

Abstract
Cardiovascular disease is the leading cause of mortality worldwide. While reperfusion therapy is vital for patient survival post-heart attack, it also causes further tissue injury, known as myocardial ischemia/reperfusion (I/R) injury in clinical practice. Exploring ways to attenuate I/R injury is of clinical interest for improving post-ischemic recovery. A platelet-inspired nanocell (PINC) that incorporates both prostaglandin E2 (PGE2)-modified platelet membrane and cardiac stromal cell-secreted factors to target the heart after I/R injury is introduced. By taking advantage of the natural infarct-homing ability of platelet membrane and the overexpression of PGE2 receptors (EPs) in the pathological cardiac microenvironment after I/R injury, the PINCs can achieve targeted delivery of therapeutic payload to the injured heart. Furthermore, a synergistic treatment efficacy can be achieved by PINC, which combines the paracrine mechanism of cell therapy with the PGE2/EP receptor signaling that is involved in the repair and regeneration of multiple tissues. In a mouse model of myocardial I/R injury, intravenous injection of PINCs results in augmented cardiac function and mitigated heart remodeling, which is accompanied by the increase in cycling cardiomyocytes, activation of endogenous stem/progenitor cells, and promotion of angiogenesis. This approach represents a promising therapeutic delivery platform for treating I/R injury.
Keywords: biomimetics, drug delivery, ischemic injury, platelets, stromal cell secretome
1. Introduction
Cardiovascular disease is the leading cause of mortality globally, accounting for over 17.3 million deaths each year. Coronary heart disease, including myocardial infarction (commonly known as heart attack), angina (chest pain), and cardiac arrest, contributes to at least 50% of deaths.[1] Coronary artery obstruction produces myocardial ischemia, which is usually treated with reperfusion therapy. While reperfusion of ischemic tissue is vital for survival, it also initiates myocardial ischemia/reperfusion (I/R) injury comprising oxidative damage, cell death, and a profound inflammatory immune response, which currently lacks an effective clinical therapy.[2]
In the past decade, the potential of using stem cells, including mesenchymal stem cells (MSCs), resident cardiac stem/stromal cells (CSCs), and induced pluripotent stem cells (iPSCs) for cardiac regenerative therapy has generated immense interest.[3] Numerous studies indicate that adult stem cells such as MSCs and CSCs have very limited, if any, ability to differentiate into cardiomyocytes. Those cells exert their functional benefits mainly through paracrine effects, i.e., secreted factors from stem cells promote cardiac regeneration and inhibit fibrosis and inflammation. However, cell therapy suffers from several limitations, such as low cellular retention and survival in the ischemic myocardium, special precautions needed during cryopreservation, and easy entrapment in the lung during intravenous delivery due to the cell size.[4,5]
Nanotechnology holds great promise to revolutionize cardiovascular therapy. In recent years, the development of nanoparticles for active targeting the heart after ischemic injury is receiving increasing attention.[6–8] The enhanced permeability and retention (EPR) effect of the leaky vasculature in the infarcted heart has been commonly used for designing targeted nanoparticles, although these nanoparticles are limited by rapid clearance within a few hours to days.[7] A recently reported matrix metalloproteinase (MMP)-responsive nanoparticle showed successful retention in the infarcted heart for up to 28 d.[8] Yet, the therapeutic efficacy of these nanoparticle systems in protecting the heart from I/R injury still remains elusive.
Recently, cell membrane-coated nanoparticles have emerged as a novel platform that can successfully combine the functionalities of various types of cells.[9–14] We previously reported that fusing platelet-derived nanovesicles onto the surface of CSCs boosts the infarct-targeting ability and functional outcomes of CSCs in rats and pigs with myocardial infarction.[15] We also fabricated therapeutic cell-mimicking microparticles by encasing CSC- or MSC-secreted factors in a biodegradable and biocompatible poly(lactic-co-glycolic acid) (PLGA) shell coated with cell membranes and tested their regenerative potential in a rodent model of heart injury.[16,17] Prostaglandin E2 (PGE2) is an FDA-approved medication (known as dinoprostone) that participates in many biological pathways. PGE2 exerts its physiologic effects via four subtypes of receptors (EPs), i.e., EP1, EP2, EP3, and EP4, among which EP2, EP3, and EP4 are overexpressed on the surface of cardiomyocytes following I/R injury.[18,19] Recent studies have identified PGE2 as an important signaling molecule that activates endogenous stem/progenitor cells for cardiac repair post-ischemic injury.[20]
Based on those concepts, we designed a platelet-inspired nanocell (PINC) that has a CSC core and a platelet membrane shell. The CSC core consists of therapeutic CSC secretome encapsulated in a PLGA nanoparticle. PLGA has been widely used in drug delivery system due to its capability of protecting growth factors from degradation while allowing for sustained release of growth factors and has been approved by the U. S. Food and Drug Administration for clinical applications.[16] In addition, recent studies showed that CSC secretome exhibited therapeutic benefits similar to CSCs for the treatment of cardiovascular disease.[17,21–23] The platelet membrane is conjugated with PGE2 which is expected to both enhance targeting to cardiovascular cells and facilitate the endogenous repair through PGE2/EP receptor signaling after I/R injury (Figure 1A). As a novel biomimetic therapeutic nanoparticle, PINC offers the following advantages compared to natural stem cells:
Figure 1.
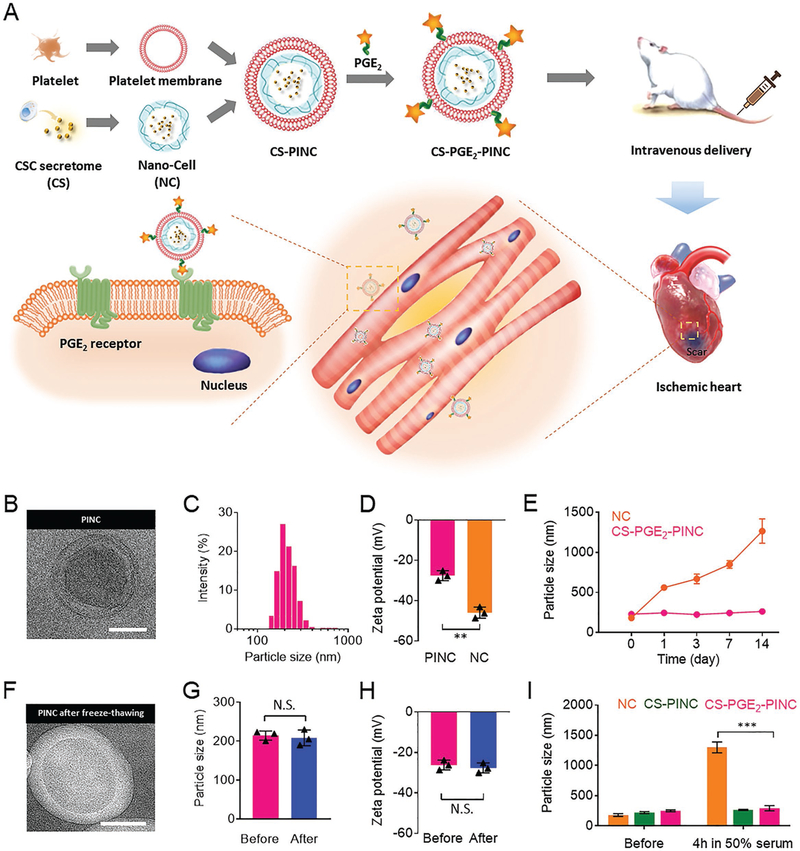
Fabrication and characterization of PINCs. A) Schematic illustration of the fabrication process of PINCs. The therapeutic effects of PINC injection were tested in mice with myocardial I/R injury. B) TEM image showing the ultrastructure of CS-PGE2-PINC. C) Size distribution of CS-PGE2-PINC measured by DLS. D) Zeta potentials of CS-PGE2-PINC and NC. E) Particle sizes of bare NC and CS-PGE2-PINC over 2 weeks in PBS. F) TEM image showing the ultrastructure of CS-PGE2-PINC after freeze-thawing. G) The comparison of particle size and H) zeta potential of CS-PGE2-PINC before and after freeze-thawing. I) In vitro stability of NC, CS-PINC, and CS-PGE2-PINC before and after incubation in 50% fetal bovine serum. Scale bars, 100 nm. All data are mean ± s.d. * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001; N.S., no statistical significance. Comparisons between any two groups were performed using two-tailed unpaired Student’s t-test. Comparisons among more than two groups were performed using one-way ANOVA followed by post hoc Bonferroni test.
Systemic administration: the nanometer size of PINC enables intravenous application; unlike stem cells, PINCs are less likely to clog the lungs.
Dual targeting: the platelet membrane on PINCs enhances targeting to the injured blood vessels while the PGE2 enhances targeting to the injured cardiomyocytes in I/R injury.
Stability: unlike real stem cells, PINCs can be readily manipulated and cryopreserved since there are no living components.
In this study, we fabricated PINCs and tested their in vitro bioactivity, biodistribution, and functional benefits in an immunocompetent mouse model of myocardial I/R injury.
