

Abstract
A series of sialosides modified at the 4- and 9-hydroxy group were synthesised and tested for inhibition of the viral haemagglutinin-esterase activity from various Orthomyxoviruses and Coronaviruses. While no inhibition of the sialate-4-O-acetylesterases from mouse hepatitis virus strain S or sialodacryoadenitis virus was found, a 9-O-methyl derivative displayed inhibitory activity against recombinant sialate-9-O-acetylesterase from influenza C virus.
Keywords: Hemagglutinin-esterase, Sialate-O-acetylesterase, Influenza C virus, Sialic acid, Sialoside, Inhibitor, Bovine coronavirus, Mouse hepatitis virus, Sialodacryoadenitis virus
Graphical abstract
A series of modified sialosides has been synthesisised and a 9-O-methyl derivative was found to inhibit the hemagglutinin-esterase from Influenza C virus but not from other selected Orthomyxoviruses and Coronaviruses.

Highlights
► The synthesis of a series of 4- and 9-O-methyl- and methylphosphono-α-sialosides has been achieved. ► A 9-O-Methyl sialic acid derivative inhibited the sialate-O-acetylesterase from Influenza C virus. ► Methylphosphonates did not inhibit the viral haemagglutinin-esterase activities investigated.
1. Introduction
Sialic acids are a group of biologically important 9-carbon sugars which decorate, ketosidically linked to cell surface glycoconjugates, the glycocalix and thus the cells of higher organisms. At this exposed position, they serve as receptors for sialic acid-recognising proteins and are consequently involved in wide variety of biological events, both pathogenic and non-pathogenic [1], [2], [3]. One of the most common modifications of sialic acids in mammals is O-acetylation. It occurs either at C-4 or at any position within the glycerol side chain of sialic acid and multiple acetylations are possible (Fig. 1 ) [4], [5].
Fig. 1.

Structures of acetylated sialic acids and action of viral sialate-O-acetylesterases (SOAEs).
Sialate-O-acetylation has attracted increased interest in recent years due its abundance and involvement in many, including pathological, biological processes. For instance, acetylation may promote or hinder recognition of sialic acid by proteins, cells or pathogens. O-acetylation may also slow down the activity of degradative enzymes such as sialate lyases or sialidases [3]. Related biological events include cell differentiation, tumor growth, immunity, apoptosis, microbial infections and in particular cancer where they are considered markers for certain skin tumors and a form of leukaemia [2], [3], [4], [5], [6], [7]. Another important function of O-acetylation is masking of siglec binding sites. CD22, a siglec regulating the activity of the B-cell receptor, binds to α2-6 linked sialic acids, and binding can be masked by 9-O-acetylation [8]. This masking is regulated by the cellular sialate-O-acetylesterase, which thereby also regulates B-cell receptor signal strength [9]. Loss of function of the esterase activity results in autoimmune disease [10], [11].
Besides these functions, sialic acids are also used as docking platforms for viral pathogens. Several RNA viruses which infect the respiratory and gastrointestinal tract utilize sialic acids as a receptor determinant. To facilitate release of progeny virus from infected cells, a number of viruses express “receptor-destroying enzymes” (RDE), which are targets for antiviral drugs. The best known are the sialidase-inhibitors Zanamivir (Relenza) and Oseltamivir (Tamiflu). Besides sialidases, the haemagglutinin-esterases (HE) of influenza C virus, isavirus, betacoronaviruses and toroviruses represent another class of RDEs. They are sialate-O-acetylesterases (SOAE) hydrolysing O-acetyl esters of O-acetylated sialic acid derivatives (Fig. 1). Two main subtypes of HEs are known: sialate-4-O-acetylesterases (4-SOAE) and sialate-9-O-acetylesterases (9-SOAE).
HE expressing viruses include human pathogens like influenza C viruses, the respiratory human coronavirus OC43 (HCoV OC43) and HKU1 (HCoV HKU1), several important animal betacorona- and toroviruses and infectious salmon anaemia virus (ISAV), a piscine orthomyxovirus. HCoVs account for 10–30% of respiratory infections, in particular, the common cold, but they can also cause gastroenteritis and neurological disorders [12], [13]. The infections are usually mild and subclinical, but strains related to OC43 and HKU1 were associated with severe human disease [14], [15]. Betacoronaviruses have attracted attention after the outbreak of SARS CoV in 2002/2003. This genus consists of human and animal viruses. Coronaviruses are able to cross the animal-to-human species barrier: bat-to-human in case of SARS CoV [16], [17] and bovine-to-human in case of BCoV leading to HCoV OC43 [18]. Toroviruses (ToV) are evolutionary related to coronaviruses [19]. ToV are associated with asymptomatic enteric infections in pigs [20]. Studies suggest that they are highly prevalent in swine populations [21], [22]. The closely related bovine toroviruses (BToV) are implicated with serious or even fatal infections [23], [24], [25]; they are found worldwide [26], [27], [28], [29], [30]. ToV are also associated with human gastroenteritis. In fecal samples from children and adults with diarrhoea, torovirus antigens were detected by ELISA [31], [32], immunoelectron microscopy [33], [34], [35] and by reverse transcription polymerase chain reaction with primers covering a highly conserved region of the ToV genomes [35].
Regarding the potential for transmission of betacoronaviruses from animal to human and the danger of the emergence of further epidemics, efficient treatment(s) would be of great interest (Table 1 ).
Table 1.
Selection of HE expressing viruses of three different taxa and their substrate specificity.
| Virus | Substrate specificity of HE |
|---|---|
| Orthomyxovirus | |
| Influenza C virus (INF-C) | Neu5,9Ac2 |
| Infectious salmon anaemia virus (ISAV) | Neu4,5Ac2 |
| Betacoronavirus | |
| Human coronavirus strain OC43 (HCoV-OC43) | Neu5,9Ac2 |
| Bovine coronavirus (BCoV) | Neu5,9Ac2 |
| Rat sialoadacryoadenitis coronavirus (SDAV) | Neu4,5Ac2 |
| Mouse hepatitis virus strain S (MHV-S) | Neu4,5Ac2 |
| Torovirus | |
| Bovine torovirus (BToV) | Neu5,7(8),9Ac3 and Neu5,9Ac2 |
| Porcine torovirus (PToV) | Neu5,9Ac2 |
Interestingly, the comparison of the crystal structures of influenza C virus HEF [36], BCoV HE [37] and two ToV HEs [38] shows highly conserved sites of the SOAE domains. Therefore, the SOAE active site is probably an excellent target for broad-spectrum antivirals against sialate-O-acetylesterases of both orthomyxo- and coronaviruses.
The successful development and introduction of the anti-influenza drugs Tamiflu and Relenza which are inhibitors of the ‘receptor-destroying’ sialidase from influenza virus has demonstrated that such an approach is promising.
Earlier we postulated two essential pharmacophoric groups of Neu5,9Ac2 in correct spatial arrangement required for strong substrate–enzyme interaction with sialate-9-O-acetylesterases: the 9-O-acetyl and the alpha-C2 carboxylate group [39]. Although no investigations about the substrate–enzyme interactions of sialate-4-O-acetylesterases are available, a similar mechanism as for 9-O-acetylesterases is suggested.
In light of these, we embarked on a study which aims to find competitive inhibitors of 9- and 4-SOAE, which are serine esterases. Enzymes from four different viruses, influenza C virus (INF-C) [36], [40], [41], [42], bovine coronavirus (BCoV) [43], [44], mouse hepatitis virus strain S (MHV-S) [45], [46] and sialodacryoadenitis virus (SDAV) [47] were investigated.
2. Inhibitor design
The allyl group was chosen as an aglycon mimetic in target allyl sialosides 1–4 and control sialoside 5 because it offers a range of selective chemical methods, such as e. g. olefin metathesis, for further functionalisation or immobilisation of the inhibitors. To probe the active sites of 4-SOAE and 9-SOAE, two types of modifications of the positions 4 and 9 of the sialosides were introduced (Fig. 2 ).
Fig. 2.
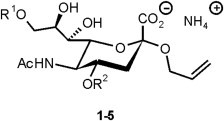
Structures of target sialosides 1–5. 1: R1 = H, R2 = CH3. 2: R1 = H, R2 = P(O) (CH3)O−NH4+. 3: R1 = CH3, R2 = H. 4: R1 = P(O) (CH3)O−NH4+, R2 = H. 5: R1 = R2 = H.
Firstly, methylation resulting in target structures 1 and 3 should yield information whether additional hydrophobic interactions could contribute to more efficient binding and about the role of the respective hydroxyl group as hydrogen bond donor. Secondly, methylphosphonate groups were introduced as mimetics of the suspected tetrahedral intermediate of acetate hydrolysis. In theory, compounds 2 and 4 could interact with active site amino acids stabilising this polar transition state, including the ‘oxyanion hole’ common in serine esterases [48].
3. Syntheses
α-Allyl sialoside 6, which serves as intermediate for all syntheses, was conveniently synthesised in high yield in 4 steps from commercially available N-acetylneuraminic acid using a well-established Koenigs-Knorr methodology (Scheme 1 ) [49], [50]. Control compound 5 was obtained from 6 through saponification.
Scheme 1.

Synthesis of reference compound 5 and intermediate 6. (a) 1. MeOH, IR-120(H+). 2. AcCl, AcOH. 3. CH2 CH2CH2OH, Ag2CO3, AgClO4. 4. NaOMe, MeOH. (b) 1. NaOH. 2. Gpc (0.1 M NH4HCO3).
