre-treatment of male ICR mice with tectorigenin or 100 mg/kg oral administration of tectoridin provided better hepatoprotection than dimethyl diphenyl bicarboxylate (DDB) and daidzein, by significantly lowering serum AST, ALT and LDH levels relative to CCl4-treated controls. The prodrug behaviour of tectoridin, identical to puerarin and daidzin above, was also established by its absence in the serum after 250 mg/kg oral administration (only tectorigenin was detected) and its failure to provide desired hepatoprotection after 100 mg/kg intraperitoneal administration.
3.1.4. Terpenoids and steroids
The triterpenoid 18β-glycyrrhetinic acid (44, Fig. 8 ) also known as enoxolone, is the aglycone metabolite by human intestinal bacteria responsible for the hepatoprotective activity exhibited by saponin – glycyrrhizinic acid 45 [151]. Glycyrrhizinic acid (45) is isolated from the rhizomes of Glycyrrhiza uralensis – the sweetening agent in SKTK. It exists as a natural glucuronide conjugate of 18β-glycyrrhetinic acid (44) hence making it (45) a substrate for βGLU-mediated hydrolysis for consequent release of active inhibitor 44. In vitro evaluation against E. coli and rat liver βGLU revealed stronger potency of glycyrrhizinic acid (IC50 = 12.15 and 97.21 μM respectively) compared to 18β-glycyrrhetinic acid (IC50 = 509.90 and 42.49 μM respectively). Moreover, at 100 mg/kg doses, oral treatment with glycyrrhizinic acid or intraperitoneal treatment with 18β-glycyrrhetinic acid, protected the hepatocytes of male Wistar rats against CCl4-induced liver injury evident by the significant reduction in AST, ALT and LDH levels compared to CCl4-controls. Intraperitoneal administration of glycyrrhizinic acid also did not provide hepatoprotection akin to tectoridin (43). In addition, structurally similar emodinol (46), isolated from the roots of perennial herb Paeonia Emodi showed stronger E. coli βGLU inhibition in vitro with IC50 = 63 μM compared to 18β-glycyrrhetinic acid [152].
Fig. 8.

Triterpenoids and steroids with promising bacterial βGLU inhibitory potency.
Microbial biotransformation of drugs and/or their precursors to new molecules with modulated pharmacological profile is considered an invaluable tool in drug design and development [153]. The natural product class – steroids, are key substrates in this regard [154]. Accordingly, naturally occurring androstane steroid hormone, 5-dehydroepiandrosterone (47, Fig. 8) and its Macrophomina phaseolina metabolites were examined for their βGLU inhibitory potentials [155]. The inferior potency/inactivity of metabolites compared to parent 5-dehydroepiandrosterone (IC50 = 77.9 μM) suggests that having an alkene unit at position-5 and cyclopentanone unit in the molecular architecture is crucial to inhibitory potency. Conversely, the synthetic anabolic steroid, dianabol and its metabolites by filamentous fungus Cunninghamella elegans or Macrophomina phaseolina were inactive against βGLU; except metabolite (48) with IC50 = 60.7 μM [156]. The E-oxime derivative (49) of an inactive metabolite however showed decent inhibitory potency with IC50 = 49.0 μM, equipotent with d-SAL and 2-fold superior than its Z-isomer. Nonetheless, the therapeutic safety of these dianabol derived βGLU inhibitors is questionable considering the increased risk of hepatotoxicity associated with oral administration of 17α-alkylated anabolic steroids [157,158].
3.1.5. Lactic acid bacteria
The fascinating ability of prebiotics and probiotics to positively influence the composition and behaviour of human intestinal microbiota, has been continuously explored with considerable success to improve human health [[159], [160], [161]]. Precisely, lactic acid bacteria (LAB) probiotics can selectively utilize non-digestible oligosaccharide prebiotics as carbon source to produce active metabolites, which modulates or stimulates the activities of certain intestinal bacteria hence, eliciting important physiological response and conferring health benefit [162].
Consequently, LAB – Lactobacillus acidophilus CSG afforded stronger inhibition of intestinal bacteria (including E. coli) producing βGLU thus providing hepatoprotection to male ICR mice, compared to Lactobacillus brevis HY7401 and Bifidobacterium Iongum HY8001 [163]. When anaerobically cocultured with E. coli (HGU-3), L. acidophilus CSG also potently inhibited βGLU productivity of E. coli compared to other LABs. As a result, oral treatment of the murine models with 500 mg/kg of L. acidophilus CSG alleviated CCl4-induced hepatotoxicity by lowering AST and ALT levels to 66% and 57% respectively, of CCl4 control group. Whereas, for t-BHP-induced hepatotoxicity, AST and ALT levels were lowered to 62% and 48% respectively, better than reference hepatoprotective agent DDB. Similarly, Lactobacillus plantarum CFR 2194 was the most effective strain of Lactobacilli metabolizing fructooligosaccharide prebiotics to short chain fatty acids, thereby altering βGLU productivity of E. coli [164]. Lactic acid (50, Fig. 9 ) and especially n-butyric acid (51) were identified as the major short chain fatty acid metabolites in the culture filtrate of Lactobacillus plantarum CFR 2194 and fructooligosaccharides responsible for the observed decrease in βGLU activity.
Fig. 9.

Simple molecules with potential chemopreventive effects via βGLU inhibition.
3.1.6. Other plant isolates
The most abundant and relatively stable thiosulfinate, S-methyl methanethiosulfinate (52, Fig. 9), found in Chinese chive (Allium tuberosum Rottier), has exhibited strong inhibitory potency (IC50 = 3.60 μM) against E. coli βGLU [165]. Compared to other disulphides viz., dimethyl, allyl methyl, and diallyl disulphides, compound 52 is more useful for alleviating drug induced toxicities or providing hepatoprotection. In parallel, a naturally occurring acetal isolated from red macroalga Neodilsea yendoana, isogloiosiphone B (53), possesses similar potential, although with weaker potency (IC50 = 57.0 μM) compared to compound 52 [166].
The adverse role of inflammatory pathways and corresponding mediators including lysosomal enzymes during the pathogenesis of myocardial infarction again implicates βGLU in the cardiovascular disease. In a study which examined the protective effects of thymol (54, Fig. 9) on inflammation in isoproterenol-induced myocardial infarction using male albino Wistar rats [167], the therapeutic benefit of inhibiting the release of βGLU, other lysosomal enzymes and pro-inflammatory cytokines was evident in the restoration of near-normal myocardial histology and function, compared to diseased controls. Rats pre and cotreated with thymol by intragastric gavage at 7.5 mg/kg body weight/day for 7 days and subcutaneous injection of 100 mg/kg isoproterenol for 2 days (day 6 and 7), showed significantly reduced levels of cardiac troponin-T (a cardiotoxicity biomarker) and high-sensitive C-reactive protein in their sera, compared to those treated with isoproterenol only. Most importantly, the activities of lysosomal enzymes i.e. βGLU, β-galactosidase, cathepsin B and D in serum and heart were also potently inhibited to near-normal. These anti-inflammatory effects of thymol afforded a well-protected myocardium with stable histological architecture.
3.1.7. Iminosugars
Polyhydroxylated piperidine and pyrrolidine alkaloids commonly referred to as iminosugars (Fig. 10 a), are a noble class of sugar mimics in which the pyrano or furano endocyclic oxygen has been replaced with a basic nitrogen atom [168]. This class also include isoiminosugars (i.e. 1-azacarbasugars or 1-N-iminosugars) having a methylene unit in place of endocyclic oxygen and the anomeric carbon replaced with nitrogen. Although these ring alterations render iminosugars metabolically inert, their protonated forms mimic the pyranosyl or furanosyl unit in GH substrates, especially the putative oxocarbenium ion-like transition state (2, Fig. 2) in GH catalysed hydrolysis [21,169]. This in turn facilitates their recognizability by GHs and other carbohydrate-recognizing proteins for corresponding enzyme inhibition. Since the isolation of nojirimycin (55, first iminosugar) and siastatin B (56, first isoiminosugar) from Streptomyces cultures in 1966 and 1974 respectively [170,171], these sugar mimics have been a recurring subject of intensive research owing to their extensive biological activities and therapeutic applications elicited majorly through potent inhibition of GHs [172]. The strongest iminosugar-based inhibitors of bovine liver-derived βGLU reported so far (Fig. 10b), with promising antitumor potentials are siastatin B derivatives 57, 58 and 59 with IC50 = 65, 62 and 68 nM respectively [173]. Therefore, this section reviews iminosugars and their analogues as natural product-inspired βGLU inhibitors, without undue repetitions of other monographs [4,21,168,169] covering them.
Fig. 10.

Iminosugars. (a) Nojirimycin and siastatin B, sugar mimics of d-glucose and d-glucuronic acid (b) Most potent siastatin B-inspired βGLU inhibitors with antitumor potentials.
Uronic analogues of nojirimycin bearing glycaro-1,5-lactams or the bicyclic analogues with imidazole and tetrazole units (i.e. tetrahydrotetrazolopyridine-5-carboxylates and imidazopyridine-5-carboxylates respectively), were prepared to examine the effect of sugar-acid configuration and presence of lactam, tetrazole or imidazole moieties on inhibitory potency against bovine liver βGLU [174,175]. Evidenced by the most potent inhibitor 60 (Fig. 11 ) with K i = 12 nM, gluco-configured units, are stronger βGLU inhibitors compared to galacto- and manno-analogues, due to their better mimicry of glucuronic acid (4). The imidazole ring also conferred stronger inhibitory potency than tetrazole or lactam units whereas glycarolactams and tetrazoles shared similar potencies; except galacto-tetrazole (64) with ∼200-fold inferior potency to galacto-lactam (62). Further, although sugar configuration at C-4 was found to be ineffective on βGLU inhibition for glycaro-1,5-lactams 61 and 62, it was very significant for tetrazole and imidazole derivatives. Gluco-tetrazole (63) was 300-fold better than galacto-tetrazole (64), while gluco-imidazole (60) was 600-fold more potent than galacto-imidazole (65) and 1200 superior to manno-imidazopyridine (66), which differs at C-2. Moreover, sugar configuration of acid moiety at C-5 also had significant influence on potency. l-configured units 67, 68 and 69 were 20-50-fold weaker than their d-isomers 63, 62 and 61 respectively. More importantly, the 2-fold increased potency of gluco-imidazole (60) compared to gluco-tetrazole (63) was attributed to stronger interaction of gluco-imidazole (60) with catalytic nucleophile Glu540 compared to gluco-tetrazole (63). Interaction with catalytic acid Glu451 was compromised due to protonation of the imidazole ring in the zwitterionic form.
Fig. 11.

Uronic analogues of nojirimycin containing lactam, tetrazole and imidazole units with most potent βGLU inhibitor, compound 60.
Conversely, similar bicyclic molecules of nojirimycin with cyclic carbamate, urea or guanidine pharmacophores are weaker βGLU inhibitors [176]. In all, only cyclic carbamates 70–73 (Fig. 12 ) showed moderate inhibitory activity against bovine liver βGLU. Cyclic carbamates with carboxylic acid unit at C-5 of nojirimycin ring were also better inhibitors than their hydroxymethyl analogues. Thus, compounds 71 and 73 were the most potent overall with IC50 = 218 and 259 μM respectively. It is noteworthy that bicyclic indolizidine iminosugar 74, with hydroxymethyl unit is also a weak inhibitor (<50% inhibition at 1 mM), although it exhibits desirable selectivity (7-fold) for E. coli βGLU [177]. Surprisingly, carbasugar 75 shared a similar fate with IC50 = 170 μM against E. coli βGLU, even though it is not an iminosugar [178]. Taken together, these results partly suggest that ability to mimic d-glucuronic acid favours βGLU inhibitory activity.
Fig. 12.

Bicyclic nojirimycin-carbamates, indolizidine iminosugar and carbasugar-based E. coli βGLU inhibitors.
Accordingly, glucuronic acid analogue of naturally occurring hemiaminal calystegine B2 (76, Fig. 13 ), uronic-noeurostegine (77), was synthesized in 27-steps from levoglucosan [179]. Compound 77 strongly inhibited both bovine liver and E. coli βGLU with K i values of 2.3 and 0.060 μM respectively; i.e. 38-fold selectivity for the bacterial ortholog. The serendipitously obtained N-alkylated derivatives of compound 77 viz., N-4-hydroxylbutyl (78) and N-ethyl (79), also showed 5 and 10-fold selectivity respectively for E. coli βGLU, although with weaker potencies compared to parent compound 77. Interestingly, uronic-noeurostegines 77–79 were selective inhibitors of βGLU as the compounds were inactive at 1 mM against other GHs examined. Molecular docking studies of 77 revealed that it binds in the central catalytic pocket of E. coli βGLU, forming H-b interactions via its NH and 2-OH units with catalytic acid Glu413 and catalytic nucleophile Glu504 in the bacterial loop. The 4-OH unit of 77 also formed H-b interaction with side chain carboxyl unit of Asp183, while the carboxyl unit at position-5 interacted similarly with phenolic OH of Tyr472, guanidine NH of Arg562, side chain NH2 of Lys568 and amide NH2 of Asn566.
Fig. 13.

Calystegine B2-inspired uronic-noeurostegines as selective E. coli βGLU inhibitors.
Iminosugar C-glycosides are a different class of iminosugars conferred with improved selectivity for E. coli βGLU. This is partly due to their improved lipophilic balance for bacterial cell penetration afforded by their C-alkyl unit and improved chemical stability of C–C bond at the pseudoanomeric C-1 centre [180]. l-iminosugar C-glycoside, (−)-adenophorine (80, Fig. 14 a), an enantiomer of natural iminosugar C-glycoside (+)-adenophorine (81), was prepared together with it analogues via skeletal rearrangement of corresponding azepanes [181]. However, compound 80 and analogues were found to be weak but selective inhibitors of E. coli βGLU. The C-propyl analogue, compound 82 with IC50 = 586 μM showed total selectivity for the bacterial ortholog. It was also superior to compound 80, its azepane precursor 83, and other iminosugars in the study which showed moderate selectivity with 3.1–18.1% inhibition at 1 mM.
Fig. 14.

Iminosugar C-glycosides with most potent E. coli βGLU inhibitor overall in blue box. (a) weakly potent (−)-adenophorine analogues (b) azepanes with markedly increased potency and high selectivity for E. coli βGLU (c) noeuromycin azepane analogues (d) l-configured iminosugar C-glycosides bearing acetamido unit. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
However, increasing the lipophilicity of azepane scaffold 84 via C-2 or N-alkylation, while tuning the configuration of alkyl and hydroxy substituents at C-2 and C-6 respectively (Fig. 14b), birthed E. coli βGLU inhibitors with markedly increased potency and highly conserved selectivity [182]. SAR analysis revealed that a combination of (6S)–OH unit and N-alkylation with hexyl, nonyl or dodecyl produced stronger inhibitors and their potency increased with increasing chain length. In contrast, (2R, 6R)-C-butyl and (2S, 6R)-C-nonyl derivatives, compounds 85 and 86 respectively, were the most active C-alkylated analogues with IC50 = 261 and 27 μM respectively. Compound 85 was stronger than its N-alkylated analogue, while compound 86 showed a compromised selectivity (IC50 = 97 μM against bovine liver βGLU). Overall, highly lipophilic compound 87, having (6S)–N-dodecyl framework was the strongest inhibitor with IC50 = 3.30 μM, 3-fold superior potency to (6S)–N-nonyl analogue 88 (IC50 = 10.0 μM) and absolute selectivity for E. coli βGLU. Furthermore, substituent type and configuration at C-6 also influenced the inhibitory potencies of noeuromycin azepane analogues [183]. (6S)-configured compound 89 and 90 were weakly potent, compared to their inactive (6R)-configured analogues. Compound 89 (IC50 = 139 μM) was 4-fold more potent than its 6-hydroxymethylated analogue compound 90.
Incorporating the acetamido unit in Siastatin B (56), while retaining the iminosugar C-glycoside scaffold via similar skeletal rearrangement of azepanes, afforded l-configured C-glycosides of 1-deoxynojirimycin (91) with weak inhibitory potencies [184]. Sugar configuration at C-1 and C-2 (Fig. 14d), significantly influenced selectivity for GHs. Hence, (2R, 3R)-configured molecules were selective inhibitors of E. coli βGLU but inactive against bovine liver βGLU, α-N-acetylgalactosaminidase and β-N-acetylgalactosaminidase. Whereas, (2S, 3S)-configuration infused selectivity for β-N-acetylgalactosaminidase. Consequently, (2R, 3R)-configured compound 92 with a rigid benzyl substituent emerged as the best E. coli βGLU inhibitor in the series with IC50 = 90.7 μM compared to 37.2% inhibition at 1 mM of compound 93 with (2S, 3S)-configuration and butyl unit. The authors attributed these inferior activities to stereochemical mismatch between the l-configured units and d-configured substrates of GHs. We also conceive that the presence of hydroxymethyl and not carboxylic unit (to mimic glucuronic acid) might also be responsible for the weak inhibitory potencies. This was indeed the case for 3,4,5-trihydroxypipecolic acids, uronic analogues of 1-deoxynojirimycin [185]. d-isomers were stronger than corresponding l-isomers, while the most active βGLU inhibitors were those with better mimicry of glucuronic acid viz. d- gluco and d- galacto configured units; compounds 94 and 95 respectively (Fig. 15 ). Compound 94 showed 3-fold selectivity for bovine liver βGLU (IC50 = 70 μM), whereas compound 95 displayed an exclusive inhibition (IC50 = 86 μM).
Fig. 15.

Potent uronic-acid analogues of 1-deoxynojirimycin.
3.2. Synthetic βGLU inhibitors
3.2.1. Azoles
Azoles are a prominent class of heterocycles with at least one nitrogen atom in their 5-membered aromatic ring. Due to their structural rigidity and amazing physicochemical properties, which confers highly coveted and broad pharmacological activity spectrum and therapeutic potentials, they have remained a targeted scaffold of many synthetic protocols. Members of this class include pyrazoles, imidazoles, thiazoles, triazoles, oxadiazoles, thiadiazoles and tetrazoles together with their benzo analogues i.e., indoles, benzimidazoles, benzothiazoles and benzotriazoles. These compounds also enjoy increased hydrogen bonding (H-b) capability furnished by ring N and/or O atoms for biomolecular targets binding. Consequently, this section is an overview of reported βGLU inhibitors bearing one or more azole nuclei. The critical focus is on the most potent inhibitor(s) in a given series, the pharmacological profile, structure-activity relationship (SAR) and molecular docking analysis.
3.2.1.1. Imidazole
Metronidazole backbone has bestowed superior βGLU inhibitory potency (IC50 = 1.20–44.16 μM) on a set of imidazolylethylaryl carboxylates compared to reference inhibitor d-SAL (IC50 = 48.38 μM) [186]. Compound 96 (Fig. 16 ) emerged as the most potent βGLU inhibitor (IC50 = 1.20 μM) with 40-fold superior potency to d-SAL. SAR analysis supported by in silico studies articulated that compounds with electron-donating groups (EDGs) displayed inferior inhibitory activities compared to those with electron-withdrawing groups (EWGs). Moreover, molecular hybridization with indole nucleus resulted in a potent βGLU inhibitor compound 97 with IC50 = 2.10 μM and 23-fold increased potency than d-SAL. Compound 96 and 97 displayed strong H-b interaction of indole NH unit and π-π interaction with active site residues of βGLU.
Fig. 16.

SAR of imidazolylethyl aryl carboxylates and most potent compounds 96 and 97.
In vitro screening of imidazolopyridines (Fig. 17 ) for their βGLU inhibitory potentials presented dihydroxy-substituted derivatives as promising inhibitors of enzyme activity [187]. The most active molecules in the series are dihydroxy substituted compounds 98 and 99 with IC50 values of 29.25 and 30.10 μM respectively, i.e., 2-fold improved potency compared to d-SAL. Docking simulations revealed the importance of adjacent OH groups to favourable binding interaction with βGLU. Position-2-OH in compound 98 interacted via H-b with both phenolic OH of Tyr504 and NH unit of Lys606, while position-3-OH interacted similarly with backbone carboxylate group of Asp207. Likewise, position-3-OH unit in compound 99 showed H-b interactions with Asn604 and Lys606, while its 4-OH unit interacted with Asp207. Conceivably, these adjacent hydroxyl groups influenced the favourable orientation of free NH unit for strong H-b interaction with catalytic acid Glu451; an interaction which was absent for weakly potent molecules such 2,4- and 2,5-dihydroxy derivatives.
Fig. 17.