2. Results and Discussion
2.1. Synthesis and Characterization of PINCs
To substantiate our design, we combine a double emulsion-based solvent evaporation/extraction process with cell membrane cloaking to prepare the PINCs (Figure 1A). The CSCs were derived from adult human hearts using the previously described cardiac explant method. Those cells consistently express CD105, partially express CD90, but do not express c-kit, CD31, CD34, or CD45, suggesting a phenotype related to cardiac stromal cells in the heart. Such cells have very limited ability to differentiate into cardiomyocytes but can secrete a variety of regenerative factors and exosomes. The conditioned medium (secretome) from CSCs was used to produce our therapeutic nanoparticles. First, the secretome derived from CSCs was incorporated into PLGA to form nanocells (NCs) through a double emulsion method. The CSC secretome loading capacity and efficiency were 2.8 wt% and 85.3%, respectively, showing that the CSC secretome was efficiently encapsulated into the hydrophilic core of NCs. Platelet membranes were then isolated and purified from the platelet-rich plasma through gradient centrifugation.[15] In order to prepare the PINC functionalized with PGE2, the amine groups of platelet membrane glycoproteins were further reacted with the terminal carboxyl group of PGE2 using N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide (EDC)/N-hydroxysuccinimide (NHS) as a linker to obtain the PGE2-platelet membrane conjugate. The content of PGE2 conjugated to the PGE2-platelet membrane conjugate was determined to be 0.68 mg g−1 dry platelet membrane by enzyme-linked immunosorbent assay (ELISA), with a high conjugation yield of over 95% (see Section 4 and Figure S1, Supporting Information). The conjugation of PGE2 onto the surface of platelet membrane was validated by confocal laser scanning microscopy (CLSM). The colocalization of the fluorescence signals from the DiI-labeled platelet membrane (red) and fluorescein isothiocyanate-tagged PGE2 (green) substantiated the successful conjugation of PGE2 onto the platelet membrane surface (Figure S2, Supporting Information). The resulting PGE2-platelet membrane conjugate was subsequently incubated with the CSC secretome-loaded NC under ultrasonication, followed by membrane extrusion to form the PGE2-plateletmembrane-coated NC (designated CS-PGE2-PINC). The PINC functionalized with only CSC secretome (designated CS-PINC) was prepared via coating the purified platelet membrane on the surface of NC. The PINC functionalized with only PGE2 (designated PGE2-PINC) was prepared via coating the PGE2platelet membrane conjugate on the surface of empty PLGA nanoparticles. Transmission electron microscopy (TEM) studies confirmed the platelet membrane coating on the CS-PGE2PINCs that appear as a core–shell structure (Figure 1B). Dynamic light scattering (DLS) analysis indicated that the CS-PGE2-PINCs had an average diameter of about 195 nm and a narrow size distribution (polydispersity index (PDI) = 0.157) (Figure 1B, C). Nanoparticle tracking analysis using NanoSight revealed that the majority of particles showed a particle size of about 205 nm, consistent with the results obtained from TEM and DLS measurements (Figure S3, Supporting Information). We employed a bicinchoninic acid (BCA)-based protein assay to determine the efficiency of membrane coating by comparing the difference between the platelet membrane protein content before membrane coating and after pelleting the platelet-membrane-coated PLGA nanoparticles from the remaining free membrane vesicles. The amount of membrane protein coated onto the PLGA core was estimated to be 0.143 pg membrane protein per nanoparticle, with a high coating efficiency of approximately 92% (see Section 4).
After cloaking with platelet membrane, the size of PINCs did not change significantly compared to the bare NC (Figure S4, Supporting Information), while the zeta potential of CS-PGE2PINC increased by about 18 mV compared with bare NC, approaching the value of −27 mV (Figure 1D and Figure S5, Supporting Information). This phenomenon is consistent with the previous analyses of nanoparticles after platelet membrane coating, which can be ascribed to the veiling of the highly negative PLGA core with the less negatively charged platelet membrane.[9,11,24] To determine the stability of different nanoformulations in solution over time, NCs and CS-PGE2-PINCs were stored in phosphate buffered saline (PBS, 1 ×, pH 7.4) at room temperature, respectively, and their size change was monitored by DLS. The PINCs exhibited stable size over a 2-week study period, while the NCs showed rapidly agglomeration in PBS (Figure 1E and Figure S6, Supporting Information). In addition, the cloaking of the platelet membrane endowed the PINCs with superior stability before and after incubation in 50% serum when compared with bare NC (Figure 1I). The small size, negatively charged cell-mimicking surface, and superior serum stability make the PINCs ideal for intravenous application. We further investigated the long-term storage stability of CS-PGE2-PINCs. After cryopreservation for over 3 months, the thawed CS-PGE2-PINCs exhibited similar morphology, size, and surface charge to those characteristics before freezing (Figure 1F–H, and Figure S7, Supporting Information). Furthermore, all the PINC formulations exhibited excellent lyophilization stability, with the size and zeta potential remained nearly identical before lyophilization and after resuspension (Figure S8, Supporting Information).
2.2. In Vitro Bioactivity of PINCs
Stem cell therapy represents a promising strategy for treating ischemic heart diseases.[25] Mounting lines of preclinical and clinical evidence indicate that stem cells, including CSCs and MSCs, exert their functional benefits through the secretion of paracrine factors, acting like “mini-drug pumps” to promote endogenous repair.[26,27] We have previously fabricated therapeutic cell-mimicking microparticles by packaging stem cell factors in a biodegradable polymeric shell and tested their regenerative potential in rodent models of heart injury.[16,17] In this study, we sought to investigate whether our PINCs could mimic CSCs by secreting regenerative growth factors. ELISA revealed that the CS-PGE2-PINCs continuously released pro-myogenic and pro-angiogenic paracrine factors, such as stromal cell-derived factor-1 (SDF-1), vascular endothelial growth factor (VEGF), and hepatocyte growth factor (HGF) for at least 14 d; the platelet membrane coating did not affect the release of stem cell factors from PINCs (Figure 2A–C). SDS-PAGE was used to run platelet membranes and all the different PINCs for protein composition analysis. As expected, all the PINCs had protein profiles that are similar to that of the platelet membrane. Western blot analysis further revealed the distinct expression of primary platelet membrane markers including CD42b (GPIba) and CD36 (GPIV) on all the PINCs, further confirming the successful platelet membrane coating onto PINCs (Figure 2D and Figure S9, Supporting Information). CD42b is one of the major adhesion molecules that regulate the binding of platelets to the injured vasculature and plays an important role in homing the platelet-nanovesicle decorated CSCs to the ischemic heart after I/R injury.[15,24] Therefore, the PINCs inherited the binding motifs of platelets. Owing to the platelet-mimicking properties, the PINCs showed robust binding to the collagen-coated surface (Figure S10, Supporting Information). In addition, DiI-labeled PINCs were plated onto green fluorescent protein-tagged human umbilical vein endothelial cells (GFP-HUVECs) cultured on the collagen-coated surface, and the selective adherence of PINCs to the collagen region was confirmed (Figure 2E, F).
Figure 2.
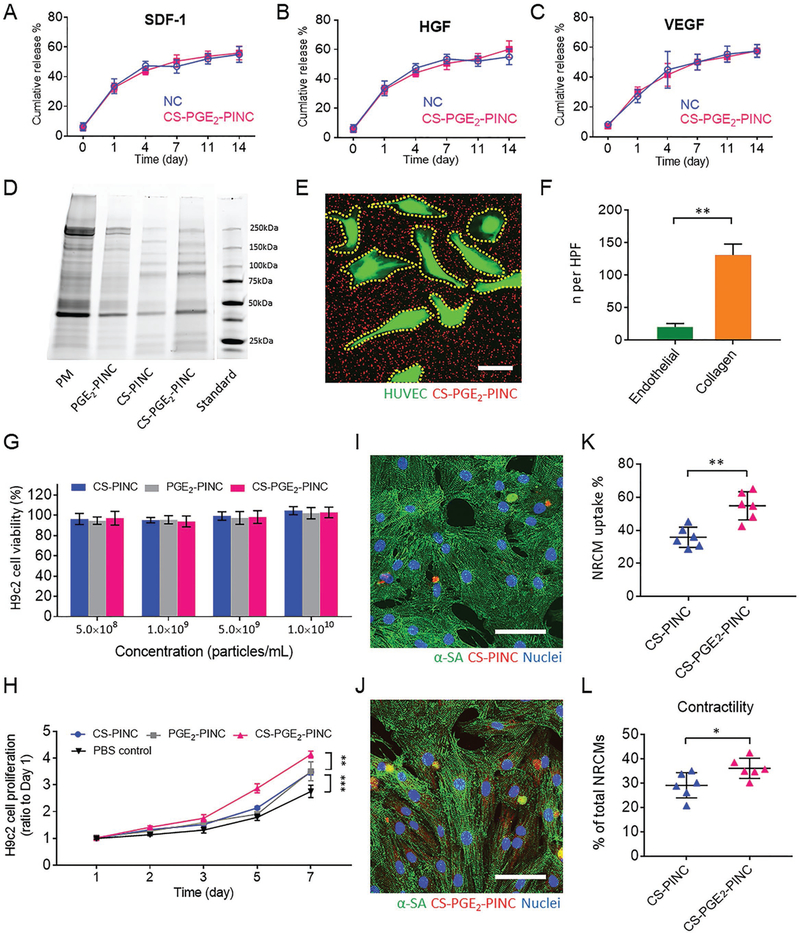
In vitro bioactivity of PINCs. A–C) Quantitative analysis of the releases of SDF-1, HGF, VEGF from NC and CS-PGE2-PINC over 2 weeks. D) Protein content visualization of platelet membrane (PM), PGE2-PINC, CS-PINC, and CS-PGE2-PINC run on SDS-PAGE at equivalent protein concentrations. E) Collagen-coated four-well slides seeded with HUVECs were incubated with CS-PGE2-PINCs for 60 s, followed by fluorescence microscopy showing selective CS-PGE2-PINC adherence to exposed collagen versus endothelial surfaces. F) Quantification of CS-PGE2-PINC in endothelial- and collagen-covered surface, respectively. G) Cytocompatibility of PINCs at various concentrations. H) The proliferation of H9c2 cells over time in the presence of different PINCs. I, J) Representative confocal image showing the internalization of I) CS-PINC and J) CS-PGE2-PINC by NRCMs. K) Quantitative analysis of the percentage of NRCMs with different nanoparticle endocytosis. L) Quantitative analysis of NRCM contractility when cocultured with different PINCs. Scale bars, E) 20 μm; I, J) 50 μm. All data are mean ± s.d. Comparisons between any two groups were performed using two-tailed unpaired Student’s t-test. Comparisons among more than two groups were performed using one-way ANOVA followed by post hoc Bonferroni test. * indicates p < 0.05, ** indicates p < 0.01, and *** indicates p < 0.001.
