

Highlights
-
•
The duplex assay monitored replication, tissue distribution, and mRNA expression.
-
•
The duplex assay monitored insert genetic stability during in vivo replication.
-
•
Primary site of CDV replication in ferrets was abdominal cavity lymphoid tissue.
-
•
CDV gRNA or mRNA was undetectable in brain tissue.
-
•
Specific primers were used in the RT step to distinguish gRNA from mRNA.
Keywords: Canine distemper virus vaccine vector, SIV gag, Duplex real-time qPCR, Ferrets
Abstract
Advancement of new vaccines based on live viral vectors requires sensitive assays to analyze in vivo replication, gene expression and genetic stability. In this study, attenuated canine distemper virus (CDV) was used as a vaccine delivery vector and duplex 2-step quantitative real-time RT-PCR (RT-qPCR) assays specific for genomic RNA (gRNA) or mRNA have been developed that concurrently quantify coding sequences for the CDV nucleocapsid protein (N) and a foreign vaccine antigen (SIV Gag). These amplicons, which had detection limits of about 10 copies per PCR reaction, were used to show that abdominal cavity lymphoid tissues were a primary site of CDV vector replication in infected ferrets, and importantly, CDV gRNA or mRNA was undetectable in brain tissue. In addition, the gRNA duplex assay was adapted for monitoring foreign gene insert genetic stability during in vivo replication by analyzing the ratio of CDV N and SIV gag genomic RNA copies over the course of vector infection. This measurement was found to be a sensitive probe for assessing the in vivo genetic stability of the foreign gene insert.
1. Introduction
Canine Distemper Virus (CDV) is a member of the Paramyxoviridae family in the Morbillivirus genus. Accordingly, it is an enveloped virus that has a single-stranded, non-segmented, negative-sense RNA genome. The genome contains six gene regions organized 3′-N-P-M-F-H-L-5′, which encode eight known viral polypeptides (McIlhatton et al., 1997). CDV, which is related to measles virus (MeV), causes systemic disease in domestic and wild dogs, foxes and wolves. CDV outbreaks also occur among small carnivores including mink and ferrets (Trebbien et al., 2014) and the virus has been linked to disease in lions, tigers, and seals (Harder and Osterhaus, 1997). Natural infection in canines can lead to central nervous system disease, and experimental infection in ferrets with some CDV strains also results in neuroinvasion and neurovirulence after intranasal inoculation (Langlois, 2005, Rudd et al., 2006). There have been recent reports of measles-like illness in macaque colonies attributed to CDV (Qiu et al., 2011, Sakai et al., 2013, Sun et al., 2010) and experimental infection of nonhuman primates has been studied recently (de Vries et al., 2014), although CDV infection has not been linked convincingly to any human diseases (Rima and Duprex, 2006). Live attenuated CDV vaccines have effectively and safely controlled the disease in domestic dog populations (Chappuis, 1995).
Paramyxoviruses are attractive vaccine vectors for a variety of reasons. Notably, systems exist for genetic manipulation, they can be delivered mucosally, and in some cases, live attenuated vaccine viruses are available that can be used as the foundation for a vaccine delivery vector (Billeter et al., 2009, Bukreyev et al., 2006). For example, attenuated MeV strains have been used to develop experimental vaccine vectors expressing antigens from hepatitis B virus (del Valle et al., 2007, Singh et al., 1999), HIV or SIV (Brandler and Tangy, 2008, Lorin et al., 2012, Tangy and Naim, 2005, Zuniga et al., 2007), West Nile virus (Despres et al., 2005), Dengue virus (Brandler et al., 2007), SARS-Coronavirus (Liniger et al., 2008), and hepatitis C virus (Satoh et al., 2010). Although MeV is an attractive vector candidate because of its long track record of use in humans, pre-existing neutralizing immunity against MeV might limit its use. Since recombinant CDV (rCDV) can express foreign proteins (Ludlow et al., 2012, Parks et al., 2002, von Messling et al., 2004, Wang et al., 2012), attenuated CDV vectors encoding SIV immunogens have been developed to evaluate whether they might provide a safe alternative to MeV for advancement of HIV vaccine candidates that are less subject to pre-existing anti-MeV antibodies (Zhang et al., 2013).
To evaluate CDV replication and gene expression in host animals, sensitive assays are required to detect and quantify virus in specimens. Typically, infectious CDV is quantified by plaque assay on susceptible cell monolayers (Bussell and Karzon, 1962), and routine diagnostic testing is conducted by immunofluorescence (Appel, 1987), serological methods, such as enzyme-linked immunosorbent (ELISA) and seroneutralization assays (Frisk et al., 1999, Gemma et al., 1995, von Messling et al., 1999), and virus isolation on canine cells (Frisk et al., 1999, Kim et al., 2001, Shin et al., 1995). A sensitive assay for virus isolation, showing cytopathic effect as early as 24 h after sample inoculation has been developed based on Vero cells expressing the canine signaling lymphocyte activation molecule (Seki et al., 2003). These assays, though proven and useful, are time consuming and require several days to weeks to obtain results. Recently, several RT-PCR and nested PCR based assays have been developed for the diagnosis of CDV nucleocapsid protein RNA in cerebrospinal fluid, nasal swabs, peripheral blood mononuclear cells, saliva, serum, urine, and whole blood of dogs (Frisk et al., 1999, Kim et al., 2001, Rzezutka and Mizak, 2002, Saito et al., 2006, Shin et al., 2004, von Messling et al., 1999). These assays are very sensitive, but sensitivity varies with sample source, viral RNA extraction methods, and choice of primers. More recently, quantitative real-time PCR (qPCR) and reverse transcription-qPCR (RT-qPCR) assays have been developed for the detection of CDV (Elia et al., 2006, Fischer et al., 2013, Meli et al., 2009, Pawar et al., 2011, Scagliarini et al., 2007, Wilkes et al., 2014) and many other viruses including rabies virus (Hughes et al., 2004), blue-tongue virus (Toussaint et al., 2007), SIV (Cline et al., 2005) and vesicular stomatitis virus (VSV) (Coleman et al., 2007). These assays have been valuable in providing estimates of the viral load in plasma, and infected tissues and organs of animals.
In this report, the development and characterization of 2-step RT-qPCR assays are described and used to evaluate rCDV and a rCDV vector encoding SIV Gag. Singleplex RT-qPCR assays were optimized for specific detection of an essential CDV gene (CDV N) or the foreign SIV gag insert sequence in gRNA or mRNA, and duplex RT-qPCR assays were developed to detect simultaneously CDV N and SIV gag gRNAs or mRNAs in homogenized tissue specimens from ferrets inoculated with CDV vector encoding Gag or the vector lacking an insert. These assays enabled rapid, reliable, and sensitive detection of CDV N and SIV gag and were used to compile in vivo data on replication, distribution, and genetic stability of an early prototype rCDV-SIV-Gag vector. Importantly, by using the gRNA duplex assay to monitor the relative quantities of CDV N and SIV gag sequences in vector genomes over the course of infection, gag insert instability was detected in the vector prototype, providing valuable information for guiding vector and gene insert improvements that stabilized foreign genes in subsequent vaccine designs (Zhang et al., 2013).
2. Materials and methods
2.1. Cell culture and viruses
Vero and 293T cells were cultured in Dulbecco's minimal essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and gentamicin. CDV was propagated in Vero cells and infectious virus was quantified by plaque assay. Recombinant CDV-SIVgag1 was constructed using procedures described in detail elsewhere (Zhang et al., 2013). Briefly, virus from a live attenuated vaccine based on the Onderstepoort strain was serially passaged in Vero cells after which a clonal isolate was derived by multiple rounds of plaque purification. The genome of the plaque isolate was sequenced and a molecular clone was constructed with genomic RNA synthesis placed under control of the T7 promoter in Bluescript II SK (+/−) plasmid (Stratagene, La Jolla, CA, USA). The molecular clone was then modified by insertion of SIVmac239 gag gene, which was codon-optimized for mammalian cell expression (Winstone et al., 2011), into the first genomic position of the CDV genome to produce the rCDV-SIVgag1 cDNA (Fig. 1A). Following gene insertion, the overall genome length followed the paramyxovirus rule-of-six (Kolakofsky et al., 1998).
Fig. 1.
The relative frequency of mRNA transcription, genome organization for each vector, the scheme for the mRNA specific RT-qPCR, and the binding sites for qPCR primers and probe for the SIV gag gRNA and mRNA assays are depicted. (A). Transcriptional attenuation resulting in a gradient of mRNA synthesis such that the gag message is more abundant than the N message in the rCDV-SIVgag1 vector; the rCDV vector is a Vero cell adapted, Onderstepoort live-attenuated CDV vaccine (Galaxy D, Schering-Plough Animal Health Corporation, Omaha, NE, USA) obtained through a commercial source; and the rCDV-SIVgag1 vector with the Gag gene in the first position of the genome and a unique UAUAAU sequence common to CDV N and SIV gag genomes. (B). Diagram of the RT step used in the singleplex or duplex assay to synthesize cDNA from mRNA. The CDV-specific mRNA RT-primer that binds to a unique UAUUA sequence common only to CDV N and SIV gag mRNA transcripts, has a 22-base adapter sequence at the 5′ end followed by a 20-base oligo-dT sequence, and a 3′-terminal 5-base anchor (ATAAT). (C) Tagged cDNA generated with this primer is then PCR amplified with CDV N and SIV-gag forward primers and labeled probes and the CDV sequence tag-reverse primer that corresponds to the adapter sequence, as described in Section 2. (E). The black box from 256–339 Nt indicates the binding site for the qPCR primers ( ) and probe (
) and probe ( ) for the Gag specific gRNA RT-qPCR assay and the 1500–1669 Nt indicates the amplicon and the binding site for qPCR forward primer (
) for the Gag specific gRNA RT-qPCR assay and the 1500–1669 Nt indicates the amplicon and the binding site for qPCR forward primer ( ) and probe () used in the Gag specific mRNA RT-qPCR assay.
) and probe () used in the Gag specific mRNA RT-qPCR assay.
