

Abstract
We report the catalytic alkylation of amide derivatives, which relies on the use of nonprecious metal catalysis. Amide derivatives are treated with organozinc reagents, utilizing nickel catalysis, to yield ketone products. The methodology is performed at ambient temperature and is tolerant of variation in both coupling partners. A precursor to a nanomolar glucagon receptor modulator was synthesized using the methodology, underscoring the mild nature of this chemistry and its potential utility in pharmaceutical synthesis. These studies are expected to further promote the use of amides as synthetic building blocks.
Keywords: nickel, catalysis, alkylation, amides, cross-coupling
Graphical Abstract
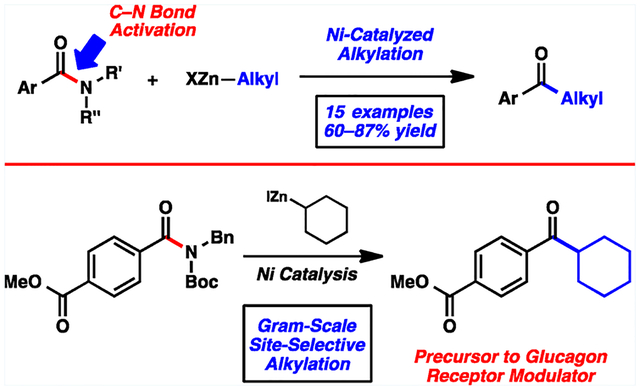
The ability to activate traditionally unreactive functional groups as synthons continues to be a vital area of research. One particularly stable functionality is the amide.1 The resonance stabilization of amides has been well understood for decades;1,2 consequently, the use of amides in C–N bond cleavage reactions has remained limited. Recently, however, there has been much interest in breaking amide C–N bonds to forge new C–heteroatom and C–C bonds.3–7 Such methodologies provide new tactics to prepare acyl derivatives, but with the key benefit of amide stability. The use of amides in multistep synthesis, followed by selective C–N bond activation and coupling, should ultimately prove advantageous in the synthesis of complex molecules.
The present study focuses on activating and coupling amides to build acyl C–C bonds in an intermolecular fashion (Figure 1). Such catalytic methodology would complement Weinreb amide chemistry, but without the use of highly basic and pyrophoric organometallic reagents.8 Prior contributions in this area include Suzuki–Miyaura couplings (1 → 2) reported by Zou (Pd),4 Szostak (Pd),5 and our laboratory (Ni).3b In each of these cases, the nucleophilic coupling partner was restricted to aryl boronate species, thus limiting the application of this methodology. The corresponding alkylative coupling (1 → 3) would be highly desirable, given the prevalence of alkyl ketones in molecules of biological importance and the versatility of alkyl ketones as synthetic building blocks. Herein, we report the first alkylative cross-coupling of amide derivatives.
Figure 1.

Nickel-catalyzed C–C bond forming reactions from amides.
Following unsuccessful attempts to couple amide derivatives with aliphatic boronic acids and esters, we opted to pursue the use of organozinc reagents as cross-coupling partners.9 Our earlier studies have relied on the use of nickel catalysis for amide C–N bond activation,3 which is notable, given that nickel is less expensive, more abundant, and displays a smaller CO2 footprint, compared to its precious metal counterpart, palladium.10 Catalytic acyl couplings11 with organozinc reagents are well-precedented using acid halides (Pd or Ni),12 anhydrides (Pd, Ni, or Rh),12a,13 and thioesters (Pd or Ni),12a,b,14 but the corresponding coupling of amides has not been reported.
To initiate our study, we examined the coupling of naphthamides 4 with benzylzinc bromide (5) in the presence of catalytic Ni(cod)2 and the NHC ligand SIPr in THF (Scheme 1). Although several amide derivatives failed to undergo the coupling (entries 1–3), we were delighted to find that N-alkyl,Boc and N-alkyl,Ts derivatives could be utilized (entries 4 and 5, respectively).15 N-Alkyl,Ts amides (e.g., 4e) are well-suited for use in multistep synthesis.16 Notably, the successful reactions of 4d and 4e proceeded at room temperature, which compares favorably to the few existing examples of catalytic amide C–N bond activation (ca. 50–160 °C),3–7 and highlights the mild nature of this coupling.