Next, we tested the effect of the PINCs on the H9c2 cardiomyoblasts, a widely used cardiac cell line isolated from the embryonic rat heart tissue. The CS-PINCs, PGE2PINCs, and CS-PGE2-PINCs have excellent cytocompatibility as confirmed by the cell viability assay. The cells maintain high viability upon exposure to the PINCs (> 95% viability), regardless of nanoparticle concentration (Figure 2G). In addition, a cell proliferation assay using a cell count kit-8 (CCK-8) revealed that the CSC secretome shows similar bioactivity before and after being encapsulated into PLGA nanoparticles (Figure S11, Supporting Information) and that PINCs promote the growth of H9c2 cardiomyoblasts, indicating that the release of therapeutic stem cell factors from the PINCs promote cell attachment and proliferation, consistent with our previous studies.[15–17,28] The H9c2 cells treated with CS-PGE2-PINCs exhibited significantly higher proliferative potential than those treated with CS-PINCs or PGE2-PINCs (Figure 2H). In contrast, the non-secretome-encapsulated PLGA nanoparticles, either with or without platelet membrane coating, were not able to promote H9c2 cardiomyoblast proliferation (Figure S12, Supporting Information). Furthermore, after cryopreservation for 3 months, the thawed CS-PGE2-PINCs exhibited comparable potency in promoting the growth of H9c2 cells to the freshly prepared controls, indicating the excellent cryostatbility of CS-PGE2-PINCs (Figure S13, Supporting Information). We further investigated the effect of PGE2 decoration on the cardiomyocyte protective ability of PINCs in vitro. Neonatal rat cardiomyocytes (NRCMs) were cocultured with DiI-labeled CS-PINCs or CS-PGE2-PINCs (red, Figure 2I, J) with equivalent concentrations. After coculturing for 3 h, the uptake of CS-PGE2-PINCs into NRCMs (stained by α-sarcomeric actinin (α-SA), green) was significantly higher than that of the nanoparticles without PGE2 decoration (Figure 2K). Furthermore, CS-PGE2-PINCs significantly promoted NRCM contractility compared with CS-PINCs (Figure 2L). Following exposure to serum-free medium supplemented with hydrogen peroxide (250 × 10−6 m) for 3 h, which simulates an ischemic microenvironment, TUNEL staining showed that the NRCMs pretreated with CS-PGE2-PINCs were less apoptotic than those pretreated with CS-PINCs (Figure S14, Supporting Information). Together, these results suggest the enhanced heart-targeting ability and regenerative potential of CS-PGE2-PINCs relative to CS-PINCs, which could be attributable to the specific interactions between CS-PGE2-PINCs and the PGE2 receptors expressed on cardiomyocyte or cardiomyoblast surface.[20,29]
2.3. In Vivo Heart Targeting and Bioactivity of PINCs in Mice with Myocardial I/R Injury
Myocardial reperfusion therapy restores blood flow and is the current standard treatment for patients after a heart attack.[30] However, it paradoxically causes further lethal tissue injury, known as myocardial I/R injury in clinical practice. Exploring ways to control or attenuate I/R injury is of clinical interest for improving post-ischemic recovery; thus we sought to test the bioactivity of PINCs in immunocompetent CD1 mice with I/R injury (Figure 3A). Following a temporary left anterior descending (LAD) coronary artery ligation for 30 min to create ischemia injury and a subsequent 24-h reperfusion, the mice were randomly divided into four groups and treated with saline (negative control), CS-PINCs, PGE2-PINCs, and CS-PGE2-PINCs via tail vein injection, respectively.
Figure 3.
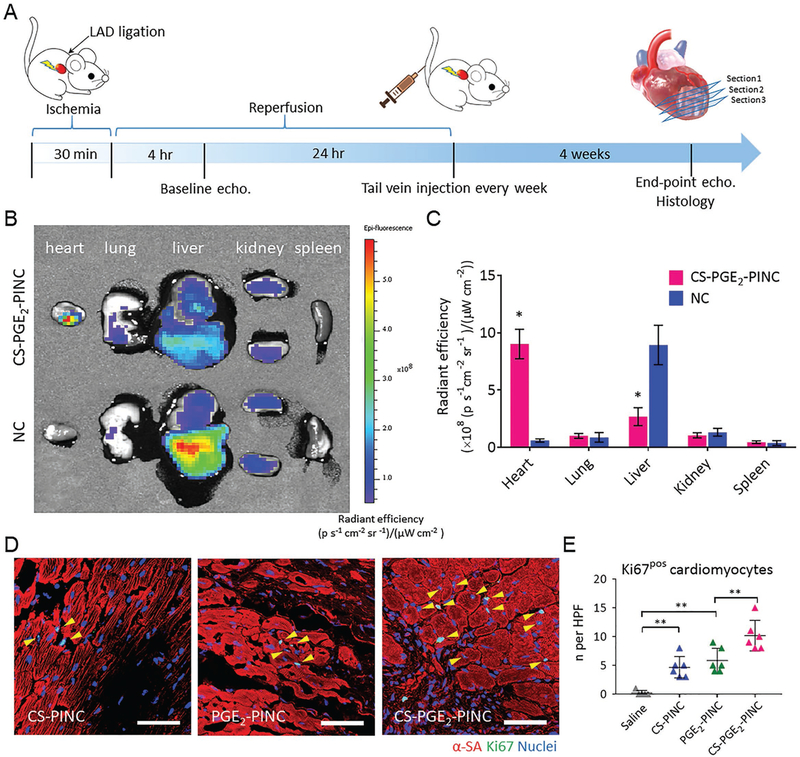
Biodistribution and in vivo bioactivity of PINCs. A) Schematic showing the animal study design. B) Biodistributions of CS-PGE2-PINCs and NCs after intravenous delivery in mice with myocardial I/R injury. Representative ex vivo fluorescent imaging of mouse organs (heart, lung, liver, kidney, and spleen) at 14 d post-intravenous injections of CS-PGE2-PINCs and NCs. C) Quantitative analysis of fluorescent intensities (n = 3 animals per group). D) Representative images showing cycling cardiomyocytes (yellow arrowheads) as indicated by α-SA and Ki67 double-positive staining in the peri-infarct regions of the hearts treated with CS-PINCs, PGE2-PINCs, and CS-PGE2-PINCs at week 4. E) Quantification of Ki67-positive cardiomyocytes at week 4 in the saline control (n = 5), CS-PINC (n = 6), PGE2-PINC (n = 6), and CS-PGE2-PINC (n = 6) groups. Scale bars, 50 μm. All data are mean ± s.d. Comparisons between any two groups were performed using two-tailed unpaired Student’s t-test. Comparisons among more than two groups were performed using one-way ANOVA followed by post hoc Bonferroni test. * indicates p < 0.05, and ** indicates p < 0.01.
To evaluate the heart targeting capability of PINC, the mice intravenously administrated with DiR-labeled CS-PGE2-PINCs or bare NCs following myocardial I/R injury were autopsied after 14 d to collect major organs for ex vivo fluorescent imaging. The infarcted hearts that received CS-PGE2-PINCs exhibited a stronger fluorescent signal than other organs as well as the hearts that received NCs (Figure 3B). The quantitative region-of-interest analysis confirmed that the CS-PGE2-PINCrecipient hearts showed 14.9-fold higher radiance efficiency (i.e., a measure of photon flux from the fluorescently labeled nanoparticle in the organ of interest, normalized by the area of emission, the exposure time, and the solid angle of the detector) than those treated with bare NCs, as well as 3.4- and 8.6-fold higher than the liver and kidney, respectively, validating the notable heart targeting ability of CS-PGE2-PINCs (Figure 3C). In contrast, greater nanoparticle accumulation was observed in the livers of animals that received the bare NCs compared to other organs, suggesting significant clearance of nanoparticles by the liver macrophages as expected. These findings were further corroborated by the quantitative analysis of DiI-labeled CS-PGE2-PINC or NC retention in different organs using high-performance liquid chromatography (HPLC).[31,32] The mice treated with DiI-labeled CS-PGE2-PINCs showed markedly higher nanoparticle retention in the injured heart than those treated with DiI-labeled NCs at 14 d post-treatment, which is consistent with the histological analysis (Figures S15 and S16, Supporting Information). The pharmacokinetics of intravenously injected CS-PGE2-PINCs was evaluated by quantitatively monitoring the human SDF-1 concentration in the blood plasma. The SDF-1 released from CS-PGE2-PINCs has a longer blood retention than that from NCs in mice with myocardial I/R injury (Figure S17, Supporting Information), which was in good accordance with previous studies and the in vitro factor release results.[13,24] We further evaluated the biocompatibility of PINCs. Negligible T cell and macrophage infiltration were confirmed by the presence of few CD3-/CD8-positive T cells and CD68-positive macrophages in the hearts that received different PINCs, indicating good biocompatibility of these nanoformulations (Figure S18, Supporting Information).