Selective methylation of the 4-OH was then made possible by blocking positions 8 and 9 as the isopropylidene ketal through acid catalysed reaction of 6 with 2,2-dimethoxypropane to give 7 in 85% yield. Alkylation of 7 under Williamson-conditions followed by acid-mediated ketal hydrolysis and basic saponification of the methyl ester gave crude inhibitor 1 which was purified by gel permeation chromatography. For the introduction of a phosphonate group at position 4, hydroxyl groups 8 and 9 were protected as the cyclic carbonate using diphosgene and dimethylaminopyridine to give 8. Introduction of the phosphonate was achieved with methyl methylphosphonyl chloride and Hünig’s base to give phosphonate diester 9. The position of the phosphonylation was confirmed by acetylation of the remaining hydroxyl group at position 7 and analysis of the product (not shown). Selective cleavage of the phosphonic acid methyl ester with thiophenol and triethylamine followed by basic hydrolysis of all other ester groups and purification furnished compound 2 in good yield. (Scheme 2 ).
Scheme 2.
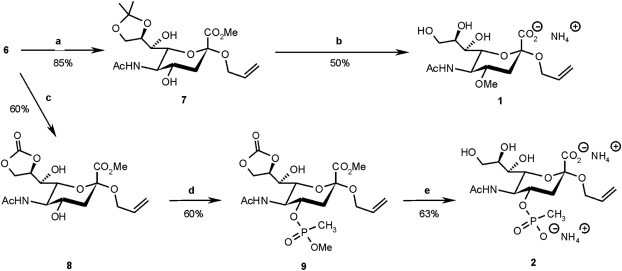
Syntheses of sialosides modified at the hydroxy group in position 4. (a) DMP, TsOH, acetone. (b) 1. DMS, NaH, acetonitrile. 2. 80% AcOH, then NaOH. 3. Gpc (0.1 M NH4HCO3). (c) Diphosgene, DMAP, CH2Cl2. (d) ClP(O) (OCH3)CH3, EtNiPr2, CH2Cl2. (e) 1. PhSH, NEt3, THF. 2. Pyridine, NEt3, H2O. 3. Gpc (0.1 M NH4HCO3).
For methylation at position 9, compound 6 was converted into the 8,9-epoxide by subsequent treatment with toluenesulfonyl chloride in pyridine and sodium methoxide in methanol to give 10. Acid-mediated opening of the epoxide in methanol gave the 9-methyl ether and, after saponification of the methyl ester and purification, target inhibitor 3. For the 9-phosphonate, direct reaction of 5 with methyl methylphosphonylchloride and Hünig’s base followed by per-O-acetylation to yield 11 proved to be the best route. Deprotection of 10 was carried out as described for 9 and thus inhibitor 4 was obtained in 20% overall yield (4 steps) (Scheme 3 ).
Scheme 3.

Syntheses of sialosides modified at the hydroxy group in position 9. (a) 1. TsCl, pyridine. 2. NaOMe, MeOH. (b) 1. MeOH, IR-120(H+). 2. 0.05 M NaOH, dioxane. 3. Gpc (0.1 M NH4HCO3). (c) 1. ClP(O) (OCH3)CH3, EtNiPr2, CH2Cl2. 2. Ac2O, pyridine. (d) 1. PhSH, NEt3, THF. 2. 0.05 M NaOH, dioxane. 3. Gpc (0.1 M NH4HCO3).
4. Inhibition of viral sialate-O-acetylesterases
4.1. General
Inhibition of the SOAE activity of three viruses, influenza C virus (INF-C), bovine coronavirus (BCoV) and mouse hepatitis virus strain S (MHV-S) and of two chimeric recombinant viral haemagglutinin esterases, from influenza C/Cal/78 virus (HE12-GFP) and sialodacryoadenitis virus (SDAV-HE) was investigated.
The inhibitory effect of compounds 3 and 4 toward the 9-SOAE activities of HE12-GFP, INF-C virus and BCoV and of compounds 1 and 2 toward the 4-SOAE activity of SDAV-HE and MHV-S were determined by pNPA assay and fluorimetric HPLC. Compound 5 was used as a negative control. No sialidase activity against any of the inhibitors was detected in the esterase preparations. The production, isolation and purification of the viruses and enzymes as well as the assays are described in the experimental section.
4.2. Inhibition results
Sialate-O-acetylesterases were incubated in the presence of different sialoside concentrations. Table 2 summarises the inhibitory effect of 9-modified sialosides 3 and 4 toward the 9-SOAE enzymes determined by pNPA assay. We identified the 9-O-methyl sialoside 3 as a potential inhibitor of HE12-GFP. At a concentration of 5 mM compound 3 the inhibition is approximately 86%. Analysis by fluorimetric HPLC revealed a 10–15% inhibition by 3 of Neu5,9Ac2-hydrolysis by HE12-GFP at a concentration of 1 mM, thus confirming the results of the pNPA assay (data not shown).
Table 2.
Inhibitory effect of 9-modified sialosides 3 and 4 on the 9-SOAE enzymes from INF-C virus and BCoV.
| Sialoside concentration | HE12-GFP |
INF-C virus |
BCoV |
|||
|---|---|---|---|---|---|---|
| 3 | 4 | 3 | 4 | 3 | 4 | |
| 0.5 mM | 22.95%a | 1.06% | 7.01% | <0% | 2.86% | 9.78% |
| 1 mM | 22.42% | 6.08% | 8.63% | 3.85% | 9.00% | 2.70% |
| 2 mM | 33.03% | 7.91% | 7.05% | 7.39% | <0% | <0% |
| 5 mM | 85.79% | – | – | – | – | – |
Inhibition (in %) of 9-SOAE-catalysed hydrolysis of p-nitrophenyl acetate.
Less inhibitory effect of 3 was detected with the whole influenza virus particles. We suggest that the different conformation of the haemagglutinin protein (recombinant HE12-GFP = monomeric protein; whole virus = trimeric protein) may contribute to the differences in the inhibitory effect of sialoside 3. The 9-SOAE of BCoV was also less effected by 3. According to [39], it seems that the BCoV is more dependent on the aglycon moiety than the influenza C virus esterase. It may be that the allyl group of the sialosides has an effect on the enzymatic reaction and consequently less inhibition was detected.
Comparison of the inhibitory activities of 9-modified sialosides 3 and 4 suggests that the negatively charged methylphosphonate does not induce detectable inhibition when compared to the unmodified sialoside 5. There is however, a significant effect of the small, hydrophobic, methyl group at the same position indicating that inhibition can be improved by further modifications at this position. This effect has so far been seen only with the recombinant esterase from the influenza C virus, not with whole virus. It can be speculated that this is a result of it being a monomer rather than the native trimer, but that will require further studies with optimised inhibitors of higher affinity.
For influenza C virus esterase, these results are in line with those from an earlier study where a Ki of 4.2 mM was determined for a 9-acetamido-9-deoxy-sialic acid derivative [51].
Table 3 summarises the inhibitory behaviour of compounds 1 and 2 toward MHV-S and SDAV-HE determined by the pNPA assay. No significant inhibition was observed even at concentrations of 5 mM, indicating that there is less scope for modification of the 4-hydroxy group. It should be kept in mind as well that the mode of action of 4-SOAE is may be different to that of 9-SOAE.
Table 3.
Inhibitory effect of 4-modified sialosides 1 and 2 on the 4-SOAE enzymes from MHV-S and SDAV.
| Sialoside concentration | MHV-S |
SDAV-HE |
||
|---|---|---|---|---|
| 1 | 2 | 1 | 2 | |
| 0.1 mM | 4.00%a | 5.00% | 2.13% | 8.30% |
| 0.5 mM | 0% | 2.36% | 2.56% | 2.89% |
| 1 mM | 8.31% | 11.89% | 0.51% | 0.65% |
| 5 mM | 7.73% | 7.81% | 8.73% | 10.96% |
Inhibition (in %) of 4-SOAE-catalysed hydrolysis of p-nitrophenyl acetate.
Unmodified control sialoside 5 (negative control) did not show detectable inhibition of any of the enzymes. 3,4-Dichloroisocoumarin (positive control) was highly reactive towards all esterase activities at 0.1 mM concentration.
5. Conclusions
We have synthesised a set of modified sialosides useful for probing the active sites of 4- and 9-sialate-O-acetylesterase enzymes. We have screened the compounds for inhibition of a set of viral SOAE’s and while no inhibition of 4-SOAE could be detected, a 9-O-methyl derivative showed inhibition of the recombinant SOAE from influenza C virus. Further studies on how his can be exploited to develop high-affinity inhibitors of the enzyme as potential lead compounds for drug development are under way.
6. Experimental section
6.1. General
Where anhydrous solvents were required for reactions, these were purchased (anhydrous) and used as received. DCM was doubly distilled (over CaH2) before use. Fine chemicals were purchased from Aldrich-, Sigma- or Acros-Chemicals and were of the highest purity available. Reactions were monitored via thin layer chromatography (TLC) using pre-coated silica sheets with fluorescent indicator UV254. Compound detection was achieved by UV absorption and by developing plates by staining with a molybdenum phosphate reagent (20 g ammonium molybdate and 0.4 g cerium(IV) sulphate in 400 mL of 10% aqueous sulphuric acid) with subsequent heating.
Chromatographic purification was performed using silica gel 60A ‘Davisil’ (particle size 35–70 μm) from Fisher Scientific, UK. Silica-based MPLC chromatography was carried out on the Büchi Sepacore system equipped with glass columns packed with LiChroprep Si 60 (15–25 μm) from Merck, Darmstadt, Germany. Solvents for chromatography were used as received except for toluene and ethyl acetate, which were distilled before use. Gel permeation chromatography was carried out in the 1–10 mg scale on an XK 16/70 column (bed volume 130 mL), from Amersham packed with Sephadex G-10 (particle size 40–120 μm) and 0.1M NH4HCO3 as buffer. Detection was achieved with a differential refractometer from Knauer, Berlin, Germany.