Imidazo[4,5-b]pyridine analogues and binding modes of most active inhibitors 98 and 99.
In another study adopting similar imidazolopyridine skeleton [188], only compound 100 (Fig. 18 ) showed appreciable activity (IC50 = 33.01 μM), compared to d-SAL (IC50 = 45.75 μM). The presence of para-methyl substituted on phenyl ring at position-2 of imidazolopyridine ring favoured inhibitory activity plausibly by increasing hydrophobicity, as the unsubstituted derivative 101 had 2-fold decreased potency. However, uninstalling trifluoromethyl (CF3) moiety at position-7 or replacing the furan ring at position-5 with Me, CF3, phenyl or thiophene rendered the resulting molecules inactive.
Fig. 18.

7-(trifluoromethyl)imidazo[4,5-b]pyridines and most active inhibitor 100.
3.2.1.2. Thiazole
SAR analysis of thiazole-Schiff bases showed the significance of OH, Cl and NO2 substituents on benzylidene fragment to inhibitory potency against bovine liver βGLU [189]. 2,3-diOH and 2-OH-5-Cl substituted compounds 102 and 103 respectively, were the strongest inhibitors in the series with IC50 values of 4.88 and 5.63 μM respectively (Fig. 19 ). However, replacing OH unit with more lipophilic MeO or EtO groups led to significant loss of activity. Molecular docking analysis also disclosed the importance of iminic nitrogen and proximity of thiazole nitrogen to catalytic acid Glu451 for strong inhibitory potency. Compound 102 with adjacent OH units had a favourable fit into βGLU catalytic pocket for stronger interactions with amino acid residues Glu540, Glu451 and Tyr508.
Fig. 19.

Most active thiazole-Schiff base-derived βGLU inhibitors.
However, installing pyren-1-ylmethylenehydrazinyl moiety on thiazole skeleton improved βGLU inhibitory potency [190]. Compounds 104 and 105 (Fig. 20 ) emerged as the most potent inhibitors with IC50 = 3.10 and 3.20 μM respectively. Interestingly, the thiosemicarbazone intermediate compound 106 was 9-fold stronger than d-SAL (IC50 = 48.38 μM), while 12-fold improved potency was observed for the thiazolone variant (107). Binding mode analysis of compounds 104 and 105 revealed hydrophobic contacts via pyrene units with Met556 and Phe557. Specifically, the hydrazine unit in compound 104 formed H-b with Arg600, arene-arene interaction with Tyr508 via its thiazole ring and arene-cation contact with Arg600 via the pyrene unit. ortho-OH unit in compound 105 also formed H-b interaction with Asp207, while both thiazole nitrogen and ortho-OH interacted with Arg600. These interactions were also observed in compounds 106 and 107 but absent in other molecules with inferior potencies.
Fig. 20.

Thiazole-pyrenes and most active inhibitors 104 and 105.
3.2.1.3. 1,3,4-Thiadiazole
SAR analysis of 1,3,4-thiadiazole-based βGLU inhibitors disclosed the strong dependence of inhibitory potency on 3,4-diCl substituent of N-phenyl ring as compounds with Br, Me and MeO substituents were weaker inhibitors [191]. As a result, compound 108 with IC50 = 2.16 μM (Fig. 21 ), was the most potent inhibitor. It was 2 and 22-fold stronger than compound 109 and d-SAL respectively. Docking studies revealed that the free amino NH unit in compound 108 formed strong H-b interaction with OH unit of catalytic acid Glu451. The dichloro substituents also aided hydrophobic interactions with Tyr504 and Tyr508, while both thiadiazole and phenyl rings interacted via π-π stacking with Asp207. On the hand, 2-fold reduced potency of compound 109 was revealed by its poorer binding modes in the enzyme’s active site.
Fig. 21.

Thiadiazole derived βGLU inhibitors and most potent compounds.
3.2.1.4. Benzimidazole-based hybrids
The inhibitory potency of benzimidazole nucleus against βGLU was investigated using substituted phenyl units at position-2 of benzimidazole core [192]. Activity was dependent on the presence and position of Cl and OH substituents hence 3,4-dichlorophenyl analogue 110 (Fig. 22 ) was found with strongest inhibitory potency (IC50 = 6.33 μM). Interestingly, introducing 5,7-dichloro unit on benzimidazole nucleus gave remarkable results as previously inactive molecules gained significant activity [193]. Compound 111 emerged as the most potent inhibitor overall with IC50 = 4.48 μM and 11-fold stronger than d-SAL. Moreover, replacing an OH unit of the dihydroxy substituent with MeO led to significant or total loss of activity. F, Me, NO2, naphthyl and anthracenyl units also gave inactive compounds. Molecular docking studies further established that Cl and OH substituents on benzimidazole and phenyl rings respectively are crucial to strong βGLU interaction. The 5,7-dichloro unit on compound 111 afforded hydrophobic interactions with phenyl rings of Tyr504, Tyr508 and side chain methyl of Asn484 and Glu451, while 2-OH substituent interacted via strong H-b with NH2 unit of Lys606 and OH unit of Tyr504. Hydrophobic interactions of phenol ring with Trp587, Asp207 and His385 also aided ligand-receptor stability in the active site.
Fig. 22.

Benzimidazole based inhibitors and most active compounds.
N-alkylation of benzimidazole with substituted phenacyl units produced βGLU inhibitors with a different trend in potency [194]. Molecules containing EWGs NO2, Cl and F groups were superior in the series (Fig. 23 ). Thus, the best activity was found with ortho-NO2 substituted compound 112 (IC50 = 4.06 μM) while meta and para-NO2 variants exhibited 5 and 2-fold reduced potency respectively. 4-Cl, 4-F and 2,4-diF substituted molecules also had appreciable potencies with IC50 = 32.11, 26.30 and 24.75 μM respectively. In contrast, EDGs, 3,4-diOH, 4-phenyl, 4-MeO and 2,4-diMeO gave inferior inhibitors compared to d-SAL (IC50 = 48.40 μM).
Fig. 23.

N-Phenacyl-2-pyridylbenzimidazole derived inhibitors and most active compound.
Akin to imidazole-indole hybrid compound 96 with IC50 = 2.10 μM (Fig. 16), molecular hybridization of benzimidazole with amide bond bioisostere, 1,3,4-oxdiazole, resulted in an isopotent hybrid 113 (Fig. 24 ) with IC50 = 2.14 μM [195]. Again, OH-substituted derivatives were stronger inhibitors compared to those containing F, Cl and NO2, Me and MeO substituents. Docking studies revealed that the molecular hybrids adopted a linear configuration in the active site of βGLU. The oxadiazole nucleus and central-phenyl ring of compound 113 formed strong H-b interaction with NH2 unit of Asn484 and phenolic OH of Tyr508 respectively. Compound 114 (IC50 = 3.14 μM) on the other hand interacted with catalytic acid Glu451 and Tyr508 via its central phenyl and phenol rings respectively.
Fig. 24.

Benzimidazole-oxadiazole hybrids and most potent βGLU inhibitors.
3.2.1.5. 1,3,4-Oxadiazole-based hybrids
In another comparative study using 113 as lead but replacing benzimidazole with benzohydrazone unit (Fig. 25 ), the importance of benzimidazole nucleus to βGLU inhibitory activity was established by the pronounced loss of activity [196]. Nevertheless, OH-substituted derivatives and benzenetriol compound 115 emerged as the strongest inhibitor with IC50 = 7.14 μM. Inhibitory potency was dependent on substituent’s position in the order; para ˃ meta ˃ ortho for OH, NO2 and Cl groups, while the presence of Me or MeO groups gave inactive compounds. Molecular docking studies showed that inhibitory activity correlated strongly with the strength of H-b interaction hence the improved potency seen with OH-substituted derivatives. Notably, the benzenetriol OH unit on compound 115 formed H-b interactions with Glu540, Asp207, Tyr508, His385, and Asn450, while the benzohydrazide unit interacted similarly with Tyr508, Glu540 and Tyr504.
Fig. 25.

Oxadiazole-benzohydrazone based βGLU inhibitors and most active inhibitor.
In like manner, replacing the pharmacophores in compound 115 i.e. hydroxyl for methoxy and imine for more polar sulfonamide unit (Fig. 26 ), led to stronger βGLU inhibitory potency for the resulting oxadiazole-benzenesulfonamides [197]. The most potent inhibitors were compounds 116 and 117 with IC50 = 2.40 and 6.34 μM respectively; compound 116 showed similar potency as compound 113. Inhibitory activity was improved for para-substituted derivatives, while EWGs produced stronger inhibitors than EDGs. Binding interactions in the active site of βGLU especially H-b interaction of benzamide NH unit with Glu451 established the improved activity of the sulfonamides compared to benzohydrazones. Compound 116 formed H-b interaction via sulfonyl oxygens with Asp207 and Asn450, while the benzamide oxygen interacted in similar fashion with Tyr508. Additional ionic bonding between NO2 groups in compound 117 and Glu451 also accounted for the improved potency.
Fig. 26.

Oxadiazole-benzenesulfonamides and most active inhibitors 116 and 117.
Molecular hybridization (Fig. 27 ) of 1,3,4-oxadiazole and its bioisostere 1,3,4-thiadiazole was attempted for βGLU inhibition [198]. However, parallel comparison with other reported oxadiazole-containing hybrids reveal that their hybridization does not produce a remarkable effect on inhibitory potency. Inhibitory potency remained with OH, F and Cl substitutions, while abolished activity persisted when OH unit is replaced with MeO. Evidently, the presence of 2,4,6-trichloro unit significantly contributed to potency. Thus, compound 118 was the most active inhibitor with IC50 = 0.96 μM, 2- and 3-fold stronger than 3,4-diOH substituted compound 119 and oxadiazole-sulfonamide hybrid 116 with similar 2,4,5-trichloro unit (IC50 = 1.40 μM) respectively. The result partly suggests that significant alteration in molecular lipophilic/hydrophobic balance and the binding potential of installed pharmacophoric units is crucial to eliciting desirable increase in βGLU inhibitory potency.
Fig. 27.

Most potent oxadiazole-thiadiazole hybrids.
3.2.1.6. Benzothiazole-based hybrids
Results of βGLU inhibition studies of 2-arylbenzothiazoles derivatives further established the significance of OH substituent to inhibitory potency, as activity was conserved to OH-substituted compounds [199]. 2,4,5-benzenetriol substituted compound 120 (IC50 = 2.26 μM) was the strongest inhibitor in the series with 2- and 21-fold improved potency compared to 2-OH analogue 121 (IC50 = 4.23 μM) and standard inhibitor d-SAL respectively (Fig. 28 ). In silico studies of these benzothiazoles revealed that they adopted a linear conformation which allowed appropriate fit into the binding groove of βGLU. Ortho-OH unit on compound 120 formed strong H-b interactions with Glu451 while the para-OH unit provided H-b interaction with Asp207. The enzyme-inhibitor complex was stabilized in the active site through hydrophobic interactions of benzenetriol ring with indole nucleus of Trp587, His385, Asn484, Tyr504, His509, Arg600, and Lys606. Most importantly, all the active inhibitors were non-cytotoxic in a cytotoxicity assay using mouse embryo fibroblasts (3T3-L1) and Wistar rat hepatocytes (CC-1).
Fig. 28.

Most active benzothiazole derived βGLU inhibitors.
Benzothiazole and benzimidazole are bioisosteres due to their structural and pharmacological similarities; therefore, bioisosteric replacement of one for the other has been consistently explored in drug development. Accordingly, bioisosteric replacement of the benzimidazole moiety in compound 113, to give new benzothiazole-oxadiazole hybrids resulted in equally potent βGLU inhibitors [200]. The most potent compound 122 (Fig. 29 ) with IC50 = 2.16 μM, was equipotent with compound 113. Fascinatingly, compound 123 (IC50 = 4.38 μM) was also isopotent with similarly substituted compounds 121 and 111. Further, docking studies of compounds 122 and 123 showed that H-b interactions of phenolic OH units with active site residues viz., Glu451, Tyr508, and Asp207, favoured their strong inhibitory potency. Moreover, compound 122 formed hydrophobic interactions with Tyr504 and Lys606 via its thiazole core and benzene ring respectively. The reduced potency of compound 123 was established in the strength of these interactions as well as the absence of additional van der Waal contacts of benzothiazole ring in compound 122 with catalytic residues Glu451 and Glu540. In silico pharmacokinetic properties modelling of these inhibitors predicted good cell permeability and solubility. The compounds were also non-cytotoxic to 3T3-L1 and CC-1 cell lines.
Fig. 29.

Benzothiazole-oxadiazole hybrids and most potent βGLU inhibitors 122 and 123.
Following a similar molecular development strategy leading to compound 122, new benzothiazole-hydrazone hybrids were assembled by deleting the 2-oxadiazolylphenol fragment in compounds 122 and 115 (Fig. 30 ) [201]. Compared to the parent compound series, the new hybrids had marked reduction in potency. The recurring pattern of reduced or abolished activity also persisted with Me and MeO substituents. Nevertheless, compound 124 was the strongest inhibitor in the library with IC50 = 16.50 μM. Its reduced activity was further established by the similar but weaker binding interactions compared to compounds 122 and 115.
Fig. 30.

Molecular design of benzothiazole-benzohydrazone hybrids and most active inhibitor 124.
3.2.1.7. Triazole
The triazole ring also affords potent βGLU inhibitors. In vitro evaluations of 1,2,4-triazole-Schiff bases [202] presented isatin-containing compound 125 (Fig. 31 ) as the strongest inhibitor with IC50 = 2.50 μM and 19-fold superior activity than d-SAL. Cl, OH and NO2 substituents at ortho-positions also gave appreciable activities whereas Me, MeO, cumyl and other aryl derivatives were inactive. Docking simulations of compound 125 in the active site of βGLU revealed strong H-b interaction between isatin NH and catalytic residues Glu540 and Tyr504. The phenyl ring also provided π-anion and π-π interactions with Glu451 and Tyr504 respectively while van der Waal contacts with Lys606, Tyr508, Val410 and Asn450 stabilized the inhibitor-βGLU complex. However, amide bond bioisostere, 1,2,3-triazole, is a stronger βGLU inhibitor compared to its 1,2,4-triazole isomer. Molecular hybridization with carbazole [203] delivered eight compounds having IC50 < 3 μM (Fig. 32 ). Compound 126 (IC50 = 0.55 μM) was the most potent hybrid in the series and 83-fold stronger than d-SAL. All the carbazole-1,2,3-triazole tethers were non-cytotoxic to 3T3 cells.
Fig. 31.

Most potent 1,2,4-Triazole Schiff base 125.
Fig. 32.

1,2,3-Triazole – carbazole tethers and most potent inhibitor 126.
3.2.1.8. Indole-based hybrids
A library of indole analogues containing the hydrazone unit in compound 115 have been examined for βGLU inhibition [204]. Evinced by the increased potency, the indole pharmacophore delivers stronger βGLU inhibitors compared to benzothiazole and oxadiazole. For instance, the most potent inhibitor 127 with IC50 = 0.50 μM (Fig. 33 ), was 100-fold more potent than d-SAL and 42-fold stronger than similarly substituted oxadiazole variant from compound 115 library. Compounds 128 (IC50 = 2.40 μM) and 129 (IC50 = 2.50 μM) were also 3-fold and 43-fold stronger respectively than their corresponding oxadiazole variants. Moreover, substitution at ortho and para-positions gave stronger inhibitors compared to meta-position, while potency in monosubstituted analogues followed the order; F ˃ OH ˃ Cl ˃ pyridyl ˃ NO2 ˃ MeO. The excellent potency of compound 127 was afforded by strong H-b interaction of position-5-OH with Glu451 and Glu540 as well as position-4-OH with Tyr508. Indole and benzohydrazone NH units formed H-b interactions with Asn502 whereas the ligand-receptor complex was stabilized by π-alkyl interaction of indole ring with Trp528. Expectedly, the reduced activity of compound 128 was seen in its weaker interactions in the active site of βGLU.
Fig. 33.

Indole-hydrazone hybrids and most active inhibitors 127–129.
Based on the pharmacological significance of thiadiazole pharmacophore and the promising inhibitory activity of indole hybrid compound 127, hybrids of indole-thiadiazole were assembled as new class of βGLU inhibitors [205]. Generally, thiadiazole nucleus infused stronger inhibitory potencies on the resulting hybrids compared to hydrazone unit. Dihydroxy substituted compound 130 emerged as the strongest inhibitor with IC50 = 0.50 μM and equipotent as compound 127. Ortho-substitution was more beneficial to potency than para and meta-substitutions, while inhibitors’ strength followed the order; OH ˃ F ˃ Me ˃ pyridyl ˃ Cl ˃ NO2 (Fig. 34 ). However, bioisosteric replacement attempt with oxadiazole proved that thiadiazole unit is preferred for stronger inhibition [206]. The strongest potency remained with 2,3-dihrdoxy substituted derivative 131 (IC50 = 0.90 μM), although 2-Cl derivative had an outlying potency.
Fig. 34.

Indole-thiadiazole/oxadiazole hybrids and most potent compounds 130 and 131.
Bazedoxifene (Fig. 35 ) is a novel indole-based inhibitor of IL-6/GP130 for the management of triple negative breast cancer [207]. It is also a selective estrogen receptor modulator administered as co-drug with conjugated estrogens (Duavee) in estrogen replacement therapy for the prevention of postmenopausal osteoporosis. Albeit, the high ratio of circulating estrogen metabolites and parent estrogen aided by βGLU deglucuronidation activity in the gut is considered a risk factor for postmenopausal estrogen receptor positive (ER+) breast cancer. Accordingly, a study on the therapeutic utility of this combination drug in an ovariectomized mouse model showed significant reduction in faecal βGLU activity without altering faecal microbiota diversity [208]. The study articulated the significance of bacterial βGLU inhibition to improving the outcome of long-term administered estrogens for postmenopausal women or breast cancer patients.
Fig. 35.

Combination drug (Duavee) as bacterial βGLU inhibitor.
Consistent with the excellent inhibitory potencies of indole-based compounds, a library of bis-indoles (Fig. 36 ) have been reported as strong βGLU inhibitors [209]. Once more, the importance of (di)hydroxy substitution to potency was prominent. Compound 132 emerged as the most active molecule with IC50 = 1.62 μM and 30-fold superior to d-SAL. In silico studies suggested that H-b donor-acceptor potential of OH units on compound 132 significantly influenced ligand-receptor interaction. Ortho-OH formed strong H-b with amino acid residues Asp207 and Tyr508, while para-OH interacted similarly with His385. Arene-arene interaction of indole ring with Tyr504 also aided the favourable binding to βGLU. In another study [210], both compound 132 and its 2,3-dihydroxy compound 133 (IC50 = 1.2 μM) were the strongest inhibitors of bacterial βGLU; albeit with moderate cytotoxicity against 3T3 mouse fibroblasts.
Fig. 36.

Bisindole based inhibitors and most active molecules 132 and 133.
The presence of N-phenyl substituted thiosemicarbazide unit however introduced marked increase in βGLU inhibition of 1H-bisindoles [211]. Inhibitory potency for monosubstituted analogues followed the order F ≈ CF3 ˃ Cl ˃ Br ˃ Me ˃ MeO and ortho-substitution produced the best result (Fig. 37 ). Accordingly, 2-F compound 134 emerged as the strongest inhibitor with IC50 = 0.1 μM; 484-fold superior potency compared to d-SAL, 2-fold stronger than 4-CF3 compound 135 and equipotent with 3,4-diCl compound 136.
Fig. 37.

Bisindole-thiosemicarbazide conjugates and strongest inhibitors 134–136.
Results from in vitro evaluation of a library of bisindole-hydrazones against βGLU activity [212], suggested a synergistic effect of two indole nuclei on enzyme inhibition (Fig. 38 ). Majority of the examined molecules showed IC50 ˂ 10 μM; the best result overall for hydrazone conjugates. Identical to compound 127 series, OH and F substituted molecules were stronger inhibitors. Therefore, compounds 137 and 138 were the strongest inhibitors with equal potency; IC50 = 0.10 μM. The presence of MeO unit was again unfavourable to activity as seen in the 2- to 11-fold decrease in activity of MeO substituted variants of di-OH derivatives. Docking studies further revealed that an extensive H-b network via OH units and hydrophobic interactions via indole nuclei fostered the favourable fit of compounds 137 and 138 in βGLU’s catalytic pocket for strong inhibition. Benzenetriol OH units of compound 137 acted as H-b donor to Asn502, Gln524, His385, Tyr508 and H-b acceptor to Glu540, Asn450, Lys606, Trp528. Tyr508 also formed H-b interaction with amide NH and arene-arene interaction with phenyl ring of tolyl unit.
Fig. 38.