To rescue rCDV-SIVgag1, 293T cells were cotransfected with the CDV-SIVgag1 genomic plasmid DNA and 7 support plasmids (Witko et al., 2006) that express CDV structural proteins (N, P, M, F, H and L) and T7 DNA-dependent RNA polymerase using calcium-phosphate reagents obtained from a commercial supplier (Clontech, Mountain View, CA, USA). Structural protein expression was controlled by the human CMV promoter in pCI-Neo (Promega, Madison, WI, USA), which had been modified by deleting the T7 RNA polymerase promoter (Zhang et al., 2013). Clonal virus isolates then were derived by plaque purification and subsequent expansion on Vero cell monolayers. To amplify large quantities of virus, Vero cell monolayers in Nunc flasks (Thermo Scientific, Waltham, MA, USA) were infected for 1 h in DMEM containing 1% FBS at multiplicity of infection (MOI) of 0.02 after which the medium was replaced with fresh DMEM containing 10% FBS. When cytopathic effect was evident in the entire monolayer, cells were scraped into culture medium and the cell suspension was subjected to two freeze/thaw cycles. The suspensions were then clarified and plaque-forming units (PFU) were determined by plaque assay conducted with Vero cells.
2.2. Specimen collection
All animal care and procedures took place at the State University of New York, Downstate Medical Center and conformed to Institutional Animal Care and Use guidelines. All in vivo studies adhered to the regulations outlined in the USDA Animal Welfare Act (9 CFR, parts 1–3) and the conditions specified in the Guide for the Care and Use of Laboratory Animals (ILAR publication, 1996, National Academy Press).
To initially study the replication time course and tissue distribution of rCDV, 10- to 12-week-old CDV-naïve ferrets were inoculated with 107 PFU of empty rCDV vector (Fig. 1A) administered by intranasal drops. Three ferrets per day were sacrificed at 1, 7, and 21 days post-inoculation (d.p.i). The animals were sedated and perfused with saline to minimize potential for contamination caused by virus in blood. Mesenteric lymph nodes, large intestine with a high density of Peyer's patches, nasal turbinates, brain, brain stem, and olfactory bulb were harvested from perfused animals.
For the study in which different routes of infection were evaluated, 10–12 week-old ferrets were inoculated by the intramuscular, intranasal, or intrarectal routes with 6 × 106 PFU of rCDV-SIVgag1 (Fig. 1A) per animal. All animals were closely monitored for symptoms of infection following inoculation and none were observed. Three ferrets after intramuscular or intranasal inoculations were sacrificed at 7, 28, and 55 d.p.i while animals inoculated by the intrarectal route were sacrificed at day 7 only, since this was a small element of the study that was included to determine if CDV could be delivered by this route. Specimens collected included large intestine with a high density of Peyer's patches, large intestine with a low density of Peyer's patches, and mesenteric lymph nodes.
2.3. Tissue sample preparation
Fresh ferret tissue specimens were weighed, suspended in 4 °C phosphate-buffered saline containing sucrose phosphate buffer with glutamate (0.2 M sucrose, 7.0 mM K2HPO4, 3.8 mM KH2PO4, 5.0 mM glutamic acid) (PBS/SPBG) to 10% weight/volume (w/v), and homogenized using an Omni PDH Programmable Digital Homogenizer (Omni, Kennesaw, GA, USA) with stainless steel autoclavable probes. For the small quantities of nasal turbinate material, the tissue was suspended in 4 °C PBS/SPBG and homogenized using an Omni THQ–Digital Tissue Homogenizer (Omni, Kennesaw, GA, USA) with plastic autoclavable probes to make a 20% (w/v) homogenate. To avoid cross contamination among tissue specimens, each tissue was homogenized with a separate probe in a sealed chamber. Homogenates were clarified by centrifugation and supernatants were frozen at −80 °C in 100 μl aliquots for subsequent RNA extraction and virus quantification by plaque assay.
2.4. Design of primers and probes
Primer and probe sets specific for CDV N, SIV gag and the ferret mitochondrial 12S rRNA (Genbank accession number-EZ466996.1), which was used as a reference gene (RG) target, were designed using the Primer Express software version 3.0 (Applied Biosystems, Foster, CA, USA). The CDV N gene was selected as an amplicon because it is an essential gene. Additionally, insertion of gag in the first position of the genome was anticipated to reduce N mRNA synthesis (Wertz et al., 1998); thus it was of interest to measure the relative quantities of N mRNA expressed from recombinant virus with or without the gag gene insert (Fig. 1A). The forward and reverse primers (Operon, Huntsville, AL, USA) and oligonucleotide probes (Applied Biosystems, Valencia, CA, USA) are shown in Table 1 . The CDV-specific mRNA RT-primer used in the singleplex or duplex assay to synthesize cDNA from mRNA was designed with a 22-base adapter sequence at the 5′ end (Craggs et al., 2001, Simon et al., 2010), followed by a 20-base oligo-dT sequence, and a 3′-terminal 5-base anchor (ATAAT) that was complimentary to a unique UAUUA sequence common to CDV N and SIV gag mRNA transcripts (Table 1 and Fig. 1B). This adapter primer was designed to overcome any signal that might be caused by gRNA self-priming and also improve qPCR specificity (Craggs et al., 2001, Lanford et al., 1994, Simon et al., 2010). In order to discriminate between the N and gag sequences in the duplex assay, the CDV N probe was labeled at the 5′ end with the reporter dye 6-carboxyrhodamine 6G (VIC) and the SIV gag probe with 6-carboxyfluorescein (FAM). The 3′ ends were labeled with a nonfluorescent black hole quencher and a minor groove binder (MGB) moiety.
Table 1.
Oligonucleotides used in singleplex and duplex RT-qPCR and traditional RT-PCR assays.
| Name | Nucleotide sequence | Nt positiona |
|---|---|---|
| CDV N-forward | ACCGGAAATCGATGGAAGC | 1457–1475 |
| CDV N-reverse | GTCCCAGGTTGACTGAGCATCT | 1502–1523 |
| CDV N-probe-VICb | CAAGATGAGGATGCTTAC | 1482–1499 |
| SIV gag-genome-forward | GTCTGCGTCATCTGGTGCAT | 256–275 |
| SIV gag-genome-reverse | CACTAGGTGTCTCTGCACTATCTGTTT | 313–339 |
| SIV gag-probe-FAMc | CTTCCTCAGTGTGTTTCA | 293–310 |
| SIV gag-mRNA-forward | AGAGGATTTGCTGCACCTCAAT | 1500–1521 |
| SIV gag-mRNA-probe-FAMc | CTCTCTTTGGAGGAGACCA | 1523–1541 |
| CDV-specific mRNA RT-primer | GGCAGTATCGTGAATTCGATGCTT20ATAAT | |
| CDV sequence tag–reverse | GGCAGTATCGTGAATTCGATGC | |
| Armored RNA-forward | ATGCGGCTAATCCCAACCT | |
| Armored RNA-reverse | CGTTACGACAGGCCAATCACT | |
| Armored RNA probe-NEDd | CAGGTGGTCACAAAC | |
| Ferret RGe-forward | CAAGTCTCTACACCCCAGTGAGAAT | |
| Ferret RG-reverse | GCTTGATACCCGCTCCTTTTAA | |
| Ferret RG-probe-Cy5f | CCTCCAAATCTATATGTTG | |
| SIV gag-forward-PCR | CCAGGGTTCAGACCTACCAA | 87–106 |
| SIV gag-reverse-PCR | TGTTGACTGATGCAAGACTGG | 1707–1727 |
Nt position-nucleotide position in CDV genome (for N primers and probes) or in rCDV-SIV gag1 (for Gag primers and probes) relative to the 3′ end of the CDV genome.
VIC: 6-carboxyrhodamine 6G.
FAM: 6-carboxyfluorescein.
NED: fluorescent dye name proprietary to Applied Biosystems.
RG: reference gene.
Cy5: Cyanine dye.
2.5. RNA extraction and cDNA synthesis
Viral nucleic acid was extracted from clarified homogenized tissues or infected Vero cells using reagents from the RNeasy Mini kit (Qiagen, Valencia, CA, USA) as described previously (Coleman et al., 2007, Johnson et al., 2009) but with minor modifications. Briefly, a master mix that included 300 μl of lysis buffer, 3 μl of 2-mercaptoethanol (Bio-Rad, Hercules, CA, USA), 16 μl of 20 mg/ml proteinase K (Qiagen, Valencia, CA, USA) and 4 μl of an encapsidated Enterovirus Armored (AR) RNA control (3 × 107 RNA copies/ml solution; Asuragen, Austin, TX, USA) was mixed with target RNA source material, which was 100 μl of tissue homogenate, pelleted infected Vero cells, a negative control (lysis buffer mix) or an aliquot of virus used for inoculation (positive control). Extracted RNA was eluted from columns in 100 μl of RNase-free water.
Subsequent cDNA synthesis to detect gRNA (negative sense) was performed in a 20-μl reaction using a Sensiscript Reverse Transcriptase kit (Qiagen, Valencia, CA, USA) as described before (Coleman et al., 2007). Briefly, the final CDV N or SIV gag gRNA singleplex reactions contained 10 μl of total RNA from tissue homogenate or infected Vero cell extracts, 400 nM of CDV N forward primer or SIV gag genome-forward primer, and reagents from the kit. To detect simultaneously CDV N and SIV gag gRNA in the duplex assay, all reagents and sample amounts remained the same except 400 nM of CDV N forward primer and SIV gag genome-forward primer were placed in the same well. Reactions that generated standard curves contained 5 μl of synthetic RNA molecules diluted serially in 10-fold increments that spanned the CDV N and SIV gag amplicons. Reverse transcription was performed at 50 °C for 45 min followed by heat inactivation of the reverse transcriptase at 95 °C for 2 min.
To detect CDV N and SIV gag mRNA transcripts in the singleplex or duplex assays, the final reactions contained 10 μM of the CDV-specific mRNA RT-primer that was substituted for the CDV N forward primer and SIV gag genome-forward primer. Otherwise, the reagents and the conditions for the reverse transcription (RT) step remained the same. To ensure that excess CDV-specific mRNA RT-primer did not carry over to the qPCR step, exonuclease 1 (2 U/sample) (New England, Ipswich, MA, USA) was added at the end of the cDNA reaction and incubated at 37 °C for 30 min followed by heat activation at 70 °C for 15 min.
2.6. Real-time quantitative PCR
To quantify gRNA, the singleplex and duplex qPCR assays were carried out with reagents from the QuantiTect Multiplex PCR Kit (Qiagen, Valencia, CA, USA) using methods previously described (Coleman et al., 2007, Johnson et al., 2009) with minor modifications. Briefly, for the singleplex assay, the 20 μl heat-inactivated RT reaction was combined with a 30-μl PCR mix such that the final reaction volume contained 1× QuantiTect Multiplex PCR Master Mix, 400 nM each of CDV N forward and reverse primers and 200 nM of the VIC-labeled probe or 400 nM each of SIV gag genome-forward and reverse primers and 200 nM of the FAM-labeled probe. For the duplex assay, primers and probe sets for both templates were combined in the same reaction.
