

Abstract
A major challenge in modern biology is to understand how naturally occurring variation in DNA sequences affects complex organismal traits through networks of intermediate molecular phenotypes. This question is best addressed in a genetic mapping population in which all molecular polymorphisms are known and for which molecular endophenotypes and complex traits are assessed on the same genotypes. Here, we performed deep RNA sequencing of 200 Drosophila Genetic Reference Panel inbred lines with complete genome sequences and for which phenotypes of many quantitative traits have been evaluated. We mapped expression quantitative trait loci for annotated genes, novel transcribed regions, transposable elements, and microbial species. We identified host variants that affect expression of transposable elements, independent of their copy number, as well as microbiome composition. We constructed sex-specific expression quantitative trait locus regulatory networks. These networks are enriched for novel transcribed regions and target genes in heterochromatin and euchromatic regions of reduced recombination, as well as genes regulating transposable element expression. This study provides new insights regarding the role of natural genetic variation in regulating gene expression and generates testable hypotheses for future functional analyses.
Understanding how naturally occurring genetic variation affects variation in organismal quantitative traits by modifying underlying molecular networks is a key challenge in modern biology. Most traits are highly polygenic (Mackay et al. 2009; Visscher et al. 2012; Mackay and Huang, 2018), and associated molecular variants have small additive effects on trait variation (Manolio et al. 2009). Most of these variants are in intergenic regions, upstream of or downstream from coding regions, or in introns and presumably play a regulatory role in modulating gene expression.
Systems genetics analysis seeks to determine how naturally occurring molecular variation gives rise to genetic variation in organismal phenotypes by examining genetic variation in gene expression (expression quantitative trait loci [eQTLs]) and other intermediate molecular phenotypes (Sieberts and Schadt 2007; Chen et al. 2008; Emilsson et al. 2008; Rockman 2008; Cookson et al. 2009; Mackay et al. 2009; Civelek and Lusis 2014; Albert and Kruglyak 2015; Gibson et al. 2015; Ogura and Busch 2016; Schughart and Williams 2017). Polymorphic variants associated with variation in gene expression are classified as cis- or trans-eQTLs depending on whether they are proximal or distal to the gene encoding the transcript, respectively. Genetic variation in gene expression is pervasive; cis-eQTLs can have large effects on gene expression that are detectable in small samples; and variants associated with human diseases and quantitative traits tend to be enriched for cis-eQTLs (Sieberts and Schadt 2007; Chen et al. 2008; Emilsson et al. 2008; Rockman 2008; Cookson et al. 2009; Mackay et al. 2009; Nicolae et al. 2010; Civelek and Lusis 2014; Albert and Kruglyak 2015; Gibson et al. 2015; Ogura and Busch 2016; Boyle et al. 2017; Schughart and Williams 2017). eQTLs with both cis- and trans- effects can be assembled into directed transcriptional networks of regulator and target genes (Liu et al. 2008; Bryois et al. 2014; Fagny et al. 2017). Elucidating such regulatory transcriptional networks will facilitate understanding how the effects of individual variants propagate through the network and how multiple variants together regulate gene expression and affect complex traits (Liu et al. 2008; Nicolae et al. 2010; Bryois et al. 2014; Fagny et al. 2017) and will improve genomic prediction (Zhou et al. 2020).
Here, we performed deep RNA sequencing of the Drosophila melanogaster Genetic Reference Panel (DGRP) of inbred lines with complete DNA sequences (Mackay et al. 2012; Huang et al. 2014). We mapped eQTLs for annotated genes, novel transcribed region (NTRs), transposable elements (TEs), and microbiome composition; constructed de novo cis-trans-eQTL gene expression networks; and evaluated associations of eQTLs and expression traits with organismal phenotypes.
Results
We collected and sequenced ribo(−) RNA from replicate pools of young flies from each of 200 DGRP lines, separately for males and females. In total, we sequenced 1.94 terabases of RNA, of which on average 13.4 million reads per sample uniquely aligned to the Drosophila melanogaster genome (Supplemental Table S1). The sequences were processed through a pipeline (Supplemental Fig. S1) that (1) removes adapter and rRNA sequences, (2) aligns and quantifies expressed TE sequences and microbial transcripts, (3) verifies the origin of each sample, and (4) quantifies known and novel D. melanogaster transcripts and corrects for potential alignment bias owing to line-specific sequence variation. We then analyzed normalized expression values for endogenous genes, TEs, and microbial species.