Scheme 1. Survey of Amide N-Substituents in the Coupling of Substrates 4 with 5a.

aConditions: Ni(cod)2, 10 mol %; SIPr, 10 mol %; substrate, 4, 1.0 equiv; benzylzinc bromide (5), 1.5 equiv; and THF, 1.0 M at 23 °C for 24 h. bYields determined by 1H NMR analysis using hexamethylbenzene as an internal standard.
Having found that the alkylative coupling of amide derivatives was indeed possible,17 we evaluated the scope of the amide substrate (Figure 2). The use of the parent naphthyl substrate gave 6 in 80% isolated yield. In addition, it was found that the methodology was not restricted to extended aromatics. For example, the substrate derived from benzoic acid coupled smoothly to furnish 7 in 74% yield. Substrates bearing electron-donating groups could also be employed, as demonstrated by the formation of 8–10. From the latter two cases, it should be emphasized that the presence of tertiary amines does not hinder catalysis. As shown by the formation of 11 and 12, the electron-withdrawing −F and −CF3 substituents were also tolerated.18
Figure 2.

Scope of the amide substrate. (Conditions, unless otherwise stated: Ni(cod)2, 10 mol %; SIPr, 10 mol %; substrate, 1.0 equiv; benzylzinc bromide (5), 1.5 equiv; and THF, 1.0 M at 23 °C for 24 h. Yields shown reflect the average of two isolation experiments. The superscripted symbol “a” in the figure denotes that the corresponding N-Bn,Boc benzamide derivative was used.)
Additionally, we examined the scope of the organozinc reagent in this methodology (Figure 3).19,20 n-Propylzinc bromide was successfully employed to furnish 13 in 80% yield. To assess the tolerance of the methodology toward β-branching, neopentylzinc iodide, a very hindered nucleophile was tested and found to undergo the desired coupling to furnish 14. α-Branched nucleophiles could also be employed, as judged by the formation of 15 and 16. Notably, couplings utilizing secondary organozinc reagents are known to be challenging.21 Finally, cyclopentyl and cyclohexyl organozinc reagents underwent the desired coupling in good yield to deliver products 17 and 18, respectively.
Figure 3.

Scope of the organozinc coupling partner. (Conditions, unless otherwise stated: Ni(cod)2, 10 mol %; SIPr, 10 mol %; substrate, 1.0 equiv; organozinc reagent, 1.5 equiv; and THF, 1.0 M at 23 °C for 24 h. Yields shown reflect the average of two isolation experiments.)
The alkylative cross-coupling methodology was further probed in a synthetic application (Scheme 2). On a gram-scale, amide derivative 19 was coupled with cyclohexylzinc iodide (20) using our optimal nickel-catalyzed reaction conditions. This transformation provided ketone 21 in 71% yield without disturbing the ester.22 Ketone 21 is an intermediate in Pfizer’s synthesis of the glucagon receptor modulator 22.23 The cross-coupling route to 21 provides a favorable alternative to the known Weinreb amide displacement chemistry described in the literature, which proceeds in 34% yield.23
Scheme 2.

Gram-Scale Coupling To Form Ketone 21
In summary, we have developed the first catalytic alkylation of amide derivatives. The transformation involves the coupling of N-alkyl,Ts or N-alkyl,Boc amides with organozinc reagents using nickel catalysis. The methodology proceeds at room temperature and is tolerant of variation in both the substrate and nucleophilic coupling partner. The synthesis of 21 underscores the mildness and scalability of this methodology, along with the applicability of this technology to pharmaceutical synthesis. As such, we expect that these studies will further promote the use of amides as synthetic building blocks for use in the synthesis of drugs and natural products.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to Boehringer Ingelheim, Bristol-Myers Squibb, the Camille and Henry Dreyfus Foundation, the A. P. Sloan Foundation, the University of California, Los Angeles (UCLA), the National Science Foundation (NSF) (N.A.W., No. DGE-1144087), the Foote Family (N.A.W. and J.E.D.), and the UCLA Gold Shield Alumnae for financial support. These studies were supported by shared instrumentation grants from the NSF (No. CHE-1048804) and the National Institutes of Health National Center for Research Resources (NIH NCRR) (No. S10RR025631).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.6b00793.