The singleplex and duplex qPCR assays for mRNA quantification contained the same reaction reagents and CDV N forward primer and probe as described above for the gRNA assays. For the gag mRNA, the SIV gag genome-forward primer and probe were replaced with the SIV gag mRNA-forward primer and probe (Table 1). This substitution was made to move the amplicon closer to the 3′ end of the mRNA transcript. The genome-specific reverse primers used to detect CDV N and SIV gag also were replaced with the 20-base CDV sequence tag – reverse primer (Table 1 and Fig. 1C). Amplification and detection were performed with a Stratagene Mx3005P Sequence Detection System using the following conditions: 15 min at 95 °C to activate the Hot-Star Taq DNA polymerase, followed by 45 cycles of 60 s at 94 °C and 90 s at 60 °C. All samples were tested in duplicate. For calculating group means, values that were below the detection limit (3.0 × 103 copies/g of tissue) were assigned a value of 3.0 × 103 copies/g of tissue.
2.7. RT-PCR to evaluate gag insert integrity
The PCR assay used to analyze gag insert integrity amplified a 1.6 kb amplicon that included the complete gag gene. The position and primers are shown in Table 1. Samples were extracted as described above and amplification was performed using the One-Step RT-PCR kit (Qiagen, Valencia, CA, USA). The final reactions contained reagents from the kit, 600 nM each of forward and reverse primers, and 1 μg of tissue extracted RNA. RT was performed at 50 °C for 45 min, and after an initial PCR activation step of 95 °C for 15 min, PCR was performed for 35 cycles of 30 s at 94 °C, 30 s at 56 °C, and 2 min at 68 °C. After a final extension at 68 °C for 10 min, the RT-PCR products were analyzed on a 1% agarose gel after staining with ethidium bromide.
2.8. Description of synthetic RNA molecule template, ferret housekeeping gene, and Armored RNA control
Two genome-sense synthetic RNA oligonucleotides were synthesized that spanned the CDV N or SIV gag amplicons. The oligoribonucleotides included two extra bases on each side of the amplicon. They were PAGE-purified and quantified (TriLink, San Diego, CA, USA) before being used as templates to develop standard curves. The copy number of each synthetic oligonucleotide was calculated as previously described (Coleman et al., 2007).
To control for sample-to-sample variation especially when analyzing different solid tissues (Bustin et al., 2009), a separate singleplex assay based on a ferret RG transcript for the mitochondrial 12S rRNA was designed and optimized. The RG-specific primers (Operon, Huntsville, AL, USA) and a Cy5 labeled probe (Applied Biosystems, Valencia, CA, USA) sequences are shown in Table 1. Samples were normalized to 1 × 105 copies of 12S rRNA but reported here as gRNA or mRNA/g tissue (data not shown).
The exogenous RNase-resistant encapsidated Enterovirus AR-RNA (Asuragen, Austin, TX, USA) control was spiked at a known concentration into the lysis buffer during RNA extraction to determine RNA extraction and amplification efficiencies (Pasloske et al., 1998). Primers (Operon, Huntsville, AL, USA) and a NED labeled probe (Applied Biosystems, Valencia, CA, USA) (Table 1) specific for the AR-RNA was designed, synthesized and used in a separate RT-qPCR reaction. Cycle Threshold (CT) values between 24 and 26 were observed for reliable extraction and amplification efficiencies (data not shown).
3. Results
Development of attenuated CDV as a live vaccine delivery vector requires study in a permissive animal host to assess important characteristics of infection like the extent of replication and dissemination in the animal host, and genetic stability of the foreign gene. In addition, monitoring infection after vaccine vector administration requires sensitive and specific assays that perform reliably when used to assess tissue specimens. To this end, three assays were developed to: (1) monitor vector replication and tissue distribution by quantifying gRNA; (2) evaluate gene expression by detecting vector-encoded mRNAs; and (3) monitor SIV gag insert stability during replication in vivo.
3.1. Development of 2-step singleplex RT-qPCR assays to detect the CDV N or SIV gag gene in vector gRNA
CDV is a paramyxovirus, and like all members of this virus family, it has a negative-sense, single-stranded, nonsegmented RNA genome (Fig. 1A). To study replication and gene expression by rCDV and the rCDV-SIV gag vector, primers and probes sets were designed to detect target sequences in CDV N, which is an essential CDV gene, and the SIV gag insert. Specifically, forward primers were designed to hybridize to the negative-sense gRNA in the RT step and to distinguish gRNA from other RNA species such as mRNA expressed by the CDV vector.
The individual N and gag 2-step singleplex RT-qPCR reaction conditions were studied to achieve optimal linearity and sensitivity using purified oligoribonucleotide templates that were equivalent to the negative-sense genome strand. Assays were conducted with 10-fold serial dilutions of template ranging from 1 × 108 to 1 × 101 copies and the data was plotted as genome copies (log10) versus CT. The results showed that both the N and gag singleplex assays were linear over this range of oligoribonucleotide template copies and that the slopes of the curves indicated that the PCR efficiencies were 97% and 92%, respectively (Supplemental Fig. 1A and B). The lower limit of detection for the synthetic RNA oligonucleotides templates was 10 copies of RNA template per reaction. The specificity of each of singleplex reaction was confirmed by switching target templates (1 × 108 copies) to observe whether cross priming amplification would occur, and in both singleplex assays, the primer-probe set detected only its specific target (data not shown).
To evaluate the reproducibility of the individual CDV N and SIV gag 2-step singleplex RT-qPCR assays, 10-fold serial dilutions of each oligoribonucleotide template ranging from 1 × 108 to 1 × 101 copies was tested repeatedly. Intra-assay variation was assessed using four replicates of each template dilution, and inter-assay variation was evaluated by testing each template dilution in four separate runs. For each oligoribonucleotide standard dilution, the mean, standard deviation (SD) and coefficient of variation (CV) were calculated based on their CT values. The CV for the intra-assay and the inter-assay variability for the CDV N singleplex RT-qPCR assay ranged between 0.22% to 1.56% and 0.93% to 1.92%, respectively (Supplemental Table 1). For the SIV gag 2-step singleplex RT-qPCR assay, the CV for the intra-assay and the inter-assay variability ranged from 0.64% to 2.88% and 0.45% to 2.19%, respectively (Supplemental Table 2). Taken together, this indicated that both assays were accurate and were capable of generating reproducible results.
3.2. Detection of CDV N and SIV gag in vector gRNA and monitoring gag insert integrity with a 2-step duplex RT-qPCR assay
After optimizing the singleplex reactions, primers and probes were used in a 2-step duplex RT-qPCR assay to detect N and gag simultaneously. When the duplex assay was optimized and tested using equimolar amounts of the N and gag oligoribonucleotide templates ranging in copy numbers from 1 × 108 to 1 × 101 of each per reaction, the linear ranges, amplification efficiencies, copy number detection limits, and the CT values of the N and gag assays in the duplex reaction were similar to those observed in the individual singleplex reactions (Supplemental Fig. 1A–C). These results indicated that an approximate 1:1 N-to-gag CT value ratio corresponded to a 1:1 molar quantity of the input templates. This result also indicated that this pair of amplicons could be used to study the genetic stability of the gag insert since their amplification efficiency was similar and both targeted single-copy genes in the CDV vector genome.
To confirm whether gag insert integrity could be studied by comparing the N-to-gag ratio, it was important to determine the response of the duplex assay when the proportions of the two target templates varied. Since N is an essential gene, changes in N-to-gag ratio during replication would likely result from deletion of gag insert sequences, thus the duplex RT-qPCR assay must be able to detect reduced quantities of the gag gRNA target under conditions when N gRNA remained abundant. This was studied by conducting the duplex reaction with the N oligoribonucleotide template held constant at 1 × 107 copies per RT-qPCR reaction while titrating serially the gag template 10-fold over a range from 1 × 107 to 1 × 103 copies, and in a similar experiment where the N template was held at 1 × 104 copies per RT-qPCR reaction while titrating serially the gag template 10-fold from 1 × 104 to 1 × 101 copies. All samples were tested in duplicate in the duplex RT-qPCR assay and the results are summarized in Supplemental Fig. 2. When the N oligoribonucleotide template was held at 1 × 107 copies per reaction, the N CT value (dark bars) remained steady as the quantity of gag template (light bars) varied (Supplemental Fig. 2A). Similar results were obtained when the N oligoribonucleotide was held constant at a lower concentration of 1 × 104 copies (Supplemental Fig. 2B). In both experiments, the measured SIV gag CT values were in accordance with the expected CT values even in the presence of a high CDV N template background of 1 × 107 or 1 × 104 copies (Table 2 ), and the SIV gag templates could be detected at a PCR efficiency of 81% or 74% respectively (Supplemental Fig. 2A and B). Importantly, this result demonstrated that the duplex assay could reliably quantify copies of N and gag sequence even when the input proportions were varied over a wide range. This result showed that the duplex assay provided the foundation for an assay to study genetic stability during in vivo replication.
Table 2.
Measured and expected CT values for SIV gag RNA in duplex RT-qPCR.
| Samplea | CDV N copies | SIV gag copies | SIV gag measured CTc | SIV gag expected CTb,c |
|---|---|---|---|---|
| 1:100 | 107 | 107 | 15.53 | 16.00 |
| 1:10−1 | 107 | 106 | 18.63 | 19.79 |
| 1:10−2 | 107 | 105 | 22.24 | 23.39 |
| 1:10−3 | 107 | 104 | 26.74 | 26.63 |
| 1:10−4 | 107 | 103 | 30.90 | 30.43 |
| 1:100 | 104 | 104 | 25.33 | 26.63 |
| 1:10−1 | 104 | 103 | 29.10 | 30.43 |
| 1:10−2 | 104 | 102 | 33.78 | 33.11 |
| 1:10−3 | 104 | 101 | 37.69 | 36.48 |
Ratios of CDV N to SIV gag.
Expected CT values were derived from standard curves based on the duplex RT-qPCR assay in Fig. 2C.
All CT values are means of duplicates.