Genetic variation in gene expression
We quantified expression levels of all RNA sequences that aligned to the reference genome in each DGRP line. After elimination of sequences with low expression, we found that 12,806 of 17,097 known D. melanogaster genes (75%) were expressed consistently in young adult males and/or females (Supplemental Table S2A). In addition, we identified 4282 NTRs (Supplemental Table S2B) that showed no overlap with exons on the same strand. A total of 3846 of the NTRs were located in introns; 290 were antisense to known genes, and 146 were intergenic. Most (95.6%) of the NTRs are ≥200 bp; the majority (4149 or 96.9%) lack protein coding potential (Supplemental Table S2C; Kang et al. 2017). These NTRs in total represent 5.61 Mb new transcribed mature RNA sequences that eluded prior annotation efforts. This increase is likely owing to the multiple genetic backgrounds profiled in this study. Although RNA-seq alignment and assembly alone are not sufficient to prove genuine transcriptional activities, our stringent expression-based filter was able to narrow down the NTRs to a subset that were similar to known genes in terms of mapping ambiguity (Supplemental Fig. S2) and expression in at least one Drosophila cell line (Supplemental Fig. S3).
Variation in gene expression among the DGRP lines may be confounded by variation in alignment rate to the reference strain owing to variation in DNA sequences between the DGRP lines and the reference. Indeed, 2735 genes (2117 known genes and 618 NTRs) were affected by alignment bias (Supplemental Table S2D). We corrected for alignment bias and partitioned variation in gene expression between males and females, DGRP lines, the sex by line interaction, and residual (environmental) terms (Supplemental Table S2D), using a false-discovery rate (FDR) of ≤0.05. Similar to previous studies (Ayroles et al. 2009; Massouras et al. 2012; Huang et al. 2015), we found that gene expression is sexually dimorphic: 98% (96%) of expressed known genes (NTRs) have a significant sex effect (Fig. 1A; Supplemental Table S2D). There is genetic variation in the magnitude of sex dimorphism: 69% (10%) of expressed known genes (NTRs) have a significant sex by line interaction (Supplemental Table S2D). Therefore, we assessed genetic variation in gene expression separately for males and females (Supplemental Table S2D,E) and found that 12,151 genes (10,354 known genes and 1797 NTRs) were genetically variable in females (Fig. 1B) and 13,819 genes (11,393 known genes and 2426 NTRs) were genetically variable in males (Fig. 1C). These numbers of genes with significant genetic variation are much higher than previously reported studies, which used microarrays (4308 in females and 5814 in males) rather than RNA-seq (Huang et al. 2015). Relative to tiling arrays, RNA-seq has a higher dynamic range and greater precision in quantifying gene expression, although the results from both analyses are positively correlated (Supplemental Fig. S4).
Figure 1.

Genetic variation of gene expression in the DGRP. (A) Sexual dimorphism of gene expression. Red indicates significant up-regulation in females; blue, in males. (B) Distribution of H2 estimates for annotated genes and NTRs in females. (C) Distribution of H2 estimates for annotated genes and NTRs in males. (D) WGCNA modules for annotated genes and NTRs in females. (E) WGCNA modules for annotated genes and NTRs in males. Heatmaps show the pairwise correlation of all genes in each module, sorted by average connectivity, with the most tightly connected module at the top left.
Broad sense heritabilities (proportion of phenotypic variance owing to genotype differences) ranged from H2 = 0.148–0.986 in females and H2 = 0.145–0.986 in males (Fig. 1B,C). A total of 472 (514) of the genetically variable genes in females (number for males in parenthesis) were located in molecularly defined heterochromatin (2LHet, 2RHet, 3LHet, 3RHet, XHet, and YHet) and Chromosome 4. Although there are 6.92× (5.52×) as many annotated genes relative to NTRs in euchromatic regions in females (males), there are 2.21 × (3.18×) as many NTRs in heterochromatin and Chromosome 4 in females (males) (Supplemental Table S2F). Thus, NTRs are highly enriched in heterochromatic regions.