1H NMR, 13C NMR, 31P NMR and all multidimensional spectra were recorded on Varian VNMRS spectrometers (600 MHz, 500 MHz or 400 MHz). Chemical shifts in 1H and 13C NMR spectra were referenced to the residual proton resonance of the respective deuterated solvents, CDCl3 (7.26 ppm), D2O (4.80 ppm) and CD3OD (3.31 ppm) respectively. For 31P NMR spectra H3PO4 was used as external standard (0 ppm).
HR-ESI-MS spectra were recorded on a Bruker Daltonics Apex III in positive mode with MeOH and/or H2O as solvent. Where possible, HR-ESI-MS has been used to characterise compounds which have been synthesised.
Abbreviations: EA: ethyl acetate; DCM: dichloromethane; Tol: toluene. THF: tetrahydrofuran.
6.2. Chemistry
6.2.1. Ammonium (allyl 5-acetamido-3,5-dideoxy-4-O-methyl-d-glycero-α-d-galacto-2-nonulopyranosidonate) (1)
Compound 7 (55.6 mg, 0.138 mmol) was dissolved in dry CH3CN (0.7 mL), under N2 (g) and was cooled to 0 °C (ice bath). DMSO4 (24.5 μL, 0.259 mmol) was added to the solution, followed by NaH(s) (6.6 mg, 0.276 mmol). The resulting suspension was allowed to stir for 4 h at 0 °C, more DMSO4 (24.5 μL, 0.259 mmol) was added and the suspension was left to stir overnight. When TLC indicated completion of the reaction, the reaction was quenched with excess NH4Cl(s) and the solution was diluted with DCM (3 mL), filtered over Celite and evaporated to dryness. After flash chromatography (Tol:Acetone 1:1) the corresponding 4-O-methyl ether was obtained as a clear oil (29.3 mg, 51%). Rf : 0.24 (Tol:Acetone 2:1). HR-ESI-MS (m/z) [M + Na]+ calculated for C19H31NO9Na: 440.1897. Found : 440.1891. 1H NMR (600 MHz, CDCl3) δ H: 1.36, 1.39 (2s, 6H, –C(CH 3)2), 1.73 (dd, 1H, J = 12.1, 12.2 Hz, H3ax), 2.05 (s, 3H, –NHCOCH3), 2.87 (dd, 1H, J = 4.5, 12.8 Hz, H3eq), 3.34 (s, 3H, –OCH3), 3.37 (d, 1H, J = 10.5 Hz, H6), 3.42 (ddd, 1H, J = 4.5, 10.7, 10.9, H4), 3.56 (dd, 1H, J = 5.6, 5.7 Hz, H7), 3.78 (s, 3H, –CO2CH 3), 3.87 (m, 1H, H5), 4.04–4.13 (m, 3H, H9, H9′, - CHH’–CH = CH2), 4.28–4.33 (m, 2H, H8, CHH’–CH = CH2), 4.42 (d, 1H, J = 4.9 Hz, –OH), 5.15 (d, 1H, J = 10.5 Hz, –CH2–CH = CH cisH), 5.25 (d, 1H, J = 17.2 Hz, –CH = CH transH), 5.39 (d, 1H, J = 7.4 Hz, –NH), 5.86 (ddd, 1H, J = 55.6, 10.7, 16.4 Hz, –CH2–CH = CH2). 13C NMR (151 MHz, CDCl3) δ C: 23.47 (–NHCOCH3), 25.75, 26.99 (–C(CH3)2), 36.42 (C3), 51.27 (C5), 52.54 (–CO2 CH3), 55.57 (–OCH3), 65.47 (–CH2CH CH2), 66.93 (C9), 69.86 (C7), 74.76 (C6), 75.44 (C8), 75.94 (C4), 99.21 (C2), 108.71 (–C(CH3)2), 117.31 (–CH2CH CH2), 134.03, (–CH2 CH CH2), 169.20 (C1), 173.85 (–NHCOCH3).
The methyl ether (19.9 mg, 0.0477 mmol) was dissolved in 80% aqueous AcOH (1.5 mL), the mixture was stirred for 2 h, the solvent was evaporated and the oily product was then co-evaporated with toluene and dried in vacuo. Aqueous NaOH (1.5 mL, 0.1 M) was added to the oil and the resulting solution was left to stir for 2 h. After completion, the solution was neutralised using Amberlite IR-120 (H+) ion-exchange resin, the resin was removed and the solvent removed in vacuo yielding 1 (17.9 mg, ∼qu). A sample of 1 (9.9 mg) was purified by gel permeation chromatography which was then lyophilised to give a white powder. HR-ESI-MS (m/z) [M + Na]+ calculated for : C15H25NO9Na: 386.1427. Found: 386.1422. 1H NMR (600 MHz, D2O) δ H: 1.65 (dd, 1H, J = 12.0, 12.1 Hz, H3ax), 2.10 (s, 3H, –NHCOCH3), 2.99 (dd, 1H, J = 4.5, 12.6 Hz, H3eq), 3.48 (s, 3H, –OCH3), 3.52 (m, 1H, H4), 3.65 (d, 1H, J = 8.9 Hz, H7), 3.72 (m, 1H, H9), 3.81 (dd, 1H, J = 1.5, 10.5 Hz, H6), 3.91–4.00 (m, 3H, H5, H8, H9’), 4.12 (ddd, 1H, J = 1.2, 5.7, 12.2 Hz, CHH’–CH = CH2), 4.33 (ddd, 1H, J = 1.1, 6.0, 12.1 Hz, CHH’–CH = CH2), 5.30 (d, 1H, J = 10.3 Hz, –CH2–CH = CH cisH), 5.41 (dd, 1H, J = 1.4, 17.3 Hz, –CH = CH transH), 6.02 (m, 1H, –CH2–CH = CH2). 13C NMR (151 MHz, D2O) δ C: 21.97 (–NHCOCH3), 36.90 (C3), 50.22 (C5), 56.34 (–OCH3), 62.68 (C9), 65.92 (–CH2CH CH2), 68.19 (C7), 71.44 (C8), 72.77 (C6), 77.20 (C4), 100.38 (C2), 118.22 (–CH2CH CH2), 133.67, (–CH2 CH CH2), 172.94 (C1), 174.93 (–NHCOCH3).
6.2.2. Diammonium (allyl 5-acetamido-3,5-dideoxy-4-O-(P-methylphosphonyl)- D-glycero-α-d-galacto-2-nonulopyranosidonate) (2)
Under an N2 (g) atmosphere, compound 9 (18 mg, 0.0305 mmol) was dissolved in anhydrous THF (0.7 mL). Triethylamine (60 μL, 0.427 mmol) was added followed by a few minutes of stirring. Thiophenol (22 μL, 0.213 mmol) was added and the solution was stirred overnight. Over the course of 4 days, gentle heating (35–40 °C) and a further 4 portions of triethylamine (60 μL, 0.427 mmol) and thiophenol (22 μL, 0.213 mmol) were added whilst maintaining the solvent volume (∼0.5–0.7 mL). Once TLC analysis had indicated that the reaction had progressed no further, the solvent was evaporated in vacuo and purified on a short silica plug (EA:MeOH; 5:1 → DCM:MeOH; 1:1 → MeOH) to give the deprotected phosphonate intermediate which was lyophilised with dioxane/H2O (9 mg, 96%). The deprotected phosphonate intermediate was then heated to 50 °C with pyridine:NEt3:H2O (1:1:1, 0.75 mL). After stirring overnight, the solvent was evaporated in vacuo and the residue was purified by gel permeation chromatography to afford compound 2 (9 mg, 76%) as a white solid after lyophilisation. HR-ESI-MS (m/z) [M + Na]+ calculated for: C15H26NOPNa: 450.1141. Found: 450.1136. 1H NMR (500 MHz, MeOH-D4) δ H: 1.29 (d, 1H, J = 16.6 Hz, P-CH 3), 1.84 (dd, 1H, 12.1, 12.3 Hz, H3ax), 2.03 (s, 3H, –NHCOCH 3), 2.91 (dd, 1H, J = 4.7, 12.6 Hz, H3eq), 3.53 (dd, 1H, J = 1.5, 9.0 Hz, H7), 3.57 (dd, 1H, J = 1.5, 10.4 Hz, H6), 3.62 (dd, 1H, J = 5.8, 11.4 Hz, H9), 3.74 (dd, 1H, J = 10.1, 10.2 Hz, H5), 3.83 (dd, 1H, J = 2.5, 11.4 Hz, H9’), 3.86 (m, 1H, H8), 4.01 (ddd, 1H, J = 1.2, 5.6, 12.6 Hz, –CHH’–CH = CH2), 4.26–4.32 (m, 2H, H4, –CHH’–CH = CH2), 5.10 (dd, 1H, J = 1.4, 10.5 Hz, –CH2–CH = CHH cis), 5.25 (dd, 1H, J = 1.7, 17.3 Hz, –CH2–CH = CHH trans), 5.90 (m, 1H, –CH2–CH = CH2). 13C NMR (126 MHz, MeOH-D4) δ C: ∼13.68 (d, J = 139.2 Hz, P–CH3), 22.94 (–NHCOCH3), 41.49 (C3), 53.69 (C5), 64.76 (C9), 66.51 (–CH2CH CH2), 70.52 (C7), 71.51 (C4), 72.92 (C8), 75.28 (C6), 100.57 (C2), 116.92 (–CH2CH CH2), 135.92 (–CH2 CH CH2), 172.77 (C1), 176.05 (–NHCOCH3). 31P NMR (400 MHz, MeOH-D4) δP: 24.73 (s).