3.3. Development of 2-step singleplex and duplex RT-qPCR assays specific for CDV N and SIV gag mRNAs
Assays were developed also to specifically detect N and gag mRNAs. These assays were necessary to study the duration of gene expression by the CDV vector in vaccinated animals because several studies with MeV indicated that genomes persisted for significantly longer periods of time than originally thought (Lin et al., 2012, Pan et al., 2005, Pan et al., 2010). A similar outcome also has been observed in animals infected with VSV, another negative-strand RNA virus, where genomes persist in infected cattle (Letchworth et al., 1996) or rodents (Barrera and Letchworth, 1996, Johnson et al., 2009, Simon et al., 2007, Turner et al., 2007). Thus it seems that negative-strand RNA virus gRNA could persist in tissues for significant periods of time after detectable shedding of infectious virus had ended, and therefore it was important to assess whether the persisting genomes expressed detectable quantities of mRNAs.
For this purpose, a RT primer was designed that would anneal to the poly-A tail and specifically prime cDNA synthesis of several CDV mRNAs. The primer was designed with a 3′ anchor sequence (ATAAT, Fig. 1B) that was specific for a sequence present at the 3′ end of both the CDV N and SIV gag mRNAs. The RT primer also contained an adapter sequence at the 5′ end, which was used for subsequent PCR amplification. The singleplex qPCR was then carried out using either the CDV N forward primer or the SIV gag mRNA-forward primer and a reverse primer (CDV sequence tag) corresponding exactly to the adapter sequence (Fig. 1B and C). The duplex assay was done with both forward primers and the reverse primer in the same reaction well.
To confirm that this primer design could detect N and gag mRNA, Vero cells were infected with rCDV-SIVgag1 and total RNA was extracted and subjected to analysis with the N or gag singleplex mRNA assays and the N/Gag duplex mRNA assay. To demonstrate that the assays were specific for the positive-sense mRNA, a negative control reaction was performed with synthetic negative-strand gRNA template. Similar CT values observed in both the singleplex and the duplex assays indicated that the mRNA RT-primer functioned equally well in both assays (Table 3 ), and was specific for CDV N and SIV gag mRNAs. No signal was detected with high levels (5 × 108 copies) of oligoribonucleotide containing the negative-strand gRNA template.
Table 3.
Evaluation of the CDV-specific mRNA RT-primer in the 2-step singleplex and duplex RT-qPCR assays.
| Samplesa | CTb for singleplex assayc |
CTb for duplex assayd |
||
|---|---|---|---|---|
| CDV N VIC | SIV gag FAM | CDV N VIC | SIV gag FAM | |
| Infected Vero cells 1 | 13.6 | 12.8 | 13.7 | 12.0 |
| Infected Vero cells 2 | 13.0 | 12.4 | 12.4 | 12.0 |
| Uninfected cells | – | – | – | – |
| 5 × 108 copies gRNA | – | – | – | – |
Vero cells infected with rCDV-SIVgag1.
CT, threshold cycle number at which fluorescence was detected above the background. Positive CT values are ≤40 while negative CT values are >40 and are shown by a dash line.
Separate singleplex reactions for CDV N and SIV-gag were carried out in different wells with specific primer-probe sets.
Duplex reactions for CDV N and SIV-gag were done in the same wells containing primer-probe sets specific for both genes.
3.4. Detection of gRNA or mRNA in tissues from ferrets infected with a CDV vector using 2-step singleplex RT-qPCR assays
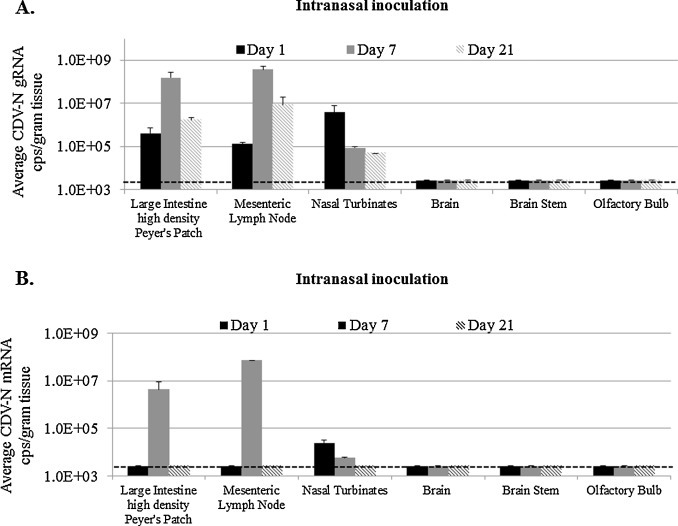
The empty rCDV vector (107 PFU) was administered to ferrets by intranasal drops after which animals were sacrificed at various times post-infection. Large intestine high density Peyer's patches, mesenteric lymph nodes, nasal turbinates, brain, brain stem, and olfactory bulb specimens were collected and processed to isolate RNA that was analyzed with two-step singleplex assays specific for N gRNA or mRNA. At 1, 7, and 21 d.p.i., gRNA was detected in the large intestine high density Peyer's patches and mesenteric lymph nodes. Genome copies increased out to day 7 (>108 copies/g) and subsequently declined by about 100-fold by day 21 (Fig. 2 A). In the nasal turbinates, gRNA peaked at day 1 (>106 copies/g) after which the copy number declined at day 7 and 21 (Fig. 2A).
Fig. 2.
Viral loads of CDV N in tissues from ferrets using the 2-step singleplex RT-qPCR assay. Detection of (A) gRNA and (B) mRNA at the indicated times post IN inoculation with rCDV. Viral loads in each tissue are displayed as either average gRNA or mRNA copies/gram of tissue. The detection limit is 3.0 × 103 copies/g of tissue (shown by dotted line).
When the mRNA-specific assay was used, N mRNA (>106 copies/g) was detected at days 1 and 7 in the nasal turbinates and at day 7 in the large intestine high density Peyer's patches and mesenteric lymph nodes. The rapid appearance of mRNA at day 1 in the nasal turbinates likely reflected the initial burst of local replication following vaccine administration. N mRNA was detected later at day 7 in the lymphoid tissues, which was the peak for gRNA. Virus titers also peaked at day 7 in the large intestine rich in Peyer's patches and mesenteric lymph nodes homogenates at 102 and 103 PFU per g tissue, respectively (data not shown). Although gRNA was detectable at days 1 and 21 in the large intestine high density Peyer's patches and mesenteric lymph nodes sites, N mRNA was below the limit of detection at these time points (Fig. 2B). The undetectable N mRNA quantities at d1 probably is indicative of a very early stage of infection in the lymphoid tissues that was initiated subsequent to spread of virus from initial foci of infection in the upper respiratory track. By d21, mRNA was undetected in nasal turbinates, large intestine high density Peyer's patches, and mesenteric lymph nodes samples indicating that the quantity of active replication had declined significantly (Fig. 2B).
There were no observable signs of illness or discomfort in ferrets that were inoculated with the empty rCDV vector. Consistent with this observation, no gRNA, mRNA, or virus was detected in brain, brainstem, or olfactory bulb. Importantly, these results indicated that even after a large intranasal dose (107 PFU) of infectious virus there was no evidence of invasion of the brain by this attenuated recombinant strain.
3.5. Simultaneous CDV N and SIV gag quantification in ferret tissues after intranasal, intramuscular, or intrarectal administration using 2-step RT-qPCR duplex assays specific for gRNA or mRNA
To determine if the N/Gag duplex assay could be used to evaluate replication, tissue distribution, gene expression, and also provide a reliable means by which to monitor gene insert stability, tissues (regions of large intestine with high or low densities of Peyer's patches and mesenteric lymph nodes) from ferrets infected with the prototype rCDV-SIVgag1 vector were analyzed. Two groups of ferrets were infected with 6 × 106 PFU administered by intranasal drops or intramuscular injection and tissues were harvested from animals at 7, 28, and 55 d.p.i. As a small element of the study, the vector also was administered by the intrarectal route to a third smaller group of animals that were sacrificed only at day 7 to determine if the vector could be delivered by this route. In the animals immunized by intranasal inoculation (Fig. 3A), the results showed that gRNA copies detected with the N amplicon were highest at day 7 in the large intestine with a high density of Peyer's patches and mesenteric lymph nodes at greater than 107 copies/g while large intestine sections with low densities of Peyer's patches contained 2 to 3-logs less genome copies at the same time point indicating that less replication had occurred in this tissue. By day 28, N gRNA declined in all tissues but remained detectable out to day 55 (Fig. 3A).
Fig. 3.
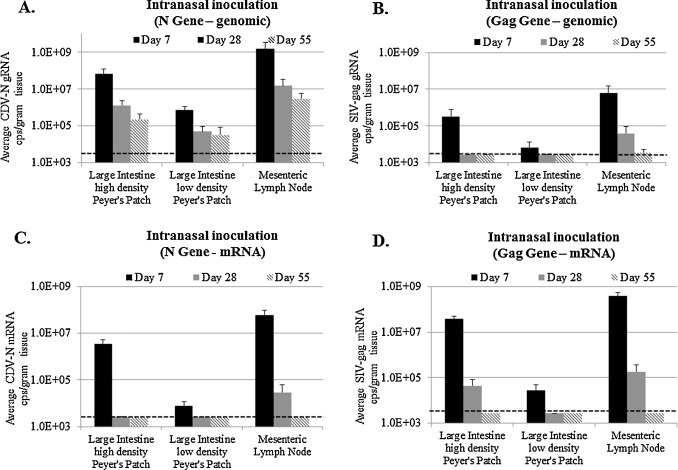
Viral loads at the indicated times in GALT tissues from ferrets after IN inoculation with rCDV-SIVgag1. Detection of (A) CDV N gRNA, (B) SIV gag gRNA, (C) CDV N mRNA and (D) SIV gag mRNA using 2-step duplex RT-qPCR assays. The detection limit is 3.0 × 103 copies/g of tissue (shown by dotted line).
When gag-specific gRNA values were compared, about 2-logs less was detected in all of the tissues at day 7 (Fig. 3B). At day 55, the quantity of gag sequence declined to near the limits of detection in mesenteric lymph nodes and was undetectable in large intestine tissues (Fig. 3B). The day 7 and 55 results showed that the expected 1:1 ratio of N to gag in the gRNA was not maintained during replication in vivo. Also, the N-to-gag ratio varied from 50 to 500:1 in the duplex gRNA assay indicating that this gag insert was unstable in the rCDV-SIV gag vector.