We used weighted gene coexpression network analysis (WGCNA) (Langfelder and Horvath 2008) to assess the extent to which gene expression levels are genetically correlated in each sex (Fig. 1D,E; Supplemental Table S3). We found 13 (15) coexpression modules in females (males). We assessed the extent to which each module was significantly (FDR ≤ 0.05) enriched for Gene Ontology (GO) terms and pathway and protein domain annotations (Supplemental Table S3; Lyne et al. 2007). For example, female module 2 (149 genes) is enriched for GO terms involved in ovary function, and male module 6 (365 genes) is enriched for biological process GO terms involved in male reproduction. Female module 12 (88 genes) and male modules 13 (35 genes) and 14 (165 genes) are enriched for GO terms affecting small-molecule metabolism. Female modules 3 (26 genes), 6 (27 genes), and 7 (21 genes) and male modules 9 (42 genes) and 12 (44 genes) are enriched for GO terms affecting innate immunity, and female module 13 (560 genes) is enriched for GO terms affecting chemosensation.
Gene eQTLs
We performed genome-wide association (GWA) eQTL analyses for each of the genetically variable genes in each sex. We used approximately 1,932,427 common (minor allele frequency > 0.05) polymorphisms and accounted for effects of Wolbachia infection, polymorphic inversions, and polygenic relatedness on gene expression (Huang et al. 2014, 2015). We mapped 90,634 eQTLs in females and 147,412 eQTLs in males (FDR ≤ 0.05). A total of 2053 genes in females (1818 known genes and 235 NTRs) and 3178 genes in males (2790 known genes and 388 NTRs) were associated with at least one significant eQTL. We defined potentially cis- and trans-regulatory eQTLs as ≤1 kb and >1 kb of their respective gene bodies. We mapped putative cis-eQTLs to 1284 (2154) genes in females (males) and trans-eQTLs to 1653 (2521) genes in females (males), of which 902 (1305) were trans-eQTLs located on different chromosomal arms (Supplemental Table S4AB).
Because of correlation between genotypes at putative eQTL positions, some genes contained a large number of eQTLs that were not independent from each other. To develop a more parsimonious model, we used forward stepwise model selection to select putative eQTLs from the significant candidates, conservatively requiring that the last eQTL entering the model had a conditional P-value < 1 × 10−5. The models contained between one and seven eQTLs, with >60% of genes containing only one eQTL (Supplemental Table S4A,B). After model selection, we visualized the significant eQTLs by plotting the polymorphism positions on the x-axis and the gene positions on the y-axis such that the diagonal corresponds to cis-eQTLs and the off-diagonal to trans-eQTLs (Fig. 2). We found the majority of eQTLs retained by model selection to be in cis with the genes they controlled, although trans-eQTLs were not uncommon (Fig. 2).
Figure 2.
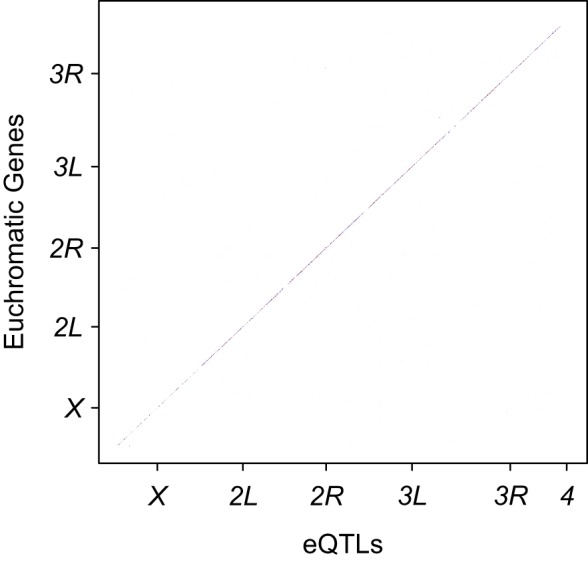
Genomic location of eQTLs for gene expression and genes they regulate. eQTL chromosome positions (bp) are given on the x-axis, and the genes with which they are associated on the y-axis. Red points denote female-specific eQTLs, blue indicates male-specific eQTLs, and black shows eQTLs shared by males and females.