6.2.3. Ammonium (allyl 5-acetamido-9-O-methyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosidonate) (3)
A stirred solution of epoxide 10 (50 mg, 0.145 mmol) in methanol (1.2 mL) was acidified (to pH 2) with Amberlite IR-120 (H+) ion-exchange resin, the resin was filtered off and the solvent evaporated from the solution to leave an oily residue. The crude product was purified by flash chromatography (EA → EA:MeOH; 5:1) to yield the 9-O-methyl ether (16.4 mg, 45%). Rf : 0.38 (EA:MeOH; 5:1). HR-ESI-MS (m/z) [M + Na]+ calculated for : C16H21NO9Na: 400.1584. Found: 400.1578. 1H NMR (500 MHz, CDCl3) δ H: 2.04 (s, 3H, -NHCOCH 3), 1.96 (dd, 1H, J = 11.7, 12.2 Hz, H3ax), 2.74 (bs, 1H, –OH), 2.81 (dd, 1H, J = 4.3, 13.3 Hz, H3eq), 3.39–3.43 (m, 1H, H6), 3.43 (s, 3H, –OCH3), 3.53 (dd, 1H, J = 3.3, 9.0 Hz, H7), 3.65–3.80 (m, 5H, H4, H5, H9, H9’, OH), 3.85 (s, 3H, –CO2CH3), 3.92 (dd, 1H, J = 5.9, 12.5 Hz, –CHH’–CH = CH2), 4.01 (dd, 1H, J = 3.5, 8.7 Hz, H8), 4.27 (dd, 1H, J = 5.4, 12.6 Hz, –CHH’–CH = CH2), 4.67 (bs, 1H, OH), 5.15 (d, 1H, J = 10.5 Hz, CH2–CH = CH cisH), 5.23 (dd, 1H, J = 1.0, 17.2 Hz, –CH = CH transH), 5.83 (ddd, 1H, J = 5.7, 10.8, 16.3 Hz, –CH2–CH = CH2), 6.73 (d, 1H, J = 6.8 Hz, –NH). 13C NMR (126 MHz, CDCl3) δ C: 23.07 (–NHCOCH3), 40.79 (C3), 53.33, 53.42 (C5, –CO2 CH3), 59.21 (–OCH3), 65.42 (–CH2–CH = CH2), 68.15 (C4), 68.79 (C7), 69.98 (C8), 73.86, 73.87 (C6, C9), 98.58 (C2), 117.46 (–CH2–CH = CH2), 133.73 (–CH2–CH = CH2), 169.82 (C1), 173.72 (–NHCOCH3).
The ether (5 mg, 0.0132 mmol) was dissolved in dioxane (0.5 mL) and NaOH (0.05M, 0.5 mL) was added with stirring. The mixture was stirred for 2 h at room temperature, neutralised with Amberlite IR-120 (H+) ion-exchange resin, filtered and the solvent was evaporated in vacuo. The residue was purified by gel permeation chromatography to afford compound 3 (5 mg, qu.) as a powder. HR-ESI-MS (m/z) [M + Na]+ calculated for: C15H25NO9Na: 386.1427. Found 386.1422. 1H NMR (500 MHz, D2O) δ H: 1.76 (dd, 1H, J = 12.1, 12.3 Hz, H3ax), 2.11 (s, 3H, –NHCOCH 3), 2.82 (dd, 1H, J = 4.6, 12.5 Hz, H3eq), 3.48 (s, 3H, –OCH 3), 3.61–3.68 (m, 2H, H7, H9), 3.72–3.83 (m, 3H, H4, H6, H9’), 3.90 (dd, 1H, J = 10.1, 10.2 Hz, H5), 4.05 (dd, 1H, J = 7.7, 7.7 Hz, H8), 4.10 (dd, 1H, J = 5.8, 12.2 Hz, –CHH’–CH = CH2), 4.32 (dd, 1H, J = 6.1, 12.1 Hz, –CHH’–CH = CH2), 5.30 (d, 1H, J = 10.4 Hz, –CH2–CH = CH cisH), 5.40 (d, 1H, J = 17.4 Hz, –CH = CH transH), 6.02 (ddd, 1H, J = 6.0, 11.2, 16.6 Hz, –CH2–CH = CH2). 13C NMR (126 MHz, D2O) δ C: 21.94 (–NHCOCH3), 40.40 (C3), 51.76 (C5), 58.36 (–OCH3), 65.96 (–CH2–CH = CH2), 68.17, 68.19 (C4, C7), 70.03 (C8), 72.46 (C6), 73.14 (C9), 100.53 (C2), 118.14 (–CH2–CH = CH2), 133.67 (–CH2–CH = CH2), 173.38 (C1), 174.94 (–NHCOCH3).
6.2.4. Diammonium (allyl 5-acetamido-3,5-dideoxy-9-O-(P-methylphosphonyl)- D-glycero-α-d-galacto-2-nonulopyranosidonate) (4)
Under an N2 (g) atmosphere, compound 11 (14 mg, 0.0240 mmol) was dissolved in anhydrous THF (0.7 mL). Triethylamine (47 μL, 0.336 mmol) was added followed by a few minutes of stirring. Thiophenol (17 μL, 0.168 mmol) was then added and the solution was stirred overnight. Over the course of 4 days, a further 4 portions of triethylamine (47 μL, 0.336 mmol) and thiophenol (17 μL, 0.168 mmol) were added whilst maintaining the THF solvent volume (∼0.5–0.7 mL). Once TLC analysis had indicated that the reaction had progressed no further, the solvent was evaporated in vacuo and purified on a short silica plug (DCM:MeOH; 5:1 → 2:1 → MeOH) to give the deprotected phosphonate intermediate which was lyophilised with dioxane/H2O (10.2 mg, 74%). The deprotected phosphonate intermediate (10 mg) was then treated with a 1:1 mixture of dioxane and 0.05 M NaOH (aq.) at approximately 4–5 °C for 2 h after which the solution was neutralised with ion-exchange resin Amberlite IR-120. After filtration, the solvent was removed and the residue was purified by gel permeation chromatography to afford compound 4 (9.6 mg, ∼qu.) after lyophilisation. HR-ESI-MS (m/z) [M + Na]+ calculated for: C15H26NOPNa: 450.1141. Found: 450.1136. 1H NMR (500 MHz, D2O) δ H: 1.41 (d, 3H, J = 16.4 Hz, P–CH 3), 1.79 (dd, 1H, J = 12.1, 12.2 Hz, H3ax), 2.11 (s, 3H, –NHCOCH 3), 2.80 (dd, 1H, J = 4.3, 12.5 Hz, H3eq), 3.74 (d, 1H, J = 8.4 Hz, H7), 3.76–3.82 (m, 1H, H4), 3.87 (d, 1H, J = 10.3 Hz, H6), 3.93 (dd, 1H, J = 9.9, 10.1 Hz, H5), 4.02–4.09 (m, 2H, H8, H9), 4.12 (dd, 1H, J = 5.7, 12.0 Hz, –CHH’–CH = CH2), 4.15–4.22 (m, 1H, H9’), 4.34 (dd, 1H, J = 6.1, 12.1 Hz, –CHH’–CH = CH2), 5.31 (d, 1H, J = 10.2 Hz, –CH2–CH = CH cisH), 5.41 (d, 1H, J = 17.3 Hz, –CH = CH transH), 6.01 (ddd, 1H, J = 5.7, 11.3, 16.1 Hz, –CH2–CH = CH2). 13C NMR (126 MHz, D2O) δ C: ∼10.80 (d, J = 137 Hz, P–CH3), 22.07 (–NHCOCH3), 40.02 (C3), 51.83 (C5), 65.04 (d, J = 5 Hz, C9), 65.90 (–CH2–CH = CH2), 67.64 (C7), 68.01 (C4), 70.13 (d, J = 7 Hz, C8), 72.62 (C6), 99.96 (C2), 118.33 (–CH2–CH = CH2), 133.57 (–CH2–CH = CH2), 172.61 (C1), 174.95 (–NHCOCH3).
31P NMR (162 MHz, D2O) δP: 28.45 (s).
6.2.5. Ammonium (allyl 5-acetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosidonate) (5)
Compound 6 (10 mg, 0.0275 mmol) was dissolved in dioxane (0.5 mL) with stirring. Aqueous NaOH (0.1 M, 0.5 mL) was added and the solution was stirred for 2 h. The solution was then neutralised with Amberlite IR-120 (H+) ion-exchange resin, filtered and the solvent was removed in vacuo. The crude residue was then purified by gel permeation chromatography and lyophilised to give a white powder (9 mg, qu). HR-ESI-MS (m/z) [M + Na]+ calculated for : C14H23NO9Na: 372.1271 Found: 372.1265. 1H NMR (500 MHz, D2O) δ H: 1.74 (dd, 1H, J = 12.1, 12.1 Hz, H3ax), 2.11 (s, 3H, –NHCOCH3), 2.83 (dd, 1H, J = 4.4, 12.4 Hz, H3eq), 3.66 (d, 1H, J = 8.9 Hz, H7), 3.69–3.82 (m, 3H, H4, H6, H9), 3.85–3.99 (m, 3H, H5, H8, H9’), 4.09 (dd, 1H, J = 5.7, 12.0 Hz, –CHH’–CH = CH2), 4.32 (dd, 1H, J = 6.2, 12.0 Hz, –CHH’–CH = CH2), 5.30 (d, 1H, J = 10.4 Hz, –CH2–CH = CH cisH), 5.40 (d, 1H, J = 17.3 Hz, –CH = CH transH), 6.02 (ddd, 1H, J = 5.9, 11.4, 16.5 Hz, –CH2–CH = CH2). 13C NMR (500 MHz, D2O) δ C: 22.01 (–NHCOCH3), 40.45 (C3), 51.88 (C5), 62.60 (C9), 65.95 (–CH2CH CH2), 68.21 (C4, C7), 71.69 (C8), 72.65 (C6), 100.59 (C2), 118.19 (–CH2CH CH2), 133.76, (–CH2 CH CH2), 173.42 (C1), 175.06 (–NHCOCH3).