N and gag mRNA abundance also was analyzed from the same tissues using the duplex RT-qPCR mRNA assay from animals infected by intranasal inoculation. It is known that the relative quantities of mRNAs expressed by negative-sense RNA viruses, like CDV, measles virus and VSV, is determined by a transcriptional gradient in which genes proximal to the single 3′ genomic promoter (Fig. 1A) are expressed in greater quantities compared to genes distal from this promoter (Abraham and Banerjee, 1976, Cattaneo et al., 1987), thus high levels of SIV gag mRNA and somewhat less N was anticipated. Quantities of N mRNA peaked at greater than 1 × 106 copies/g in the mesenteric lymph nodes and large intestine high density Peyer's patches at day 7 and were low but detectable at 1 × 104 copies/g in large intestine low density Peyer's patches (Fig. 3C). N mRNA remained detectable at day 28 in mesenteric lymph nodes, but was below the limit of detection in large intestine sections. At day 55, N mRNA was undetectable in all tissues. Early after infection at day 7, the gag mRNA was more abundant (Fig. 3D) than N in all tissues as expected based on its placement in a highly transcribed region of the CDV genome. Gag mRNA quantities also exceeded N at day 28 even though results with the genomic qPCR assay described above predicted that gag mRNA would decline more rapidly compared to N (see Sections 3.6, 4). Taken together, these results showed that gRNA persisted for at least 55 days, but that mRNA levels were undetectable by this time point suggesting that the persisting genomes might be defective or that transcription and replication is greatly reduced in the population of cells that harbor persisting CDV gRNA.
Similar patterns of genome and mRNA abundance were observed following intramuscular injection of rCDV-SIVgag1 (Fig. 4 ). As in animals inoculated by intranasal inoculation, gRNA copies containing N sequence were highest in large intestine with a high density of Peyer's patches and in mesenteric lymph nodes (>107 copies/g) at day 7 with about 1-log less in large intestine tissue that had low densities of Peyer's patches (Fig. 4A). gRNA detected with the N amplicon persisted but slowly decreased out to day 55 in all tissues. Comparatively, gRNA copies of gag were decreased approximately 2-logs at day 7 and were only just above the detection limit by day 55 in the mesenteric lymph nodes (Fig. 4B). No SIV gag gRNA copies were detected at any time point in large intestine that has low Peyer's patch densities (Fig. 4B). Ratios greater than 100 and 300:1 CDV N to SIV gag gRNA copies were observed in large intestine containing high amounts of Peyer's patches and in mesenteric lymph nodes, respectively, consistent with results in the intranasal study, which indicated that the gag insert was unstable during replication in vivo.
Fig. 4.
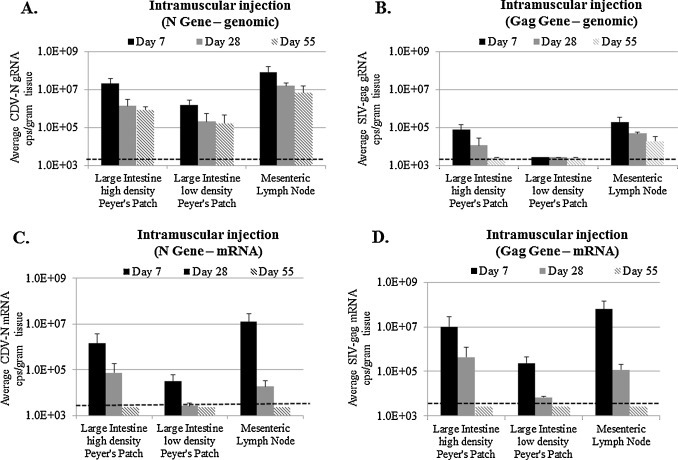
Viral loads in GALT tissues from ferrets after IM injection with rCDV-SIVgag1. Detection of (A) CDV N gRNA, (B) SIV gag gRNA, (C) CDV N mRNA and (D) SIV gag mRNA using 2-step duplex RT-qPCR assays. The detection limit is 3.0 × 103 copies/g of tissue (shown by dotted line).
The mRNA profiles obtained following intramuscular injection also were similar to the results in the intranasal study. CDV N mRNA was detected at the highest level in the large intestine tissue that contained higher densities of Peyer's patches and in the mesenteric lymph nodes at day 7 (>106 copies/g) and became undetectable between days 28 and 55. Less CDV N mRNA was detected in the large intestine samples in which Peyer's patches were less abundant (>103 copies/g) at day 7, and was undetectable at later time points (Fig. 4C). At day 7 and 28, SIV-gag mRNA copies (Fig. 4D) were 5–7 times more abundant than CDV N mRNA copies (Fig. 4C), which reflected the fact that there was increased transcriptional activity of the Gag gene in position 1 of the CDV genome (Fig. 1A).
Finally, following intrarectal inoculation, 2 out of the 3 animals were infected with rCDV-SIVgag1 at day 7, which was the only time point taken. A similar distribution and quantity of gRNA and mRNA was observed as seen in animals inoculated by intranasal drops and intramuscular injection indicating that regardless of the route of inoculation, the vector spread to the lymphoid tissues and amplified. The day 7 results following intrarectal inoculation also showed that gag mRNA was more abundant than N mRNA at this time point and that the N-to-gag gRNA ratio was indicative of gag insert instability (data not shown).
Overall, analysis of genomic RNA was indicative of gag insert instability. Despite this, when mRNA was detectable, the gag mRNA signal was consistently greater than N. In part, this probably was due to the gag gene being transcribed more actively since it was in position 1 in the genome and that the gag mutant emerged gradually over the course of replication in vivo and was a subpopulation of the total pool of replicating virus. Furthermore, as described below, the gag sequence deletion affected the gRNA amplicon target but not sequences detected by the mRNA amplicon.
3.6. Confirmation of gag insert deletion during in vivo replication in ferret tissues by traditional RT-PCR and sequencing
To confirm that the change in N-to-gag gRNA ratio was indicative of gag insert instability during replication in ferrets, traditional Gag RT-PCR to analyze insert integrity was performed and the gag gene isolated from a subset of the day 7 and day 28 samples was subsequently sequenced. Instead of the expected 1.6 kb RT-PCR product indicative of an intact gag gene, a small fragment of about 200 bases was detected (Fig. 5A). When the smaller PCR product was sequenced, the results showed that there was a deletion starting near the 5′ end of the Gag coding sequence (3′ end with respect to the negative-sense viral genome), which extended through most of the gag gene (the first 1416 nucleotides from the 1542 genome) leaving only 126 nucleotides (Fig. 5B) corresponding to the c-terminal coding sequence of Gag. Since the deleted region contained the annealing sites (nucleotides 256–339; Fig. 1D and 5B) for the Gag-specific qPCR primers and probe for the gRNA assay, the changing N-to-gag gRNA ratio did reflect emergence of a deletion mutant (Fig. 3B and 4B). The remaining 126 nucleotides, which is just before the AUUAU sequence adjoining the poly(A) tail, maintained the annealing sites for the SIV gag forward primer and the probe used in the SIV gag mRNA RT-qPCR assay (Fig. 1D); thus, this truncated gag sequence which includes the gag mRNA amplicon contributed to the high mRNA-specific signals seen at day 7 following IN (Fig. 3D) and IM (Fig. 4D) vaccination.
Fig. 5.
Confirmations of gag insert deletion during in vivo replication of the rCDV-SIVgag1 in ferret tissues by traditional RT-PCR and sequencing. (A) RNA extracted from ferret tissues was RT-PCR amplified and run on a 1% agarose gel. Lane 1, TrackIt™ 1 kb Plus DNA Ladder; lane 2, mesenteric lymph nodes 7 days after intramuscular injection; lane 3, mesenteric lymph nodes 7 days after intranasal inoculation; lane 4, large intestines high density Peyer's patches 28 days after intramuscular injection; lane 5, RNA positive control; lane 6, plasmid DNA positive control; lane 7, no template negative control. (B) The solid line indicates the 1.6 kb gag mRNA with the solid box showing the amplicon used in the SIV gag specific gRNA RT-qPCR (see Fig. 1D). The dotted line indicates the position of the 1416 Nt deletion as determined by sequencing.
4. Discussion
Sensitive and specific TaqMan-based 2-step duplex RT-qPCR assays were developed to analyze CDV vector replication, tissue distribution, mRNA abundance, and foreign gene insert stability in infected ferrets. The assays worked effectively with RNA isolated from various tissue homogenates and provided detection down to about 10 copies per PCR assay. The assays were used to show that a primary site of CDV vector replication in infected ferrets was abdominal cavity lymphoid tissue, and importantly, that CDV gRNA or mRNA was undetectable in brain tissue.
TaqMan-based 1-step (Meli et al., 2009, Wilkes et al., 2014) and 2-step singleplex RT-qPCR assays (Elia et al., 2006, Fischer et al., 2013, Scagliarini et al., 2007) have been described before using either random hexamers or gene-specific primers in the RT step mainly for the purpose of monitoring CDV outbreaks by detecting any CDV RNA species in specimens. The assays described in this report differ because they are designed to monitor the course of an infection in an animal model, study gag gene delivery by a CDV-SIV gag vector, and differentiate between genomic and mRNA species. In addition, for mRNA analysis, a primer for the RT-step was designed that included an adapter sequence and a 5-nucleotide sequence at the 3′ end that was specific for SIV gag and CDV N mRNAs, which significantly reduced background from cellular RNA and positive-sense vector genomic RNA and also helped to eliminate amplification of DNA caused by RNA self-priming (Craggs et al., 2001, Simon et al., 2010). More recently, an RT-qPCR assay was reported (Pawar et al., 2011) based on SYBR Green I chemistry for detection of the hemagglutinin gene in samples from infected dogs. This assay used oligo-dT primers to synthesize cDNA from mRNA in the RT step and a relative ΔΔCT quantification method to determine fold changes in CDV gene expression. Using the TaqMan system described here, permitted absolute quantification of gRNA and mRNA and also multiplexing of CDV N and SIV gag amplicons.