eQTL regulatory networks
The existence of eQTLs that are cis-eQTL for gene X and also trans-eQTL for gene Y (Supplemental Table S5A,B) enables us to construct gene regulatory networks based on multifactorial variation in a natural population. Although significant putative eQTLs may not remain in the selected models, we still considered them when constructing regulatory networks because we could not genetically distinguish them and their associations with gene expression when all P-values were highly significant. We identified 408 (794) such regulatory interactions supported by at least one cis-trans-eQTL connecting 257 (471) regulatory genes (cis-end) to 251 (447) target genes (trans-end) in females (males) (Supplemental Table S5C,D). There are two or three large regulatory networks in each sex, as well as many smaller networks (Supplemental Figs. S5–S7). The regulatory genes are largely distinct between the two sexes, although many target genes are in common between males and females (Fig. 3; Supplemental Fig. S5; Supplemental Table S5E). Genes from the sex-specific regulatory networks or from the common networks are not enriched for any GO terms. It is not clear from their anatomical gene expression patterns how the sex specificity could arise, because the majority of these genes are expressed in multiple tissues, including the reproductive tissues of both sexes (Gramates et al. 2017).
Figure 3.
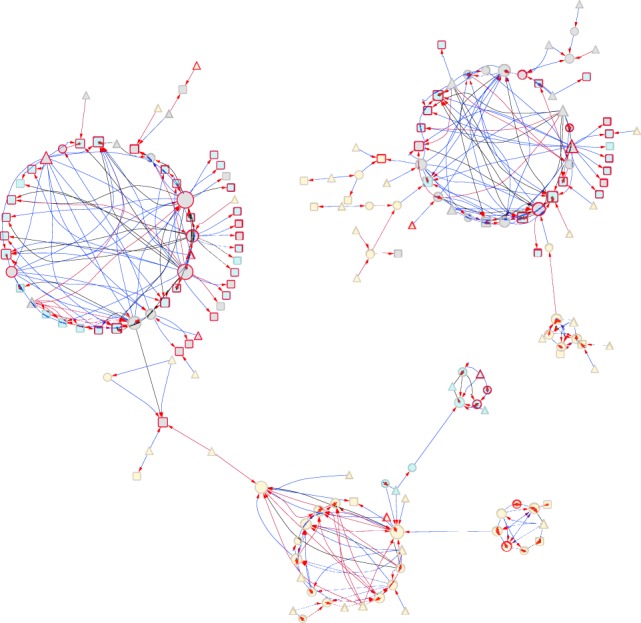
Large cis-trans-eQTL genetic network in females and males. Node interior colors indicate genomic location of genes: yellow, euchromatic regions with normal recombination; gray, euchromatic regions with reduced recombination; and blue, heterochomatin. Node border colors denote annotated gene (gray) or NTR (red). Node shape indicates whether a gene is a regulator and/or target: triangles, regulator only; squares, target only; and circles, both regulator and target. The node size indicates the number of node connections. Arrows on the edges point to the target. Edges are color-coded to show female-specific regulation (red), male-specific regulation (blue), and regulation common to both sexes (black).