Most potent inhibitors, SAR analysis and binding interaction of bis-indole-hydrazone tethers.
3.2.2. Chalcones
Chalcones are both a class of naturally occurring flavonoid family and synthetically obtainable compounds with extensive therapeutic applications [213]. The presence of a highly reactive α,β-unsaturated ketone unit and delocalized electrons makes them coveted precursors for many synthetic manipulations and pharmacological explorations.
The pro-inflammatory role of βGLU in inflammatory disorders motivated the design of a series of (di)hydroxychalcones (Fig. 39 ), to inhibit the release of βGLU from stimulated rat mast cells and neutrophils [[214], [215], [216]]. It was established that hydroxychalcones were stronger and selective inhibitors of neutrophil-derived βGLU over mast cell-derived βGLU. The best inhibitors were compounds 139 (IC50 = 0.6 μM; ˃ 160-fold selectivity) and 140 (IC50 = 1.3 μM; 7-fold selectivity). SAR analysis showcased the importance of α,β-unsaturation to βGLU inhibition as dihydro derivatives were 20 to 30-fold less potent than parent chalcones. The OH unit at position-5 of ring A was crucial to the inhibition of mast cell degranulation and release of βGLU from neutrophils. O-alkylation of hydroxyl units on phenyl rings particularly ring A also reduced potency while abolished activity occurred with increased alkyl size.
Fig. 39.

Dihydroxychalcones and strongest βGLU inhibitors.
Compound 141 (Fig. 40 ) has emerged as the most potent inhibitor with 75% inhibition at 10 μM and only 3-fold stronger than standard salicylic acid, amongst a series of chalcone-Mannich adducts [217]. Adopting a similar molecular design [218], a library of chalcone-benzamides were found with slightly improved potency and SAR analysis established the preference of EWGs over EDGs for better enzyme inhibition. Compound 142 containing similar 3-Br substituent as compound 141, emerged as the most potent molecule (84.68% inhibition at 1 mM); 3-fold stronger potency than salicylic acid (24.77% at 1 mM). It was also non-cytotoxic in CCK-8 assay in contrast to 2-fold increased cytotoxicity of equally potent 3-CF3 analogue. Notably, activity modulation by substituting trimethoxy unit for indole was unsuccessful [219], leading to prominent reduction in percentage inhibitory activity of the chalcones (˃ 8-fold).
Fig. 40.

Chalcone-benzamide tethers and most active inhibitors 141 and 142.
3.2.3. Coumarins and azacoumarins
Structural development of a coumarin (chromen-2-one) compound 143 (Fig. 41 ) recovered from virtual screening delivered a set of moderately potent inhibitors of βGLU activity [220]. Superior activity compared to d-SAL (IC50 = 48.40 μM) was recorded compounds 144–147 with IC50 values of 9.90, 11.70, 21.40 and 34.20 μM respectively. In another attempt [221], despite different molecular tunings including polar OH and Cl groups, the coumarin pharmacophore remained inactive against bacterial βGLU. Conversely, structurally isomeric flavenone (chromen-4-one) bearing a pyranose appendage, showed stronger inhibition of bacterial βGLU [210]. Compound 148 (Fig. 42 ), the most active inhibitor thereof with IC50 = 4.50 μM and 10-fold superior potency to d-SAL, was totally non-cytotoxic against 3T3 mouse fibroblasts. In silico studies further disclosed the importance of pyranose ring in H-b interactions with active site residues Glu503, Asp161 and Glu413.
Fig. 41.

Most potent chromen-2-one analogues 144 to 147 for βGLU inhibition.
Fig. 42.

Most potent chromen-4-one as inhibitor of E. coli βGLU activity.
Fascinatingly, introducing 1,3,4-oxadiazole pharmacophore to a chromen-4-one scaffold (Fig. 43 ), led to significant increase in potency [222]. The new hybrids exhibited consistent trend in their potency based on substituent type and position in the order; F > Cl > Me > NO2 > Pyridyl > MeO and ortho > para > meta respectively. Thus, fluoro substituted derivatives 149 (IC50 = 0.80 μM) and 150 (IC50 = 1.10 μM) were the strongest inhibitors overall with 60-fold and 44-fold stronger potencies respectively than d-SAL. The binding modes of 149 reiterated the importance of H-b atoms and 1,3,4-oxadiazole’s multiple-binding potential to molecular activity. Ortho-fluorine atom was a H-b acceptor to Tyr508 and Lys606 whereas Asn484 formed carbon to H-b interaction with chromenone endocyclic oxygen. Hydrophobic interactions of oxadiazole core with Glu451 and Tyr508 as well as 2-fluorophenyl ring with Asp207, Glu540, Trp597, Lys606 stabilized βGLU-inhibitor complex in the active site.
Fig. 43.

Most potent chromen-4-one – oxadiazole hybrids for βGLU inhibition.
On the other hand, azacoumarins (1H-quinolin-2-ones) having a basic nitrogen atom in place of endocyclic coumarin oxygen, are conferred with stronger and selective inhibitory potencies against bacterial βGLU. Plausibly, their admirable activity is provided by the ability to mimic or interfere the oxocarbenium ion-like transition state akin to iminosugars; thus, allowing the alleviation of drug-induced toxicities and prevention of colon carcinomas.
Renowned examples in this class of βGLU inhibitors are the strongly potent azacoumarins 151 and 152 (Fig. 44 ), identified from high-throughput screening [19,223]. The compounds exhibited in vitro IC50 values of 0.28 and 0.37 μM respectively, over 1000-fold selectivity for E. coli βGLU and 100-fold increased potency than reference inhibitor glucaro-δ-lactam 61. The inhibitors also maintained their potency in living bacterial cells with EC50 = 0.018 and 0.028 μM. Other similarly active compounds (IC50 ≤ 5 μM) from the screening are presented Table SI. The high selectivity of compounds 151 and 152 for E. coli βGLU was established via crystal structure resolution of enzyme-inhibitor complex of compound 152, which showed binding interactions at the entrance of the active site cavity with catalytic residue Glu413, as well as Leu361 and Phe365 in the bacterial loop. No inhibition was observed with an engineered mutant form of E. coli βGLU lacking residues 360 to 376.
Fig. 44.

Azacoumarins and strongest E. coli βGLU inhibitor 151 and 152.
More importantly, in vivo studies of these azacoumarins expresses their remarkable therapeutic potentials. Co-administration of compound 151 at 10 μg (twice daily) with 50 mg/kg of CPT-11 (once daily), alleviated Irinotecan (CPT-11) induced diarrhoea evident by a protected GI epithelium of BALB/cJ mice compared to bloody diarrhoea due to damaged tissues and glandular structure in those receiving CPT-11 alone [19]. Pre-treating C57BL/6J mice with 10 μg of inhibitor 151 before intraperitoneal administration of ulcerogenic dose of NSAIDs – diclofenac (60 mg/kg), indomethacin (10 mg/kg) and ketoprofen (100 mg/kg), also successfully protected the mice models against NSAID-induced small intestine mucosal damage [224,225]. Moreover, compound 151 reduced all enteropathy parameters by inhibiting GI bacterial βGLU-mediated hydrolysis of NSAID-acyl glucuronide in a concentration-dependent manner (IC50 ≈ 0.164 μM). Interestingly, the compound did not alter drug’s systemic exposure, hepatobiliary excretion of glucuronides and GI microbiota diversity. Although it possesses a short half-life (˂1 h) and pan-cytochrome P450-mediated poor bioavailability (21%). However, an attempt to reduce the inhibitor’s serum exposure for increased GI residence by replacing ethoxy unit with hydroxyl or morpholinyl groups led to inferior pharmacological profile compared to parent compound 151 [226]. Further, compound 152 exerted 2-fold reduction in the severity of diclofenac-induced anastomotic leakage, without affecting drug’s plasma concentration in Wistar rats receiving 0.8 μg of the inhibitor and 3 mg/kg of diclofenac [227]. In contrast, rats receiving diclofenac only suffered severe leakage thus suggesting the inhibitor’s potential clinical utility to improve anastomotic healing.
3.2.4. Piperazines
Emerging from the screening protocol which birthed compound 151, three piperazine-containing compounds 153, 154 and 155 (Fig. 45 ) showed promising inhibitory potentials against bacterial βGLU [228]. Though with slightly weaker inhibitory strengths (IC50 = 0.54, 8.89, 6.43 μM respectively), their pharmacological profile viz., potency in living bacterial cells, selectivity for E. coli βGLU over mammalian βGLU or other related glycosidases and non-cytotoxicity to bacterial or human epithelial cells, were identical to compound 151. X-ray crystallography of compound 155-E. coli βGLU complex revealed similar binding interactions with active sites as compound 151. The most active compound 153, equally alleviated CPT-11 induced GI toxicity (bloody diarrhoea) in BALB/cJ mice when co-administered (10 μg/day) by oral gavage with CPT-11, without affecting the anticancer drug’s systemic pharmacodynamic properties measured by weight loss.
Fig. 45.

Piperazine-containing scaffolds as potent E. coli βGLU inhibitors.
Detailed kinetic analysis, enzyme inhibition and X-ray crystallography studies of piperazines 156 (IC50 = 0.12 μM) and its synthetic analogue 157 (IC50 = 0.080 μM) [229], disclosed their mechanistic behaviour as substrate-selective, slow-binding inhibitors of gut bacterial βGLU with similar pharmacological profile as compound 151. Compounds 156 and 157 both exerted their inhibitory activities by targeting βGLU-glucuronic acid intermediate to form inhibitor-glucuronide conjugate even in the presence of CPT-11 glucuronide hence disrupting the enzyme’s catalytic cycle. SAR probing of compound 156 revealed the importance of nucleophilic NH on piperazine unit to potency. Both mono and dimethylated analogues were 80 to 120-fold weaker respectively than parent compounds 156 and 157. The piperidine derivative having methylene unit for NH was also totally inactive. Moreover, evaluating βGLU inhibitory potentials of clinically available drugs containing piperazine or piperidine units viz., Palbociclib, Crizotinib, Vortioxetine, Amoxapine and Ciprofloxacin also established a similar substrate-dependent, high in vitro and cell-based potency against enterobacterial βGLU. Amoxapine was the most potent inhibitor with IC50 = 0.53 μM. The results consequently rearticulates similar studies [[230], [231], [232], [233]] proposing the repurposing of approved drugs (Fig. 46 ) as potent inhibitors of βGLU in GI microbiota, to improve the therapeutic efficacy of CPT-11.
Fig. 46.

Repurposable drugs for E. coli βGLU inhibition.
3.2.5. Pyridinone, pyrimidinones and quinazolinone
The therapeutic significance of fused pyridinone-furans as anti-inflammatory drug candidates via inhibition of neutrophil-derived βGLU has been reported [234]. Compounds bearing MeO and CF3 substituents on ring B (Fig. 47 ) showed over 70% inhibition at 1 μM with no cytotoxic effects (<32% at 10 μM) in CCK-8 assay as compared to reference salicylic acid (25% inhibition). Replacing ring B with furan, thiophene or pyridine however gave poor inhibitors (<45% inhibition). Compound 158 was the most potent with 75% inhibition.
Fig. 47.

Fused pyridinone-furans as anti-inflammatory agents via βGLU inhibition.
The inhibitory potencies of dihydropyrimidinone carboxylates (Fig. 48 ), against bovine liver-derived βGLU has reiterated that small polar substituents are necessary for strong enzyme inhibition [235]. Appreciable potency (IC50 ˂ 20 μM) was exclusive to CF3 ˃ F ˃ OH ˃ thienyl ˃ Cl substituted compounds in that order. Whereas, MeO, Br, benzyloxy, furanyl, aliphatic alkyl or aryl units either reduced potency when co-substituted with polar F or OH groups, gave inferior activity with monosubstitution or rendered the molecule totally inactive. Therefore, the most active compound 159 with IC50 = 9.38 μM, found favourable fit in the active site of βGLU to form strong H-b interactions with key residues. NH of Asp207 was both a H-b donor and acceptor to pyrimidinone carbonyl oxygen and free NH at position-1 respectively. Carboxylate carbonyl oxygen interacted similarly with Arg600 whereas position-3 NH interacted with catalytic residues Glu451 and Tyr508. However, pyrimidinetriones (barbituric acid) [236,237], exhibited inferior potencies compared to standard d-SAL (IC50 = 45.75 μM), despite their similar binding interactions as compound 159 with active site residues and non-cytotoxicity to 3T3 cells.
Fig. 48.

Pyrimidinone-based inhibitors and most active compound.
In a predating study, the structural homolog of pyrimidinone, quinazolinone [238], exhibited a unique trend in inhibitory potency against E.Coli βGLU with significant tolerance for lipophilic alkoxy groups, although poor activity with methyl substituent persisted. The most active compound 160 (Fig. 49 ) bearing 3,4-diMeO unit had IC50 = 0.60 μM, 76-fold superior potency to d-SAL and equipotent with 2-EtO substituted analogue 161 (IC50 = 0.70 μM). Intriguingly, replacing alkoxy units with more polar OH, NO2 and Cl substituents led to significant reduction in potency. Inhibitory potency therefore followed the order; MeO ˃ EtO ˃ N(Me)2 ˃ NO2 ˃ OH ˃ Cl and ortho ˃ para ˃ meta. Importantly, the quinazolinone-based inhibitors were all non-cytotoxic to 3T3 cells. It is also noteworthy that a quinazolinone compound (Table SI) from Ref. [223], also showed strong inhibitory potency (IC50 = 1.90 μM) against bacterial βGLU.
Fig. 49.

Quinazolinone-based E. coli βGLU inhibitors and most active inhibitors 160 and 161.
3.2.6. Quinolines
The quinoline nucleus can be regarded as a household name in medicinal chemistry, due to its inexhaustible pharmacological application and occurrence in many clinically important natural products and synthetic molecules. The desirable pharmacokinetic profile and ease of accessibility with wide substrate tolerance for different synthetic protocols, also encourages its utility in countless synthetic explorations. Research endeavours aimed at quinoline-based βGLU inhibitors are therefore presented in this section.
Quinoline-3-carbohydrazides [239], prepared analogously as hydrazone tethers above also holds promising inhibitory activities. Similarly, OH unit at ortho-position yielded stronger inhibitors than F > Cl > NO2 > Pyridyl > thiophenyl > furanyl ≈ Me > MeO, in that order. Moreover, ortho-substitution consistently gave superior inhibitors compared to para-position, while meta-substitution conferred the weakest potency. 2 to 3-fold reduction in potency persisted when OH unit is substituted for MeO unit (Fig. 50 ). Hence, compound 162 (IC50 = 2.11 μM) and 163 (IC50 = 2.60 μM) emerged as the most potent inhibitors overall. Docking studies revealed that OH unit at position-4 of quinoline ring fostered important H-b interaction with catalytic acid Glu451, while carbonyl unit of hydrazone moiety formed H-b interaction with NH of Tyr504.
Fig. 50.

Quinoline-hydrazone tethers and most active compounds 162 and 163.
In another quest for new anti-inflammatory agents, quinoline-furan hybrids (Fig. 51 ) were designed as inhibitors of βGLU release from activated neutrophils [240]. SAR analysis, established that 2-(furan-2-yl)quinolines (Type A) were stronger inhibitors of inflammatory mediators than tricyclic furo[2,3-b]quinolines (Type B). Whereas, formyl unit favoured potency compared to acetyl, oxime, methyl oxime as well as both α,β-unsaturated butan-2-one and its saturated variant. Therefore, non-cytotoxic compound 164 (IC50 = 5.0 μM) was the strongest inhibitor of βGLU release with 3-fold superiority to reference trifluoperazine. Acetyl isomer compound 165, also had similar non-cytotoxic and strong inhibitory potency (IC50 = 7.5 μM).
Fig. 51.

SAR and most active fused quinoline-furan hybrids 164 and 165.
Akin to azacoumarins (1H-quinolin-2-ones) 151 and 152, new quinoline-pyrazole conjugates were prepared via hit development protocol of compound 166 (Fig. 52 ) identified from high-throughput screening, for the inhibition of intestinal bacterial βGLU’s hydrolytic activity on SN-38G fo alleviate CPT-11 drug-induced toxicity [241,242]. Interestingly, all the resulting derivatives showed good selectivity for E. coli βGLU except 2-Cl, 3-OH, 3-Me and 4-NH2 substituted derivatives. The trend in inhibitory potency, based on substituent’s position varied in the order; meta < ortho < para, while the presence of strongly hydrophilic OH, NO2 and NH2 units at para-position was detrimental to potency. Consequently, 4-CF3, 4-Cl, 4-F, 4-COMe and 4-Me substituted derivatives with IC50 = 98, 35, 140, 130 and 37 nM respectively, exhibited the highest selectivity and % inhibition of E. coli βGLU. Although installing F, Cl, Br or Me substituents on quinoline pharmacophore increased potency for 4-Cl and 4-Me compounds, cytotoxicity at 100 μM to E. coli cells thwarted their therapeutic utility as well as other potent inhibitors (4-CF3, 4-Cl and 4-COMe) in the study. Only 4-F and 4-Me substituted compounds 167 and 168 respectively, remained non-lethal to E. coli cells. In addition, compound 167 showed greater inhibition of intestinal βGLU (75%) following oral administration to BALB/cJ mice for 5 consecutive days compared to compound 168 (40%). Whereas, pharmacokinetic profiling of compound 168 revealed its lower plasma concentration and increased GI residence for improved intestinal βGLU inhibition compared to azacoumarin compound 151. Most importantly, oral co-administration of compounds 167 or 168 at 3 mg kg−1 day−1 for 10 consecutive days with 50 mg kg−1 day−1 intraperitoneal injection of CPT-11, protected BALB/cJ mice models against CPT-11 drug-induced intestinal mucosal injury, without altering drug’s therapeutic efficacy. Furthermore, mechanistic studies established that the presence of electronegative and electroneutral surface charges on E. coli active site pocket and the protonation of N-1 and N-5 atoms avails a pH-dependent and selective inhibition for the compounds (Fig. 52). Consequently, compounds 167 and 168 are clinically viable agents for protecting against GI E. coli βGLU-mediated mucosal damage and preventing the release of chemical carcinogens crucial to the development of precancerous lesions for colon carcinogenesis.
Fig. 52.

Hit development, SAR and clinically useful quinoline-pyrazole conjugates 167 and 168.
3.2.7. Other synthetic inhibitors
The 200-fold increased inhibitory potency of benzohydrazide–N-cyanoethyl tethers [243], compared to parent compounds articulates the significance of a balanced molecular lipophilicity to βGLU inhibition. O-alkylation with benzyl, benzoyl or tosyl groups slightly influenced potency for OH-substituted units; benzylation produced the best result (Fig. 53 ). Hence, 169 (IC50 = 1.60 μM) and 170 (IC50 = 2.20 μM) were the most active conjugates in the series. Similarly, phenoxyacetohydrazones [244] have displayed 2 to 5-fold improved potency compared to d-SAL (IC50 = 48.40 μM). Compounds 171 and 172 (Fig. 54 ) emerged as the strongest inhibitors with IC50 values of 9.20 and 9.47 μM respectively. However, parallel comparison with other hydrazone tethers revealed that the phenoxyacetohydrazones are generally weaker βGLU inhibitors.
Fig. 53.

Most potent benzohydrazide – N-cyanoethyl tethers against βGLU.
Fig. 54.

Strongest phenoxyacetohydrazone-based βGLU inhibitors.
The desirable therapeutic significance of fused thienothiophenes has stimulated the synthetic exploration of a thienothiophene skeleton 173 (Fig. 55 ) for bacterial βGLU inhibition [245]. From the resulting thienothiophene library with interesting structural diversity, only compound 174 showed inhibitory activity against bacterial βGLU with IC50 value of 0.9 μM; 51-fold superior potency to d-SAL. Notably, the predicted pharmacokinetic properties using Molinspiration and Osiris calculations suggested that the inhibitor possesses good bioavailability and no toxicity risk as bacterial βGLU inhibitor.
Fig. 55.

Thienothiophene-based bacterial βGLU inhibitor.
In vitro study of phenyl disulphide–benzenesulfonamide tethers [246], disclosed compounds 175 and 176 (Fig. 56 ) as potent inhibitors of βGLU (IC50 = 2.20 and 3.34 μM respectively), analogous to similarly substituted oxadiazole-benzenesulfonamide compounds 116 and 117 (Fig. 28). Interestingly, bulkier phenyl disulfides 175 and 176 shared similar potency with lower molecular mass thiosulfinate 52, Fig. 9 (IC50 = 3.60 μM). However, substitution at ortho-position gave stronger inhibitors than para- and meta-positions, in contrast to compounds 116 and 117 series. Docking studies further revealed that the strong inhibitory potency of compound 175 is aided by the H-b interactions of sulfonamide NH and oxygen with Tyr205 and Asp207 respectively.
Fig. 56.