6.2.6. Methyl (allyl 5-acetamido-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosid)onate (6)
Compound 6 was synthesised as described in the literature [20]. Rf : 0.16 (DCM:MeOH 7:1). HR-ESI-MS (m/z) [M + Na]+ calculated for C15H25NO9Na : 386.1427. Found: 386.1421.
1H NMR (500 MHz, MeOH-D4) δ H: 1.76 (dd, 1H, J = 12.5, 12.5 Hz, H3ax), 2.00 (s, 3H, –NHCOCH 3), 2.70 (dd, 1H, J = 4.6, 12.8 Hz, H3eq), 3.51 (d, 1H, J = 9.1 Hz, H7), 3.58 (d, 1H, J = 10.5 Hz, H6), 3.61–3.68 (m, 2H, H4, H9), 3.77 (dd, 1H, J = 10.0, 10.2 Hz, H5), 3.83 (s, 3H, –CO2CH 3), 3.84–3.87 (m, 2H, H8, H9’), 3.99 (dd, 1H, J = 5.4, 12.6 Hz, –CHH’–CH = CH2), 4.29 (dd, 1H, J = 5.2, 12.9 Hz, –CHH’–CH = CH2), 5.12 (d, 1H, J = 10.3 Hz, –CHH’–CH = CHH cis), 5.23 (d, 1H, J = 17.3 Hz, –CHH’–CH = CHH trans), 5.87 (m, 1H, –CHH’–CH = CH2). 13C NMR (126 MHz, CDCl3) δ C: 22.82 (–NHCOCH3), 41.86 (C3), 53.50 (–CO2 CH3), 53.99 (C5), 64.88 (C9), 66.37 (–CH2CH CH2), 68.67 (C4), 70.38 (C7), 72.60 (C8), 75.11 (C6), 100.16 (C2), 117.09 (–CH2CH CH2), 135.58 (–CH2 CH CH2), 171.15 (C1), 175.35 (–NHCOCH3).
6.2.7. Methyl (allyl 5-acetamido-3,5-dideoxy-8,9-O-isopropylidene-d-glycero-α-d-galacto-2-nonulopyranosid)onate (7)
Compound 6 (56 mg, 0.154 mmol) was dissolved in dry acetone (1.85 mL) under a atmosphere of N2(g). 2,2-Dimethoxypropane (200 μL) was added to the solution, followed by p-TsOH·H2O (7 mg). The solution was left to stir at room temperature, under N2, for 2 h. Once complete, NEt3 (few drops) was added to the solution to neutralise the reaction and the solvent was evaporated to dryness. After flash chromatography (Tol:Acetone 1:1), a clear oil was obtained, which after further drying in vacuo yielded 7 as a white foam (53 mg, 85%). Rf : 0.52 (Tol:Acetone 1:1). HR-ESI-MS (m/z) [M + Na]+ calculated for C18H29NO9Na : 426.1735. Found : 426.1734. 1H NMR (500 MHz, CDCl3) δ H: 1.34, 1.38 (2s, 6H, –C(CH 3)2), 1.79 (dd, 1H, J = 12.0, 12.2 Hz, H3ax), 2.02 (s, 3H, –NHCOCH3), 2.69 (dd, 1H, J = 4.7, 12.8 Hz, H3eq), 3.43 (d, 1H, J = 10.4 Hz, H6), 3.60 (dd, 1H, J = 5.6, 5.7 Hz, H7), 3.69 (m, 1H, H4), 3.74–3.81 (m, 4H, H5, –CO2CH3), 3.98–4.11 (m, 4H, H9, H9′, –OH, –CHH’–CH = CH2), 4.14 (d, 1H, J = 5.7 Hz, –OH), 4.21–4.27 (m, 2H, –CHH’–CH = CH2, H8), 5.13 (ddd, 1H, J = 1.1, 1.4, 10.5 Hz, –CH2–CH = CH cisH), 5.23 (ddd, 1H, J = 1.4, 1.6, 17.2 Hz, –CH = CH transH), 5.83 (ddd, 1H, J = 5.6, 10.6, 16.3 Hz, –CH2–CH = CH2), 6.59 (d, 1H, J = 8.0 Hz, –NH). 13C NMR (126 MHz, CDCl3) δ C: 23.30 (–NHCOCH3), 25.64, 26.93 (–C(CH3)2), 40.62 (C3), 52.67 (–CO2 CH3), 53.19 (C5), 65.53 (–CH2CH CH2), 66.55 (C9), 67.94 (C4), 69.79 (C7), 74.66 (C6), 75.88 (C8), 99.15 (C2), 108.75 (–C(CH3)2), 117.27 (–CH2CH CH2), 134.02, (–CH2 CH CH2), 169.17 (C1), 173.50 (–NHCOCH3).
6.2.8. Methyl (allyl 5-acetamido-3,5-dideoxy-8,9-O-carbonyl-d-glycero-α-d-galacto-2-nonulopyranosid)onate (8)
Compound 6 (345 mg, 0.949 mmol) and 4-dimethylaminopyridine (278 mg, 2.28 mmol) were dried in vacuo for approximately 1 h and then placed under an atmosphere of N2. Dry DCM (20 mL) was added via syringe and the resulting suspension formed was ultrasonicated for a few minutes then cooled to approximately 0 °C. Diphosgene (152 μL, 1.23 mmol) was added via microsyringe to the white suspension with vigourous stirring. When TLC indicated completion of the reaction, the reaction was quenched by pouring the solution into 40 mL of cold 10% KH2PO4. The DCM layer was separated and the aqueous layer was extracted with ethyl acetate (4 × 25 mL). The organic phases were then dried over MgSO4 and the solvents were evaporated. After flash chromatography (EA:MeOH; 15:1), compound 8 was obtained as a clear oil (220 mg, 60%). Rf : 0.63 (EA:MeOH; 5:1). HR-ESI-MS (m/z) [M + Na]+ calculated for C16H23NO10Na : 412.1220. Found: 412.1214. 1H NMR (500 MHz, MeOH-D4) δ H: 1.74 (dd, 1H, J = 12.3, 12.3 Hz, H3ax), 2.01 (s, 3H, –NHCOCH 3), 2.66 (dd, 1H, J = 4.6, 12.8 Hz, H3eq), 3.53 (d, 1H, J = 10.5 Hz, H6), 3.61 (ddd, 1H, J = 4.5, 10.6, 12.9 Hz, H4), 3.75–3.79 (m, 1H, H5), 3.80 (s, 3H, –CO2CH3), 3.93 (m, 1H, H7), 3.97 (dd, 1H, J = 5.3, 12.6 Hz, –CHH’–CH = CH2), 4.19 (dd, J = 4.2, 12.8 Hz, –CHH’–CH = CH2), 4.55 (dd, 1H, J = 8.4, 8.4 Hz, H9), 4.70 (dd, 1H, J = 7.2, 7.9 Hz, H9’), 4.95 (m, 1H, H8), 5.14 (d, 1H, J = 10.5 Hz, –CHH’–CH = CHH cis), 5.26 (d, 1H, J = 17.4 Hz, –CHH’–CH = CHH trans), 5.86 (dddd, 1H, J = 1.08, 5.2, 10.5, 16.8, –CHH’–CH = CH2). 13C NMR (126 MHz, MeOH–D4) δ C: 22.91 (–NHCOCH3), 41.61 (C3), 53.25 (C5), 53.81 (–CO2CH3), 66.44 (–CH2CH CH2), 67.39 (C9), 68.32 (C4), 69.37 (C7), 76.35 (C6), 79.78 (C8), 100.64 (C2), 117.09 (–CH2CH CH2), 135.36 (–CH2 CH CH2), 157.53 (–O–C(O)–O–), 170.05 (C1), 175.25 (–NHCOCH3).