Examination of cis- and trans-eQTLs (Supplemental Table S5) showed that there are more NTRs than expected among genes with cis-trans-eQTLs based on the total number of NTRs with eQTLs among the target genes ( 29.74, P = 4.95 × 10−8 in females; 60.54, P = 7.20 × 10−15 in males) but not the regulatory genes ( 1.54, P = 0.21 in females; 1.49, P = 0.22 in males). The regulatory genes tend to be located in pericentromeric regions of reduced recombination ( 17.28, P = 3.23 × 10−5 in females; 120.28, P < 2.2 × 10−16 in males) (Fiston-Lavier et al. 2010), and target gene locations are enriched for heterochromatin and pericentromeric regions of reduced recombination ( 28.53, P = 9.21 × 10−8 in females; 147.78, P < 2.2 × 10−16 in males). Regulatory genes with many target genes thus tend to have multiple cis-eQTLs in linkage disequilibrium (LD) near the centromere and regulate other NTRs both in heterochromatic regions across the genome and euchromatic regions on other chromosomes (Fig. 3; Supplemental Figs. S5–S7). The smaller networks with fewer regulators and targets tend to consist of genes in euchromatin in regions of normal recombination (Fig. 3; Supplemental Figs. S5–S7; Supplemental Table S5C,D). Regulatory genes often have many cis-eQTLs; a single cis-eQTL can regulate multiple target genes; and multiple cis-eQTLs (which may or may not be in LD) within a gene can regulate different target genes. It is possible that multiple cis-eQTLs in LD can be classified as a trans-eQTL for different target genes owing to differences in thresholding and ranking of eQTLs among the target genes. Each gene with at least one cis-eQTL may itself be regulated in trans by cis-eQTLs in one or more upstream genes, and the genes regulated by a focal cis-eQTL may themselves have cis-eQTLs regulating other genes.
Genetic variation in TE expression
A total of 9% of the D. melanogaster genome contains TEs spanning multiple families (Spradling and Rubin 1981). Active retrotransposon sequences are present in our RNA-seq libraries. We aligned reads to the Repbase database of known repetitive elements (Jurka et al. 2005), and quantified TE RNA levels based on normalized read counts. Overall, 1.3% of the RNA-seq reads align to Repbase. The most abundant families of TE sequences were gypsy, copia, BEL, jockey, and Mariner/Tc1 elements, but all TE families represented in Repbase were detected (Fig. 4A; Supplemental Table S6A).
Figure 4.

Genetic variation of TE expression in the DGRP. (A) Total signal for each TE family, summed over all individual transposon sequences and averaged across all DGRP lines, sex, and replicates. (B) Distribution of copy number independent H2 estimates for TE sequences in females. (C) Distribution of copy number independent H2 estimates for TE sequences in males. (D) WGCNA modules of TEs for females. (E) WGCNA modules of TEs for males. Heatmaps are depicted as in Figure 1. TE sequences not assigned to any module are included at the bottom right.
Line-specific differences in TE RNA levels can be driven by both differences in underlying copy number (Lee and Langley 2010) and differences in the rate of transcription per genomic copy. We quantified DNA copy variation for each TE sequence (Supplemental Table S6B) and used linear models to estimate the percentage of variation in TE expression that arises from differences in copy number (Supplemental Table S6C). We then partitioned the remaining copy number–independent variation in TE expression between sexes, DGRP lines, the line by sex interaction, and residual terms (Supplemental Table S6C), using FDR ≤ 0.05 as the significance threshold for each term in the analysis. Because the majority (153, 79%) of TEs had a significant sex by line interaction effect, we assessed genetic variation in TE expression for each transposon sequence separately for each sex (Supplemental Table S6D,E). We observed significant genetic variation in expression for 187 (97%) TE sequences in females (Fig. 4B) and 186 (96%) TE sequences in males (Fig. 4C). Broad sense heritabilities of TE expression ranged from H2 = 0.15–0.99 in females and H2 = 0.15–0.98 in males (Fig. 4B,C). Thus, there is host genetic control of expression for most D. melanogaster TEs.
We assessed whether different TE sequences had similar patterns of expression across the DGRP lines (Langfelder and Horvath 2008), separately for males and females (Fig. 4D,E; Supplemental Table S6F,G). We found minimal correlation structure in the activity scores of different TEs (Supplemental Table S6H), with the strongest correlations between pairs of TE sequences from the same family. This suggests that host genetic factors independently affect variation in expression of each TE family.