Most active disulfide-benzenesulfonamide based βGLU inhibitor.
The efficacy of urea unit for βGLU inhibition was also examined using a library of NO2 substituted N-phenyl ureas [247]. Therefrom, appropriate positioning of polar NO2 unit was important to inhibitory potency of examined molecules. Compound 177 (Fig. 57 ), emerged as the only inhibitor with promising activity with IC50 value of 3.38 μM. However, thiourea analogues bearing meta-Cl substituent on N-phenyl ring [248], were stronger inhibitors compared to those with unsubstituted N-phenyl ring or NO2 substituted ureas (Fig. 58 ). Akin to compound 141, thioureas containing piperazine unit also exhibited improved potency compared to their morpholine and piperidine analogues, while N-alkylation of piperazine with N-(o or p-methoxyphenyl) or N-(2-pyridyl), abolished potency. Compounds 178 (IC50 = 0.86 μM) and 179 (IC50 = 1.24 μM) were the most potent inhibitors overall. In addition, 8-aminoquinoline derivatives 180 and 181 are also promising inhibitors with IC50 = 1.64 and 2.12 μM respectively.
Fig. 57.
Most potent urea based βGLU inhibitor.
Fig. 58.

SAR analysis of thioureas and most active βGLU inhibitors.
3.2.8. Metal complexes and glycopolymers
Extant empirical data identified bacterial βGLU as a chelating agent, since enzyme activity was totally lost in the presence of Cu2+, Hg2+ and Ag+ metal ions and restored in the presence of other chelating agents – Versene and cysteine [249]. Therefore, metal complexes with coordinatable metal centres exhibit significant bacterial βGLU inhibitory activities. To this end, Pd(II) complexes containing aniline and triphenylphosphine ligands were synthesized [250]. Para-chloroaniline derived metal complex 182 (Fig. 59 ), displayed the strongest activity against bacterial βGLU with 98.5% inhibition and IC50 = 15.40 μM. It was also 2- and 5-fold superior to meta-chloroaniline derived and N-methyl substituted metal complexes respectively. Similarly, N-heterocyclic carbene (NHC) complexes of Au(I) [251], as well as bis (NHC) complexes of Au(I) and Au(III) [252], containing hydroxy, chloride, acetoxy and acetonyl ligands were equally potent bacterial βGLU inhibitors with IC50 ranging from 0.14 to 2.60 μM. The strongest Au(I) NHC complex 183 (IC50 = 0.14 μM) was 327-fold more potent than reference d-SAL and 7-fold superior to strongest bis (NHC) complexes 184. Nonetheless, undesirable cytotoxicity against 3T3 cells discomfits the therapeutic significance of these metal complexes.
Fig. 59.

Metal complexes for bacterial βGLU inhibition.
Parallel to iminosugars 60–74, the inhibitory potencies of structurally similar glycopolymers (Fig. 60 ) was dependent on the presence of carboxyl unit, type and configuration of sugar unit. Hence, glycopolymers with d-glucaric pendants linked to the polymer frame at C-1 (185), were stronger inhibitors compared to glycopolymers linked at C-5 (186) or those bearing d-gluconic (187) [253], d-mannaric (188) [254] and shorter chain d,l-xylaric (189) and l-tartaric (190) [255] pendants. However, these glycopolymers only show >60% inhibition at millimolar concentrations.
Fig. 60.