It has been established that adult cardiomyocytes have extremely limited capacity to proliferate in vivo. To test the bioactivity of PINCs in adult mouse cardiomyocytes, we first assessed the cardiomyocyte proliferation 4 weeks after treatment by α-SA and Ki67 expressions. The number of Ki67-positive cardiomyocytes in the peri-infarct region of both the CS-PINC-recipient and the PGE2-PINC-recipient hearts was significantly higher than that of the control hearts treated with a saline injection, although the difference between the two groups was indiscernible (Figure 3D and Figure S19, Support Information). Notably, the highest number of cycling cardiomyocytes was found in the peri-infarct region of the CS-PGE2-PINC-recipient hearts among all the groups (Figure 3D, E). We further stained the hearts for a specific marker of late G2/ mitosis, phosphorylated histone H3 (pH3), and a marker of cytokinesis, Aurora B kinase (AURKB). Remarkably, CS-PGE2PINCs induced robust mitotic activity of cardiomyocytes in the injured hearts after 4 weeks of treatment as evidenced by the elevated number of pH3-positive cardiomyocytes at the peri-infarct zone compared to other PINC treatment (Figure 4A, B). Expression of AURKB suggested that the cardiomyocytes not only entered the cell cycle but also were undergoing cytokinesis (Figure 4C, D). We further performed the Langendorff perfusion to isolate the cardiac cells from the saline-recipient or the CS-PGE2-PINC-recipient hearts collected 2 weeks post-treatment. Flow cytometry analysis revealed an increase in the percentage of Ki67-positive cycling cardiomyocytes in the CS-PGE2-PINC-recipient hearts relative to the saline-treated control hearts, which corroborates the previous findings of the immunohistochemical study (Figure S20, Supporting Information).
Figure 4.
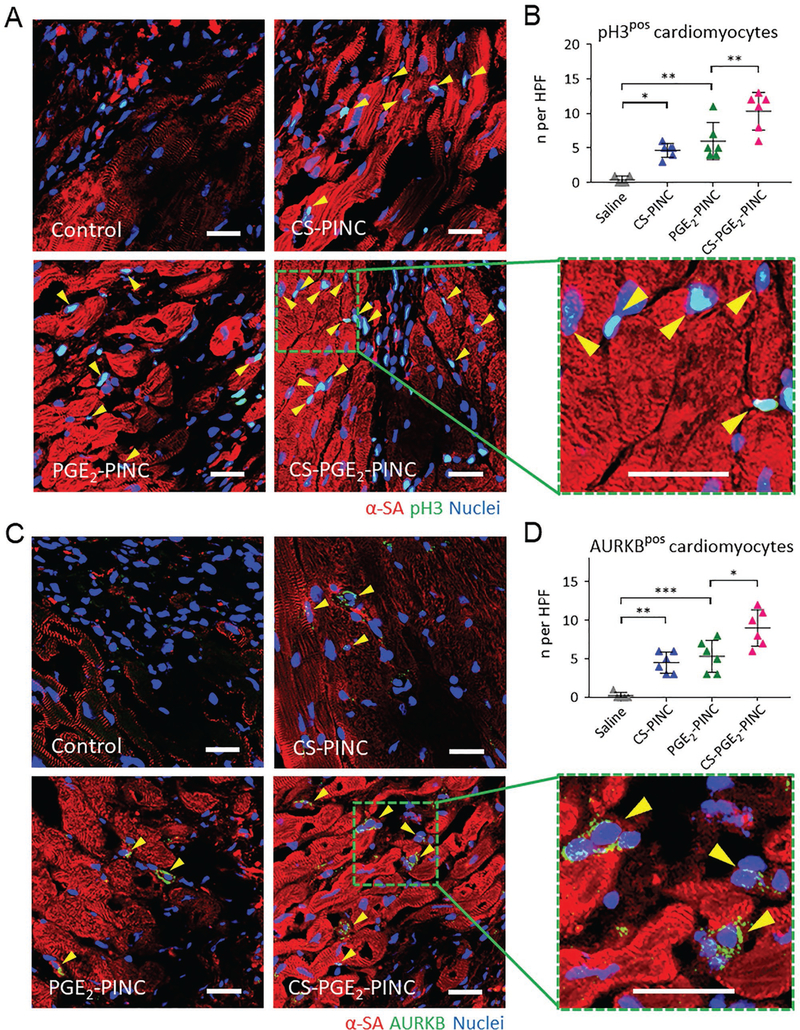
In vivo mitotic activities of cardiomyocytes. A, B) Visualization of phospho-histone H3 phosphorylation in cardiomyocytes (yellow arrowheads) in the peri-infarct regions of saline control-, CS-PINC-, PGE2-PINC-, and CS-PGE2-PINC-treated hearts at week 4. Representative images are in (A) (blue: DAPI, staining nuclei; red: α-SA, staining cardiomyocytes; green: pH3, indicating the cells that are in late G2/mitosis phase; the green square highlights the localization of pH3 (yellow arrowheads) in the nuclei of cycling cardiomyocytes). Quantification in (B) shows pH3-positive cardiomyocytes at week 4 in the saline control (n = 5), CS-PINC (n = 6), PGE2-PINC (n = 6), and CS-PGE2-PINC (n = 6) groups. C, D) Visualization of AURKB in cardiomyocytes (yellow arrowheads) in the peri-infarct regions of saline control-, CS-PINC-, PGE2-PINC-, and CS-PGE2-PINC-treated hearts at week 4. Representative images are in (C) (blue: DAPI, staining nuclei; red: α-SA, staining cardiomyocytes; green: AURKB, marking the cells in karyokinesis and cytokinesis; the green square highlights the localization of AURKB in mid-bodies (yellow arrowheads). Quantification in (D) shows AURKB-positive cardiomyocytes at week 4 in the saline control (n = 5), CS-PINC (n = 6), PGE2-PINC (n = 6), and CS-PGE2-PINC (n = 6) groups. Scale bars, 20 μm. All data are mean ± s.d. Comparisons among more than two groups were performed using one-way ANOVA followed by post hoc Bonferroni test. * indicates p < 0.05; ** indicates p < 0.01; and *** indicates p < 0.001.
2.4. Functional Benefits of PINC Therapy in a Mouse Model of I/R
To test the potency of PINCs for the treatment of heart injury after I/R, adult mice were randomized to intravenously receive CS-PINCs, PGE2-PINCs, CS-PGE2-PINCs, or saline injection after myocardial I/R injury. After 4 weeks of treatment, heart morphometry imaged by Masson’s trichrome staining revealed the protective effects of PINC injections on heart morphometry (Figure 5A; blue, scar tissue; red, viable myocardium). When quantified, CS-PGE2-PINC injection resulted in the highest amount of viable myocardium (Figure 5B) with the smallest scar size (Figure 5C). The reduced cardiac remodeling of CS-PGE2PINC-treated mice was further demonstrated by the reductions in left ventricular end diastolic volume (LVEDV) and end systolic volume (LVESV), respectively, compared with CS-PINCs, PGE2PINCs, or saline controls (Figure 5D, E). Left ventricular ejection fraction (LVEFs), LVEDVs, and LVESVs were similar at baseline among all the groups, indicating a similar degree of initial injury (Figure 5F and Figure S21, Supporting Information). After 4 weeks, the LVEF of saline-treated animals evidently declined, while LVEF was well preserved in the CS-PINC and PGE2-PINC treatment groups. The hearts of CS-PGE2-PINCtreated animals showed the highest LVEF (Figure 5F). When we calculated the treatment effects (i.e., change of LVEFs from baseline), it was clear that saline injection had negative treatment effect; CS-PINCs and PGE2-PINC treatments preserved cardiac functions, and CS-PGE2-PINC injection robustly boosted cardiac functions (Figure 5G).
Figure 5.

Functional benefits of PINC therapy in mice with myocardial I/R injury. A) Representative Masson’s trichrome-stained sections showing scar tissue (blue) and viable myocardium (red) from the basal, mid-left ventricular (LV), and apical regions of the hearts 4 weeks after treatment with saline (n = 5), CS-PINCs (n = 6), PGE2-PINCs (n = 6), and CS-PGE2-PINCs (n = 6), respectively. B, C) Quantitative analyses of B) viable myocardium and C) scar size from the Masson’s trichrome images. D) LVEDV and E) LVESV measured by echocardiography at 4 weeks after I/R in mice treated with saline, CS-PINCs, PGE2-PINCs, and CS-PGE2-PINCs, respectively. F) LVEF measured by echocardiography at baseline (4 h post-I/R) and 4 weeks afterward in the saline, CS-PINC, PGE2-PINC, and CS-PGE2-PINC groups. Scale bar, 2 mm. All data are mean ± s.d. * indicates p < 0.05; ** indicates p < 0.01; and *** indicates p < 0.001. G) Treatment effects were assessed by the change in LVEF over the 4-week time course relative to baseline. # indicates p < 0.05 when compared with saline control group; and † indicates p < 0.05 when compared with any other groups.