TE eQTLs
We mapped eQTLs for each of the TEs with genetically variable expression in females and males (Supplemental Table S7). We found 54 TEs with significant eQTLs (FDR ≤ 0.05): 36 in females and 39 in males. A total of 20 TE sequences were expressed in both males and females; 16 (18) TE sequences were expressed only in females (males). The number of eQTLs per TE sequence ranged from one to 1020 with, on average, more eQTL associations for TEs in males than females (Supplemental Table S7A–C). However, forward model selection retained between one and four eQTLs associated with TE activity, suggesting substantial LD among the eQTLs. Indeed, the large numbers of eQTLs associated with some TEs were located in LD blocks in pericentromeric regions and on the fourth chromosome (Supplemental Fig. S8; Supplemental Table S7D,E). Many eQTLs for TEs expressed in both males and females overlapped between the sexes, but typically additional eQTLs were present in males. Although there was little clustering of expression patterns of different TE sequences, 202 (1032) eQTLs were associated with two or more sequences in females (males) (Supplemental Table S7F,G).
Many eQTLs associated with TE expression were within 1 kb of annotated genes and NTRs. Indeed, 19.8% (17.7%) of TE eQTLs were within 1 kb of NTRs in females (males). Known genes near TE eQTLs were enriched (FDR < 0.05) for GO categories related to regulation of gene expression and protein binding (Supplemental Table S7H). We next asked to what extent eQTLs associated with gene expression were also associated with expression of TE sequences. We found 1206 eQTLs associated with 85 genes (37 known genes and 48 NTRs) and 23 TEs in females and found 3656 eQTLs associated with 166 genes (79 known genes and 87 NTRs) and 30 TEs in males (Supplemental Fig. S9; Supplemental Table S8). We could thus incorporate variation in TE expression into the cis-trans gene regulatory network via shared eQTLs (Fig. 5). These eQTLs are predominantly located in pericentromeric regions, and the genes they regulate are in pericentromeric regions as well as heterochromatin.
Figure 5.
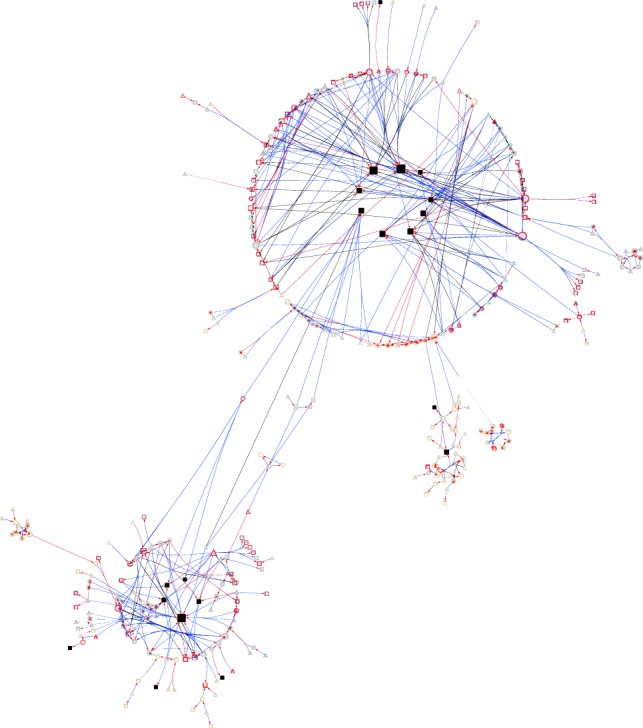
TE genetic regulatory network. Symbols and color-coding are as for Figure 3. Black squares denote TE sequences.