Glycopolymer-based βGLU inhibitors and most active analogue (185) bearing pendant d-glucaric acid units.
4. Patents describing the therapeutic significance and inhibition of βGLU activity
Patents and patent applications disclosing potent inhibition of βGLU particularly bacterial βGLU and the therapeutic significance thereof, have been documented. We therefore review in this section, patents applications filed in the present millennium relevant to βGLU inhibition. We have also included the only biomarker application of the enzyme filed within the period [256]. However, some of the patents and applications [[257], [258], [259], [260], [261], [262], [263]] emanated from published papers [19,210,228,229,241,262], reviewed in previous sections; therefore, only their summary is presented in Table 3 .
Table 3.
Filed patents (2000–2019) describing the therapeutic significance of βGLU activity and its inhibition.
| Patent Number | Invention | Most active βGLU inhibitor | IC50 (nM) | Therapeutic significance |
|---|---|---|---|---|
| US 6,277,587 B1 | Simple method for quantifying the elevated levels of βGLU in saliva sample of periodontally diseased patients by adding glucuronide substrates or labelled βGLU-selective antibody | – | – | Biomarker of periodontal disease risk and status |
| US 7,294,330 B2 | Deodorant and antiperspirant formulations containing compounds which inhibits odour-producing skin bacterial βGLU-mediated hydrolysis of steroid glucuronides, without affecting skin microbiota composition |
|
99%a 99%a 97%a |
Non-therapeutic cosmetic formulations |
| US 9.200,269 B2 | Incorporating potent inhibitors into hygiene and sanitary products to suppress the generation of urine or other excreta odour due to bacterial βGLU activity on the body wastes |  |
99%a | Non-therapeutic products to suppress urine odour |
| US 9617.239 B2 | Phenoxy thiophene sulfonamides as CPT-11 co-drugs for potent and selective inhibition of intestinal bacterial βGLU activity |
|
20 | Alleviation of drug induced dose-limiting toxicities to improve therapeutic efficacy |
| US 2015/0011542 A1 | Safe and effective methods for alleviating NSAID-induced GI tract injuries via therapeutic doses of potent and selective bacterial βGLU inhibitors | 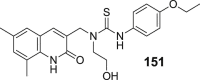 |
140 | |
| WO 2019/051185 Al | Compounds, compositions and methods for selective inhibition of intestinal bacterial βGLU to reduce the side effects (diarrhoea) of camptothecin-derived antineoplastic agents or other drugs or metabolites or compounds that are βGLU substrates, used in treating neoplasms or other conditions. | 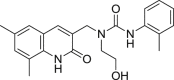 |
118 | |
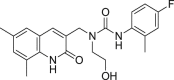 |
142 | |||
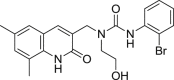 |
347 | |||
 |
468 | |||
 |
759 | |||
| US 9334.288 B2 | 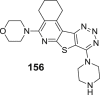 |
1.17 | ||
| WO 2018/017874 Al |  |
Ec = 70 Sa = 39 Cp = 180 |
||
 |
Ec = 130 Sa = 690 Cp = 53 |
|||
 |
Ec = 30 Sa = NA Cp = NA |
|
||
| US 2018/0214426 A1 | Methods for preparing pyrazolo[4,3-c]quinoline derivatives for selective inhibition of microbiota βGLU and their therapeutic applications as; (1) chemotherapy adjuvants to reduce chemotherapy-induced diarrhoea and enhance chemotherapy efficiency (2) health-food supplements to prevent colon carcinoma | 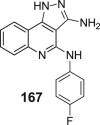 |
120 | |
| US 2014/0256754 Al | Preparation of quinazolin-4(3H)-ones-based βGLU inhibitors | 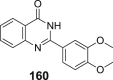 |
600 | Therapeutic intervention for health disorders stimulated by high βGLU activity |
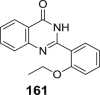 |
700 | |||
| US 2017/0009272 A1 | Virtual screening methods for identifying potent inhibitors of βGLU |  |
450 |
% inhibition at test concentration; Ec = E. coli βGLU; Sa = S. agalactiae βGLU; Cp = C. perfringens βGLU; NA - not active.
Considering the limitations associated with traditional clinical assessments for periodontal diseases diagnosis and the improved reliability of βGLU as a biomarker, a simple method requiring less expertise has been claimed to quantify elevated levels of the enzyme in the saliva of diseased patients relative to healthy ones, as a biomarker of disease risk and status [256]. The invention involves adding known β-d-glucuronide substrates (4-methylumbelliferone or phenolphthalein β-d-glucuronides) to a sample of patient’s saliva in βGLU activity assay and thereafter quantifying the amount of aglycone produced via fluorometry, colorimetry or spectrophotometry. The addition of a labelled polyclonal or monoclonal antibody specific for βGLU to saliva sample and subsequent estimation of the amount of labelled antibody forming βGLU-antibody complex was also claimed, as well as a test-kit for claimed methods.
Bacterial βGLU-mediated hydrolysis of steroid glucuronides on the skin leads to body odours hence inhibitors of the process were developed and applied in deodorant and antiperspirant formulations [264]. The invention claimed that deodorant formulations with described compounds only inhibits βGLU activity without altering the natural composition of skin microbiota; in contrast to bacteriostatic or bactericidal mode of action of prior arts. Amongst the claimed inhibitors of different class, the strongest inhibition of E. coli βGLU was found with galactaric acid (99% inhibition at 0.1% test concentration in water), as well as zinc glycinate and zinc gluconate; 99% and 97% inhibition respectively at 0.25% test concentrations.
In the same vein, potent inhibitors of bacterial βGLU activity on urine have found non-therapeutic application in hygiene and sanitary products for suppressing the generation of urine odour [265]. At 0.1 wt% in dipropylene glycol, all the claimed inhibitors viz. macrocyclic ketones, ketones, macrocyclic lactones, macrocyclic oxalactones, esters, aldehydes, alcohols, ethers and terpenes, showed over 60% inhibition of E. coli βGLU. Macrocyclic ketones were stronger inhibitors than others. Consequently, the invention’s most active inhibitor was compound 191 with 100%, 99.9% and 95.8% inhibition at 0.1, 0.01 and 0.001 wt% respectively. It was also effective after 48 h in suppressing the increase in urine odour when incorporated into different hygiene and sanitary products.
Phenoxy thiophene sulfonamides [266] were designed as adjuvants to eliminate the dose-limiting GI toxicity (diarrhoea) of CPT-11, through potent inhibition of bacterial βGLU-mediated deconjugation of active metabolite’s glucuronide (SN-38G) in the intestine; thus, improving the drug’s therapeutic efficacy. The synthesis of 76 βGLU inhibitors and 18 analogues of BRITE-355252 (compound 192), the most active compound (IC50 = 20 nM), together with their therapeutic application was claimed. SAR study of compound 192 analogues revealed that inhibitory potency was markedly dependent on N-piperazinyl pendant at meta-position of phenoxy ring. Over 500-fold reduced inhibitory potency occurred with N-methylation (akin to compound 156), 15-fold reduction when appended at para-position and total loss of activity when removed. Uninstalling the chloro unit on thiophene ring also slightly diminished potency by 5-fold. Whereas, replacing naphthyl with substituted phenyl units afforded inhibitors with similar potencies i.e. IC50 < 50 nM (Table SI); as a result, 193 was equipotent with 192.
5. Concluding remarks
βGLU is a physiologically important lysosomal glycosyl hydrolase with appreciable therapeutic potentials such as biomarker for disease diagnosis, endogenous bioactivator in prodrug monotherapy and enzyme replacement therapy. The enzyme’s role in cell proliferation and inflammation also renders it a potential target for anticancer and anti-inflammatory drugs development respectively. Moreover, since a significant number of commercially available drugs are metabolised by glucuronidation, hydrolytic activity (i.e. deglucuronidation) by βGLU in human intestinal microbiota has been linked to colon carcinogenesis and genotoxicity as well as drug-induced dose-limiting toxicities of anticancer agents and NSAIDs, which thwarts their therapeutic potential and clinical utility. Therefore, potent inhibition of βGLU holds enormous clinical importance.
Generally, our literature survey found that a significant number of natural products-derived inhibitors display moderate inhibition of βGLU, and increased potency is found with inhibitors containing a flavonoid skeleton. The physiological tolerance, acceptable toxicity and favourable pharmacodynamic profiles of these natural inhibitors, posits them as worthy scaffolds warranting witty molecular developments. In addition, iminosugars distinguish themselves as a unique class of natural product-inspired molecules with promising potentials regardless of their usually multiple synthetic steps. Notable amongst these are the nojirimycin analogues and iminosugar C-glycosides, both conferred with highly selective inhibition of bacterial βGLU. However, the inhibitory potency of these carbohydrate mimics relies on several factors such as sugar configuration, mimicry of glucuronic acid substrate and correct lipophilic balance. Hence, to harness their remarkable potential, ingenious manipulations of the synthetic medicinal chemist interested in exploring their chemical space is expressly required.
On the other hand, purely synthetic inhibitors show fascinating diversity in their inhibitory potencies and pattern, due to the plethora of pharmacophoric enrichments possible for a single molecular design. Based on the foregoing, a comprehensive list of potent synthetic inhibitors (IC50 ≤ 5 μM) in this review is provided in Table SI of supporting information. Evinced by their nanomolar IC50 values, quinoline-pyrazoles (167 and 168), piperazine-containing compounds (156 and 157; 192 and analogues) as well as the indole nucleus, are conferred with the strongest inhibition of βGLU activity overall. The order of potency for hydrazone tethers was found as; bisindole ˃ indole > quinoline > thiazole ˃ benzothiazole ≈ oxadiazole. Whereas, NO2, Cl, particularly F and OH substituents at ortho- or para-positions usually produce stronger βGLU inhibitors compared to their meta-substitutions.
Taken together, these suggests that the presence of polar groups with strong hydrogen bonding potential is crucial to inhibitory potency, while an indiscriminate increase in molecular lipophilicity is detrimental to strong βGLU inhibition. Consequently, we envisage that the model βGLU inhibitor will be that which is appositely tuned in its lipophilicity and polarity for easy cell penetration, favourable fit into the binding/catalytic pocket of βGLU for energetically stable binding and strong enzyme inhibition. We also perceive that rationale molecular hybridization of those active class of natural products and synthetic molecules will furnish stronger inhibitors of βGLU with improved selectivity and acceptable toxicity profile.
Furthermore, we found that due to the mixture of electroneutral and electronegative spots on the surface of bacterial βGLU binding pocket at physiological pH, in contrast to the electropositive spots for human βGLU, the presence of a protonatable N-atom confers improved selectivity for bacterial βGLU inhibition. This was particularly true for piperazine based inhibitors. Therefore, we believe further probing of this phenomenon will allow structure-activity guided molecular tuning to afford stronger βGLU inhibitors.
To the best of our knowledge, mechanistic studies on the behaviour of these inhibitors is scanty in literature and only found in two articles describing gluco-imidazole (60) and piperazine (156) based inhibitors. This investigative approach provides insight to the behaviour of these inhibitors in the catalytic cycle of βGLU thereby allowing deeper understanding of important structural features crucial to inhibitory activity beyond SAR and molecular docking studies. Finally, we hope this review will inspire medicinal chemists to direct their search light towards this molecular target, for the development of new therapeutic agents with improved efficacy and betterment of human health in consequential.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgement
The National Research Foundation (NRF) of South Africa is appreciated for KIC grant and other financial support.
List of abbreviations
- AChE
Acetylcholinesterase
- AcP
Acid phosphatase
- ADP
Adenosine diphosphate
- ALT
Alanine aminotransferase
- ABL
Alveolar bone loss
- AST
Aspartate aminotransferase
- BOP
Bleeding on probing
- βGLU
beta-glucuronidase
- BALF
Bronchoalveolar lavage fluid
- BuChE
Butyrylcholinesterase
- CPT-11
Irinotecan – an analogue of Camptothecin
- CSF
Cerebrospinal fluid
- CAL
Clinical attachment level
- DMBA
7,12-dimethylbenz[α]anthracene
- DMH
1,2-dimethylhydrazine
- d-SAL
d-saccharic acid-1,4-lactone
- d-SDL
2,5-di-O-acetyl-d-glucaro-1,4:6,3-dilactone
- d-GL
d-glucurono-γ-lactone
- GCF
Gingival crevicular fluid
- GH
Glycosyl hydrolase
- GI
Gastrointestinal
- Gg-I
Gingival index
- GPCR
G protein-coupled receptors
- H-b
Hydrogen bonding
- IC50
Half maximal inhibitory concentration
- IL-1β
Interleukin-1 beta
- IL-8
Interleukin-8
- kcat
Rate constant of catalysed reaction
- kDa
kilodalton
- Ki
Inhibition constant
- kuncat
Rate constant of uncatalyzed reaction
- LAB
Lactic acid bacteria
- LDH
Lactic acid dehydrogenase
- MERS-CoV
Middle East respiratory syndrome coronavirus
- MMPs
Matrix metalloproteinases
- NSAID
Nonsteroidal anti-inflammatory drug
- OP
Organophosphorus compounds
- PARP
Poly (ADP-ribose) polymerase
- PD
Probing depth
- SAR
Structure-activity relationship
- SN2
Nucleophilic substitution 2
- SN-38
Active metabolite of CPT-11
- SN-38G
Glucuronide conjugate of SN-38
- TNF-α
Tumour necrosis factor-α
- UDP
Uridine-5′-diphosphate
- UGT
UDP-glucuronosyltransferase
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2019.111921.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Kiser A.K., Pronovost P.J. Management of diseases without current treatment options: something can be done. J. Am. Med. Assoc. 2009;301:1708–1709. doi: 10.1001/jama.2009.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morens D.M., Fauci A.S. Emerging infectious diseases: threats to human health and global stability. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asano N. Glycosidase inhibitors: update and perspectives on practical use. Glycobiology. 2003;13:93R–104R. doi: 10.1093/glycob/cwg090. [DOI] [PubMed] [Google Scholar]
- 4.Gruner S.A., Locardi E., Lohof E., Kessler H. Carbohydrate-based mimetics in drug design: sugar amino acids and carbohydrate scaffolds. Chem. Rev. 2002;102:491–514. doi: 10.1021/cr0004409. [DOI] [PubMed] [Google Scholar]
- 5.Leiria Campo V., Aragão-Leoneti V., Carvalho I. In: Carbohydrate Chemistry. Rauter A.P., Lindhorst T.K., editors. The Royal Society of Chemistry; Cambridge, UK: 2013. Glycosidases and diabetes: metabolic changes, mode of action and therapeutic perspectives; pp. 181–203. [DOI] [Google Scholar]
- 6.AFMB . 2019. Glycoside Hydrolase Family Classification.http://www.cazy.org/Glycoside-Hydrolases.html [Accessed 30th January 2019]; Available on: [Google Scholar]
- 7.Lombard V., Golaconda Ramulu H., Drula E., Coutinho P.M., Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henrissat B., Callebaut I., Fabrega S., Lehn P., Mornon J.P., Davies G. Conserved catalytic machinery and the prediction of a common fold for several families of glycosyl hydrolases. Proc. Natl. Acad. Sci. U. S. A. 1995;92:7090–7094. doi: 10.1073/pnas.92.15.7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fishman W.H. In: Adv. Enzymol. Relat. Areas Mol. Biol. Nord F.F., editor. Interscience Publishers, Inc.; New York, London: 1955. β-Glucuronidase; pp. 361–409. [Google Scholar]
- 10.Brot F.E., Bell C.E., Jr., Sly W.S. Purification and properties of β-glucuronidase from human placenta. Biochemistry. 1978;17:385–391. doi: 10.1021/bi00596a001. [DOI] [PubMed] [Google Scholar]
- 11.Oshima A., Kyle J.W., Miller R.D., Hoffmann J.W., Powell P.P., Grubb J.H., Sly W.S., Tropak M., Guise K.S., Gravel R.A. Cloning, sequencing, and expression of cDNA for human β-glucuronidase. Proc. Natl. Acad. Sci. U. S. A. 1987;84:685–689. doi: 10.1073/pnas.84.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Islam M.R., Grubb J., Sly W. C-terminal processing of human β-glucuronidase. The propeptide is required for full expression of catalytic activity, intracellular retention, and proper phosphorylation. J. Biol. Chem. 1993;268:22627–22633. [PubMed] [Google Scholar]
- 13.Shipley J.M., Grubb J.H., Sly W.S. The role of glycosylation and phosphorylation in the expression of active human β-glucuronidase. J. Biol. Chem. 1993;268:12193–12198. [PubMed] [Google Scholar]
- 14.Jain S., Drendel W.B., Chen Z.W., Mathews F.S., Sly W.S., Grubb J.H. Structure of human β-glucuronidase reveals candidate lysosomal targeting and active-site motifs. Nat. Struct. Biol. 1996;3:375–381. doi: 10.1038/nsb0496-375. [DOI] [PubMed] [Google Scholar]
- 15.Islam M.R., Tomatsu S., Shah G.N., Grubb J.H., Jain S., Sly W.S. Active site residues of human β-glucuronidase. Evidence for Glu(540) as the nucleophile and Glu(451) as the acid-base residue. J. Biol. Chem. 1999;274:23451–23455. doi: 10.1074/jbc.274.33.23451. [DOI] [PubMed] [Google Scholar]
- 16.Haq I.U., Akram F. Enhanced production, overexpression and characterization of a hyperthermophilic multimodular GH family 2 β-glucuronidase (TpGUS) cloned from Thermotoga petrophila RKU-1(T) in a mesophilic host. Int. J. Biol. Macromol. 2019;123:1132–1142. doi: 10.1016/j.ijbiomac.2018.11.189. [DOI] [PubMed] [Google Scholar]
- 17.Kayahan S. Atherosclerosis and β-glucuronidase. Lancet. 1960;2:667–669. doi: 10.1016/s0140-6736(60)91744-x. [DOI] [PubMed] [Google Scholar]
- 18.Khan F.I., Shahbaaz M., Bisetty K., Waheed A., Sly W.S., Ahmad F., Hassan M.I. Large scale analysis of the mutational landscape in β-glucuronidase: a major player of mucopolysaccharidosis type VII. Gene. 2016;576:36–44. doi: 10.1016/j.gene.2015.09.062. [DOI] [PubMed] [Google Scholar]
- 19.Wallace B.D., Wang H., Lane K.T., Scott J.E., Orans J., Koo J.S., Venkatesh M., Jobin C., Yeh L.A., Mani S., Redinbo M.R. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330:831–835. doi: 10.1126/science.1191175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies G.J., Williams S.J. Carbohydrate-active enzymes: sequences, shapes, contortions and cells. Biochem. Soc. Trans. 2016;44:79–87. doi: 10.1042/BST20150186. [DOI] [PubMed] [Google Scholar]
- 21.Lillelund V.H., Jensen H.H., Liang X., Bols M. Recent developments of transition-state analogue glycosidase inhibitors of non-natural product origin. Chem. Rev. 2002;102:515–553. doi: 10.1021/cr000433k. [DOI] [PubMed] [Google Scholar]
- 22.Whitaker B.L. Plasma β-glucuronidase levels in breast cancer. Br. J. Canc. 1960;14:471–477. doi: 10.1038/bjc.1960.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Odell L., Burt J., Bethea R. β-Glucuronidase activity. Sci. Total Environ. 1949;109:564–565. doi: 10.1126/science.109.2840.564. [DOI] [PubMed] [Google Scholar]
- 24.Kim D.H., Jin Y.H. Intestinal bacterial β-glucuronidase activity of patients with colon cancer. Arch Pharm. Res. (Seoul) 2001;24:564–567. doi: 10.1007/BF02975166. [DOI] [PubMed] [Google Scholar]
- 25.Schumacher U., Adam E., Zangemeister-Wittkel U., Gossrau R. Histochemistry of therapeutically relevant enzymes in human tumours transplanted into severe combined immunodeficient (sCID) mice: nitric oxide synthase—associated diaphorase, β-D-glucuronidase and nonspecific alkaline phosphatase. Acta Histochem. 1996;98:381–387. doi: 10.1016/s0065-1281(96)80004-3. [DOI] [PubMed] [Google Scholar]
- 26.Vlacha V., Eliopoulou M., Haidas S., Beratis N.G. Correlation of cerebrospinal fluid β-glucuronidase activity with plasma methotrexate concentrations in leukemic children receiving high-dose methotrexate. Pediatr. Blood Cancer. 2004;42:350–356. doi: 10.1002/pbc.20002. [DOI] [PubMed] [Google Scholar]
- 27.Lu S., Li G., Lv Z., Qiu N., Kong W., Gong P., Chen G., Xia L., Guo X., You J., Wu Y. Facile and ultrasensitive fluorescence sensor platform for tumor invasive biomaker β-glucuronidase detection and inhibitor evaluation with carbon quantum dots based on inner-filter effect. Biosens. Bioelectron. 2016;85:358–362. doi: 10.1016/j.bios.2016.05.021. [DOI] [PubMed] [Google Scholar]
- 28.Gonick H.C., Kramer H.J., Schapiro A.E. Urinary β-glucuronidase activity in renal disease. Arch. Intern. Med. 1973;132:63–69. [PubMed] [Google Scholar]
- 29.Saha A.K., Glew R.H., Kotler D.P., Omene J.A. Elevated serum β-glucuronidase activity in acquired immunodeficiency syndrome. Clin. Chim. Acta. 1991;199:311–316. doi: 10.1016/0009-8981(91)90125-v. [DOI] [PubMed] [Google Scholar]
- 30.Miller B.F., Keyes F.P., Curreri P.W. Increase of serum β-glucuronidase activity in human diabetes mellitus. J. Am. Med. Assoc. 1966;195:189–192. [PubMed] [Google Scholar]