Detailed experimental and compound characterization data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science; Greenberg A, Breneman CM, Liebman JF, Eds. Wiley: New York, 2003; pp 1–672. [Google Scholar]
- (2).Pauling L; Corey RB Proc. Natl. Acad. Sci. U. S. A 1951, 37, 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Hie L; Fine Nathel NF; Shah TK; Baker EL; Hong X; Yang Y-F; Liu P; Houk KN; Garg NK Nature 2015, 524, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weires NA; Baker EL; Garg NK Nat. Chem 2015, 8, 75–79. [DOI] [PubMed] [Google Scholar]
- (4).Li X; Zou G Chem. Commun 2015, 51, 5089–5092. [DOI] [PubMed] [Google Scholar]
- (5).(a) Meng G; Szostak M Org. Lett 2015, 17, 4364–4367. [DOI] [PubMed] [Google Scholar]; (b) Meng G; Szostak M Org. Biomol. Chem 2016, DOI: 10.1039/C6OB00084C. [DOI] [PubMed] [Google Scholar]
- (6).For the activation of strained β-lactams using Pd, see:; Yada A; Okajima S; Murakami MJ Am. Chem. Soc 2015, 137, 8708–8711. [DOI] [PubMed] [Google Scholar]
- (7).For amide activation, accompanied by decarbonylation, see:; (a) Meng G; Szostak M Angew. Chem., Int. Ed 2015, 54, 14518–14522. [DOI] [PubMed] [Google Scholar]; (b) Meng G; Szostak M Org. Lett 2016, 18, 796–799. [DOI] [PubMed] [Google Scholar]
- (8).Nahm S; Weinreb SM Tetrahedron Lett. 1981, 22, 3815–3818. [Google Scholar]
- (9).Organozinc halides are functional group tolerant and are not known to undergo nucleophilic attack on ketones, esters, or amides (including Weinreb amides). For pertinent discussions, see:; (a) Cárdenas DJ Angew. Chem., Int. Ed 2003, 42, 384–387. [DOI] [PubMed] [Google Scholar]; (b) Knochel P; Singer RD Chem. Rev 1993, 93, 2117–2188. [Google Scholar]
- (10).(a) Rosen BM; Quasdorf KW; Wilson DA; Zhang N; Resmerita A-M; Garg NK; Percec V Chem. Rev 2011, 111, 1346–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tasker SZ; Standley EA; Jamison TF Nature 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mesganaw T; Garg NK Org. Process Res. Dev 2013, 17, 29–39. [Google Scholar]; (d) Ananikov VP ACS Catal. 2015, 5, 1964–1971. [Google Scholar]
- (11).For the coupling of acid chlorides with alkyl Grignard reagents, see:; Scheiper B; Bonnekessel M; Krause H; Fürstner AJ Org. Chem 2004, 69, 3943–3949. [DOI] [PubMed] [Google Scholar]
- (12).(a) Zhang Y; Rovis TJ Am. Chem. Soc 2004, 126, 15964–15965. [DOI] [PubMed] [Google Scholar]; (b) Cherney AH; Reisman SE Tetrahedron 2014, 70, 3259–3265. [Google Scholar]; (c) Harada T; Kotani Y; Katsuhira T; Oku A Tetrahedron Lett. 1991, 32, 1573–1576. [Google Scholar]; (d) Negishi E.-i.; Bagheri V; Chatterjee S; Luo F-T; Miller JA; Stoll AT Tetrahedron Lett. 1983, 24, 5181–5184. [Google Scholar]; (e) Iwai T; Nakai T; Mihara M; Ito T; Mizuno T; Ohno T Synlett 2009, 1091–1094. [Google Scholar]; (f) Grey RA J. Org. Chem 1984, 49, 2288–2289. [Google Scholar]; (g) Sato T; Naruse K; Enokiya M; Fujisawa T Chem. Lett 1981, 10, 1135–1138. [Google Scholar]