Genetic variation in microbiome composition
RNA samples extracted from pools of whole flies contain RNA from gut microbial communities, as well as from microbes on their exoskeleton. We assessed the contribution of microbial sequences to the RNA-seq libraries by aligning reads to a database of candidate microbial genomes (Supplemental Table S9). Wolbachia pipientis, a bacterial endosymbiont that infects ∼50% of the DGRP lines (Mackay et al. 2012), is the most abundant source of expressed sequence, followed by multiple Acetobacter species and genome assemblies (Fig. 6A; Supplemental Table S9). We estimated the total gene expression from each microbial species in all samples (Supplemental Table S10A) and partitioned variation in microbial gene expression between sexes, DGRP lines, the sex by line interaction, and residual terms, using FDR ≤ 0.05 as the significance threshold (Supplemental Table S10B). The H2 of W. pipientis abundance is extremely high (H2 = 0.972), as expected. We next assessed whether the sum of all non-Wolbachia microbial species is genetically variable after accounting for any Wolbachia effects, and estimated H2 = 0.595 (Fig. 6B; Supplemental Table S10B). The sex by line interaction for total microbial gene expression was not significant, indicating that total microbial RNA is highly correlated between males and females. We estimated the heritability of gene expression for the 122 non-Wolbachia microbial species and found that 84 microbial species had significant genetic variation in RNA abundance, with broad sense heritabilities ranging from H2 = 0.07–0.90 (Fig. 6C; Supplemental Table S10B). Microbial species that are likely to colonize the Drosophila gut (Acetobacter and Lactobacillus species) were among those with the highest H2.
Figure 6.

Genetic variation of microbiome composition. (A) The proportion of microbiome signal in RNA-seq libraries aligned to species in each genus or viral group. (B) Line means of total microbial signal (excluding Wolbachia). (C) Distribution of H2 estimates for individual microbe species. (D) WGCNA modules for microbial species. Heatmaps are depicted as in Figure 1. Species not assigned to any module are included at the bottom right.
We used WGCNA (Langfelder and Horvath 2008) to group species with similar abundance patterns based on the average of male and female line means (Fig. 6D; Supplemental Table S10C,D). We found three groups of strongly correlated species, consisting primarily of the gut-related microbes (Acetobacter and Lactobacillus species), and two additional clusters of microbes primarily consisting of viral and fungal species that are strongly anticorrelated with the abundances of species in the first three clusters. Thus, there is line-specific variation in the microbial communities living in and on DGRP flies. Species that most plausibly colonize the Drosophila gut are largely correlated across lines, with some fluctuation in the relative abundance of Acetobacter versus Lactobacillus species.
eQTLs for microbiome composition
There was little genetic variation in sexual dimorphism for microbial gene expression; therefore, we performed eQTL mapping using the average expression of males and females for each microbial species. Four microbial species and total microbial sequence expression were associated with significant eQTLs (FDR ≤ 0.05) (Supplemental Table S11A). The sum of all microbial species is associated with one eQTL that maps to an NTR; the expression of Borrelia coriaceae, Acidovorax temperans, and Podospora anserine map, respectively, to single eQTLs in CG2616 and CG46301 and to cic and an NTR, and Leuconostoc pseudomesenteroides expression maps to 39 variants in or near GC and nSyb (Supplemental Table S11A).
We lowered the significance threshold to P < 10−5 to explore the extent to which common eQTLs may control the expression of multiple microbial species that cluster together based on the WGCNA analysis (Fig. 6D). At this threshold, 1455 eQTLs are associated with 88 microbial species and the sum of all species (Supplemental Table S11B); 268 variants were associated with expression of more than one microbial species, and five eQTLs were associated with expression of 10 or more microbial species (Supplemental Table S11C). These data suggest that there is genetic variation in host control of microbial gene expression and that some variants have pleiotropic effects on multiple microbial species.
We assessed whether the genes to which the eQTLs associated with variation in microbial gene expression were enriched for GO categories (FDR ≤ 0.05). The most highly enriched biological process GO terms were related to development and morphogenesis, including development and function of the nervous system (Supplemental Table S11D).
Gene expression and complex traits
To examine the relationship between variation in gene expression and variation in organismal quantitative trait phenotypes, we chose 11 quantitative traits with published phenotypic data (chill coma recovery time and startle response (Mackay et al. 2012); starvation resistance (Huang et al. 2014); day and night sleep bout number, day and night total sleep duration, and total waking activity (Harbison et al. 2013); food consumption (Garlapow et al. 2015); male aggression (Shorter et al. 2015); phototaxis (Carbone et al. 2016)); and additionally measured five metabolic traits (levels of free glucose, glycogen, free glycerol, triglyceride, and protein) and three metrics of body size (body weight, thorax length, thorax width). All traits were quantified in the same laboratory under the same culture conditions used in this study. The line means for all traits are given in Supplemental Table S12; quantitative genetic analyses of the metabolic and body size traits are given in Supplemental Table S13; and the most significant associations (P < 10−5) from GWA analyses (separately for males and females) for these quantitative traits based on the 200 lines for which we have gene expression data are in Supplemental Table S14.