- 31.Rose G.P., Dewar A.J., Stratford I.J. A biochemical method for assessing the neurotoxic effects of misonidazole in the rat. Br. J. Canc. 1980;42:890–899. doi: 10.1038/bjc.1980.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ganguly N.K., Kingham J.G., Lloyd B., Lloyd R.S., Price C.P., Triger D.R., Wright R. Acid hydrolases in monocytes from patients with inflammatory bowel disease, chronic liver disease, and rheumatoid arthritis. Lancet. 1978;1:1073–1075. doi: 10.1016/s0140-6736(78)90917-0. [DOI] [PubMed] [Google Scholar]
- 33.BRENDA . 2019. Disease On EC 3.2.1.31 - β-glucuronidase.https://www.brenda-enzymes.org/enzyme.php?ecno=3.2.1.31&showtm=0&onlyTable=Disease [Accessed 28 May 2019]; Available on: [Google Scholar]
- 34.Caira M.R., Ionescu C. vol. 7. Springer Science & Business Media; 2006. (Drug Metabolism: Current Concepts). [Google Scholar]
- 35.Compain G., Oumata N., Clarhaut J., Peraudeau E., Renoux B., Galons H., Papot S. A β-glucuronidase-responsive albumin-binding prodrug for potential selective kinase inhibitor-based cancer chemotherapy. Eur. J. Med. Chem. 2018;158:1–6. doi: 10.1016/j.ejmech.2018.08.100. [DOI] [PubMed] [Google Scholar]
- 36.Di Stanislao C., Angelini F., Gagliardi M.C., Di Bernardino L., Fundaro C., Galli E., Rossi P. Beta glucuronidase short-term immunotherapy. Allergy. 2003;58:459. doi: 10.1034/j.1398-9995.2003.00133.x. [DOI] [PubMed] [Google Scholar]
- 37.Galli E., Bassi M., Mora E., Martelli M., Gianni S., Auricchio G., Arabito E., Rossi P. A double-blind randomized placebo-controlled trial with short-term β-glucuronidase therapy in children with chronic rhinoconjunctivitis and/or asthma due to dust mite allergy. J Investig. Allergol. Clin. Immunol. 2006;16:345–350. [PubMed] [Google Scholar]
- 38.Bramwell K.K., Ma Y., Weis J.H., Chen X., Zachary J.F., Teuscher C., Weis J.J. Lysosomal β-glucuronidase regulates Lyme and rheumatoid arthritis severity. J. Clin. Investig. 2014;124:311–320. doi: 10.1172/JCI72339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drummer O.H., Di Rago M., Gerostamoulos D. LC-MS in Drug Analysis. Springer; 2019. Analysis of benzodiazepines for drug-facilitated assaults and abuse settings (urine) pp. 23–39. [DOI] [PubMed] [Google Scholar]
- 40.Stadler P., Loken L.C., Crawford J.T., Schramm P.J., Sorsa K., Kuhn C., Savio D., Striegl R.G., Butman D., Stanley E.H. Spatial patterns of enzymatic activity in large water bodies: ship-borne measurements of β-D-glucuronidase activity as a rapid indicator of microbial water quality. Sci. Total Environ. 2019;651:1742–1752. doi: 10.1016/j.scitotenv.2018.10.084. [DOI] [PubMed] [Google Scholar]
- 41.Humblot C., Murkovic M., Rigottier-Gois L., Bensaada M., Bouclet A., Andrieux C., Anba J., Rabot S. β-glucuronidase in human intestinal microbiota is necessary for the colonic genotoxicity of the food-borne carcinogen 2-amino-3-methylimidazo [4, 5-f] quinoline in rats. Carcinogenesis. 2007;28:2419–2425. doi: 10.1093/carcin/bgm170. [DOI] [PubMed] [Google Scholar]
- 42.Takasuna K., Kasai Y., Kitano Y., Mori K., Kobayashi R., Hagiwara T., Kakihata K., Hirohashi M., Nomura M., Nagai E., et al. Protective effects of kampo medicines and baicalin against intestinal toxicity of a new anticancer camptothecin derivative, irinotecan hydrochloride (CPT-11), in rats. Jpn. J. Cancer Res. 1995;86:978–984. doi: 10.1111/j.1349-7006.1995.tb03010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhong Z.Y., Sun B.B., Shu N., Xie Q.S., Tang X.G., Ling Z.L., Wang F., Zhao K.J., Xu P., Zhang M., Li Y., Chen Y., Liu L., Xia L.Z., Liu X.D. Ciprofloxacin blocked enterohepatic circulation of diclofenac and alleviated NSAID-induced enteropathy in rats partly by inhibiting intestinal β-glucuronidase activity. Acta Pharmacol. Sin. 2016;37:1002–1012. doi: 10.1038/aps.2016.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boyland E., Wallace D.M., Williams Enzyme activity in relation to cancer: inhibition of urinary β-glucuronidase of patients with cancer of the bladder by oral administration of 1:4-saccharolactone and related compounds. Br. J. Canc. 1957;11:578–589. doi: 10.1038/bjc.1957.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sloane B.F., Dunn J.R., Honn K.V. Lysosomal cathepsin B: correlation with metastatic potential. Science. 1981;212:1151–1153. doi: 10.1126/science.7233209. [DOI] [PubMed] [Google Scholar]
- 46.Poland R.L., Odell G.B. Physiologic jaundice: the enterohepatic circulation of bilirubin. N. Engl. J. Med. 1971;284:1–6. doi: 10.1056/NEJM197101072840101. [DOI] [PubMed] [Google Scholar]
- 47.Gourley G.R. Breast-feeding, neonatal jaundice and kernicterus. Semin. Neonatol. 2002:135–141. doi: 10.1053/siny.2002.0101. [DOI] [PubMed] [Google Scholar]
- 48.Southerland J.H., Taylor G.W., Offenbacher S. Diabetes and periodontal infection: making the connection. Clin. Diabetes. 2005;23:171–178. doi: 10.2337/diaclin.23.4.171. [DOI] [Google Scholar]
- 49.Llambes F., Arias-Herrera S., Caffesse R. Relationship between diabetes and periodontal infection. World J. Diabetes. 2015;6:927–935. doi: 10.4239/wjd.v6.i7.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henson P.M. Pathologic mechanisms in neutrophil-mediated injury. Am. J. Pathol. 1972;68:593–612. [PMC free article] [PubMed] [Google Scholar]
- 51.Hsieh H.K., Lee T.H., Wang J.P., Wang J.J., Lin C.N. Synthesis and anti-inflammatory effect of chalcones and related compounds. Pharm. Res. 1998;15:39–46. doi: 10.1023/a:1011940401754. [DOI] [PubMed] [Google Scholar]
- 52.Vasella A., Davies G.J., Bohm M. Glycosidase mechanisms. Curr. Opin. Chem. Biol. 2002;6:619–629. doi: 10.1016/s1367-5931(02)00380-0. [DOI] [PubMed] [Google Scholar]
- 53.Stütz A.E., Wrodnigg T.M. In: Carbohydrate Chemistry. Rauter A.P., Lindhorst T.K., editors. The Royal Society of Chemistry; Cambridge, UK: 2013. Positive attitude, shape, flexibility, added-value accessories or “just being different”: how to attract a glycosidase; pp. 120–149. [DOI] [Google Scholar]
- 54.Singh P., Ngcoya N., Kumar V. A review of the recent developments in synthetic anti-breast cancer agents. Anti Cancer Agents Med. Chem. 2016;16:668–685. doi: 10.2174/1871520616666151120122120. [DOI] [PubMed] [Google Scholar]
- 55.Kerru N., Singh P., Koorbanally N., Raj R., Kumar V. Recent advances (2015-2016) in anticancer hybrids. Eur. J. Med. Chem. 2017;142:179–212. doi: 10.1016/j.ejmech.2017.07.033. [DOI] [PubMed] [Google Scholar]
- 56.Singh P., Jaiyeola B., Kerru N., Ebenezer O., Bissessur A. A review of recent advancements in anti-tubercular molecular hybrids. Curr. Med. Chem. 2017;24:4180–4212. doi: 10.2174/0929867324666170712164400. [DOI] [PubMed] [Google Scholar]
- 57.Kerru N., Singh-Pillay A., Awolade P., Singh P. Current anti-diabetic agents and their molecular targets: a review. Eur. J. Med. Chem. 2018;152:436–488. doi: 10.1016/j.ejmech.2018.04.061. [DOI] [PubMed] [Google Scholar]
- 58.Singh P., Mothilal S., Kerru N., Singh-Pillay A., Gummidi L., Erukainure O.L., Islam M.S. Comparative α-glucosidase and α-amylase inhibition studies of rhodanine–pyrazole conjugates and their simple rhodanine analogues. Med. Chem. Res. 2018:1–17. [Google Scholar]
- 59.Naz H., Islam A., Waheed A., Sly W.S., Ahmad F., Hassan I. Human β-glucuronidase: structure, function, and application in enzyme replacement therapy. Rejuvenation Res. 2013;16:352–363. doi: 10.1089/rej.2013.1407. [DOI] [PubMed] [Google Scholar]
- 60.Tranoy-Opalinski I., Legigan T., Barat R., Clarhaut J., Thomas M., Renoux B., Papot S. β-Glucuronidase-responsive prodrugs for selective cancer chemotherapy: an update. Eur. J. Med. Chem. 2014;74:302–313. doi: 10.1016/j.ejmech.2013.12.045. [DOI] [PubMed] [Google Scholar]
- 61.Zhang X., Li X., You Q., Zhang X. Prodrug strategy for cancer cell-specific targeting: a recent overview. Eur. J. Med. Chem. 2017;139:542–563. doi: 10.1016/j.ejmech.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 62.Knas M., Choromanska M., Karaszewska K., Dudzik D., Waszkiel D., Borzym-Kluczyk M., Zaniewska A., Zwierz K. Activity of lysosomal exoglycosidases in saliva of patients with HIV infection. Adv. Med. Sci. 2007;52:186–190. [PubMed] [Google Scholar]
- 63.Wallace M.A., Kormos T.M., Pleil J.D. Blood-borne biomarkers and bioindicators for linking exposure to health effects in environmental health science. J. Toxicol. Environ. Health B Crit. Rev. 2016;19:380–409. doi: 10.1080/10937404.2016.1215772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Camilleri M., Halawi H., Oduyebo I. Biomarkers as a diagnostic tool for irritable bowel syndrome: where are we? Expert Rev. Gastroenterol. Hepatol. 2017;11:303–316. doi: 10.1080/17474124.2017.1288096. [DOI] [PubMed] [Google Scholar]
- 65.Hemalatha T., UmaMaheswari T., Krithiga G., Sankaranarayanan P., Puvanakrishnan R. Enzymes in clinical medicine: an overview. Indian J. Exp. Biol. 2013;51:777–788. [PubMed] [Google Scholar]
- 66.Taba M., Jr., Kinney J., Kim A.S., Giannobile W.V. Diagnostic biomarkers for oral and periodontal diseases. Dent. Clin. N. Am. 2005;49:551–571. doi: 10.1016/j.cden.2005.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Preshaw P.M., Alba A.L., Herrera D., Jepsen S., Konstantinidis A., Makrilakis K., Taylor R. Periodontitis and diabetes: a two-way relationship. Diabetologia. 2012;55:21–31. doi: 10.1007/s00125-011-2342-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haffajee A.D., Socransky S.S., Goodson J.M. Clinical-parameters as predictors of destructive periodontal-disease activity. J. Clin. Periodontol. 1983;10:257–265. doi: 10.1111/j.1600-051X.1983.tb01274.x. [DOI] [PubMed] [Google Scholar]
- 69.Lamster I.B., Kaufman E., Grbic J.T., Winston L.J., Singer R.E. β-Glucuronidase activity in saliva: relationship to clinical periodontal parameters. J. Periodontol. 2003;74:353–359. doi: 10.1902/jop.2003.74.3.353. [DOI] [PubMed] [Google Scholar]
- 70.Prabhahar C.S., Niazi K.T.M., Prakash R., Yuvaraj A., Goud S., Ravishekar P. Estimation of salivary β-glucuronidase activity as a marker of periodontal disease: a case control study. J. Int. Soc. Prev. Community Dent. 2014;4:S193. doi: 10.4103/2231-0762.149039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ali S.A., Telgi R.L., Tirth A., Tantry I.Q., Aleem A. Lactate dehydrogenase and β-glucuronidase as salivary biochemical makers of periodontitis among smokers and non-smokers. Sultan Qaboos Univ. Med. J. 2018;18:e318–e323. doi: 10.18295/squmj.2018.18.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Buchmann R., Hasilik A., Van Dyke T.E., Lange D.E. Resolution of crevicular fluid leukocyte activity in patients treated for aggressive periodontal disease. J. Periodontol. 2002;73:995–1002. doi: 10.1902/jop.2002.73.9.995. [DOI] [PubMed] [Google Scholar]
- 73.Koss M.A., Castro C.E., Guarnieri S.M., Hermosilla D. Determination of salivary alkaline phosphatase and β-glucuronidase in treated periodontal disease patients. EC Dental Science. 2019;18:1225–1231. [Google Scholar]
- 74.Pietruska M., Bernaczyk A., Knas M., Pietruski J., Zwierz K. Assessment of salivary levels of the chosen exoglycosidases in patients with aggressive periodontitis after treatment with doxycycline. Adv. Med. Sci. 2006;51(Suppl 1):158–161. [PubMed] [Google Scholar]
- 75.Donath M.Y., Storling J., Maedler K., Mandrup-Poulsen T. Inflammatory mediators and islet β-cell failure: a link between type 1 and type 2 diabetes. J. Mol. Med. (Berl.) 2003;81:455–470. doi: 10.1007/s00109-003-0450-y. [DOI] [PubMed] [Google Scholar]
- 76.Donath M.Y., Ehses J.A., Maedler K., Schumann D.M., Ellingsgaard H., Eppler E., Reinecke M. Mechanisms of β-cell death in type 2 diabetes. Diabetes. 2005;54(Suppl 2):S108–S113. doi: 10.2337/diabetes.54.suppl_2.s108. [DOI] [PubMed] [Google Scholar]
- 77.Eizirik D.L., Colli M.L., Ortis F. The role of inflammation in insulitis and β-cell loss in type 1 diabetes. Nat. Rev. Endocrinol. 2009;5:219–226. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- 78.Engebretson S.P., Hey-Hadavi J., Ehrhardt F.J., Hsu D., Celenti R.S., Grbic J.T., Lamster I.B. Gingival crevicular fluid levels of interleukin-1β and glycemic control in patients with chronic periodontitis and type 2 diabetes. J. Periodontol. 2004;75:1203–1208. doi: 10.1902/jop.2004.75.9.1203. [DOI] [PubMed] [Google Scholar]
- 79.Shimada Y., Tabeta K., Sugita N., Yoshie H. Profiling biomarkers in gingival crevicular fluid using multiplex bead immunoassay. Arch. Oral Biol. 2013;58:724–730. doi: 10.1016/j.archoralbio.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Q., Chen B., Zhu D., Yan F. Biomarker levels in gingival crevicular fluid of subjects with different periodontal conditions: a cross-sectional study. Arch. Oral Biol. 2016;72:92–98. doi: 10.1016/j.archoralbio.2016.08.020. [DOI] [PubMed] [Google Scholar]
- 81.Wu Y.C., Ning L., Tu Y.K., Huang C.P., Huang N.T., Chen Y.F., Chang P.C. Salivary biomarker combination prediction model for the diagnosis of periodontitis in a Taiwanese population. J. Formos. Med. Assoc. 2018;117:841–848. doi: 10.1016/j.jfma.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 82.Miranda T.S., Heluy S.L., Cruz D.F., da Silva H.D.P., Feres M., Figueiredo L.C., Duarte P.M. The ratios of pro-inflammatory to anti-inflammatory cytokines in the serum of chronic periodontitis patients with and without type 2 diabetes and/or smoking habit. Clin. Oral Investig. 2019;23:641–650. doi: 10.1007/s00784-018-2471-5. [DOI] [PubMed] [Google Scholar]
- 83.Grossi S.G., Skrepcinski F.B., DeCaro T., Robertson D.C., Ho A.W., Dunford R.G., Genco R.J. Treatment of periodontal disease in diabetics reduces glycated hemoglobin. J. Periodontol. 1997;68:713–719. doi: 10.1902/jop.1997.68.8.713. [DOI] [PubMed] [Google Scholar]
- 84.Stewart J.E., Wager K.A., Friedlander A.H., Zadeh H.H. The effect of periodontal treatment on glycemic control in patients with type 2 diabetes mellitus. J. Clin. Periodontol. 2001;28:306–310. doi: 10.1034/j.1600-051x.2001.028004306.x. [DOI] [PubMed] [Google Scholar]
- 85.Sun W.L., Chen L.L., Zhang S.Z., Wu Y.M., Ren Y.Z., Qin G.M. Inflammatory cytokines, adiponectin, insulin resistance and metabolic control after periodontal intervention in patients with type 2 diabetes and chronic periodontitis. Intern. Med. 2011;50:1569–1574. doi: 10.2169/internalmedicine.50.5166. [DOI] [PubMed] [Google Scholar]
- 86.Ramamurthy J., Nd J., Varghese S. Comparison of salivary β-glucuronidase activity in chronic periodontitis patients with and without diabetes mellitus. J. Clin. Diagn. Res. 2014;8:ZC19–ZC21. doi: 10.7860/JCDR/2014/8713.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pushparani D.S., Nirmala S. Comparison of acid phosphatase and β D-glucuronidase enzyme levels in type 2 diabetes mellitus with and without periodontitis. Int. J. Sci. Eng. Res. 2013;4:1164–1168. [Google Scholar]
- 88.Surna A., Sakalauskiene J., Gleiznys A., Ivanauskiene E., Saferis V. Activity of neutrophil β-glucuronidase in diabetic and nondiabetic patients with chronic generalized periodontitis and healthy subjects. Medicina. 2011;47:91–97. [PubMed] [Google Scholar]
- 89.Yoon A.J., Cheng B., Philipone E., Turner R., Lamster I.B. Inflammatory biomarkers in saliva: assessing the strength of association of diabetes mellitus and periodontal status with the oral inflammatory burden. J. Clin. Periodontol. 2012;39:434–440. doi: 10.1111/j.1600-051X.2012.01866.x. [DOI] [PubMed] [Google Scholar]
- 90.Engebretson S.P., Vossughi F., Hey-Hadavi J., Emingil G., Grbic J.T. The influence of diabetes on gingival crevicular fluid β-glucuronidase and interleukin-8. J. Clin. Periodontol. 2006;33:784–790. doi: 10.1111/j.1600-051X.2006.00984.x. [DOI] [PubMed] [Google Scholar]
- 91.Liang S.L., Chan D.W. Enzymes and related proteins as cancer biomarkers: a proteomic approach. Clin. Chim. Acta. 2007;381:93–97. doi: 10.1016/j.cca.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hayes D.F. Biomarker validation and testing. Mol. Oncol. 2015;9:960–966. doi: 10.1016/j.molonc.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Coussens L.M., Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Francuz T., Czajka-Francuz P., Cison-Jurek S., Wojnar J. The role of inflammation in colon cancer pathogenesis. Postepy Hig. Med. Dosw. 2016;70:360–366. doi: 10.5604/17322693.1200551. [DOI] [PubMed] [Google Scholar]
- 95.Jin Y., Tian X., Jin L., Cui Y., Liu T., Yu Z., Huo X., Cui J., Sun C., Wang C., Ning J., Zhang B., Feng L., Ma X. Highly specific near-infrared fluorescent probe for the real-time detection of β-glucuronidase in various living cells and animals. Anal. Chem. 2018;90:3276–3283. doi: 10.1021/acs.analchem.7b04813. [DOI] [PubMed] [Google Scholar]
- 96.Waszkiewicz N., Szajda S.D., Konarzewska-Duchnowska E., Zalewska-Szajda B., Gałązkowski R., Sawko A., Nammous H., Buko V., Szulc A., Zwierz K., Ładny J.R. Serum β-glucuronidase as a potential colon cancer marker: a preliminary study. Adv. Hyg. Exp. Med. 2015;69:436–439. doi: 10.5604/17322693.1148704. [DOI] [PubMed] [Google Scholar]
- 97.Beratis N.G., Kaperonis A., Eliopoulou M.I., Kourounis G., Tzingounis V.A. Increased activity of lysosomal enzymes in the peritoneal fluid of patients with gynecologic cancers and pelvic inflammatory disease. J. Cancer Res. Clin. Oncol. 2005;131:371–376. doi: 10.1007/s00432-004-0649-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Beratis N.G., Georgiou G., Eliopoulou M. Increased activity of lysosomal enzymes in the peritoneal fluid of bacterial peritonitis. Pediatrics. 2002;109:e44. doi: 10.1542/peds.109.3.e44. [DOI] [PubMed] [Google Scholar]
- 99.Beratis N.G., Eliopoulou M.I., Syrogiannopoulos G.A. β-Glucuronidase in the diagnosis of bacterial meningitis and response to treatment. Acta Paediatr. 2003;92:1272–1276. doi: 10.1080/08035250310004342. [DOI] [PubMed] [Google Scholar]
- 100.Eliopoulou M.I., Georgakopoulos C.D., Beratis N.G. β-Glucuronidase activity in cerebrospinal fluid pleocytosis due to urinary tract infection. Acta Pædiatrica. 2007;96:1053–1058. doi: 10.1111/j.1651-2227.2007.00346.x. [DOI] [PubMed] [Google Scholar]
- 101.Panagiotopoulou E.C., Fouzas S., Douros K., Triantaphyllidou I.E., Malavaki C., Priftis K.N., Karamanos N.K., Anthracopoulos M.B. Increased β-glucuronidase activity in bronchoalveolar lavage fluid of children with bacterial lung infection: a case–control study. Respirology. 2015;20:1248–1254. doi: 10.1111/resp.12596. [DOI] [PubMed] [Google Scholar]
- 102.Abdollahi M. In: Anticholinesterase Pesticides: Metabolism, Neurotoxicity, and Epidemiology. Satoh T., Gupta R.C., editors. John Wiley & Sons, Inc; 2010. Poisoning with anticholinesterase insecticides in Iran; pp. 433–446. [DOI] [Google Scholar]
- 103.Swank R.T., Paigen K. Biochemical and genetic evidence for a macromolecular, β-glucuronidase complex in microsomal membranes. J. Mol. Biol. 1973;77:371–389. doi: 10.1016/0022-2836(73)90445-2. [DOI] [PubMed] [Google Scholar]
- 104.Medda S., Swank R.T. Egasyn, a protein which determines the subcellular distribution of β-glucuronidase, has esterase activity. J. Biol. Chem. 1985;260:15802–15808. [PubMed] [Google Scholar]
- 105.Medda S., Stevens A.M., Swank R.T. Involvement of the esterase active site of egasyn in compartmentalization of β-glucuronidase within the endoplasmic reticulum. Cell. 1987;50:301–310. doi: 10.1016/0092-8674(87)90225-X. [DOI] [PubMed] [Google Scholar]
- 106.Satoh T., Suzuki S., Kawai N., Nakamura T., Hosokawa M. Toxicological significance in the cleavage of esterase-β-glucuronidase complex in liver microsomes by organophosphorus compounds. Chem. Biol. Interact. 1999;119-120:471–478. doi: 10.1016/S0009-2797(99)00060-5. [DOI] [PubMed] [Google Scholar]
- 107.Fujikawa Y., Satoh T., Suganuma A., Suzuki S., Niikura Y., Yui S., Yamaura Y. Extremely sensitive biomarker of acute organophosphorus insecticide exposure. Hum. Exp. Toxicol. 2005;24:333–336. doi: 10.1191/0960327105ht532oa. [DOI] [PubMed] [Google Scholar]
- 108.Ueyama J., Satoh T., Kondo T., Takagi K., Shibata E., Goto M., Kimata A., Saito I., Hasegawa T., Wakusawa S., Kamijima M. β-Glucuronidase activity is a sensitive biomarker to assess low-level organophosphorus insecticide exposure. Toxicol. Lett. 2010;193:115–119. doi: 10.1016/j.toxlet.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 109.Hernández A.F., Gómez M.A., Pena G., Gil F., Rodrigo L., Villanueva E., Pla A. Effect of long-term exposure to pesticides on plasma esterases from plastic greenhouse workers. J. Toxicol. Environ. Health. 2004;67:1095–1108. doi: 10.1080/15287390490452371. [DOI] [PubMed] [Google Scholar]
- 110.Soltaninejad K., Shadnia S., Afkhami-Taghipour M., Saljooghi R., Mohammadirad A., Abdollahi M. Blood β-glucuronidase as a suitable biomarker at acute exposure of severe organophosphorus poisoning in human. Hum. Exp. Toxicol. 2007;26:963–966. doi: 10.1177/0960327107085349. [DOI] [PubMed] [Google Scholar]
- 111.Beltagy D.M., Sadek K.M., Hafez A.S. Serum β-glucuronidase activity as a biomarker for acute cholinesterase inhibitor pesticide poisoning. Toxicol. Ind. Health. 2018;34:891–897. doi: 10.1177/0748233718802068. [DOI] [PubMed] [Google Scholar]