2.5. PINC Injection Promotes Endogenous Repair in the Infarcted Heart
To reveal the mechanisms underlying the functional effects of PINCs, we performed immunostaining analysis with Nkx2.5 (Figure 6A), CD34 (Figure 6B), and von Willibrand Factor (vWF, Figure 6C) in the infarcted hearts of mice treated with saline, CS-PINCs, PGE2-PINCs, and CS-PGE2-PINCs. Previous studies have confirmed the important roles of Nkx2.5-positive cells in cardiomyogenesis and heart repair.[33,34] Compared with control, PINC treatments remarkably increased the recruitment of Nkx2.5-positive cardiac progenitor cells to the infarcted heart, with the highest number of cardiac progenitor cells entering the peri-infarct region of the CS-PGE2-PINC-treated hearts (Figure 6D). In addition, a greater number of CD34-positive cells were found in the CS-PGE2-PINC-treated hearts compared with other treatment groups (Figure 6E), suggesting the homing of endothelial progenitor cells may also be elicited after PINC injection. This was corroborated by the enhanced vWF-positive vessel density found in the PINC-treated hearts compared with the control. Together, these results suggest that the therapeutic effects of PINCs may be through activation of Nkx2.5-positive cells, endothelial progenitor cells, and promotion of neovascularization.
Figure 6.
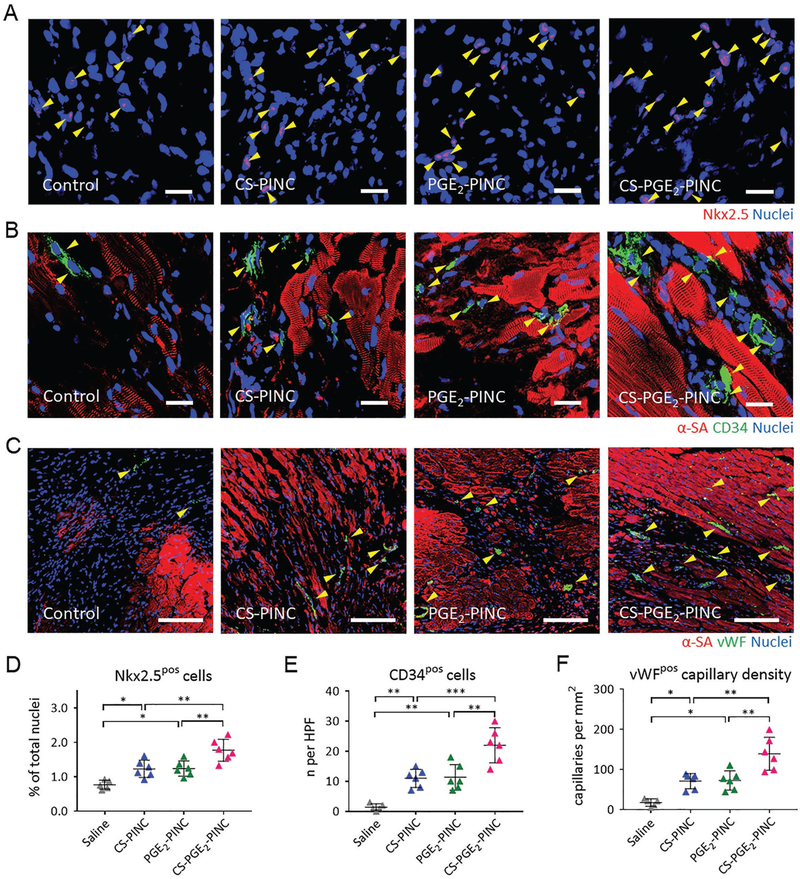
PINC injection promotes endogenous repair in the infarcted heart. A–C) Representative images showing Nkx2.5-positive cells, CD34-positive cells, and vWF-positive capillaries in the infarcted hearts 4 weeks after saline (n = 5), CS-PINC (n = 6), PGE2-PINC (n = 6), or CS-PGE2-PINC (n = 6) treatment. Yellow arrowheads indicate the positively stained cells. D–F) Quantification of the number of D) Nkx2.5-positive cells, E) CD34-positive cells, and F) vWF-positive capillary density in the infarcted hearts 4 weeks after saline (n = 5), CS-PINC (n = 6), PGE2-PINC (n = 6), or CS-PGE2-PINC (n = 6) treatment. Scale bars, A, B) 20 μm; and C) 100 μm. Comparisons among more than two groups were performed using one-way ANOVA followed by post hoc Bonferroni test. * indicates p < 0.05; ** indicates p < 0.01; and *** indicates p < 0.001.
To date, most nanoformulations aiming at treating myocardial infarction employed the passive targeting mechanism (i.e., EPR effect of the leaky vasculature in the acutely ischemic heart)), thus were limited by the short-term retention from a few hours to days in the injured heart.[7,35] Christman and coworkers reported an enzyme-responsive nanoparticle that can form a network-like structure like structure infarction for realizing long-term nanoparticle retention.[8] Recently, we reported that the decoration of CSCs with platelet membrane nanovesicles boosts CSC retention in the infarcted myocardium and therapeutic outcomes in rats and pigs with ischemic heart injury.[15] In addition, it has been established that the expression levels of three subtypes of EPs, EP2, EP3, and EP4, are remarkably upregulated in the heart after myocardial I/R injury.[36,37] PGE2 not only activated the endogenous stem/progenitor cells to replenish cardiomyocytes after ischemic injury, but also enhanced the recruitment of CD34-positive hematopoietic stem cells after xenotransplantation through PGE2/EP2/EP4 signaling.[20,38] The CS-PGE2-PINCs reported here harness both the natural myocardial infarction-homing ability of platelet membrane and the upregulation of PGE2 receptors in the cardiac microenvironment after I/R injury, resulting in prolonged retention in the infarcted heart after minimally invasive intravenous delivery. Our findings showed that the released regenerative factors and PGE2/EPs signaling could synergistically boost the therapeutic efficacy of CS-PGE2-PINCs. As a result, the animals that received intravenous injection with CS-PGE2-PINCs exhibited significantly augmented cardiac function and mitigated heart remodeling compared to other treatment groups. Such functional benefit was accompanied by the increase in cycling cardiomyocytes, activation of endogenous progenitor cells, and promotion of angiogenesis. Nevertheless, our study has several limitations. First, we investigated the cardiac repair through immunohistochemical analysis. Lineage tracing using transgenic mice will allow for more insightful analysis of the origin of newly formed blood vessels and cardiomyocytes. Second, our study did not include an empty (no secretome) nanoparticle control in the animal studies. Nonetheless, based on the in vitro data (Figure S12, Supporting Information), these control nanoparticles are not expected to improve cardiac functions in the animal model. In addition, for clinical applicability, a large animal model of I/R injury and longer study duration will be needed to fully characterize the efficacy and safety of the PINC therapy.
3. Conclusions
In summary, we reported a novel nanoparticle that incorporates both PGE2-modified platelet membrane and regenerative factors to target the heart after ischemic injury. The CS-PGE2PINCs were fabricated by functionalizing a CSC factor-releasing core with a platelet membrane shell with PGE2 decoration. By taking advantage of the natural infarct-homing ability of platelet membrane and the overexpression of EPs in the pathological cardiac microenvironment, the CS-PGE2-PINC can realize targeted delivery of the therapeutic payload to the injured heart. Furthermore, the synergistic treatment efficacy can be achieved by CS-PGE2-PINC, which combines the paracrine mechanism of stem cell therapy with the PGE2/EP receptor signaling that is involved in the repair and regeneration of multiple tissues.[19,38,39] This PINC can be applied as a promising therapeutic delivery platform for treating multiple diseases.
4. Experimental Section
Materials:
All chemical reagents were purchased from Sigma–Aldrich or Thermo Fisher Scientific and were used as received unless specifically noted.
Preparation of CSC Secretome-Loaded PLGA Nanoparticles (NCs):
Human CSCs were derived from donor human hearts according to the previously published method.[17] Heart tissues from patients 2–4 weeks after myocardial infarction (with LVEF of 25%–45%) were used. All procedures were approved by the institutional review board and written informed consent was obtained from all patients. The CSCs were used at passage 2–4. Briefly, the CSCs were cultured in serum-free media for 3 d and then the supernatant was collected to harvest conditioned media. The collected conditioned media was filtered through a 0.22 μM filter into a sterile 50 mL conical to remove any cell debris and contaminants. Sterile conditioned media was then lyophilized by a LABCONCO FreeZone 2.5 Liter Freeze Dry System to produce the purified CSC secretome. The CSC secretome-loaded PLGA cores were fabricated through a double emulsion method followed by membrane extrusion. In brief, concentrated CSC secretome aqueous solution was prepared as the internal aqueous phase in polyvinyl alcohol (PVA, MW: 30 000–50 000, 0.1% w/v), and then injected into dichloromethane (DCM) containing PLGA (MW: 7000–17 000) as the oil phase. The whole content was sonicated on ice for emulsification using a Misonix XL2020 sonicator. Afterward, the emulsion was immediately transferred into water containing 0.7% (w/v) PVA. The secondary emulsion was emulsified again to produce the final water/oil/water PLGA nanoparticle emulsion. The w/o/w emulsion was continuously stirred overnight to promote solvent evaporation. The nanoparticles were then sequentially extruded 19 times through polycarbonate membranes with pore sizes of 400 and 200 nm, respectively, using an extruder (Avanti Polar Lipids, USA). To determine the secretome loading capacity and efficiency, the nanoparticles were pelleted by centrifugation at 20 000 × g and then the non-encapsulated amount of secretome in the supernatant was measured using a BCA protein assay.