Recombinant CDV is being investigated as a delivery vector for HIV vaccines because it has the potential to specifically deliver vaccine immunogens to lymphoid tissues that are susceptible to HIV infection (Parks et al., 2013, Zhang et al., 2013). Previous studies showed that virulent CDV replicated in the ferret gut-associated lymphoid tissues (GALT) (Pillet et al., 2009, von Messling et al., 2006), and this work extends those results by showing that a recombinant vector based on an attenuated vaccine strain also targeted the GALT. After intranasal inoculation with the empty rCDV vector, abundant quantities of gRNA (>108 copies/g) were detected in ferret GALT at day 7 that persisted out to day 21. Viral mRNA similarly was detected and was highest at day 7. Genomes also were evident at day 1 in large intestine high density Peyer's patches and in mesenteric lymph nodes, but mRNA was not detectable at this early timepoint. The lack of an mRNA signal at day 1 probably was due to this being an early time point during virus replication at this secondary site of infection. CDV N gRNA was detectable out to day 55 in ferret tissues, but mRNA was out to day 28 (Fig. 3, Fig. 4) were observed. This result might indicate that persisting genomes were defective or that their transcriptional activity was low, which was consistent with the lack of detectable infectious virus in tissues harvested at day 55. Interestingly, results from a number of studies indicate that negative-strand RNA virus genomes persist for considerable periods of time post-infection. Genomic RNA from measles virus (Lin et al., 2012, Pan et al., 2005, Pan et al., 2010, Riddell et al., 2007) and vesicular stomatitis virus (Barrera and Letchworth, 1996, Cooper et al., 2008, Johnson et al., 2009, Johnson et al., 2007, Letchworth et al., 1996, Simon et al., 2007, Simon et al., 2010, Turner et al., 2007) could be detected in tissues well after shedding of infectious virus had ended.
The CDV vector described in this report was developed from a live attenuated canine vaccine, which is used to safely and effectively control distemper in domestic dogs (Chappuis, 1995). Consistent with the canine vaccine safety record, the recombinant vector also was safe for use in ferrets, which are a permissive species that is susceptible to disease caused by natural CDV infections. Importantly, nucleic acid or virus was not detected in the brain, brain stem, or olfactory bulb following intranasal inoculation with a high dose (107 PFU) indicating that the attenuated CDV vector did not replicate in the brain. This conclusion also was consistent with visual observations, which showed that there were no signs of illness.
Interestingly, the duplex assay indicated that SIV gag signal from gRNA decreased significantly compared to N over the course of infection indicating that all or part of the gag insert deleted when this prototype vector replicated in vivo. Traditional RT-PCR and genome sequencing using a subset of day-7 and day-28 samples from the intranasal and intramuscular studies confirmed that the gag gene was unstable and that vector variants emerged with most of the insert deleted during replication in vivo. The deleted sequences included the target RT-qPCR amplicon used to detect gag in the gRNA assay, which explained reduction in signal compared to the N amplicon. This result also demonstrated that the duplex qPCR assay and gene quantity ratios could be used to effectively monitor gene insert stability in vivo, which provided valuable practical data for guiding future vector development by specifically showing that for some foreign genes alternative gene optimization approaches were needed to generate genetically stable negative-strand RNA virus vectors (Rabinovich et al., 2014).
Although these results demonstrated the feasibility of using a duplex assay as a sensitive monitor of genome stability, it should be noted that the genome deletion observed was relatively large and resulted in excision of an amplicon target. Thus the assay can be improved if multiple amplicons are developed and spaced across the foreign gene insert, which would increase the probability of detecting smaller deletions and observing a shift in ratio gene insert to genome copy ratio.
Nucleotide sequencing also demonstrated that a 126-nucleotide gag fragment remained after the 1.416 kb sequence was deleted, and as expected, the genome of the deletion mutant conformed to the paramyxovirus rule-of-six (Kolakofsky et al., 1998), which states that the total number of nucleotides in the genomic RNA must be a multiple of 6 to replicate efficiently. Because this truncated gag gene contained the target amplicon sequence for the gag mRNA-specific RT-qPCR assay, abundant mRNA signal was observed even as the mutant virus emerged and became a dominant species in the infected ferrets. This was consistent with results that showed SIV gag mRNA was generally 3–10-fold more abundant than CDV N mRNA as anticipated based on the promoter-proximal placement of the gag gene (Wertz et al., 1998).
In summary, the results presented here show that the duplex RT-qPCR assays described here are rapid and sensitive tools for analysis of CDV vector replication in vivo and that they can be used to quantify genome and mRNA species extracted from a variety of tissues. Furthermore, in these initial studies conducted in ferrets with a prototype CDV-SIV Gag vector, it has been demonstrated that these qPCR procedures provide the foundation for developing future assays that can be used to analyze important biological properties of new replicating viral vectors including replication, tissue distribution, mRNA expression, and genetic stability.
Acknowledgements
The authors thank Drs. Beth Rasmussen, C. Richter King, Wayne Koff, Ross Lindsay and Eddy Sayeed for helpful advice on the research and comments on the manuscript. IAVI's work is made possible by generous support from many donors including: the Bill & Melinda Gates Foundation; the Ministry of Foreign Affairs of Denmark; Irish Aid; the Ministry of Finance of Japan; the Ministry of Foreign Affairs of the Netherlands; the Norwegian Agency for Development Cooperation (NORAD); the United Kingdom Department for International Development (DFID), and the United States Agency for International Development (USAID). The full list of IAVI donors is available at www.iavi.org. The Bill and Melinda Gates Foundation Collaboration for AIDS Vaccine Discovery (CAVD) supported this research. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of donors or funding agencies.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jviromet.2014.11.015.
Appendix A. Supplementary data
The following are the supplementary data to this article:
Supplemental Fig. 1.
The linearity, amplification efficiency, sensitivity, and specificity of singleplex RT-qPCR assays to detect CDV N and SIV-gag genomic RNA and of the 2-step duplex assay to detect CDV N and SIV-gag genomic RNA. (A) Ten-fold serial dilutions of a synthetic RNA oligonucleotide spanning the CDV N amplicon were made and 1 × 108 to 1 × 101 copies were used to initiate the CDV N gRNA RT-qPCR assay. (B) Similarly, 10-fold serial dilutions of a SIV-gag specific synthetic RNA oligonucleotide were made and 1 × 108 to 1 × 101 copies of the amplicon assayed. (C) Also, 10-fold serial dilutions from 1 × 108 to 1 × 101 copies of synthetic RNA oligonucleotides of (A) and (B) were assayed in a duplex RT-qPCR to detect CDV N and SIV-gag simultaneously. Standard curves were generated by plotting the log of genome copies against the cycle threshold (CT). For singleplex assays, gene specific primer-probe sets were used in separate wells and in the duplex assay, gene specific primer-probe sets for both templates were combined in one well. Positive CT values are ≤40 while negative CT values are >40. All CT values are means of duplicates.
Supplemental Fig. 2.
Response of the duplex RT-qPCR assay on the CT values of different ratios of CDV N-to-gag RNA concentrations in a sample. (A) N oligoribonucleotide template held constant at 1 × 107 copies per RT-qPCR while serially titrating the gag template 10-fold over a range from 1 × 107 to 1 × 103 copies. (B). N oligoribonucleotide template held at 1 × 104 copies per RT-qPCR while serially titrating the gag template 10-fold from 1 × 104 to 1 × 101 copies. CT values (vertical axis) for each target RNA and ratios of CDV N: SIV copies (horizontal axis). Standard curves were generated by plotting the log of gag genome copies against the measured cycle threshold (CT). All CT values are means of duplicates.
References
- Abraham G., Banerjee A.K. Sequential transcription of the genes of vesicular stomatitis virus. Proc. Natl. Acad. Sci. U.S.A. 1976;73:1504–1508. doi: 10.1073/pnas.73.5.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel M.J.G. Elsevier Science; Distributors for the U. S. and Canada Elsevier Science Pub. Co; Amsterdam; New York, NY: 1987. Virus Infections of Carnivores. [Google Scholar]
- Barrera J.C., Letchworth G.J. Persistence of vesicular stomatitis virus New Jersey RNA in convalescent hamsters. Virology. 1996;219:453–464. doi: 10.1006/viro.1996.0271. [DOI] [PubMed] [Google Scholar]
- Billeter M.A., Naim H.Y., Udem S.A. Reverse genetics of measles virus and resulting multivalent recombinant vaccines: applications of recombinant measles viruses. Curr. Top. Microbiol. Immunol. 2009;329:129–162. doi: 10.1007/978-3-540-70523-9_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandler S., Lucas-Hourani M., Moris A., Frenkiel M.P., Combredet C., Fevrier M., Bedouelle H., Schwartz O., Despres P., Tangy F. Pediatric measles vaccine expressing a dengue antigen induces durable serotype-specific neutralizing antibodies to dengue virus. PLoS Negl. Trop. Dis. 2007;1:e96. doi: 10.1371/journal.pntd.0000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandler S., Tangy F. Recombinant vector derived from live attenuated measles virus: potential for flavivirus vaccines. Comp. Immunol. Microbiol. Infect. Dis. 2008;31:271–291. doi: 10.1016/j.cimid.2007.07.012. [DOI] [PubMed] [Google Scholar]