- 112.Ruíz-Arias M.A., Herrera-Moreno J.F., Medina-Díaz I.M., Bernal-Hernández Y.Y., González-Arias C.A., Rojas-García A.E. β-Glucuronidase and its relationship with clinical parameters and biomarkers of pesticide exposure. J. Occup. Environ. Med. 2018;60:e602–e609. doi: 10.1097/JOM.0000000000001460. [DOI] [PubMed] [Google Scholar]
- 113.Inayat-Hussain S.H., Lubis S.H., Sakian N.I.M., Ghazali A.R., Ali N.S., el Sersi M., Toong L.M., Zainal A.M., Hashim S., Ghazali M.S., Saidin M.N., Rahman A.R.A., Rafaai M.J.M., Omar S., Rapiai R., Othman R., Chan L.T., Johari A., Soon W.H., Salleh A.R., Satoh T. Is plasma β-glucuronidase a novel human biomarker for monitoring anticholinesterase pesticides exposure? A Malaysian experience. Toxicol. Appl. Pharmacol. 2007;219:210–216. doi: 10.1016/j.taap.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 114.Sabbe M.B., Desruelles D., Lissens W. Is β-glucuronidase a clinical useful biomarker for an acute organophosphorus poisoning? Hum. Exp. Toxicol. 2008;27:431–433. doi: 10.1177/0960327108094614. [DOI] [PubMed] [Google Scholar]
- 115.Islam M.R., Waheed A., Shah G.N., Tomatsu S., Sly W.S. Human egasyn binds β-glucuronidase but neither the esterase active site of egasyn nor the C terminus of β-glucuronidase is involved in their interaction. Arch. Biochem. Biophys. 1999;372:53–61. doi: 10.1006/abbi.1999.1449. [DOI] [PubMed] [Google Scholar]
- 116.Rodrigues T., Reker D., Schneider P., Schneider G. Counting on natural products for drug design. Nat. Chem. 2016;8:531–541. doi: 10.1038/nchem.2479. [DOI] [PubMed] [Google Scholar]
- 117.Levvy G. The preparation and properties of β-glucuronidase. 4. Inhibition by sugar acids and their lactones. Biochem. J. 1952;52:464. doi: 10.1042/bj0520464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Walaszek Z., Hanausek-Walaszek M., Webb T.E. Inhibition of 7,12-dimethylbenzanthracene-induced rat mammary tumorigenesis by 2,5-di-O-acetyl-D-glucaro-1,4: 6,3-dilactone, an in vivo β-glucuronidase inhibitor. Carcinogenesis. 1984;5:767–772. doi: 10.1093/carcin/5.6.767. [DOI] [PubMed] [Google Scholar]
- 119.O’Leary K.A., Day A.J., Needs P.W., Sly W.S., O’Brien N.M., Williamson G.J.F.l. Flavonoid glucuronides are substrates for human liver β-glucuronidase. FEBS Lett. 2001;503:103–106. doi: 10.1016/s0014-5793(01)02684-9. [DOI] [PubMed] [Google Scholar]
- 120.Gourley G.R., Kreamer B.L., Cohnen M. Inhibition of β-glucuronidase by casein hydrolysate formula. J. Pediatr. Gastroenterol. Nutr. 1997;25:267–272. doi: 10.1097/00005176-199709000-00005. [DOI] [PubMed] [Google Scholar]
- 121.Xie B., Zhang Z., Gong T., Zhang N., Wang H., Zou H. Application of metabonomic strategy to discover an unreported active ingredient in LiuWeiDiHuang pills suppressing beta-glucuronidase. Anal. Bioanal. Chem. 2015;407:609–614. doi: 10.1007/s00216-014-8253-2. [DOI] [PubMed] [Google Scholar]
- 122.Fittkau M., Voigt W., Holzhausen H.-J., Schmoll H.-J., oncology c. Saccharic acid 1.4-lactone protects against CPT-11-induced mucosa damage in rats. J. Cancer Res. Clin. Oncol. 2004;130:388–394. doi: 10.1007/s00432-004-0557-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Morita N., Walaszek Z., Kinjo T., Nishimaki T., Hanausek M., Slaga T.J., Mori H., Yoshimi N. Effects of synthetic and natural in vivo inhibitors of β-glucuronidase on azoxymethane-induced colon carcinogenesis in rats. Mol. Med. Rep. 2008;1:741–746. doi: 10.3892/mmr_00000022. [DOI] [PubMed] [Google Scholar]
- 124.Iida R., Nagata S., Kakimoto M., Akaike H., Watanabe H., Shioya A. 2,5-di-O-acetyl-D-glucosaccharo-1,4:6,3-dilactone, a new potent β-glucuronidase inhibitor. Jpn. J. Pharmacol. 1965;15:88–90. doi: 10.1254/jjp.15.88. [DOI] [PubMed] [Google Scholar]
- 125.Kowalczyk M.C., Spears E., Narog M., Zoltaszek R., Kowalczyk P., Hanausek M., Yoshimi N., Slaga T.J., Walaszek Z. Modulation of biomarkers related to tumor initiation and promotion in mouse skin by a natural β-glucuronidase inhibitor and its precursors. Oncol. Rep. 2011;26:551–556. doi: 10.3892/or.2011.1351. [DOI] [PubMed] [Google Scholar]
- 126.Haroon M.H., Premaratne S.R., Choudhry M.I., Dharmaratne H.R.W. A new β-glucuronidase inhibiting butyrolactone from the marine endophytic fungus Aspergillus terreus. Nat. Prod. Res. 2013;27:1060–1066. doi: 10.1080/14786419.2012.708659. [DOI] [PubMed] [Google Scholar]
- 127.Kreamer B.L., Siegel F.L., Gourley G.R. A novel inhibitor of β-glucuronidase: L-Aspartic acid. Pediatr. Res. 2001;50:460–466. doi: 10.1203/00006450-200110000-00007. [DOI] [PubMed] [Google Scholar]
- 128.Gourley G.R., Li Z., Kreamer B.L., Kosorok M.R. A controlled, randomized, double-blind trial of prophylaxis against jaundice among breastfed newborns. Pediatrics. 2005;116:385–391. doi: 10.1542/peds.2004-1807. [DOI] [PubMed] [Google Scholar]
- 129.Cushnie T.P., Lamb A.J. Antimicrobial activity of flavonoids. Int. J. Antimicrob. Agents. 2005;26:343–356. doi: 10.1016/j.ijantimicag.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Singh M., Kaur M., Silakari O. Flavones: an important scaffold for medicinal chemistry. Eur. J. Med. Chem. 2014;84:206–239. doi: 10.1016/j.ejmech.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 131.Wang T.Y., Li Q., Bi K.S. Bioactive flavonoids in medicinal plants: structure, activity and biological fate. Asian J. Pharm. Sci. 2018;13:12–23. doi: 10.1016/j.ajps.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Manju V., Nalini N. Protective role of luteolin in 1,2-dimethylhydrazine induced experimental colon carcinogenesis. Cell Biochem. Funct. 2007;25:189–194. doi: 10.1002/cbf.1305. [DOI] [PubMed] [Google Scholar]
- 133.Kavak D.D., Altıok E., Bayraktar O., Ülkü S. Pistacia terebinthus extract: as a potential antioxidant, antimicrobial and possible β-glucuronidase inhibitor. J. Mol. Catal. B Enzym. 2010;64:167–171. [Google Scholar]
- 134.Weng Z.-M., Wang P., Ge G.-B., Dai Z.-R., Wu D.-C., Zou L.-W., Dou T.-Y., Zhang T.-Y., Yang L., Hou J. Structure-activity relationships of flavonoids as natural inhibitors against E. coli β-glucuronidase. Food Chem. Toxicol. 2017;109:975–983. doi: 10.1016/j.fct.2017.03.042. [DOI] [PubMed] [Google Scholar]
- 135.Acharya J., De B. Bioactivity-guided fractionation to identify β-glucuronidase inhibitors in Nymphaea pubescens flower extract. Cogent Food. Agric. 2016;2:1134379. [Google Scholar]
- 136.Wei B., Yang W., Yan Z.-X., Zhang Q.-W., Yan R. Prenylflavonoids sanggenon C and kuwanon G from mulberry (Morus alba L.) as potent broad-spectrum bacterial β-glucuronidase inhibitors: biological evaluation and molecular docking studies. J. Funct. Foods. 2018;48:210–219. [Google Scholar]
- 137.Chung M.I., Weng J.R., Wang J.P., Teng C.M., Lin C.N. Antiplatelet and anti-inflammatory constituents and new oxygenated xanthones from Hypericum geminiflorum. Planta Med. 2002;68:25–29. doi: 10.1055/s-2002-19871. [DOI] [PubMed] [Google Scholar]
- 138.Wang L., Yang Y., Liu C., Chen R.Y. Three new compounds from Morus nigra L. J. Asian Nat. Prod. Res. 2010;12:431–437. doi: 10.1080/10286020.2010.489824. [DOI] [PubMed] [Google Scholar]
- 139.Han Q.T., Ren Y., Li G.S., Xiang K.L., Dai S.J. Flavonoid alkaloids from Scutellaria moniliorrhiza with anti-inflammatory activities and inhibitory activities against aldose reductase. Phytochemistry. 2018;152:91–96. doi: 10.1016/j.phytochem.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 140.Han Q.T., Xiao K., Xiang K.L., Li G.S., Dai S.J. New flavonoid glucuronate esters with anti-inflammatory activities from Scutellaria regeliana. Chem. Biodivers. 2018;15 doi: 10.1002/cbdv.201800038. [DOI] [PubMed] [Google Scholar]
- 141.Wang Q., Li Z., Yang Z., Fang Y., Ouyang H., Liao H., Feng Y., Yang S. New alkaloids with anti-inflammatory activities from Corydalis decumbens. Phytochem Lett. 2016;18:83–86. doi: 10.1016/j.phytol.2016.09.003. [DOI] [Google Scholar]
- 142.Remirez D., Gonzalez R., Merino N., Rodriguez S., Ancheta O. Inhibitory effects of Spirulina in zymosan-induced arthritis in mice. Mediat. Inflamm. 2002;11:75–79. doi: 10.1080/09629350220131917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nile S.H., Park S.W. HPTLC analysis, antioxidant, anti-inflammatory and antiproliferative activities of Arisaema tortuosum tuber extract. Pharm. Biol. 2014;52:221–227. doi: 10.3109/13880209.2013.831110. [DOI] [PubMed] [Google Scholar]
- 144.Nile S.H., Park S.W. Chromatographic analysis, antioxidant, anti-inflammatory, and xanthine oxidase inhibitory activities of ginger extracts and its reference compounds. Ind. Crops Prod. 2015;70:238–244. doi: 10.1016/j.indcrop.2015.03.033. [DOI] [Google Scholar]
- 145.Karak S., Acharya J., Begum S., Mazumdar I., Kundu R., De B. Essential oil of Piper betle L. leaves: chemical composition, anti-acetylcholinesterase, anti-β-glucuronidase and cytotoxic properties. J. Appl. Res. Med. Aromatic Plants. 2018;10:85–92. [Google Scholar]
- 146.Karak S., Nag G., De B. Metabolic profile and β-glucuronidase inhibitory property of three species of Swertia. Rev. Bras. Farmacognosia. 2017;27:105–111. [Google Scholar]
- 147.Yoo B.H., Lee B.H., Kim J.S., Kim N.J., Kim S.H., Ryu K.W. Effects of Shikunshito-Kamiho on fecal enzymes and formation of aberrant crypt foci induced by 1,2-dimethylhydrazine. Biol. Pharm. Bull. 2001;24:638–642. doi: 10.1248/bpb.24.638. [DOI] [PubMed] [Google Scholar]
- 148.Devasena T., Menon V.P. Fenugreek affects the activity of β-glucuronidase and mucinase in the colon. Phytother Res. 2003;17:1088–1091. doi: 10.1002/ptr.1331. [DOI] [PubMed] [Google Scholar]
- 149.Bae H.-S., Kim Y.-S., Cho K.-H., Lee K.-S., Kim J.-J., Lee H.-U., Kim D.-H. Hepatoprotective activity of reduohanxiao-tang (Yuldahanso-tang) is related to the inhibition of β-glucuronidase. Am. J. Chin. Med. 2003;31:111–117. doi: 10.1142/S0192415X03000722. [DOI] [PubMed] [Google Scholar]
- 150.Lee H.W., Choo M.K., Bae E.A., Kim D.H. β-glucuronidase inhibitor tectorigenin isolated from the flower of Pueraria thunbergiana protects carbon tetrachloride-induced liver injury. Liver Int. 2003;23:221–226. doi: 10.1034/j.1600-0676.2003.00830.x. [DOI] [PubMed] [Google Scholar]
- 151.Shim S.-B., Kim N.-J., Kim D.-H.J.P.m. β-Glucuronidase inhibitory activity and hepatoprotective effect of 18β-glycyrrhetinic acid from the rhizomes of Glycyrrhiza uralensis. 2000;66:40–43. doi: 10.1055/s-2000-11109. [DOI] [PubMed] [Google Scholar]
- 152.Riaz N., Anis I., Aziz ur R., Malik A., Ahmed Z., Muhammad P., Shujaat S., Atta ur R. Emodinol, β-glucuronidase inhibiting triterpene from Paeonia Emodi. Nat. Prod. Res. 2003;17:247–251. doi: 10.1080/1057563021000060103. [DOI] [PubMed] [Google Scholar]
- 153.Pervaiz I., Ahmad S., Madni M.A., Ahmad H., Khaliq F.H. Microbial biotransformation: a tool for drug designing. Appl. Biochem. Microbiol. 2013;49:437–450. doi: 10.1134/s0003683813050098. [DOI] [PubMed] [Google Scholar]
- 154.Sultana N. Microbial biotransformation of bioactive and clinically useful steroids and some salient features of steroids and biotransformation. Steroids. 2018;136:76–92. doi: 10.1016/j.steroids.2018.01.007. [DOI] [PubMed] [Google Scholar]
- 155.Choudhary M.I., Zafar S., Khan N.T., Ahmad S., Noreen S., Marasini B.P., Al-Khedhairy A.A., Atta Ur R. Biotransformation of dehydroepiandrosterone with Macrophomina phaseolina and β-glucuronidase inhibitory activity of transformed products. J. Enzym. Inhib. Med. Chem. 2012;27:348–355. doi: 10.3109/14756366.2011.590804. [DOI] [PubMed] [Google Scholar]
- 156.Khan N.T., Zafar S., Noreen S., Al Majid A.M., Al Othman Z.A., Al-Resayes S.I. Atta ur, R., and Choudhary, M.I., Biotransformation of dianabol with the filamentous fungi and β-glucuronidase inhibitory activity of resulting metabolites. Steroids. 2014;85:65–72. doi: 10.1016/j.steroids.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 157.Nieschlag E., Vorona E. Doping with anabolic androgenic steroids (AAS): adverse effects on non-reproductive organs and functions. Rev. Endocr. Metab. Disord. 2015;16:199–211. doi: 10.1007/s11154-015-9320-5. [DOI] [PubMed] [Google Scholar]
- 158.Wecker L. Elsevier Health Sciences; 2018. Brody’s Human Pharmacology: Mechanism-Based Therapeutics. [Google Scholar]
- 159.Devine D.A., Marsh P.D. Prospects for the development of probiotics and prebiotics for oral applications. J. Oral Microbiol. 2009;1:1949. doi: 10.3402/jom.v1i0.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Panebianco C., Andriulli A., Pazienza V. Pharmacomicrobiomics: exploiting the drug-microbiota interactions in anticancer therapies. Microbiome. 2018;6:92. doi: 10.1186/s40168-018-0483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Quigley E.M.M. Prebiotics and probiotics in digestive health. Clin. Gastroenterol. Hepatol. 2019;17:333–344. doi: 10.1016/j.cgh.2018.09.028. [DOI] [PubMed] [Google Scholar]
- 162.Gibson G.R., Hutkins R., Sanders M.E., Prescott S.L., Reimer R.A., Salminen S.J., Scott K., Stanton C., Swanson K.S., Cani P.D., Verbeke K., Reid G. Expert consensus document: the International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017;14:491–502. doi: 10.1038/nrgastro.2017.75. [DOI] [PubMed] [Google Scholar]
- 163.Han S.Y., Huh C.S., Ahn Y.T., Lim K.S., Baek Y.J., Kim D.H. Hepatoprotective effect of lactic acid bacteria, inhibitors of β-glucuronidase production against intestinal microflora. Arch Pharm. Res. (Seoul) 2005;28:325–329. doi: 10.1007/BF02977800. [DOI] [PubMed] [Google Scholar]
- 164.Arenahalli Ningegowda M., Siddalingaiya Gurudutt P. In vitro fermentation of prebiotics by Lactobacillus plantarum CFR 2194: selectivity, viability and effect of metabolites on beta-glucuronidase activity. World J. Microbiol. Biotechnol. 2012;28:901–908. doi: 10.1007/s11274-011-0887-z. [DOI] [PubMed] [Google Scholar]
- 165.Watanabe Y., Muroi R., Tsuchiya H., Uda Y., Hashimoto K. Inhibitory effect of methyl methanethiosulfinate on β-glucuronidase activity. Biosci. Biotechnol. Biochem. 2013;77:2325–2327. doi: 10.1271/bbb.130510. [DOI] [PubMed] [Google Scholar]
- 166.Zhang D., Kurihara H. Isogloiosiphone B, a novel acetal, and hydrophobic compounds as β-glucuronidase inhibitors derived from the red alga Neodilsea yendoana. Biosci. Biotechnol. Biochem. 2018;82:46–48. doi: 10.1080/09168451.2017.1403885. [DOI] [PubMed] [Google Scholar]
- 167.Nagoor Meeran M.F., Jagadeesh G.S., Selvaraj P. Thymol attenuates inflammation in isoproterenol induced myocardial infarcted rats by inhibiting the release of lysosomal enzymes and downregulating the expressions of proinflammatory cytokines. Eur. J. Pharmacol. 2015;754:153–161. doi: 10.1016/j.ejphar.2015.02.028. [DOI] [PubMed] [Google Scholar]
- 168.Compain P., Martin O.R., editors. Iminosugars: from Synthesis to Therapeutic Applications. John Wiley & Sons Ltd; 2007. [Google Scholar]
- 169.Nishimura Y. Gem-diamine 1-N-iminosugars as versatile glycomimetics: synthesis, biological activity and therapeutic potential. J. Antibiot. (Tokyo) 2009;62:407. doi: 10.1038/ja.2009.53. [DOI] [PubMed] [Google Scholar]
- 170.Ishida N., Kumagai K., Niida T., Shomura T., Yumoto H. Nojirimycin, a new antibiotic. II Isolation, characterization and biological activity. J. Antibiotics. 1967;20:66–71. [PubMed] [Google Scholar]
- 171.Umezawa H., Aoyagi T., Komiyama T., Morishima H., Hamada M., Takeuchi T. Purification and characterization of a sialidase inhibitor, siastatin, produced by Streptomyces. J. Antibiotics. 1974;27:963–969. doi: 10.7164/antibiotics.27.963. [DOI] [PubMed] [Google Scholar]
- 172.Asano N., Nash R.J., Molyneux R.J., Fleet G.W.J. Sugar-mimic glycosidase inhibitors: natural occurrence, biological activity and prospects for therapeutic application. Tetrahedron Alert. 2000;11:1645–1680. doi: 10.1016/S0957-4166(00)00113-0. [DOI] [Google Scholar]
- 173.Nishimura Y. gem-Diamine 1-N-iminosugars and related iminosugars, candidate of therapeutic agents for tumor metastasis. Curr. Top. Med. Chem. 2003;3:575–591. doi: 10.2174/1568026033452492. [DOI] [PubMed] [Google Scholar]
- 174.Pabba J., Rempel B.P., Withers S.G., Vasella A. Synthesis of glycaro-1,5-lactams and tetrahydrotetrazolopyridine-5-carboxylates: inhibitors of β-D-glucuronidase and α-L-Iduronidase. Helv. Chim. Acta. 2006;89:635–666. [Google Scholar]
- 175.Pabba J., Mohal N., Vasella A. Synthesis of glucuronic, mannuronic, and galacturonic acid-derived imidazoles as inhibitors of bovine liver β-glucuronidase. Helv. Chim. Acta. 2006;89:1373–1386. [Google Scholar]
- 176.Cipolla L., Fernandes M.R., Gregori M., Airoldi C., Nicotra F.J.C.r. Synthesis and biological evaluation of a small library of nojirimycin-derived bicyclic iminosugars. Carbohydr. Res. 2007;342:1813–1830. doi: 10.1016/j.carres.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 177.Chronowska A., Gallienne E., Nicolas C., Kato A., Adachi I., Martin O.R. An expeditious synthesis of an analogue of (−)-steviamine by way of the 1, 3-dipolar cycloaddition of a nitrile oxide with a 1-C-allyl iminosugar. Tetrahedron Lett. 2011;52:6399–6402. doi: 10.1016/j.tetlet.2011.09.065. [DOI] [Google Scholar]
- 178.Narayana C., Kumari P., Ide D., Hoshino N., Kato A., Sagar R. Design and synthesis of N-acetylglucosamine derived 5a-carbasugar analogues as glycosidase inhibitors. Tetrahedron. 2018;74:1957–1964. doi: 10.1016/j.tet.2018.02.063. [DOI] [Google Scholar]
- 179.Rasmussen T.S., Koldsø H., Nakagawa S., Kato A., Schiøtt B., Jensen H.H. Synthesis of uronic-Noeurostegine–a potent bacterial β-glucuronidase inhibitor. Org. Biomol. Chem. 2011;9:7807–7813. doi: 10.1039/c1ob06038d. [DOI] [PubMed] [Google Scholar]
- 180.Compain P., Chagnault V., Martin O.R. Tactics and strategies for the synthesis of iminosugar C-glycosides: a review. Tetrahedron Alert. 2009;20:672–711. doi: 10.1016/j.tetasy.2009.03.031. [DOI] [Google Scholar]
- 181.Mondon M., Lecornué F., Guillard J., Nakagawa S., Kato A., Blériot Y. Skeletal rearrangement of seven-membered iminosugars: synthesis of (−)-adenophorine(−)-1-epi-adenophorine and derivatives and evaluation as glycosidase inhibitors. Biorg. Med. Chem. 2013;21:4803–4812. doi: 10.1016/j.bmc.2013.03.035. [DOI] [PubMed] [Google Scholar]
- 182.Désiré J., Mondon M., Fontelle N., Nakagawa S., Hirokami Y., Adachi I., Iwaki R., Fleet G., Alonzi D., Twigg G. N-and C-alkylation of seven-membered iminosugars generates potent glucocerebrosidase inhibitors and F508del-CFTR correctors. Org. Biomol. Chem. 2014;12:8977–8996. doi: 10.1039/c4ob00325j. [DOI] [PubMed] [Google Scholar]
- 183.Deschamp J., Mondon M., Nakagawa S., Kato A., Alonzi D.S., Butters T.D., Zhang Y., Sollogoub M., Blériot Y. Towards a stable noeuromycin analog with a D-manno configuration: synthesis and glycosidase inhibition of D-manno-like tri- and tetrahydroxylated azepanes. Biorg. Med. Chem. 2012;20:641–649. doi: 10.1016/j.bmc.2010.09.053. [DOI] [PubMed] [Google Scholar]
- 184.Fontelle N., Yamamoto A., Arda A., Jiménez-Barbero J., Kato A., Désiré J., Blériot Y. 2-Acetamido-2-deoxy-L-iminosugar C-alkyl and C-aryl glycosides: synthesis and glycosidase inhibition. Eur. J. Org. Chem. 2018:5477–5488. 2018. [Google Scholar]
- 185.Yoshimura Y., Ohara C., Imahori T., Saito Y., Kato A., Miyauchi S., Adachi I., Takahata H.J.B., chemistry M. Synthesis of both enantiomers of hydroxypipecolic acid derivatives equivalent to 5-azapyranuronic acids and evaluation of their inhibitory activities against glycosidases. Biorg. Med. Chem. 2008;16:8273–8286. doi: 10.1016/j.bmc.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 186.Salar U., Khan K.M., Taha M., Ismail N.H., Ali B., Qurat Ul A., Perveen S., Ghufran M., Wadood A. Biology-oriented drug synthesis (BIODS): in vitro β-glucuronidase inhibitory and in silico studies on 2-(2-methyl-5-nitro-1H-imidazole-1-yl)ethyl aryl carboxylate derivatives. Eur. J. Med. Chem. 2017;125:1289–1299. doi: 10.1016/j.ejmech.2016.11.031. [DOI] [PubMed] [Google Scholar]
- 187.Taha M., Ismail N.H., Imran S., Rashwan H., Jamil W., Ali S., Kashif S.M., Rahim F., Salar U., Khan K.M. Synthesis of 6-chloro-2-Aryl-1H-imidazo [4,5-b] pyridine derivatives: antidiabetic, antioxidant, β-glucuronidase inhibiton and their molecular docking studies. Bioorg. Chem. 2016;65:48–56. doi: 10.1016/j.bioorg.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 188.Ali I., Khan A., Hussain A., Farooq U., Ismail M., Hyder V., Ahmad V.U., Iaroshenko V.O., Hussain H., Langer P. Comparative enzyme inhibition study of 1-deazapurines. Med. Chem. Res. 2016;25:2599–2606. doi: 10.1007/s00044-016-1700-1. [DOI] [Google Scholar]
- 189.Khan K.M., Karim A., Saied S., Ambreen N., Rustamova X., Naureen S., Mansoor S., Ali M., Perveen S., Choudhary M.I. Evaluation of the thiazole Schiff bases as β-glucuronidase inhibitors and their in silico studies. Mol. Divers. 2014;18:295–306. doi: 10.1007/s11030-013-9500-8. [DOI] [PubMed] [Google Scholar]
- 190.Salar U., Khan K.M., Syed S., Taha M., Ali F., Ismail N.H., Perveen S., Wadood A., Ghufran M. Synthesis, in vitro β-glucuronidase inhibitory activity and in silico studies of novel (E)-4-Aryl-2-(2-(pyren-1-ylmethylene)hydrazinyl)thiazoles. Bioorg. Chem. 2017;70:199–209. doi: 10.1016/j.bioorg.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 191.Salar U., Taha M., Ismail N.H., Khan K.M., Imran S., Perveen S., Wadood A., Riaz M. 2016. Thiadiazole Derivatives as New Class of β-glucuronidase Inhibitors. [DOI] [PubMed] [Google Scholar]