- (13).(a) Wang D; Zhang Z Org. Lett 2003, 5, 4645–4648. [DOI] [PubMed] [Google Scholar]; (b) Cook MJ; Rovis T Synthesis 2009, 335–338. [Google Scholar]; (c) Bercot EA; Rovis TJ Am. Chem. Soc 2002, 124, 174–175. [DOI] [PubMed] [Google Scholar]; (d) Bercot EA; Rovis TJ Am. Chem. Soc 2005, 127, 247–254. [DOI] [PubMed] [Google Scholar]; (e) Johnson JB; Cook MJ; Rovis T Tetrahedron 2009, 65, 3202–3210. [Google Scholar]; (f) Rogers RL; Moore JL; Rovis T Angew. Chem., Int. Ed 2007, 46, 9301–9304. [DOI] [PubMed] [Google Scholar]; (g) Bercot EA; Rovis TJ Am. Chem. Soc 2004, 126, 10248–10249. [DOI] [PubMed] [Google Scholar]; (h) Johnson JB; Yu RT; Fink P; Bercot EA; Rovis T Org. Lett 2006, 8, 4307–4310. [DOI] [PubMed] [Google Scholar]; (i) Johnson JB; Bercot EA; Rowley JM; Coates GW; Rovis TJ Am. Chem. Soc 2007, 129, 2718–2725. [DOI] [PubMed] [Google Scholar]; (j) Cook MJ; Rovis TJ Am. Chem. Soc 2007, 129, 9302–9303. [DOI] [PubMed] [Google Scholar]
- (14).(a) Tokuyama H; Yokoshima S; Yamashita T; Fukuyama T Tetrahedron Lett. 1998, 39, 3189–3192. [Google Scholar]; (b) Mori Y; Seki M Tetrahedron Lett. 2004, 45, 7343–7345. [Google Scholar]; (c) Shimizu T; Seki M Tetrahedron Lett. 2002, 43, 1039–1042. [Google Scholar]; (d) Miyazaki T; Han-ya Y; Tokuyama H; Fukuyama T Synlett 2004, 477–480. [Google Scholar]; (e) Shimizu T; Seki M Tetrahedron Lett. 2001, 42, 429–432. [Google Scholar]; (f) Mori Y; Seki M Adv. Synth. Catal 2007, 349, 2027–2038. [Google Scholar]
- (15).The role of the N-substituents in amide C–N bond cleavage reactions is currently under investigation and will be described elsewhere in due course.
- (16).N-Alkyl,Ts amides can be readily prepared by sulfonamide coupling of the corresponding carboxylic acid or acid halide (see the Supporting Information). For a discussion of the robustness of sulfonamides and their stability, see:; Searles S; Nukina S Chem. Rev 1959, 59, 1077–1103. [Google Scholar]
- (17).Substrates derived from aliphatic carboxylic acids do not couple under the reported reaction conditions; studies to overcome this limitation are currently underway.
- (18).Lower yields of 12 were obtained using the corresponding N-Me,Ts benzamide substrate. Generally, amides derived from electron-poor arenes were found to couple in higher yields when the N-Bn,Boc derivatives were employed.
- (19).The organozinc bromide or iodide was used in accord with literature precedent for the formation of each organozinc species. Generally, alkyl bromides and iodides are known to undergo organozinc formation more readily than alkyl chlorides; see ref 9b.
- (20).Although primary and secondary organozinc species were well tolerated in the coupling, it was found that couplings with tertiary organozinc halides and organozinc reagents bearing heterocycles, acetals, and esters gave only trace amounts of product.
- (21).Han C; Buchwald SL J. Am. Chem. Soc 2009, 131, 7532–7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).For the Ni-catalyzed activation of methyl esters, see:; Hie L; Fine Nathel NF; Hong X; Yang Y-F; Houk KN; Garg NK Angew. Chem., Int. Ed 2016, 55, 2810–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]; (see ref 10 for alternative examples of ester activation using nickel catalysis).
- (23).Apnes GE; Didluk MT; Filipski KJ; Guzman-Perez A; Lee ECY; Pfefferkorn JA; Stevens BD; Tu MM Glucagon Receptor Modulators. U.S. Patent 20120202834, August 9, 2012.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.