Isolation of Platelet Membrane:
The platelet membrane was isolated from human platelet-rich plasma (PRP, ZenBio, USA) through gradient centrifugation as previously described with modification.[15] Briefly, the PRP was centrifuged at 100 × g for 20 min. PBS (1 ×, pH 7.4) containing 1 × 10−3 M of ethylenediaminetetraacetic acid and 2 × 10−6 M of prostaglandin E1 was added to keep platelets inactivated. The isolated platelets were then pelleted by centrifugation at 800 × g for 20 min at room temperature. The platelet membrane was obtained by three freeze-thaw cycles, followed by sonication. The protein content in the purified platelet membrane was determined by the BCA protein assay for further preparation of PINCs.
Preparation of PGE2-Platelet Membrane Conjugate:
PGE2-platelet membrane conjugate was synthesized using EDC/NHS coupling chemistry. Briefly, PGE2 (0.52 μg, 1.5 nmol) dissolved in anhydrous ethanol was reacted with EDC (58 μg, 0.37 μmol) and NHS (14 μg, 0.12 μmol) in 15 mL 2-(4-Morpholino) ethane sulfonic acid (MES) buffer (100 × 10−3 M, pH 6.0). After 30 min of carboxylate activation, platelet membrane containing 0.36 mg protein was added. The pH of the reaction mixture was adjusted to 7.2 by the addition of 1 M sodium bicarbonate. The mixture was stirred overnight at room temperature. The resulting product was dialyzed (MWCO 1000) for 24 h against PBS. The amount of unconjugated PGE2 was measured by ELISA (R&D Systems, USA) to determine the conjugation yield and the content of PGE2 conjugated to the PGE2-platelet membrane conjugate.
Fabrication and Characterization of PINCs:
To cloak the platelet membrane or PGE2-platelet membrane conjugate onto the surface of CSC secretome-loaded NCs, 0.5 mL of NCs (5 × 109 particles mL−1) were incubate with 0.5 mL of platelet membrane or PGE2-platelet membrane conjugate containing 0.36 mg membrane protein under ultrasonication for 5 min, and then extruded 19 times as previously described to prepare the PINCs. The CS-PGE2-PINCs were prepared via coating the PGE2-platelet membrane conjugate on the surface of NCs. The CS-PINCs were prepared via coating the platelet membrane on the surface of NCs while the PGE2-PINCs were prepared via coating the PGE2-platelet membrane conjugate on the surface of empty PLGA nanoparticles. A BCA-based protein assay was performed to estimate the membrane coating efficiency and the amount of membrane protein coated onto each PLGA nanoparticle. Briefly, 0.5 mL of platelet membrane solution containing 0.36 mg membrane protein was mixed with 0.5 mL of empty PLGA nanoparticles (5 × 109 particles mL−1) to fabricate the membrane-coated nanoparticles. After pelleting the membrane-coated nanoparticles from the remaining free platelet membrane by centrifugation at 20 000 × g, the amount of membrane protein in the remaining solution was measured using a BCA protein assay. The membrane coating efficiency was determined by comparing the difference between the amount of total platelet membrane protein before the membrane coating and that of the remaining free platelet membrane. The pelleted nanoparticles were then resuspended back to their original volume, followed by examination using NanoSight NS300 to determine the total number of membrane-coated nanoparticles for calculating the amount of membrane protein coated onto each PLGA nanoparticle.
Physicochemical Characterization:
Nanoparticle size and surface zeta potential were measured by DLS using a Malvern ZEN 3600 Zetasizer. Nanoparticle concentration was examined by NanoSight NS300. The nanoparticle structure was visualized using a JEOL JEM-2000FX transmission electron microscope after negative staining with vanadium (Abcam). To assess the stability of the different nanoformulations over time, the bare NCs, CS-PINCs, PGE2-PINCs, and CS-PGE2-PINCs were suspended in PBS (1 ×, pH 7.4) at a concentration of 109 particles mL−1. The change of particle size was measured by DLS at pre-determined time-points over the course of 2 weeks. To evaluate the stability in serum, the different nanoformulations were incubated with 50% fetal bovine serum (Hyclone, USA). The change of particle sizes within 4 h was determined by DLS. Long-term stability was assessed by the particle size change measured by DLS before lyophilization in 10 wt% sucrose and after resuspension in PBS (1 ×, pH 7.4) back to the original volume. SDS-PAGE was performed to examine the protein components of the platelet membrane and the different PINCs. Western blotting was performed to assess the presence of specific platelet membrane markers using rabbit anti-human CD42b (Santa Cruz, sc-292722) and rabbit anti-human CD36 (Santa Cruz, sc-9154) antibodies, respectively, along with a goat anti-rabbit HRP-conjugated secondary antibody.
Collagen Surface Binding Assay:
GFP-tagged HUVECs (Angio-Proteomie, USA) were seeded on collagen-coated four-well culture chamber slides and cultured for 24 h. The cells were then incubated with DiI-labeled CS-PGE2-PINCs in PBS (1 ×, pH 7.4) at 4 °C for 60 s. Then the cells were washed with PBS twice. Images were taken on an Olympus IX81 fluorescent microscope and analyzed using NIH ImageJ software.
Growth Factor Release Study:
Total protein and growth factor releases from PINCs were determined as previously described.[16,17] In brief, freeze-dried PINCs were dissolved in DCM. After that, PBS was added to the solution. The sample was subjected to vortex for 5 min to isolate proteins from the oil phase to the water phase. After centrifugation, the protein concentration in the water phase was measured by BCA protein assay. For growth factor release studies, nanoparticles were incubated in PBS at 37 °C. The supernatant was collected at various time points (day 3, 7, 11, and 14) after centrifugation at 20 000 × g for 30 min to pellet the nanoparticles. The concentrations of various growth factors were measured using ELISA kits (R&D Systems, USA) according to the manufacturer’s instructions. The data were averaged from three independent measurements.
Cell Viability and Proliferation Assay:
The H9c2 cardiomyoblasts derived from embryonic rat heart (Sigma–Aldrich) were incubated with different concentrations of PINCs. PBS (1 ×, pH 7.4) was used as a control. The cell viability and proliferation were assessed by using a Cell Counting Kit-8 (CCK-8) according to the manufacturer’s instructions.
NRCM Uptake Assay:
NRCMs were derived from SD rats and cultured on four-well chamber slides for 3 d, followed by co-incubation with DiI-labeled CS-PINCs or CS-PGE2-PINCs (1.5 × 109 particles mL−1) in an incubator for 3 h. The beating cardiomyocytes were counted under a white-light microscope. Then, the cells were washed with PBS twice, fixed, permeabilized, and stained for α-sarcomeric actinin (α-SA, 1:200, ab9465, Abcam), followed by DAPI staining for nucleus visualization. For assessment of cell apoptosis, the cells were cocultured with CS-PINCs or CS-PGE2-PINCs (1.5 × 109 particles mL−1) for 48 h. Subsequently, the cells were washed with PBS twice and then exposed to serum-free medium supplemented with hydrogen peroxide (250 × 10−6 M) for 3 h, followed by incubated with TUNEL solution (Roche Diagnostics GmbH, Germany) and counter-stained with DAPI. Images were taken using a Zeiss LSM 710 confocal microscope (Carl Zeiss, Germany) and analyzed using NIH ImageJ software.
Mouse Model of Myocardial I/R Injury:
All animal work was compliant with the Institutional Animal Care and Use Committee (IACUC) of the University of North Carolina at Chapel Hill and North Carolina State University. Briefly, female immunocompetent CD1 mice (8–10 weeks old, Charles River Laboratories) were anesthetized with an intraperitoneal injection of ketamine (100 mg kg−1 mouse) plus xylazine (10 mg kg−1 mouse). Under sterile condition, the heart was exposed by a left thoracotomy and a 30 min ischemia was achieved by temporal ligation of the LAD coronary artery. After 24 h of reperfusion, animals were randomized to receive intravenous injection of CS-PINCs (CS dose: 1.2 mg kg−1 mouse), PGE2-PINCs (PGE2 dose: 0.053 mg kg−1 mouse), CS-PGE2-PINCs (CS dose: 1.2 mg kg−1 mouse, PGE2 dose: 0.053 mg kg−1 mouse), and saline (control) every 7 d for 4 weeks, respectively. Therefore, there were total four injections in 4 weeks.