6.2.9. Methyl (allyl 5-acetamido-8,9-O-carbonyl-3,5-dideoxy-4-O-(O,P-dimethylphosphonyl)- D-glycero-α-d-galacto-2-nonulopyranosid)onate (9)
Compound 8 (75 mg, 0.193 mmol) was placed in a flask under an N2 (g) atmosphere. Dry DCM (2 mL) was added via syringe resulting in a suspension which required ultrasonication. N,N-diisopropylethylamine (67 μL, 0.386 mmol) was added to the suspension followed by further ultrasonication. With continuous stirring, the suspension was cooled to approximately −15 °C. Methyl methylphosphonyl chloride (27 μL, 0.270 mmol) was added dropwise to the solution via microsyringe. The reaction was allowed to warm to room temperature and was stirred overnight. The reaction was again then cooled to −15 °C, where a further portion of N,N-diisopropylethylamine (67 μL, 0.386 mmol) and Methyl methylphosphonyl chloride (27 μL, 0.270 mmol) was added. Once TLC indicated completion, the reaction was quenched by the addition of MeOH (1 mL) and a small spatula of NaHCO3 (s). After stirring for 10 min, the solvent was removed in vacuo and the product was purified by flash chromatography (EA:MeOH; 7:1 → 5:1) to give 9 (57 mg, 60%). Rf : 0.21 (EA:MeOH; 10:1). HR-ESI-MS (m/z) [M + Na]+ calculated for C18H28NO12P Na : 504.1247. Found 504.1241. 1H NMR (500 MHz, CDCl3) δ H: 1.53, 1.47 (2d, 3H, J = 17.7, 17.8 Hz, P–CH 3), 2.01 (m, 1H, H3ax), 2.06 (s, 3H, –NHCOCH 3), 2.73, 2.78 (2dd, 1H, J = 5.0, 12.8, 4.9, 12.9 Hz, H3eq), 3.25 (d, 1H, J = 10.3 Hz, H6), 3.66, 3.75 (2d, 3H, J = 11.5, 11.2 Hz, P–OCH 3), 3.81, 3.82 (2s, 3H, –CO2CH 3), 3.83–3.92 (m, 1H, H5), 4.02 (dd, 1H, J = 5.4, 12.4 Hz, –CHH’–CH2 = CH2), 4.22 (dd, 1H, J = 5.1, 12.6 Hz, CHH’–CH2 = CH2), 4.40, 4.53 (m, 1H, H4), 4.49 (dd, 1H, J = 8.5 Hz, H9), 4.65 (m, 1H, H9’), 4.74 (bs, 1H, –OH), 4.87 (m, 1H, H8), 5.16 (d, 1H, J = 10.5 Hz, –CH2–CH = CHH cis), 5.24 (d, 1H, J = 17.3 Hz, –CH2–CH = CHH trans), 5.82 (ddd, 1H, J = 5.4, 10.5, 16.1 Hz, –CH2–CH = CH2), 7.43 (m, 1H, –NH). 13C NMR (126 MHz, CDCl3) δ C: ∼10.85, ∼11.18 (2d, J = 143.41, 146.06 Hz, P–CH3), 23.14 (2s, –NHCOCH3), 38.95 (C3), 52.00 (C5), 52.27 (d, J = 7.4 Hz, P–OCH3), 53.09, 53.16 (–CO2CH3), 65.65 (–CH2–CH = CH2), 66.85, 66.89 (C9), 69.03, 69.05 (C7), 71.68, 71.72 (C4), 74.99 (C6), 75.86, 75.91 (C8), 99.04, 99.13 (C2), 117.55 (–CH2–CH = CH2), 133.57, 133.59 (–CH2–CH = CH2), 155.15 (–O–C(O)–O–), 168.38, 168.41 (C1), 174.23, 174.32 (–NHCOCH3).
31P NMR (126 MHz, CDCl3) δP: 34.72 (bs).
6.2.10. Methyl (allyl 5-acetamido-8,9-anhydro-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosid)onate (10)
Compound 6 (560 mg, 1.54 mmol) was co-evaporated with dry pyridine and was then suspended in dry pyridine (12 mL) under N2 (g). The resulting suspension was cooled to to 0 °C (ice bath) and toluenesulfonyl chloride (293 mg, 1.54 mmol) was quickly added to the stirred solution.
After 20 min, the solution was allowed to warm up to room temperature and was subsequently stirred for a further 5 h after which another portion of toluenesulfonyl chloride (293 mg, 1.54 mmol) was added. The solution was left to stir overnight. Once completed, solvent was removed in vacuo and the crude product was purified by flash chromatography (EA → EA:MeOH; 5:1) to yield the 9-O-tosylate as a white foam (470 mg, 59%). Rf : 0.70 (EA:methanol; 4:1). HR-ESI-MS (m/z) [M + Na]+ calculated for C22H31NO11SNa : 540.1516. Found: 540.1510. 1H NMR (500 MHz, CDCl3) δ H: 1.81 (dd, 1H, J = 11.7, 11.9 Hz, H3ax), 2.01 (s, 3H, –NHCOCH 3), 2.41 (s, 3H, –(Ar)C–CH3), 2.74 (dd, 1H, J = 3.8, 13.8 Hz, H3eq), 3.40 (d, 1H, J = 9.2 Hz, H6), 3.45 (d, 1H, J = 8.8 Hz, H7), 3.64–3.75 (m, 2H, H4, H5), 3.65–3.77 (bs, 1H, –OH), 3.77 (s, 3H, –CO2CH3), 3.86 (dd, 1H, J = 5.7, 12.6 Hz, –CHH’–CH = CH2), 3.91 (bs, 1H, –OH), 4.04 (m, 1H, H8), 4.12–4.20 (m, 2H, H9, –CHH’–CH = CH2), 4.29 (m, 1H, H9’), 4.68 (bs, 1H, –OH), 5.10 (dd, 1H, J = 0.8, 10.5 Hz, –CH2–CH = CHH cis), 5.18 (dd, 1H, J = 1.1, 17.3 Hz, –CH2–CH = CHH trans), 5.77 (ddd, 1H, J = 5.6, 10.8, 16.2 Hz, –CH2–CH = CH2), 6.61 (d, 1H, J = 7.0 Hz, –NH), 7.32 (d, 2H, J = 8.2 Hz, –(Ar)C–H), 7.75 (d, 2H, J = 8.3 Hz, –S–C(Ar)C–H). 13C NMR (126 MHz, CDCl3) δ C: 21.83 ((Ar)C–CH3), 23.22 (–NHCOCH3), 40.59 (C3), 52.99 (C5), 53.56 (–CO2 CH3), 65.49 (–CH2CH CH2), 67.69 (C4), 68.70 (C7), 69.29 (C8), 72.22 (C9), 73.72 (C6), 98.76 (C2), 117.50 (–CH2CH CH2), 128.13, 130.19 (H3C(Ar)C(Ar)C(H)–, H3C(Ar)C(Ar)C(H)–), 132.61 (–S(Ar)C(Ar)C(H)–), 133.68 (–CH2 CH CH2), 145.28 (–S(Ar)C(Ar)C(H)–), 169.89 (C1), 174.22 (–NHCOCH3).
The tosylate (194 mg, 0.375 mmol) was dissolved in in anhydrous MeOH (2.4 mL). Freshly prepared NaOMe (in anhydrous MeOH) (0.3 mL, 0.5 M) was added dropwise to the stirring solution. Further 0.1 mL portions of NaOMe (0.5 M) were added as required (a total of 0.7 mL NaOMe was used). The reaction was monitored by TLC. On completion, the solution was neutralised with Amberlite IR-120 (H+) ion-exchange resin. The resin was removed by filtration and the solvent was removed in vacuo. Flash chromatography of the crude material (EA:MeOH; 15:1 + 0.5% NEt3) gave epoxide 10 as a white foam (87 mg, 68%). Rf : 0.34 (EA:methanol; 10:1). HR-ESI-MS (m/z) [M + Na]+ calculated for C15H23NO8Na : 368.1321. Found: 368.1316. 1H NMR (500 MHz, CDCl3) δ H: 1.81 (dd, 1H, J = 12.1, 12.3 Hz, H3ax), 2.00 (s, 3H, –NHCOCH 3), 2.71 (dd, 1H, J = 4.6, 12.7 Hz, H3eq), 2.77 (dd, 1H, J = 2.5, 5.1 Hz, H9), 2.83 (dd, 1H, J = 4.3, 4.6 Hz, H9’), 3.24 (m, 1H, H8), 3.38 (dd, 1H, J = 4.8, 4.8 Hz, H7), 3.55 (dd, 1H, J = 0.9, 10.4 Hz, H6), 3.65 (m, 1H, H4), 3.76–3.82 (m, 1H, H5), 3.93 (bs, 1H, –OH), 3.96 (dd, 1H, J = 5.9, 12.6 Hz, –CHH’–CH = CH2), 4.25 (m, 1H, –CHH’–CH = CH2), 4.27 (d, 1H, J = 5.7 Hz, –OH), 5.12 (dd, 1H, J = 1.3, 10.5 Hz, –CH2–CH = CHH cis), 5.21 (dd, 1H, J = 1.5, 17.2 Hz, –CH2–CH = CHH trans), 5.82 (ddd, 1H, J = 5.6, 10.8, 16.1 Hz, –CH2–CH = CH2), 6.57 (d, 1H, J = 7.7 Hz, –NH). 13C NMR (126 MHz, CDCl3) δ C: 23.31 (–NHCOCH3), 40.90 (C3), 46.15 (C9), 52.06 (C8), 52.92 (C5), 53.10 (–CO2 CH3), 65.64 (–CH2CH CH2), 67.91 (C4), 69.55 (C7), 75.44 (C6), 99.11 (C2), 117.35 (–CH2CH CH2), 134.07 (–CH2 CH CH2), 169.29 (C1), 173.72 (–NHCOCH3).