- 192.Mohammed Khan K., Khan M., Ambreen N., Rahim F., Naureen S., Perveen S., Iqbal Choudhary M., Voelter W. Synthesis and β-glucuronidase inhibitory potential of benzimidazole derivatives. Med. Chem. 2012;8:421–427. doi: 10.2174/1573406411208030421. [DOI] [PubMed] [Google Scholar]
- 193.Taha M., Ismail N.H., Imran S., Selvaraj M., Rashwan H., Farhanah F.U., Rahim F., Kesavanarayanan K.S., Ali M. Synthesis of benzimidazole derivatives as potent β-glucuronidase inhibitors. Bioorg. Chem. 2015;61:36–44. doi: 10.1016/j.bioorg.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 194.Kamil A., Akhtar S., Noureen S., Saify Z.S., Jahan S., Khan K.M., Rahim F., Mushtaq N., Arif M., Perveen S. 2-(2’-Pyridyl) benzimidazole analogs and their β-glucuronidase inhibitory activity. J. Chem. Soc. Pak. 2015;37 [Google Scholar]
- 195.Zawawi N.K., Taha M., Ahmat N., Wadood A., Ismail N.H., Rahim F., Ali M., Abdullah N., Khan K.M. Novel 2,5-disubtituted-1,3,4-oxadiazoles with benzimidazole backbone: a new class of β-glucuronidase inhibitors and in silico studies. Bioorg. Med. Chem. 2015;23:3119–3125. doi: 10.1016/j.bmc.2015.04.081. [DOI] [PubMed] [Google Scholar]
- 196.Taha M., Ismail N.H., Imran S., Selvaraj M., Rahim A., Ali M., Siddiqui S., Rahim F., Khan K.M. Synthesis of novel benzohydrazone–oxadiazole hybrids as β-glucuronidase inhibitors and molecular modeling studies. Bioorg. Med. Chem. Lett. 2015;23:7394–7404. doi: 10.1016/j.bmc.2015.10.037. [DOI] [PubMed] [Google Scholar]
- 197.Taha M., Baharudin M.S., Ismail N.H., Selvaraj M., Salar U., Alkadi K.A., Khan K.M. Synthesis and in silico studies of novel sulfonamides having oxadiazole ring: as β-glucuronidase inhibitors. Bioorg. Chem. 2017;71:86–96. doi: 10.1016/j.bioorg.2017.01.015. [DOI] [PubMed] [Google Scholar]
- 198.Taha M., Imran S., Alomari M., Rahim F., Wadood A., Mosaddik A., Uddin N., Gollapalli M., Alqahtani M.A., Bamarouf Y.A. Synthesis of oxadiazole-coupled-thiadiazole derivatives as a potent β-glucuronidase inhibitors and their molecular docking study. Bioorg. Med. Chem. 2019 doi: 10.1016/j.bmc.2019.05.049. [DOI] [PubMed] [Google Scholar]
- 199.Khan K.M., Rahim F., Halim S.A., Taha M., Khan M., Perveen S., Mesaik M.A., Choudhary M.I. Synthesis of novel inhibitors of β-glucuronidase based on benzothiazole skeleton and study of their binding affinity by molecular docking. Bioorg. Med. Chem. 2011;19:4286–4294. doi: 10.1016/j.bmc.2011.05.052. [DOI] [PubMed] [Google Scholar]
- 200.Taha M., Ismail N.H., Imran S., Selvaraj M., Rahim F. Synthesis of novel inhibitors of β-glucuronidase based on the benzothiazole skeleton and their molecular docking studies. RSC Adv. 2016;6:3003–3012. [Google Scholar]
- 201.Taha M., Arbin M., Ahmat N., Imran S., Rahim F. Synthesis: small library of hybrid scaffolds of benzothiazole having hydrazone and evaluation of their β-glucuronidase activity. Bioorg. Chem. 2018;77:47–55. doi: 10.1016/j.bioorg.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 202.Jamil W., Kumari D., Taha M., Khan M.N., Baharudin M.S., Ali M., Kanwal M., Lashari M.S., Khan K.M. Synthesis, β-glucuronidase inhibition, and molecular docking studies of 1, 2, 4-triazole hydrazones. J. Iran. Chem. Soc. 2018:1–14. [Google Scholar]
- 203.Shaikh N.N., Iqbal S., Syed N., Khan M.A., Moin S.T., Choudhary M.I., Basha F.Z. Carbazole-linked 1,2,3-triazoles: in vitro β-glucuronidase inhibitory potential, kinetics, and molecular docking studies. Chemistry. 2019;4:6181–6189. doi: 10.1002/slct.201900647. [DOI] [Google Scholar]
- 204.Baharudin M.S., Taha M., Imran S., Ismail N.H., Rahim F., Javid M.T., Khan K.M., Ali M. Synthesis of indole analogs as potent β-glucuronidase inhibitors. Bioorg. Chem. 2017;72:323–332. doi: 10.1016/j.bioorg.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 205.Almandil N.B., Taha M., Gollapalli M., Rahim F., Ibrahim M., Mosaddik A., Anouar E.H. Indole bearing thiadiazole analogs: synthesis, β-glucuronidase inhibition and molecular docking study. BMC Chemistry. 2019;13 doi: 10.1186/s13065-019-0522-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Anouar E.H., Moustapha M.E., Taha M., Geesi M.H., Farag Z.R., Rahim F., Almandil N.B., Farooq R.K., Nawaz M., Mosaddik A. Synthesis, molecular docking and β-glucuronidase inhibitory potential of indole base oxadiazole derivatives. Molecules. 2019;24:963. doi: 10.3390/molecules24050963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Tian J.L., Chen X., Fu S.L., Zhang R.J., Pan L., Cao Y., Wu X.J., Xiao H., Lin H.J., Lo H.W., Zhang Y., Lin J.Y. Bazedoxifene is a novel IL-6/GP130 inhibitor for treating triple-negative breast cancer. Breast Canc. Res. Treat. 2019;175:553–566. doi: 10.1007/s10549-019-05183-2. [DOI] [PubMed] [Google Scholar]
- 208.Chen K.L.A., Liu X., Zhao Y.C., Hieronymi K., Rossi G., Auvil L.S., Welge M., Bushell C., Smith R.L., Carlson K.E., Kim S.H., Katzenellenbogen J.A., Miller M.J., Madak-Erdogan Z. Long-term administration of conjugated estrogen and bazedoxifene decreased murine fecal β-Glucuronidase activity without impacting overall microbiome community. Sci. Rep. 2018;8:8166. doi: 10.1038/s41598-018-26506-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Khan K.M., Rahim F., Wadood A., Taha M., Khan M., Naureen S., Ambreen N., Hussain S., Perveen S., Choudhary M.I. Evaluation of bisindole as potent β-glucuronidase inhibitors: synthesis and in silico based studies. Bioorg. Med. Chem. Lett. 2014;24:1825–1829. doi: 10.1016/j.bmcl.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 210.Yousuf M., Shaikh N.N., Ul-Haq Z., Choudhary M.I. Bioinformatics: a rational combine approach used for the identification and in-vitro activity evaluation of potent β-Glucuronidase inhibitors. PLoS One. 2018;13 doi: 10.1371/journal.pone.0200502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Taha M., Ismail N.H., Imran S., Rahim F., Wadood A., Khan H., Ullah H., Salar U., Khan K.M. Synthesis, β-glucuronidase inhibition and molecular docking studies of hybrid bisindole-thiosemicarbazides analogs. Bioorg. Chem. 2016;68:56–63. doi: 10.1016/j.bioorg.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 212.Taha M., Ullah H., Al Muqarrabun L.M.R., Khan M.N., Rahim F., Ahmat N., Ali M., Perveen S. Synthesis of bis-indolylmethanes as new potential inhibitors of β-glucuronidase and their molecular docking studies. Eur. J. Med. Chem. 2018;143:1757–1767. doi: 10.1016/j.ejmech.2017.10.071. [DOI] [PubMed] [Google Scholar]
- 213.Singh P., Anand A., Kumar V. Recent developments in biological activities of chalcones: a mini review. Eur. J. Med. Chem. 2014;85:758–777. doi: 10.1016/j.ejmech.2014.08.033. [DOI] [PubMed] [Google Scholar]
- 214.Hsieh H.K., Tsao L.T., Wang J.P., Lin C.N. Synthesis and anti-inflammatory effect of chalcones. J. Pharm. Pharmacol. 2000;52:163–171. doi: 10.1211/0022357001773814. [DOI] [PubMed] [Google Scholar]
- 215.Ko H.H., Tsao L.T., Yu K.L., Liu C.T., Wang J.P., Lin C.N. Structure-activity relationship studies on chalcone derivatives. the potent inhibition of chemical mediators release. Bioorg. Med. Chem. 2003;11:105–111. doi: 10.1016/s0968-0896(02)00312-7. [DOI] [PubMed] [Google Scholar]
- 216.Won S.J., Liu C.T., Tsao L.T., Weng J.R., Ko H.H., Wang J.P., Lin C.N. Synthetic chalcones as potential anti-inflammatory and cancer chemopreventive agents. Eur. J. Med. Chem. 2005;40:103–112. doi: 10.1016/j.ejmech.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 217.Bandgar B.P., Patil S.A., Gacche R.N., Korbad B.L., Hote B.S., Kinkar S.N., Jalde S.S. Synthesis and biological evaluation of nitrogen-containing chalcones as possible anti-inflammatory and antioxidant agents. Bioorg. Med. Chem. Lett. 2010;20:730–733. doi: 10.1016/j.bmcl.2009.11.068. [DOI] [PubMed] [Google Scholar]
- 218.Bandgar B.P., Hote B.S., Jalde S.S., Gacche R.N. Synthesis and biological evaluation of novel curcumin analogues as anti-inflammatory, anti-cancer and anti-oxidant agents. Med. Chem. Res. 2012;21:3006–3014. doi: 10.1007/s00044-011-9834-7. [DOI] [Google Scholar]
- 219.Bandgar B.P., Kinkar S.N., Chavan H.V., Jalde S.S., Shaikh R.U., Gacche R.N. Synthesis and biological evaluation of asymmetric indole curcumin analogs as potential anti-inflammatory and antioxidant agents. J. Enzym. Inhib. Med. Chem. 2014;29:7–11. doi: 10.3109/14756366.2012.743536. [DOI] [PubMed] [Google Scholar]
- 220.Khan K.M., Ambreen N., Taha M., Halim S.A., Zaheer ul H., Naureen S., Rasheed S., Perveen S., Ali S., Choudhary M.I. Structure-based design, synthesis and biological evaluation of β-glucuronidase inhibitors. J. Comput. Aided Mol. Des. 2014;28:577–585. doi: 10.1007/s10822-014-9745-z. [DOI] [PubMed] [Google Scholar]
- 221.Mohammed Khan K., Imran Fakhri M., Naveed Shaikh N., Muhammad Saad S., Hussain S., Perveen S., Iqbal Choudhary M. β-Glucuronidase inhibitory studies on coumarin derivatives. Med. Chem. 2014;10:778–782. doi: 10.2174/1573406410666140311093352. [DOI] [PubMed] [Google Scholar]
- 222.Taha M., Rahim F., Ali M., Khan M.N., Alqahtani M.A., Bamarouf Y.A., Gollapalli M., Farooq R.K., Shah S.A.A., Ahmed Q.U., Zakaria Z.A. Synthesis of chromen-4-one-oxadiazole substituted analogs as potent β-glucuronidase inhibitors. Molecules. 2019;24:1528. doi: 10.3390/molecules24081528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ahmad S., Hughes M.A., Lane K.T., Redinbo M.R., Yeh L.A., Scott J.E. A high throughput assay for discovery of bacterial β-glucuronidase inhibitors. Curr. Chem. Genom. 2011;5:13–20. doi: 10.2174/1875397301105010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.LoGuidice A., Wallace B.D., Bendel L., Redinbo M.R., Boelsterli U.A. Pharmacologic targeting of bacterial β-glucuronidase alleviates nonsteroidal anti-inflammatory drug-induced enteropathy in mice. J. Pharmacol. Exp. Ther. 2012;341:447–454. doi: 10.1124/jpet.111.191122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Saitta K.S., Zhang C., Lee K.K., Fujimoto K., Redinbo M.R., Boelsterli U.A. Bacterial β-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: mode of action and pharmacokinetics. Xenobiotica. 2014;44:28–35. doi: 10.3109/00498254.2013.811314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Wallace B.D., Roberts A.B., Pollet R.M., Ingle J.D., Biernat K.A., Pellock S.J., Venkatesh M.K., Guthrie L., O’Neal S.K., Robinson S.J., Dollinger M., Figueroa E., McShane S.R., Cohen R.D., Jin J., Frye S.V., Zamboni W.C., Pepe-Ranney C., Mani S., Kelly L., Redinbo M.R. Structure and inhibition of microbiome β-Glucuronidases essential to the alleviation of cancer drug toxicity. Chem. Biol. 2015;22:1238–1249. doi: 10.1016/j.chembiol.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Yauw S.T.K., Arron M., Lomme R., van den Broek P., Greupink R., Bhatt A.P., Redinbo M.R., van Goor H. Microbial glucuronidase inhibition reduces severity of diclofenac-induced anastomotic leak in rats. Surg. Infect. 2018;19:417–423. doi: 10.1089/sur.2017.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Roberts A.B., Wallace B.D., Venkatesh M.K., Mani S., Redinbo M.R. Molecular insights into microbial β-glucuronidase inhibition to abrogate CPT-11 toxicity. Mol. Pharmacol. 2013;84:208–217. doi: 10.1124/mol.113.085852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Pellock S.J., Creekmore B.C., Walton W.G., Mehta N., Biernat K.A., Cesmat A.P., Ariyarathna Y., Dunn Z.D., Li B., Jin J., James L.I., Redinbo M.R. Gut microbial β-glucuronidase inhibition via catalytic cycle interception. ACS Cent. Sci. 2018;4:868–879. doi: 10.1021/acscentsci.8b00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ahmad S., Hughes M.A., Yeh L.-A., Scott J.E. Potential repurposing of known drugs as potent bacterial β-glucuronidase inhibitors. J. Biomol. Screen. 2012;17:957–965. doi: 10.1177/1087057112444927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Kodawara T., Masuda S., Yano Y., Matsubara K., Nakamura T., Masada M. Inhibitory effect of ciprofloxacin on β-glucuronidase-mediated deconjugation of mycophenolic acid glucuronide. Biopharm Drug Dispos. 2014;35:275–283. doi: 10.1002/bdd.1894. [DOI] [PubMed] [Google Scholar]
- 232.Kong R., Liu T., Zhu X., Ahmad S., Williams A.L., Phan A.T., Zhao H., Scott J.E., Yeh L.-A., Wong S.T. Old drug new use-Amoxapine and its metabolites as potent bacterial β-glucuronidase inhibitors for alleviating cancer drug toxicity. Clin. Cancer Res. 2014 doi: 10.1158/1078-0432.CCR-14-0395. clincanres. 0395.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Yang W., Wei B., Yan R. Amoxapine demonstrates incomplete inhibition of β-glucuronidase activity from human gut microbiota. SLAS Discov. 2018;23:76–83. doi: 10.1177/2472555217725264. [DOI] [PubMed] [Google Scholar]
- 234.Tangeti V.S., Reddy B.H., Evangeline K.S. One pot multicomponent synthesis of anti-inflammatory active tetrahydrofuro[3,2-c]pyridinone-2-carboxylate derivatives. Asian J. Chem. 2018;30:403–410. doi: 10.14233/ajchem.2018.21039. [DOI] [Google Scholar]
- 235.Ali F., Khan K.M., Salar U., Iqbal S., Taha M., Ismail N.H., Perveen S., Wadood A., Ghufran M., Ali B. Dihydropyrimidones: as novel class of β-glucuronidase inhibitors. Bioorg. Med. Chem. 2016;24:3624–3635. doi: 10.1016/j.bmc.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 236.Barakat A., Islam M.S., Al-Majid A.M., Ghabbour H.A., Yousuf S., Ashraf M., Shaikh N.N., Iqbal Choudhary M., Khalil R., Ul-Haq Z. Synthesis of pyrimidine-2,4,6-trione derivatives: anti-oxidant, anti-cancer, α-glucosidase, β-glucuronidase inhibition and their molecular docking studies. Bioorg. Chem. 2016;68:72–79. doi: 10.1016/j.bioorg.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 237.Barakat A., Ghabbour H.A., Al-Majid A.M., Qurat-ul-ain, Imad R., Javaid K., Shaikh N.N., Yousuf S., Choudhary M.I., Wadood A. Synthesis, X-ray crystal structures, biological evaluation, and molecular docking studies of a series of barbiturate derivatives. J. Chem. 2016 doi: 10.1155/2016/8517243. 2016. [DOI] [Google Scholar]
- 238.Khan K.M., Saad S.M., Shaikh N.N., Hussain S., Fakhri M.I., Perveen S., Taha M., Choudhary M.I. Synthesis and β-glucuronidase inhibitory activity of 2-arylquinazolin-4(3H)-ones. Bioorg. Med. Chem. 2014;22:3449–3454. doi: 10.1016/j.bmc.2014.04.039. [DOI] [PubMed] [Google Scholar]
- 239.Taha M., Sultan S., Nuzar H.A., Rahim F., Imran S., Ismail N.H., Naz H., Ullah H. Synthesis and biological evaluation of novel N-arylidenequinoline-3-carbohydrazides as potent β-glucuronidase inhibitors. Bioorg. Med. Chem. 2016;24:3696–3704. doi: 10.1016/j.bmc.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 240.Chen Y.L., Zhao Y.L., Lu C.M., Tzeng C.C., Wang J.P. Synthesis, cytotoxicity, and anti-inflammatory evaluation of 2-(furan-2-yl)-4-(phenoxy)quinoline derivatives. Part 4. Bioorg. Med. Chem. 2006;14:4373–4378. doi: 10.1016/j.bmc.2006.02.039. [DOI] [PubMed] [Google Scholar]
- 241.Cheng K.W., Tseng C.H., Yang C.N., Tzeng C.C., Cheng T.C., Leu Y.L., Chuang Y.C., Wang J.Y., Lu Y.C., Chen Y.L., Cheng T.L. Specific inhibition of bacterial β-glucuronidase by pyrazolo[4,3-c]quinoline derivatives via a pH-dependent manner to suppress chemotherapy-induced intestinal toxicity. J. Med. Chem. 2017;60:9222–9238. doi: 10.1021/acs.jmedchem.7b00963. [DOI] [PubMed] [Google Scholar]
- 242.Cheng K.-W., Tseng C.-H., Tzeng C.-C., Leu Y.-L., Cheng T.-C., Wang J.-Y., Chang J.-M., Lu Y.-C., Cheng C.-M., Chen I.-J. Pharmacological inhibition of bacterial β-glucuronidase prevents irinotecan-induced diarrhea without impairing its antitumor efficacy in vivo. Pharmacol. Res. 2019;139:41–49. doi: 10.1016/j.phrs.2018.10.029. [DOI] [PubMed] [Google Scholar]
- 243.Khan K.M., Shujaat S., Rahat S., Hayat S. Atta ur, R., and Choudhary, M.I., Beta-N-cyanoethyl acyl hydrazide derivatives: a new class of β-glucuronidase inhibitors. Chem. Pharm. Bull. (Tokyo) 2002;50:1443–1446. doi: 10.1248/cpb.50.1443. [DOI] [PubMed] [Google Scholar]
- 244.Jamil W., Perveen S., Shah S.A., Taha M., Ismail N.H., Perveen S., Ambreen N., Khan K.M., Choudhary M.I. Phenoxyacetohydrazide Schiff bases: β-glucuronidase inhibitors. Molecules. 2014;19:8788–8802. doi: 10.3390/molecules19078788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Mabkhot Y.N., Barakat A., Yousuf S., Choudhary M.I., Frey W., Ben Hadda T., Mubarak M.S. Substituted thieno[2,3-b]thiophenes and related congeners: synthesis, β-glucuronidase inhibition activity, crystal structure, and POM analyses. Bioorg. Med. Chem. 2014;22:6715–6725. doi: 10.1016/j.bmc.2014.08.014. [DOI] [PubMed] [Google Scholar]
- 246.Taha M., Ismail N.H., Imran S., Wadood A., Rahim F., Al Muqarrabin L.M.R., Zaki H.M., Ahmat N., Nasir A., Khan F. Synthesis of novel disulfide and sulfone hybrid scaffolds as potent β-glucuronidase inhibitor. Bioorg. Chem. 2016;68:15–22. doi: 10.1016/j.bioorg.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 247.Perveen S., Mustafa S., Qamar K., Dar A., Khan K.M., Choudhary M.I., Khan A., Voelter W. Antiproliferative effects of novel urea derivatives against human prostate and lung cancer cells; and their inhibition of β-glucuronidase activity. Med. Chem. Res. 2014;23:1099–1113. doi: 10.1007/s00044-013-0702-5. [DOI] [Google Scholar]
- 248.Taha M., Ismail N.H., Jamil W., Khan K.M., Salar U., Kashif S.M., Rahim F., Latif Y. Synthesis and evaluation of unsymmetrical heterocyclic thioureas as potent β-glucuronidase inhibitors. Med. Chem. Res. 2015;24:3166–3173. [Google Scholar]
- 249.Doyle M.L., Katzman P.A., Doisy E. Production and properties of bacterial β-glucuronidase. J. Biol. Chem. 1955;217:921–930. [PubMed] [Google Scholar]
- 250.Asma M., Badshah A., Ali S., Sohail M., Fettouhi M., Ahmad S., Malik A. Synthesis, characterization of mixed ligand palladium (II) complexes of triphenylphosphine and anilines and their enzyme inhibition studies against β-glucuronidase. The crystal structure of trans-dichloro-(m-chloroaniline)(triphenylphosphine) palladium (II) Transition Met. Chem. 2006;31:556–559. [Google Scholar]
- 251.Al-Majid A.M., Yousuf S., Choudhary M.I., Nahra F., Nolan S.P. Gold-NHC complexes as potent bioactive compounds. Chemistry. 2016;1:76–80. doi: 10.1002/slct.201600009. [DOI] [Google Scholar]
- 252.Al-Majid A.M., Choudhary M.I., Yousuf S., Jabeen A., Imad R., Javeed K., Shaikh N.N., Collado A., Sioriki E., Nahra F., Nolan S.P. In vitro biological activities of gold(I) and gold(III) bis(N-heterocyclic carbene) complexes. Chemistry. 2017;2:5316–5320. doi: 10.1002/slct.201700795. [DOI] [Google Scholar]
- 253.Hashimoto K., Saito H., Ohsawa R. Glycopolymeric inhibitors of β-glucuronidase. III. Configurational effects of hydroxy groups in pendant glyco-units in polymers upon inhibition of β-glucuronidase. J. Polym. Sci., Part A: Polym. Chem. 2006;44:4895–4903. [Google Scholar]
- 254.Kawaguchi A.W., Okawa H., Hashimoto K. Synthesis of glycopolymers bearing mannaric pendants as inhibitors on the β-glucuronidase activity: the inhibition mechanisms of mannaric-and glucaric-compounds. J. Polym. Sci., Part A: Polym. Chem. 2009;47:2032–2042. [Google Scholar]
- 255.Kawaguchi A.W., Kaida T., Okawa H., Hashimoto K. The synthesis of the glycopolymers containing pendant D, L-xylaric and L-tartaric moieties and their inhibition behavior on the β-glucuronidase activity. Polym. J. 2008;40:944. [Google Scholar]
- 256.Lamster I.B. 2001. Method of Testing for Periodontal Disease. US 6,277,587 B1. [Google Scholar]
- 257.Boelsterli U.A. 2015. Methods of Treating Adverse Intestinal Effects of Non-sterodal Anti-inflammatory Drugs. US 2015/0011542 A1. [Google Scholar]
- 258.Tavares F.X., Wallace B.D., Petterson W. 2019. Compounds, Compositions, and Methods for Selectively Inhibiting β-glucuronidases and Alleviating Side Effects Associated with Drug Treatment Induced Diarrhea. WO 2019/051185 A1. [Google Scholar]
- 259.Redinbo M.R. 2016. Selective β-glucuronidase Inhibitors as a Treatment for Side Effects of Camptothecin Antineoplasticagents. US 9,334.288 B2. [Google Scholar]
- 260.Redinbo M.R., Jin J., James L., Pellock S. 2018. Inhibitors of Microbial β-glucuronidase Enzymes and Uses Thereof. WO 2018/017874 Al. [Google Scholar]
- 261.Chen Y.-L., Cheng T.-L., Tzeng C.-C., Tseng C.-H., Cheng T.-C., Cheng K.-W., Luo W.-F. 2018. Pyrazolo [4,3-c]quinoline Derivatives for Inhibition of β-glucuronidase. US 2018/0214426 A1. [Google Scholar]
- 262.Choudhary M.I., Khan K.M., Shaikh N.N., Saad S.M., Yousuf S., Rahman A.-u. 2014. Quinazolines as β-glucuronidase Novel Inhibitors. US 2014/0256754 A1. [Google Scholar]
- 263.Yousuf M., Haq Z., Shaikh N.N., Choudhary M.I. 2017. In-silico Based Techniques in the Identification of Potent β-glucoronidase Inhibitors. US 2017/0009272 A1. [Google Scholar]
- 264.Banowski B., Hoffmann D., Wadle A., Siegert P., Saettler A., Gerke T. 2007. β-Glucuronidase Inhibitors for Use in Deodorants and Antiperspirants. US 7,294,330 B2. [Google Scholar]
- 265.Mori I., Akiba S. 2015. β-Glucuronidase Inhibitor. US 9.200,269 B2. [Google Scholar]
- 266.Williams A.L., Scott J., Yeh L.-A., Redinbo M.R. 2017. Phenoxy Thiophene Sulfonamides and Their Use as Inhibitors of Glucuronidase. US 9,617.239 B2. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.