- Bukreyev A., Skiadopoulos M.H., Murphy B.R., Collins P.L. Nonsegmented negative-strand viruses as vaccine vectors. J. Virol. 2006;80:10293–10306. doi: 10.1128/JVI.00919-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussell R.H., Karzon D.T. Canine distemper virus in chick embryo cell culture. Plaque assay, growth, and stability. Virology. 1962;18:589–600. doi: 10.1016/0042-6822(62)90062-4. [DOI] [PubMed] [Google Scholar]
- Bustin S.A., Benes V., Garson J.A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M.W., Shipley G.L., Vandesompele J., Wittwer C.T. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- Cattaneo R., Rebmann G., Schmid A., Baczko K., ter Meulen V., Billeter M.A. Altered transcription of a defective measles virus genome derived from a diseased human brain. EMBO J. 1987;6:681–688. doi: 10.1002/j.1460-2075.1987.tb04808.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappuis G. Control of canine distemper. Vet. Microbiol. 1995;44:351–358. doi: 10.1016/0378-1135(95)00028-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline A.N., Bess J.W., Piatak M., Jr., Lifson J.D. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J. Med. Primatol. 2005;34:303–312. doi: 10.1111/j.1600-0684.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- Coleman J.W., Ogin-Wilson E., Johnson J.E., Nasar F., Zamb T.P., Clarke D.K., Hendry R.M., Udem S.A. Quantitative multiplex assay for simultaneous detection of the Indiana serotype of vesicular stomatitis virus and HIV gag. J. Virol. Methods. 2007;143:55–64. doi: 10.1016/j.jviromet.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Cooper D., Wright K.J., Calderon P.C., Guo M., Nasar F., Johnson J.E., Coleman J.W., Lee M., Kotash C., Yurgelonis I., Natuk R.J., Hendry R.M., Udem S.A., Clarke D.K. Attenuation of recombinant vesicular stomatitis virus-human immunodeficiency virus type 1 vaccine vectors by gene translocations and g gene truncation reduces neurovirulence and enhances immunogenicity in mice. J. Virol. 2008;82:207–219. doi: 10.1128/JVI.01515-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craggs J.K., Ball J.K., Thomson B.J., Irving W.L., Grabowska A.M. Development of a strand-specific RT-PCR based assay to detect the replicative form of hepatitis C virus RNA. J. Virol. Methods. 2001;94:111–120. doi: 10.1016/s0166-0934(01)00281-6. [DOI] [PubMed] [Google Scholar]
- de Vries R.D., Ludlow M., Verburgh R.J., van Amerongen G., Yuksel S., Nguyen D.T., McQuaid S., Osterhaus A.D., Duprex W.P., de Swart R.L. Measles vaccination of non-human primates provides partial protection against infection with canine distemper virus. J. Virol. 2014;88:4423–4433. doi: 10.1128/JVI.03676-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Valle J.R., Devaux P., Hodge G., Wegner N.J., McChesney M.B., Cattaneo R. A vectored measles virus induces hepatitis B surface antigen antibodies while protecting macaques against measles virus challenge. J. Virol. 2007;81:10597–10605. doi: 10.1128/JVI.00923-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despres P., Combredet C., Frenkiel M.P., Lorin C., Brahic M., Tangy F. Live measles vaccine expressing the secreted form of the West Nile virus envelope glycoprotein protects against West Nile virus encephalitis. J. Infect. Dis. 2005;191:207–214. doi: 10.1086/426824. [DOI] [PubMed] [Google Scholar]
- Elia G., Decaro N., Martella V., Cirone F., Lucente M.S., Lorusso E., Di Trani L., Buonavoglia C. Detection of canine distemper virus in dogs by real-time RT-PCR. J. Virol. Methods. 2006;136:171–176. doi: 10.1016/j.jviromet.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Fischer C.D., Ikuta N., Canal C.W., Makiejczuk A., Allgayer M.D., Cardoso C.H., Lehmann F.K., Fonseca A.S., Lunge V.R. Detection and differentiation of field and vaccine strains of canine distemper virus using reverse transcription followed by nested real time PCR (RT-nqPCR) and RFLP analysis. J. Virol. Methods. 2013;194:39–45. doi: 10.1016/j.jviromet.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisk A.L., Konig M., Moritz A., Baumgartner W. Detection of canine distemper virus nucleoprotein RNA by reverse transcription-PCR using serum, whole blood, and cerebrospinal fluid from dogs with distemper. J. Clin. Microbiol. 1999;37:3634–3643. doi: 10.1128/jcm.37.11.3634-3643.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemma T., Miyashita N., Shin Y.S., Okita M., Mori T., Iwatsuki K., Mikami T., Kai C. Serological survey of canine distemper virus infection using enzyme-linked immunosorbent assay. J. Vet. Med. Sci. 1995;57:761–763. doi: 10.1292/jvms.57.761. [DOI] [PubMed] [Google Scholar]
- Harder T.C., Osterhaus A.D. Canine distemper virus – a morbillivirus in search of new hosts? Trends Microbiol. 1997;5:120–124. doi: 10.1016/S0966-842X(97)01010-X. [DOI] [PubMed] [Google Scholar]
- Hughes G.J., Smith J.S., Hanlon C.A., Rupprecht C.E. Evaluation of a TaqMan PCR assay to detect rabies virus RNA: influence of sequence variation and application to quantification of viral loads. J. Clin. Microbiol. 2004;42:299–306. doi: 10.1128/JCM.42.1.299-306.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J.E., Coleman J.W., Kalyan N.K., Calderon P., Wright K.J., Obregon J., Ogin-Wilson E., Natuk R.J., Clarke D.K., Udem S.A., Cooper D., Hendry R.M. In vivo biodistribution of a highly attenuated recombinant vesicular stomatitis virus expressing HIV-1 Gag following intramuscular, intranasal, or intravenous inoculation. Vaccine. 2009;27:2930–2939. doi: 10.1016/j.vaccine.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J.E., Nasar F., Coleman J.W., Price R.E., Javadian A., Draper K., Lee M., Reilly P.A., Clarke D.K., Hendry R.M., Udem S.A. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology. 2007;360:36–49. doi: 10.1016/j.virol.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.H., Cho K.W., Youn H.Y., Yoo H.S., Han H.R. Detection of canine distemper virus (CDV) through one step RT-PCR combined with nested PCR. J. Vet. Sci. 2001;2:59–63. [PubMed] [Google Scholar]
- Kolakofsky D., Pelet T., Garcin D., Hausmann S., Curran J., Roux L. Paramyxovirus RNA synthesis and the requirement for hexamer genome length: the rule of six revisited. J. Virol. 1998;72:891–899. doi: 10.1128/jvi.72.2.891-899.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford R.E., Sureau C., Jacob J.R., White R., Fuerst T.R. Demonstration of in vitro infection of chimpanzee hepatocytes with hepatitis C virus using strand-specific RT/PCR. Virology. 1994;202:606–614. doi: 10.1006/viro.1994.1381. [DOI] [PubMed] [Google Scholar]
- Langlois I. Viral diseases of ferrets. Vet. Clin. N. Am. Exot. Anim. Pract. 2005;8:139–160. doi: 10.1016/j.cvex.2004.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letchworth G.J., Barrera J.C., Fishel J.R., Rodriguez L. Vesicular stomatitis New Jersey virus RNA persists in cattle following convalescence. Virology. 1996;219:480–484. doi: 10.1006/viro.1996.0275. [DOI] [PubMed] [Google Scholar]
- Lin W.H., Kouyos R.D., Adams R.J., Grenfell B.T., Griffin D.E. Prolonged persistence of measles virus RNA is characteristic of primary infection dynamics. Proc. Natl. Acad. Sci. U.S.A. 2012;109:14989–14994. doi: 10.1073/pnas.1211138109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liniger M., Zuniga A., Tamin A., Azzouz-Morin T.N., Knuchel M., Marty R.R., Wiegand M., Weibel S., Kelvin D., Rota P.A., Naim H.Y. Induction of neutralising antibodies and cellular immune responses against SARS coronavirus by recombinant measles viruses. Vaccine. 2008;26:2164–2174. doi: 10.1016/j.vaccine.2008.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorin C., Segal L., Mols J., Morelle D., Bourguignon P., Rovira O., Mettens P., Silvano J., Dumey N., Le Goff F., Koutsoukos M., Voss G., Tangy F. Toxicology, biodistribution and shedding profile of a recombinant measles vaccine vector expressing HIV-1 antigens, in cynomolgus macaques. Naunyn-Schmiedeberg's Arch. Pharmacol. 2012;385:1211–1225. doi: 10.1007/s00210-012-0793-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow M., Nguyen D.T., Silin D., Lyubomska O., de Vries R.D., von Messling V., McQuaid S., De Swart R.L., Duprex W.P. Recombinant canine distemper virus strain Snyder Hill expressing green or red fluorescent proteins causes meningoencephalitis in the ferret. J. Virol. 2012;86:7508–7519. doi: 10.1128/JVI.06725-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlhatton M.A., Curran M.D., Rima B.K. Nucleotide sequence analysis of the large (L) genes of phocine distemper virus and canine distemper virus (corrected sequence) J. Gen. Virol. 1997;78(Pt 3):571–576. doi: 10.1099/0022-1317-78-3-571. [DOI] [PubMed] [Google Scholar]
- Meli M.L., Cattori V., Martinez F., Lopez G., Vargas A., Simon M.A., Zorrilla I., Munoz A., Palomares F., Lopez-Bao J.V., Pastor J., Tandon R., Willi B., Hofmann-Lehmann R., Lutz H. Feline leukemia virus and other pathogens as important threats to the survival of the critically endangered Iberian lynx (Lynx pardinus) PLoS ONE. 2009;4:e4744. doi: 10.1371/journal.pone.0004744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan C.H., Greer C.E., Hauer D., Legg H.S., Lee E.Y., Bergen M.J., Lau B., Adams R.J., Polo J.M., Griffin D.E. A chimeric alphavirus replicon particle vaccine expressing the hemagglutinin and fusion proteins protects juvenile and infant rhesus macaques from measles. J. Virol. 2010;84:3798–3807. doi: 10.1128/JVI.01566-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan C.H., Valsamakis A., Colella T., Nair N., Adams R.J., Polack F.P., Greer C.E., Perri S., Polo J.M., Griffin D.E. Modulation of disease, T cell responses, and measles virus clearance in monkeys vaccinated with H-encoding alphavirus replicon particles. Proc. Natl. Acad. Sci. U.S.A. 2005;102:11581–11588. doi: 10.1073/pnas.0504592102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks C.L., Picker L.J., King C.R. Development of replication-competent viral vectors for HIV vaccine delivery. Curr. Opin. HIV AIDS. 2013;8:402–411. doi: 10.1097/COH.0b013e328363d389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks C.L., Wang H.P., Kovacs G.R., Vasilakis N., Kowalski J., Nowak R.M., Lerch R.A., Walpita P., Sidhu M.S., Udem S.A. Expression of a foreign gene by recombinant canine distemper virus recovered from cloned DNAs. Virus Res. 2002;83:131–147. doi: 10.1016/s0168-1702(01)00430-0. [DOI] [PubMed] [Google Scholar]