6.2.11. Methyl (allyl 5-acetamido-4,7,8-tri-O-acetyl-3,5-dideoxy-9-O-(O,P-dimethylphosphonyl)-d-glycero-α-d-galacto-2-nonulopyranosid)onate (11)
Under an N2 (g) atmosphere, compound 6 (77 mg, 0.212 mmol) was suspended in dry DCM (2 mL) and the mixture was ultrasonicated. Diisopropylethylamine (81 μL, 0.466 mmol) was added to the suspension followed by further ultrasonication. With continuous stirring, the suspension was cooled to approximately −15 °C. Once acclimatised to temperature, methyl methylphosphonyl chloride (29 μL, 0.297 mmol) was added dropwise to the solution via microsyringe. After 5 h the reaction was again cooled to −15 °C, where a further portion of N,N-diisopropylethylamine (81 μL, 0.466 mmol) and methyl methylphosphonyl chloride (29 μL, 0.297 mmol) was added. The reaction was left to stir overnight. Once TLC indicated completion, the reaction was quenched by the addition of MeOH (1 mL) and a small spatula of NaHCO3 (s). After stirring for 10 min, the solvent was removed in vacuo and the product was purified by flash chromatography (EA:MeOH; 10:1 → 5:1) to give the 9-O-phosphonate (27 mg, 30%). Rf : 0.25 (EA:MeOH; 10:1). HR-ESI-MS (m/z) [M + Na]+ calculated for C17H30NO11PNa : 478.1454. Found 478.1449. 1H NMR (500 MHz, CDCl3) δ H: 1.54 (dd, 3H, J = 2.2, 17.6 Hz, P–CH 3), 1.75 (dd, 1H, J = 12.2, 12.3 Hz, H3ax), 2.00 (s, 3H, –NHCOCH 3), 2.69 (dd, 1H, J = 4.6, 12.8 Hz, H3eq), 3.52 (ddd, 1H, J = 1.4, 9.9, 10.1 Hz, H7), 3.62 (dd, 1H, J = 1.4, 10.5 Hz, H6), 3.66 (m, 1H, H4), 3.75 (dd, 3H, J = 1.7, 11.2 Hz, P–OCH 3), 3.75–3.80 (m, 1H, H5), 3.98–4.02 (m, 1H, H8), 3.98–4.02 (m, 1H, –CHH’–CH = CH2), 4.15 (m, 1H, H9), 4.26–4.33 (m, 1H, –CHH’–CH = CH2), 4.26–4.33 (m, 1H, H9’), 5.12 (dd, 1H, J = 1.1, 10.5 Hz, –CH2–CH = CHH cis), 5.24 (dd, 1H, J = 1.7, 17.2 Hz, –CH2–CH = CHH trans), 5.87 (ddd, 1H, J = 5.5, 10.6, 16.2 Hz, –CH2–CH = CH2). 13C NMR (126 MHz, CDCl3) δ C: ∼9.15, ∼10.29 (2d, J = 144 Hz, P–CH3), 22.68 (–NHCOCH3), 41.69 (C3), ∼52.91, 53.83, 53.85 (s, m, C5, P–OCH3), 53.28 (–CO2CH3), 66.26 (–CH2–CH = CH2), 68.46 (C4), 69.12, 69.30 (2d, J = 6.3, 6.4 Hz, C9), 69.80, 69.94 (C7), 70.80, 70.90 (2d, J = 6.4, 6.8 Hz, C8), 74.70 (C6), 100.12 (C2), 116.94 (–CH2–CH = CH2), 135.47 (–CH2–CH = CH2), 170.84 (C1), 175.16 (–NHCOCH3). 31P NMR (162 MHz, CDCl3) δP: 34.24 (s), 34.34 (s).
The phosphonate (23 mg, 0.0520 mmol) was dissolved in pyridine (0.5 mL) and acetic anhydride (0.5 mL). The flask was stoppered and allowed to stir overnight. Once the reaction was complete, the solvent was removed in vacuo. Repeated co-evaporation with toluene, followed by flash chromatography (EA:MeOH 20:1 → 10:1) gave 11 (26 mg, 90%). Rf : 0.45 (EA:MeOH; 10:1). HR-ESI-MS (m/z) [M + Na]+ calculated for : C23H36NO14PNa : 604.1771. Found: 604.1766. 1H NMR (500 MHz, CDCl3) δ H: 1.45 (dd, 1H, J = 3.4, 17.6 Hz, P-CH 3), 1.85, 2.12–2.14 (s,m, 9H, 3 × –OCOCH 3), 2.00 (s, 3H, –NHCOCH 3), 1.95 (dd, 1H, J = 12.4, 12.6 Hz, H3ax), 2.60 (dd, 1H, J = 4.6, 12.8 Hz, H3eq), 3.68, 3.72 (2d, 3H, J = 11.1, 11.2 Hz, P–OCH 3), 3.87 (dd, 1H, J = 5.9, 12.8 Hz, –CHH’–CH = CH2), 3.95–4.13 (m, 3H, H5, H6, H9), 4.21–4.33 (m, 2H, H9’, –CHH’–CH = CH2), 4.87 (m, 1H, H4), 5.14 (dd, 1H, J = 1.4, 10.4 Hz, –CH2–CH = CHH cis), 5.22 (d, 1H, J = 10.0 Hz, –NH), 5.26 (d, 1H, J = 17.2 Hz, –CH2–CH = CHH trans), 5.30 (m, 1H, H7), 5.37 (m, 1H, H8), 5.84 (ddd, 1H, J = 5.5, 10.7, 16.1 Hz, –CH2–CH = CH2). 13C NMR (126 MHz, CDCl3) δ C: ∼10.49, ∼10.57 (2d, J = 145.1, 145.4 Hz, P–CH3), 21.00, 21.27, 23.35 (3 × –OCOCH3, –NHCOCH3), 38.17 (C3), 49.69, 49.73 (C5), ∼52.31 (m, P–OCH3), 52.83 (–CO2 CH3), 63.88 (m, C9), 66.02 (–CH2–CH = CH2), 67.54 (C7), 69.18, 69.23 (C4), ∼69.60, ∼69.82 (2d, J = 6.5, 6.5 Hz, C8), 72.65, 72.68 (C6), 98.70 (C2), 117.38 (–CH2–CH = CH2), 133.76 (–CH2–CH = CH2), 168.49, 168.52 (C1), 170.16, 170.25, 170.35, 171.06 (3 × –OCOCH3, –NHCOCH3). 31P NMR (162 MHz, CDCl3) δP: 32.51 (s), 32.73 (s).
6.3. SOAE assays
6.3.1. Abbreviations
INF-C: influenza C; HEF: haemagglutinin esterase fusion; BCoV: bovine coronavirus; SDAV-HE: sialodacryoadenitis virus haemagglutinin esterase; MHV-S: mouse hepatitis virus strain S; BSM: bovine submaxillary gland mucine.
6.3.2. Viruses and recombinant viral sialate-O-acetylesterases
Influenza C virus C/JJ/50 was grown in embryonated chicken eggs. Bovine coronavirus (BCoV) and mouse hepatitis virus strain S (MHV-S) were grown in Madin–Darby bovine kidney (MDBK) cells and mouse L cells, respectively.
The INF-C virus HEF and SDAV-HE were expressed as chimeric recombinant influenza C/Cal/78 virus and sialodacryoadenitis virus haemagglutinin esterase, respectively, fused in frame to enhanced green fluorescent protein (HE12-GFP and SDAV-HE). Enzymes were expressed in insect Sf9 cells in serum-free media by recombinant baculovirus [47].
6.3.3. Enzymes and inhibitors
The three different viruses (INF-C virus, BCoV and MHV-S) were concentrated and purified by ultracentrifugation through a 3 ml cushion of 20% sucrose for 1.5 h at 4 °C at 26,000 rpm (110.000 × g) in a Beckman SW 41 rotor. The virus pellets were resuspended in 500 μl PBS (phosphate buffered saline) pH 7.4.
The recombinant enzymes (HE12-GFP and SDAV-HE) were recovered from cell culture supernatants by ultracentrifugation for 1.5 h at 4 °C at 26,000 rpm.
The five sialosides were dissolved in ddH2O to a final concentration of 10 mM. Stock solutions of p-nitrophenyl acetate (100 mM) were prepared in acetonitrile, and 3,4-dichloroisocoumarin was dissolved in dimethylsulfoxide to a final concentration of 10 mM. The esterase activities of all viruses and recombinant proteins were determined with p-nitrophenyl acetate (pNPA) as described previously [42], [47]. One milliunit of viral esterase activity was defined as the amount of enzymatic activity resulting in the hydrolysis of 1 nmol of pNPA per minute.
6.3.4. pNPA assay
An esterase activity was incubated in the presence of a sialoside at room temperature for 30 min. For control inhibition reactions, 100 μM 3,4-dichloroisocoumarin was incubated with the different esterases [52]. 10 μl pNPA and PBS pH 7.4 was added to 1 ml and the A400 was monitored.
6.3.5. Fluorimetric HPLC analysis
Reverse-phase high-pressure liquid chromatography (HPLC) analysis of sialic acids was performed as described previously [45]. 10 mU esterase and 1 mM sialosides were added to PBS pH 7.4 and incubated at room temperature for 30 min. 10 μg glycosidically bound sialic acids (BSM) or free O-acetylated sialic acids (synthesised in our lab) were added and incubated at 37 °C for various time periods (20–240 min). Samples containing glycosidically bound sialic acids were first hydrolysed with 2 M propionic acid for 2 h at 80 °C. The further processing of the samples was described elsewhere [53].
Footnotes
Supplementary data related to this article can be found online at doi:10.1016/j.ejmech.2011.04.008.