In Vivo Targeting Ability Study:
Immunocompetent CD1 mice with myocardial I/R injury were randomized into two groups (n = 3 per group) to receive intravenous injections with DiR-labeled NCs or CS-PGE2-PINCs (CS dose: 1.2 mg kg−1 mouse and PGE2 dose: 0.053 mg kg−1 mouse) every 7 d for 2 weeks. After that, the mice were euthanized and the major organs (heart, lung, liver, kidney, and spleen) were carefully collected. The fluorescence signals of each organ were recorded using an IVIS Spectrum imaging system (Caliper Lifesciences, USA) for quantification. For HPLC analysis, the major organs were harvested after 2 weeks from the mice treated with DiI-labeled NCs or CS-PGE2-PINCs. The organs were washed with PBS, dried, and then minced into small pieces using a surgical scalpel and weighted prior to homogenization. The homogenized samples were thoroughly mixed with 500 μL of chloroform/methanol (1:1, v/v) for fluorescent dye extraction. After 10-min ultrasonication and three freeze-thaw cycles, the samples were centrifuged at 14 000 × g for 30 min and the supernatants containing the extracted fluorescent dye were subjected to HPLC analysis. All chromatographic experiments were conducted using an Agilent 1290 Infinity system (Agilent, USA) equipped with an Agilent 1290 diode array detector measured at the wavelength of 551 nm. The samples were eluted using a gradient method starting at 90/10 acetonitrile/water (v/v) with 0.1% trifluoroacetic acid to 100% acetonitrile with 0.1% trifluoroacetic acid in 10 min using a Zorbax RRHD Eclipse Plus-C18 column at 40 °C with a flow rate of 0.5 mL min−1. The quantities of fluorescent dye which were in proportional to the number of nanoparticles retained in different tissue samples were quantified using a standard curve created from known concentrations of fluorescent dye diluted in the mobile phase.
In Vivo Pharmacokinetic Study.
Six CD1 mice were randomized into two groups (n = 3 per group) and intravenously injected with CS-PGE2-PINCs or NCs (CS dose: 1.2 mg kg−1 mouse, and PGE2 dose: 0.053 mg kg−1 mouse) 24 h after myocardial I/R injury. At pre-determined time intervals (0.5, 1, 2, 4, 12, 24, and 48 h), 20 μL of whole blood was collected and centrifuged at 800 × g for 10 min. The plasma level of human SDF-1 was analyzed using an ELISA kit (R&D Systems, USA) according to the manufacturer’s instruction. The data were averaged from three independent measurements.
Cardiac Function Assessment:
All animals underwent transthoracic echocardiography under anesthesia at 4 h post-I/R injury and 4 weeks after treatment using a VisualSonics Vevo 2100 Imaging System. Hearts were imaged 2D in long-axis views at the level of the greatest left ventricular diameter. LVEDV and LVESV were measured. LVEF was determined by measurement from views taken from the infarcted area. All measurements were done in random order, with the surgeon and echocardiographer being blind to the treatment groups.
Heart Morphometry:
Hearts were harvested and cut into 10 μm-thick tissue sections. Masson’s trichrome staining was performed, and images were acquired with a PathScan Enabler IV slide scanner (Advanced Imaging Concepts, USA). Image analysis related to viable myocardium and scar size was performed using NIH ImageJ software. Three selected sections were quantified for each animal.
Immunohistochemistry Assessment:
Heart cryosections were fixed with 4% paraformaldehyde in PBS for 30 min, permeabilized and blocked with Protein Block Solution (DAKO) containing 0.1% saponin for 1 h at room temperature. For immunostaining, the samples were incubated overnight at 4 °C with the following primary antibodies diluted in the blocking solution: rabbit anti-mouse α-SA (1:200, ab137346, Abcam) was used to identify cardiomyocytes; rat anti-mouse Ki67 antibody (1:200, 151 202, Biolegend), rabbit anti-mouse histone H3 phosphorylated at serine 10 (pH3, 1:200, ab5176, Abcam), and rabbit anti-mouse Aurora B kinase (AURKB, 1:200, ab2254, Abcam) antibodies were used to analyze cell-cycle re-entry, karyokinesis, and cytokinesis, respectively; sheep anti-mouse vWF (1:200, ab11713, Abcam) antibody was used to detect myocardial capillaries in the peri-infarct regions; goat anti-mouse Nkx2.5 (1:200, ab106923, Abcam), and rat anti-mouse CD34 (1:200, MA1–22646, Thermo Fisher Scientific) antibodies were used to examine endothelial progenitor cell recruitment; rabbit anti-mouse CD3 (1:200, ab16669, Abcam), rat anti-mouse CD8 (1:200, ab22378, Abcam), and rabbit anti-mouse CD68 (1:200, ab125212, Abcam) antibodies were used to detect immune response. After three 10-min washes with PBS, samples were stained for 1.5 h at room temperature with fluorescent secondary antibodies including goat anti-rabbit IgG-Alexa Fluor 594 conjugate (1:400, ab150080, Abcam), goat anti-rat IgG-Alexa Fluor 488 conjugate (1:400, ab150157, Abcam), donkey anti-rabbit IgG-Alexa Fluor 488 conjugate (1:400, ab150073, Abcam), donkey anti-goat IgG-Alexa Fluor 594 conjugate (1:400, ab150136, Abcam), donkey anti-sheep IgG-Alexa Fluor 488 conjugate (1:400, ab150177, Abcam), goat anti-rabbit IgG-Alexa Fluor 488 conjugate (1:400, ab150077, Abcam), and goat anti-rabbit IgG-Alexa Fluor 594 conjugate (1:400, ab150080, Abcam), and goat anti-rat IgG-Cy5 conjugate (1:400, ab6563, Abcam) based on the isotopes of the primary antibodies. This was followed by 10 min of 4, 6-diamidino-2-phenylindole dihydrochloride (DAPI) staining for nucleus visualization. Slides were mounted with ProLong Gold mountant (Thermo Fisher Scientific) and viewed under a Zeiss LSM 710 confocal microscope (Carl Zeiss). Images were analyzed using NIH ImageJ software.
Flow Cytometric Analysis of Isolated Cardiomyocytes:
At 2 weeks posttreatment, the mouse hearts that received saline or CS-PGE2-PINC injection were carefully excised. The cardiomyocytes were isolated using a Langendorff perfusion system according to the previously published method.[40,41] Flow cytometry was performed on a CytoFlex (Beckman Coulter, USA) and the data were analyzed using the FCS Express software (De Novo, USA). The isolated cells were labeled with rabbit anti-mouse α-SA (1:200, ab137346, Abcam) was used to identify cardiomyocytes and rat anti-mouse Ki67 antibody (1:200, 151202, Biolegend) to analyze cell-cycle re-entry.
Statistics:
All experiments were performed independently at least three times, and the results were presented as mean ± s.d. Comparisons between any two groups were performed using two-tailed unpaired Student’s t-test. Comparisons among more than two groups were performed using one-way ANOVA followed by post-hoc Bonferroni test. Single, double, and triple asterisks represent p < 0.05, 0.01, and 0.001, respectively; p < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgements
This work was supported by the grants from the National Institutes of Health (R01 HL123920 and HL137093) to K.C., American Heart Association (18TPA34230092) to K.C., North Carolina State University Chancellor’s Innovation Fund (to K.C.), and UNC General Assembly Research Opportunities Initiative award (to K.C. and F.L.). We appreciate Dr. Eva Johannes and Dr. Marius Zareba at the Cellular and Molecular Imaging Facility (CMIF) at North Carolina State University for their assistance. This work was performed in part at the Analytical Instrumentation Facility (AIF) at North Carolina State University, which is supported by the State of North Carolina and the National Science Foundation (award number ECCS-1542015). The AIF is a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), a site in the National Nanotechnology Coordinated Infrastructure (NNCI).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Teng Su, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North, Carolina State University, Raleigh, NC 27695, USA; Department of Molecular Biomedical Sciences and Comparative, Medicine Institute, North Carolina State University, Raleigh, NC 27607, USA.
Ke Huang, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North, Carolina State University, Raleigh, NC 27695, USA; Department of Molecular Biomedical Sciences and Comparative, Medicine Institute, North Carolina State University, Raleigh, NC 27607, USA.
Hong Ma, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Hongxia Liang, Department of Molecular Biomedical Sciences and Comparative, Medicine Institute, North Carolina State University, Raleigh, NC 27607, USA; Department of Infectious Diseases and Hepatology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450052, China.
Phuong-Uyen Dinh, Department of Molecular Biomedical Sciences and Comparative, Medicine Institute, North Carolina State University, Raleigh, NC 27607, USA.
Justin Chen, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North, Carolina State University, Raleigh, NC 27695, USA.
Deliang Shen, Department of Cardiology, The First Affiliated Hospital of Zhengzhou University Zhengzhou, Henan 450052, China.
Tyler A. Allen, Department of Molecular Biomedical Sciences and Comparative, Medicine Institute, North Carolina State University, Raleigh, NC 27607, USA
Li Qiao, Department of Molecular Biomedical Sciences and Comparative, Medicine Institute, North Carolina State University, Raleigh, NC 27607, USA; Department of Cardiology, the Second Hospital of Hebei Medical University, Shijiazhuang, Hebei, 050000, China.
Zhenhua Li, Department of Molecular Biomedical Sciences and Comparative, Medicine Institute, North Carolina State University, Raleigh, NC 27607, USA.
Shiqi Hu, Department of Molecular Biomedical Sciences and Comparative, Medicine Institute, North Carolina State University, Raleigh, NC 27607, USA.
Jhon Cores, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North, Carolina State University, Raleigh, NC 27695, USA; Department of Molecular Biomedical Sciences and Comparative, Medicine Institute, North Carolina State University, Raleigh, NC 27607, USA.