- Pasloske B.L., Walkerpeach C.R., Obermoeller R.D., Winkler M., DuBois D.B. Armored RNA technology for production of ribonuclease-resistant viral RNA controls and standards. J. Clin. Microbiol. 1998;36:3590–3594. doi: 10.1128/jcm.36.12.3590-3594.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar R.M., Raj G.D., Gopinath V.P., Ashok A., Raja A. Isolation and molecular characterization of canine distemper virus from India. Trop. Anim. Health Prod. 2011;43:1617–1622. doi: 10.1007/s11250-011-9880-7. [DOI] [PubMed] [Google Scholar]
- Pillet S., Svitek N., von Messling V. Ferrets as a model for morbillivirus pathogenesis, complications, and vaccines. Curr. Top. Microbiol. Immunol. 2009;330:73–87. doi: 10.1007/978-3-540-70617-5_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W., Zheng Y., Zhang S., Fan Q., Liu H., Zhang F., Wang W., Liao G., Hu R. Canine distemper outbreak in rhesus monkeys, China. Emerg. Infect. Dis. 2011;17:1541–1543. doi: 10.3201/eid1708.101153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich S., Powell R.L., Lindsay R.W., Yuan M., Carpov A., Wilson A., Lopez M., Coleman J.W., Wagner D., Sharma P., Kemelman M., Wright K.J., Seabrook J.P., Arendt H., Martinez J., DeStefano J., Chiuchiolo M.J., Parks C.L. A novel, live-attenuated vesicular stomatitis virus vector displaying conformationally intact, functional HIV-1 envelope trimers that elicits potent cellular and humoral responses in mice. PLOS ONE. 2014;9:e106597. doi: 10.1371/journal.pone.0106597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell M.A., Moss W.J., Hauer D., Monze M., Griffin D.E. Slow clearance of measles virus RNA after acute infection. J. Clin. Virol. 2007;39:312–317. doi: 10.1016/j.jcv.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Rima B.K., Duprex W.P. Morbilliviruses and human disease. J. Pathol. 2006;208:199–214. doi: 10.1002/path.1873. [DOI] [PubMed] [Google Scholar]
- Rudd P.A., Cattaneo R., von Messling V. Canine distemper virus uses both the anterograde and the hematogenous pathway for neuroinvasion. J. Virol. 2006;80:9361–9370. doi: 10.1128/JVI.01034-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzezutka A., Mizak B. Application of N-PCR for diagnosis of distemper in dogs and fur animals. Vet. Microbiol. 2002;88:95–103. doi: 10.1016/s0378-1135(02)00097-4. [DOI] [PubMed] [Google Scholar]
- Saito T.B., Alfieri A.A., Wosiacki S.R., Negrao F.J., Morais H.S., Alfieri A.F. Detection of canine distemper virus by reverse transcriptase-polymerase chain reaction in the urine of dogs with clinical signs of distemper encephalitis. Res. Vet. Sci. 2006;80:116–119. doi: 10.1016/j.rvsc.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Sakai K., Nagata N., Ami Y., Seki F., Suzaki Y., Iwata-Yoshikawa N., Suzuki T., Fukushi S., Mizutani T., Yoshikawa T., Otsuki N., Kurane I., Komase K., Yamaguchi R., Hasegawa H., Saijo M., Takeda M., Morikawa S. Lethal canine distemper virus outbreak in cynomolgus monkeys in Japan in 2008. J. Virol. 2013;87:1105–1114. doi: 10.1128/JVI.02419-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M., Saito M., Tanaka K., Iwanaga S., Ali S.N., Seki T., Okada S., Kohara M., Harada S., Kai C., Tsukiyama-Kohara K. Evaluation of a recombinant measles virus expressing hepatitis C virus envelope proteins by infection of human PBL-NOD/Scid/Jak3null mouse. Comp. Immunol. Microbiol. Infect. Dis. 2010;33:e81–e88. doi: 10.1016/j.cimid.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scagliarini A., Dal Pozzo F., Gallina L., Vaccari F., Morganti L. TaqMan based real time PCR for the quantification of canine distemper virus. Vet. Res. Commun. 2007;31(Suppl. 1):261–263. doi: 10.1007/s11259-007-0020-9. [DOI] [PubMed] [Google Scholar]
- Seki F., Ono N., Yamaguchi R., Yanagi Y. Efficient isolation of wild strains of canine distemper virus in Vero cells expressing canine SLAM (CD150) and their adaptability to marmoset B95a cells. J. Virol. 2003;77:9943–9950. doi: 10.1128/JVI.77.18.9943-9950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y., Mori T., Okita M., Gemma T., Kai C., Mikami T. Detection of canine distemper virus nucleocapsid protein gene in canine peripheral blood mononuclear cells by RT-PCR. J. Vet. Med. Sci. 1995;57:439–445. doi: 10.1292/jvms.57.439. [DOI] [PubMed] [Google Scholar]
- Shin Y.J., Cho K.O., Cho H.S., Kang S.K., Kim H.J., Kim Y.H., Park H.S., Park N.Y. Comparison of one-step RT-PCR and a nested PCR for the detection of canine distemper virus in clinical samples. Aust. Vet. J. 2004;82:83–86. doi: 10.1111/j.1751-0813.2004.tb14651.x. [DOI] [PubMed] [Google Scholar]
- Simon I.D., Publicover J., Rose J.K. Replication and propagation of attenuated vesicular stomatitis virus vectors in vivo: vector spread correlates with induction of immune responses and persistence of genomic RNA. J. Virol. 2007;81:2078–2082. doi: 10.1128/JVI.02525-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon I.D., van Rooijen N., Rose J.K. Vesicular stomatitis virus genomic RNA persists in vivo in the absence of viral replication. J. Virol. 2010;84:3280–3286. doi: 10.1128/JVI.02052-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M., Cattaneo R., Billeter M.A. A recombinant measles virus expressing hepatitis B virus surface antigen induces humoral immune responses in genetically modified mice. J. Virol. 1999;73:4823–4828. doi: 10.1128/jvi.73.6.4823-4828.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z., Li A., Ye H., Shi Y., Hu Z., Zeng L. Natural infection with canine distemper virus in hand-feeding Rhesus monkeys in China. Vet. Microbiol. 2010;141:374–378. doi: 10.1016/j.vetmic.2009.09.024. [DOI] [PubMed] [Google Scholar]
- Tangy F., Naim H.Y. Live attenuated measles vaccine as a potential multivalent pediatric vaccination vector. Viral Immunol. 2005;18:317–326. doi: 10.1089/vim.2005.18.317. [DOI] [PubMed] [Google Scholar]
- Toussaint J.F., Sailleau C., Breard E., Zientara S., De Clercq K. Bluetongue virus detection by two real-time RT-qPCRs targeting two different genomic segments. J. Virol. Methods. 2007;140:115–123. doi: 10.1016/j.jviromet.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Trebbien R., Chriel M., Struve T., Hjulsager C.K., Larsen G., Larsen L.E. Wildlife reservoirs of canine distemper virus resulted in a major outbreak in Danish farmed mink (Neovison vison) PLOS ONE. 2014;9:e85598. doi: 10.1371/journal.pone.0085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner D.L., Cauley L.S., Khanna K.M., Lefrancois L. Persistent antigen presentation after acute vesicular stomatitis virus infection. J. Virol. 2007;81:2039–2046. doi: 10.1128/JVI.02167-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Messling V., Harder T.C., Moennig V., Rautenberg P., Nolte I., Haas L. Rapid and sensitive detection of immunoglobulin M (IgM) and IgG antibodies against canine distemper virus by a new recombinant nucleocapsid protein-based enzyme-linked immunosorbent assay. J. Clin. Microbiol. 1999;37:1049–1056. doi: 10.1128/jcm.37.4.1049-1056.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Messling V., Milosevic D., Cattaneo R. Tropism illuminated: lymphocyte-based pathways blazed by lethal morbillivirus through the host immune system. Proc. Natl. Acad. Sci. U.S.A. 2004;101:14216–14221. doi: 10.1073/pnas.0403597101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Messling V., Svitek N., Cattaneo R. Receptor (SLAM [CD150]) recognition and the V protein sustain swift lymphocyte-based invasion of mucosal tissue and lymphatic organs by a morbillivirus. J. Virol. 2006;80:6084–6092. doi: 10.1128/JVI.00357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Feng N., Ge J., Shuai L., Peng L., Gao Y., Yang S., Xia X., Bu Z. Recombinant canine distemper virus serves as bivalent live vaccine against rabies and canine distemper. Vaccine. 2012;30:5067–5072. doi: 10.1016/j.vaccine.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Wertz G.W., Perepelitsa V.P., Ball L.A. Gene rearrangement attenuates expression and lethality of a nonsegmented negative strand RNA virus. Proc. Natl. Acad. Sci. U.S.A. 1998;95:3501–3506. doi: 10.1073/pnas.95.7.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkes R.P., Sanchez E., Riley M.C., Kennedy M.A. Real-time reverse transcription polymerase chain reaction method for detection of Canine distemper virus modified live vaccine shedding for differentiation from infection with wild-type strains. J. Vet. Diagn. Investig.: Off. Publ. Am. Assoc. Vet. Lab. Diagn. Inc. 2014;26:27–34. doi: 10.1177/1040638713517232. [DOI] [PubMed] [Google Scholar]
- Winstone N., Wilson A.J., Morrow G., Boggiano C., Chiuchiolo M.J., Lopez M., Kemelman M., Ginsberg A.A., Mullen K., Coleman J.W., Wu C.D., Narpala S., Ouellette I., Dean H.J., Lin F., Sardesai N.Y., Cassamasa H., McBride D., Felber B.K., Pavlakis G.N., Schultz A., Hudgens M.G., King C.R., Zamb T.J., Parks C.L., McDermott A.B. Enhanced control of pathogenic Simian immunodeficiency virus SIVmac239 replication in macaques immunized with an interleukin-12 plasmid and a DNA prime-viral vector boost vaccine regimen. J. Virol. 2011;85:9578–9587. doi: 10.1128/JVI.05060-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witko S.E., Kotash C.S., Nowak R.M., Johnson J.E., Boutilier L.A., Melville K.J., Heron S.G., Clarke D.K., Abramovitz A.S., Hendry R.M., Sidhu M.S., Udem S.A., Parks C.L. An efficient helper-virus-free method for rescue of recombinant paramyxoviruses and rhadoviruses from a cell line suitable for vaccine development. J. Virol. Methods. 2006;135:91–101. doi: 10.1016/j.jviromet.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Zhang X., Wallace O., Wright K.J., Backer M., Coleman J.W., Koehnke R., Frenk E., Domi A., Chiuchiolo M.J., Destefano J., Narpala S., Powell R., Morrow G., Boggiano C., Zamb T.J., Richter King C., Parks C.L. Membrane-bound SIV envelope trimers are immunogenic in ferrets after intranasal vaccination with a replication-competent canine distemper virus vector. Virology. 2013;446:25–36. doi: 10.1016/j.virol.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Zuniga A., Wang Z., Liniger M., Hangartner L., Caballero M., Pavlovic J., Wild P., Viret J.F., Glueck R., Billeter M.A., Naim H.Y. Attenuated measles virus as a vaccine vector. Vaccine. 2007;25:2974–2983. doi: 10.1016/j.vaccine.2007.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.