Appendix. Supplementary material
References
- 1.Angata T., Varki A. Chem. Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- 2.Schauer R. Glycoconj J. 2000;17:485–499. doi: 10.1023/A:1011062223612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schauer R., Kamerling J.P. In: Glycoproteins II. Montreuil J., Vliegenthart J.F.G., Schachter H., editors. Elsevier; Amsterdam: 1997. Chemistry, biochemistry and biology of sialic acids; pp. 243–402. [Google Scholar]
- 4.Schauer R. Zoology (Jena) 2004;107:49–64. doi: 10.1016/j.zool.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Tiralongo J., Schauer R. Trends Glycosci. Glycotechnol. 2004;16:1–15. [Google Scholar]
- 6.Fahr C., Schauer R. J. Invest. Dermatol. 2001;116:254–260. doi: 10.1046/j.1523-1747.2001.01237.x. [DOI] [PubMed] [Google Scholar]
- 7.Hubl U., Ishida H., Kiso M., Hasegawa A., Schauer R. J. Biochem. (Tokyo) 2000;127:1021–1031. doi: 10.1093/oxfordjournals.jbchem.a022693. [DOI] [PubMed] [Google Scholar]
- 8.Sjoberg E.R., Powell L.D., Klein A., Varki A. J. Cell Biol. 1994;126:549–562. doi: 10.1083/jcb.126.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cariappa A., Takematsu H., Liu H., Diaz S., Haider K., Boboila C., Kalloo G., Connole M., Shi H.N., Varki N., Varki A., Pillai S. J. Exp. Med. 2008;206:125–138. doi: 10.1084/jem.20081399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Surolia I., Pirnie S.P., Chellappa V., Taylor K.N., Cariappa A., Moya J., Liu H., Bell D.W., Driscoll D.R., Diederichs S., Haider K., Netravali I., Le S., Elia R., Dow E., Lee A., Freudenberg J., De Jager P.L., Chretien Y., Varki A., Macdonald M.E., Gillis T., Behrens T.W., Bloch D., Collier D., Korzenik J., Podolsky D.K., Hafler D., Murali M., Sands B., Stone J.H., Gregersen P.K., Pillai S. Nature. 2010;466:243–247. doi: 10.1038/nature09115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pillai S., Cariappa A., Pirnie S.P. Trends Immunol. 2009;30:488–493. doi: 10.1016/j.it.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arbour N., Day R., Newcombe J., Talbot P.J. J. Virol. 2000;74:8913–8921. doi: 10.1128/jvi.74.19.8913-8921.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.St-Jean J.R., Jacomy H., Desforges M., Vabret A., Freymuth F., Talbot P.J. J. Virol. 2004;78:8824–8834. doi: 10.1128/JVI.78.16.8824-8834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sloots T.P., McErlean P., Speicher D.J., Arden K.E., Nissen M.D., Mackay I.M. J. Clin. Virol. 2006;35:99–102. doi: 10.1016/j.jcv.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vijgen L., Keyaerts E., Lemey P., Moës E., Li S., Vandamme A.M., Van Ranst M. Virology. 2005;337:85–92. doi: 10.1016/j.virol.2005.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H., Wang H., Crameri G., Hu Z., Zhang H., Zhang J., McEachern J., Field H., Daszak P., Eaton B.T., Zhang S., Wang L.F. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 17.Wang L.F., Shi Z., Zhang S., Field H., Daszak P., Eaton B.T. Emerg. Infect. Dis. 2006;12:1834–1840. doi: 10.3201/eid1212.060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vijgen L., Keyaerts E., Lemey P., Maes P., Van Reeth K., Nauwynck H., Pensaert M., Van Ranst M. J. Virol. 2006;80:7270–7274. doi: 10.1128/JVI.02675-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smits S.L., Lavazza A., Matiz K., Horzinek M.C., Koopmans M.P., de Groot R.J. J. Virol. 2003;77:9567–9577. doi: 10.1128/JVI.77.17.9567-9577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kroneman A., Cornelissen L.A., Horzinek M.C., de Groot R.J., Egberink H.F. Identification and characterization of a porcine torovirus. J. Virol. 1998;72:3507–3511. doi: 10.1128/jvi.72.5.3507-3511.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matiz K., Kecskemeti S., Kiss I., Adam Z., Tanyi J., Nagy B. Acta Vet. Hung. 2002;50:293–296. doi: 10.1556/AVet.50.2002.3.5. [DOI] [PubMed] [Google Scholar]
- 22.Pignatelli J., Grau-Roma L., Jimenez M., Segales J., Rodriguez D. Vet. Microbiol. 2010 doi: 10.1016/j.vetmic.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woode G.N., Pohlenz J.F., Gourley N.E., Fagerland J.A. J. Clin. Microbiol. 1984;19:623–630. doi: 10.1128/jcm.19.5.623-630.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woode G.N., Reed D.E., Runnels P.L., Herrig M.A., Hill H.T. Vet. Microbiol. 1982;7:221–240. doi: 10.1016/0378-1135(82)90036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woode G.N., Saif L.J., Quesada M., Winand N.J., Pohlenz J.F., Gourley N.K. Am. J. Vet. Res. 1985;46:1003–1010. [PubMed] [Google Scholar]
- 26.Hoet A.E., Saif L.J. Anim. Health Res. Rev. 2004;5:157–171. doi: 10.1079/ahr200498. [DOI] [PubMed] [Google Scholar]
- 27.Ito T., Okada N., Fukuyama S. Virus Res. 2007;126:32–37. doi: 10.1016/j.virusres.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito T., Okada N., Okawa M., Fukuyama S., Shimizu M. Vet. Microbiol. 2009;136:366–371. doi: 10.1016/j.vetmic.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirisawa R., Takeyama A., Koiwa M., Iwai H. J. Vet. Med. Sci. 2007;69:471–476. doi: 10.1292/jvms.69.471. [DOI] [PubMed] [Google Scholar]
- 30.Park S.J., Oh E.H., Park S.I., Kim H.H., Jeong Y.J., Lim G.K., Hyun B.H., Cho K.O. Vet. Microbiol. 2008;126:364–371. doi: 10.1016/j.vetmic.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koopmans M., Petric M., Glass R.I., Monroe S.S. J. Clin. Microbiol. 1993;31:2738–2744. doi: 10.1128/jcm.31.10.2738-2744.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koopmans M.P., Goosen E.S., Lima A.A., McAuliffe I.T., Nataro J.P., Barrett L.J., Glass R.I., Guerrant R.L. Pediatr. Infect. Dis. J. 1997;16:504–507. doi: 10.1097/00006454-199705000-00010. [DOI] [PubMed] [Google Scholar]
- 33.Beards G.M., Brown D.W., Green J., Flewett T.H. J. Med. Virol. 1986;20:67–78. doi: 10.1002/jmv.1890200109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beards G.M., Hall C., Green J., Flewett T.H., Lamouliatte F., Du Pasquier P. Lancet. 1984;1:1050–1052. doi: 10.1016/S0140-6736(84)91454-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duckmanton L., Luan B., Devenish J., Tellier R., Petric M. Virology. 1997;239:158–168. doi: 10.1006/viro.1997.8879. [DOI] [PubMed] [Google Scholar]
- 36.Rosenthal P.B., Zhang X., Formanowski F., Fitz W., Wong C.H., Meier-Ewert H., Skehel J.J., Wiley D.C. Nature. 1998;396:92–96. doi: 10.1038/23974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeng Q., Langereis M.A., van Vliet A.L., Huizinga E.G., de Groot R.J. Proc. Natl. Acad. Sci. U S A. 2008;105:9065–9069. doi: 10.1073/pnas.0800502105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langereis M.A., Zeng Q., Gerwig G.J., Frey B., von Itzstein M., Kamerling J.P., de Groot R.J., Huizinga E.G. Proc. Natl. Acad. Sci. U S A. 2009;106:15897–15902. doi: 10.1073/pnas.0904266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mayr J., Haselhorst T., Langereis M.A., Dyason J.C., Huber W., Frey B., Vlasak R., de Groot R.J., von Itzstein M. Glycoconj J. 2008;25:393–399. doi: 10.1007/s10719-007-9094-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herrler G., Rott R., Klenk H.D., Muller H.P., Shukla A.K., Schauer R. The receptor-destroying enzyme of influenza C virus is neuraminate-O-acetylesterase. Embo J. 1985;4:1503–1506. doi: 10.1002/j.1460-2075.1985.tb03809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rogers G.N., Herrler G., Paulson J.C., Klenk H.D. J. Biol. Chem. 1986;261:5947–5951. [PubMed] [Google Scholar]
- 42.Vlasak R., Krystal M., Nacht M., Palese P. Virology. 1987;160:419–425. doi: 10.1016/0042-6822(87)90013-4. [DOI] [PubMed] [Google Scholar]
- 43.Schultze B., Wahn K., Klenk H.D., Herrler G. Virology. 1991;180:221–228. doi: 10.1016/0042-6822(91)90026-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vlasak R., Luytjes W., Spaan W., Palese P. Proc. Natl. Acad. Sci. U S A. 1988;85:4526–4529. doi: 10.1073/pnas.85.12.4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Regl G., Kaser A., Iwersen M., Schmid H., Kohla G., Strobl B., Vilas U., Schauer R., Vlasak R. J. Virol. 1999;73:4721–4727. doi: 10.1128/jvi.73.6.4721-4727.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wurzer W.J., Obojes K., Vlasak R. J. Gen. Virol. 2002;83:395–402. doi: 10.1099/0022-1317-83-2-395. [DOI] [PubMed] [Google Scholar]
- 47.Strasser P., Unger U., Strobl B., Vilas U., Vlasak R. Glycoconj J. 2004;20:551–561. doi: 10.1023/B:GLYC.0000043292.64358.f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Groot R.J. Glycoconj J. 2006;23:59–72. doi: 10.1007/s10719-006-5438-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McAuliffe J.C., Rabuka D., Hindsgaul O. Org. Lett. 2002;4:3067–3069. doi: 10.1021/ol026317g. [DOI] [PubMed] [Google Scholar]
- 50.Roy R., Laferriere C. Can. J. Chem. 1990;68:2045–2054. [Google Scholar]
- 51.Fitz W., Rosenthal P.B., Wong C.H. Bioorg. Med. Chem. 1996;4:1349–1353. doi: 10.1016/0968-0896(96)00123-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vlasak R., Muster T., Lauro A.M., Powers J.C., Palese P. J. Virol. 1989;63:2056–2062. doi: 10.1128/jvi.63.5.2056-2062.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hellebo A., Vilas U., Falk K., Vlasak R. J. Virol. 2004;78:3055–3062. doi: 10.1128/JVI.78.6.3055-3062.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.