Brianna N. Frame, Division of Cardiothoracic Surgery, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Ashlyn T. Young, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North, Carolina State University, Raleigh, NC 27695, USA
Qi Yin, Department of Molecular Biomedical Sciences and Comparative, Medicine Institute, North Carolina State University, Raleigh, NC 27607, USA.
Jiandong Liu, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Li Qian, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Thomas G. Caranasos, Division of Cardiothoracic Surgery, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
Yevgeny Brudno, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North, Carolina State University, Raleigh, NC 27695, USA.
Frances S. Ligler, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North, Carolina State University, Raleigh, NC 27695, USA
Ke Cheng, Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North, Carolina State University, Raleigh, NC 27695, USA; Department of Molecular Biomedical Sciences and Comparative, Medicine Institute, North Carolina State University, Raleigh, NC 27607, USA; Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
References
- [1].Townsend N, Wilson L, Bhatnagar P, Wickramasinghe K, Rayner M, Nichols M, Eur. Heart J 2016, 37, 3232. [DOI] [PubMed] [Google Scholar]
- [2].Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu C-H, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP, Nature 2014, 515, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bolli R, Ghafghazi S, Nat. Rev. Cardiol 2017, 14, 257. [DOI] [PubMed] [Google Scholar]
- [4].Lin Z, Pu WT, Sci. Transl. Med 2014, 6, 239rv1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fischer UM, Harting MT, Jimenez F, Monzon-Posadas WO, Xue H, Savitz SI, Laine GA, Cox CS, Stem Cells Dev. 2009, 18, 683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dvir T, Bauer M, Schroeder A, Tsui JH, Anderson DG, Langer R, Liao R, Kohane DS, Nano Lett. 2011, 11, 4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Paulis LE, Geelen T, Kuhlmann MT, Coolen BF, Schäfers M, Nicolay K, Strijkers GJ, Control J. Release 2012, 162, 276. [DOI] [PubMed] [Google Scholar]
- [8].Nguyen MM, Carlini AS, Chien MP, Sonnenberg S, Luo C, Braden RL, Osborn KG, Li Y, Gianneschi NC, Christman KL, Adv. Mater 2015, 27, 5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang F, Fang RH, Luk BT, Hu CJ, Thamphiwatana S, Dehaini D, Angsantikul P, Kroll AV, Pang Z, Gao W, Lu W, Zhang L, Adv. Funct. Mater 2016, 26, 1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Anselmo AC, Modery-Pawlowski CL, Menegatti S, Kumar S, Vogus DR, Tian LL, Chen M, Squires TM, Sen Gupta A, Mitragotri S, ACS Nano 2014, 8, 11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hu Q, Qian C, Sun W, Wang J, Chen Z, Bomba HN, Xin H, Shen Q, Gu Z, Adv. Mater 2016, 28, 9573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dehaini D, Wei X, Fang RH, Masson S, Angsantikul P, Luk BT, Zhang Y, Ying M, Jiang Y, Kroll AV, Gao W, Zhang L, Adv. Mater 2017, 29, 1606209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hu Q, Sun W, Qian C, Wang C, Bomba HN, Gu Z, Adv. Mater 2015, 27, 7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hickman DA, Pawlowski CL, Sekhon UDS, Marks J, Sen Gupta A, Adv. Mater 2018, 30, 1700859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tang J, Su T, Huang K, Dinh PU, Wang Z, Vandergriff A, Hensley MT, Cores J, Allen T, Li T, Sproul E, Mihalko E, Lobo LJ, Ruterbories L, Lynch A, Brown A, Caranasos TG, Shen D, Stouffer GA, Gu Z, Zhang J, Cheng K, Nat. Biomed. Eng 2018, 2, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Luo L, Tang J, Nishi K, Yan C, Dinh PU, Cores J, Kudo T, Zhang J, Li TS, Cheng K, Circ. Res 2017, 120, 1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tang J, Shen D, Caranasos TG, Wang Z, Vandergriff AC, Allen TA, Hensley MT, Dinh PU, Cores J, Li TS, Zhang J, Kan Q, Cheng K, Nat. Commun 2017, 8, 13724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kim SH, Jeong JH, Ou M, Yockman JW, Kim SW, Bull DA, Biomaterials 2008, 29, 4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang Y, Desai A, Yang SY, Bae KB, Antczak MI, Fink SP, Tiwari S, Willis JE, Williams NS, Dawson DM, Wald D, Chen W-D, Wang Z, Kasturi L, Larusch GA, He L, Cominelli F, Di Martino L, Djuric Z, Milne GL, Chance M, Sanabria J, Dealwis C, Mikkola D, Naidoo J, Wei S, Tai H-H, Gerson SL, Ready JM, Posner B, V Willson JK, Markowitz SD, Science 2015, 348, aaa2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hsueh YY, Wu JMF, Yu CC, Wu KK, Hsieh PCH, EMBO Mol. Med 2014, 6, 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Garikipati VN, Kishore R, Circ. Res 2017, 120, 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tang J, Cores J, Huang K, Cui X, Luo L, Zhang J, Li T, Qian L, Cheng K, STEM CELLS Transl. Med 2018, 7, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Torán JL, Aguilar S, López JA, Torroja C, Quintana JA, Santiago C, Abad JL, Gomes-Alves P, Gonzalez A, Bernal JA, Jiménez-Borreguero LJ, Sci. Rep 2017, 7, 12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hu CMJ, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu J, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L, Nature 2015, 526, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Menasché P, Vanneaux V, Hagège A, Bel A, Cholley B, Cacciapuoti I, Parouchev A, Benhamouda N, Tachdjian G, Tosca L, Trouvin J-H, Fabreguettes J-R, Bellamy V, Guillemain R, Suberbielle Boissel C, Tartour E, Desnos M, Larghero J, Eur. Heart J 2015, 36, 2011. [DOI] [PubMed] [Google Scholar]
- [26].Li T, Cheng K, Malliaras K, Smith RR, Zhang Y, Sun B, Matsushita N, Blusztajn A, Terrovitis J, Kusuoka H, Marbán L, Marbán E, J. Am. Coll. Cardiol 2012, 59, 942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Barry F, Murphy M, Nat. Rev. Rheumatol 2013, 9, 584. [DOI] [PubMed] [Google Scholar]
- [28].Tang J, Cui X, Caranasos TG, Hensley MT, Vandergriff AC, Hartanto Y, Shen DL, Zhang H, Zhang JY, Cheng K, ACS Nano 2017, 11, 9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li C, Wang J, Wang Q, Zhang Y, Zhang N, Lu L, Wu Y, Zhang Q, Wang W, Wang Y, Tu P, Sci. Rep 2016, 6, 36949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vicencio JM, Yellon DM, Sivaraman V, Das D, Boi-Doku C, Arjun S, Zheng Y, Riquelme JA, Kearney J, Sharma V, Multhoff G, Hall AR, Davidson SM, J. Am. Coll. Cardiol 2015, 65, 1525. [DOI] [PubMed] [Google Scholar]
- [31].Lundy DJ, Chen K-H, Toh EK-W, Hsieh PC-H, Sci. Rep 2016, 6, 25613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen K-H, Lundy DJ, Toh EK-W, Chen C-H, Shih C, Chen P, Chang H-C, Lai JJ, Stayton PS, Hoffmand AS, Hsieh PC-H, Nanoscale 2015, 7, 15863. [DOI] [PubMed] [Google Scholar]
- [33].Chen W-P, Liu Y-H, Ho Y-J, Wu SM, Cardiovasc. Res 2015, 105, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ye L, Chang Y-H, Xiong Q, Zhang PY, Zhang LY, Somasundaram P, Lepley M, Swingen C, Su LP, Wendel JS, Guo J, Jang A, Rosenbush D, Greder L, Dutton JR, Zhang JH, Kamp TJ, Kaufman DS, Ge Y, Zhang JY, Cell Stem Cell 2014, 15, 750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chang M-Y, Yang Y-J, Chang C-H, Tang ACL, Liao W-Y, Cheng F-Y, Yeh C-S, Lai JJ, Stayton PS, Hsieh PC-H, J. Control. Release 2013, 170, 287. [DOI] [PubMed] [Google Scholar]
- [36].Hishikari K, Suzuki J, Ogawa M, Isobe K, Takahashi T, Onishi M, Takayama K, Isobe M, Cardiovasc. Res 2009, 81, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Calabresi L, Rossoni G, Gomaraschi M, Sisto F, Berti F, Franceschini G, Circ. Res 2003, 92, 330. [DOI] [PubMed] [Google Scholar]
- [38].Goessling W, Allen RS, Guan X, Jin P, Uchida N, Dovey M, Harris JM, Metzger ME, Bonifacino AC, Stroncek D, Stegner J, Armant M, Schlaeger T, Tisdale JF, Zon LI, Donahue RE, North TE, Cell Stem Cell 2011, 8, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].North TE, Goessling W, Walkley CR, Lengerke C, Kopani KR, Lord AM, Weber GJ, V Bowman T, Jang I-H, Grosser T, FitzGerald GA, Daley GQ, Orkin SH, Zon LI, Nature 2007, 447, 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu J, Srivastava D, Nature 2012, 485, 593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Malliaras K, Zhang Y, Seinfeld J, Galang G, Tseliou E, Cheng K, Sun B, Aminzadeh M, Marbán E, EMBO Mol. Med 2013, 5, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.