We first assessed whether variants associated with all organismal traits were enriched for eQTLs, as found in human studies (Chen et al. 2008; Emilsson et al. 2008; Cookson et al. 2009; Nicolae et al. 2010; Boyle et al. 2017). Of all the eQTLs (before model selection) and GWA study (GWAS) hits, only 26 in males and eight in females were common between eQTLs and GWAS hits, with clear patterns of clustering. We found no enrichment of cis-eQTLs (P = 0.13 in females and P = 0.71 in males), trans-eQTLs (P = 0.98 in females and P = 0.28 in males), or all eQTLs (P = 0.94 in females and P = 0.23 in males) among top GWA hits in either sex. Many top GWA hits as well as eQTLs map to regions >1 kb from any gene and may indicate novel regulatory regions. To exclude the possibility that the lack of overlap was owing to using different mapping procedures, we performed QTL mapping for the organismal traits using the same procedure as the eQTL mapping. At an empirical FDR = 0.05, we found four SNPs associated with three traits (chill coma recovery in females, day sleep duration, and free glucose level in males), and none was an eQTL.
We next performed transcriptome-wide association studies (TWASs) for individual genetically variable transcripts for gene expression, TE sequences, and microbial species for each of the 18 (19) genetically variable organismal phenotypes in females (males). We found several significant (Benjamini–Hochberg FDR < 0.05) associations of transcripts with organismal phenotypes (Supplemental Table S15). These associations include a known noncoding RNA (CR46032) with male aggression, two NTRs with male waking activity, Gbs-70E with free glucose in both sexes, AkhR with starvation resistance in males and females, and A. temperans with male aggression (Supplemental Table S15).
Discussion
Deep RNA sequencing gives accurate estimates of gene expression of annotated genes and can implicate novel noncoding RNAs and their regulatory interactions with annotated genes. We have identified 4282 NTRs, which are unlikely to be artifacts because the majority are genetically variable, and they are not randomly distributed in the genome but are preferentially located in heterochromatic regions and in pericentromeric euchromatin bordering heterochromatin. Thus, there is genetic variation in heterochromatic gene expression, thought to be largely transcriptionally silent (Riddle et al. 2011). These heterochromatic and pericentromeric NTRs are regulated by pericentromeric cis-eQTLs as well as trans-eQTLs dispersed throughout the euchromatic genome. Genes associated with eQTLs with both cis- and trans-effects form sex-specific networks of regulator and target genes, the largest of which is enriched for NTR target genes in heterochromatin and regulator and target genes in pericentromeric euchromatin. The considerable overlap between eQTLs associated with NTRs in the large networks and eQTLs associated with TE expression recruits TEs to the network. We do not know where the TE sequences with genetically variable expression are integrated in the genome; however, heterochromatin is composed of largely silenced TE repeats (Riddle et al. 2011), raising the possibility that TEs in heterochromatin are subject to the same regulation as other heterochromatic genes. Further work is needed to confirm the regulatory networks derived from naturally occurring genetic variation and determine the regulatory mechanism(s) through which the NTRs act. We speculate that many of the NTRs may be long noncoding RNAs, operationally defined as encoding transcripts >200 bp with no significant protein-coding potential, but further work is needed to establish whether this is true (Khalil et al. 2009; Wang et al. 2011; Hacisuleyman et al. 2014; Rogoyski et al. 2017; Wang et al. 2017; Ransohoff et al. 2018).
The first step in systems genetic analysis is to identify eQTLs associated with both gene expression and organismal quantitative traits, for which variation in gene expression is correlated with variation in the organismal phenotypes (Sieberts and Schadt 2007; Rockman 2008; Mackay et al. 2009). We did not find any such trios, although we did find interesting transcript–trait associations. This may be because our sample size is adequate to detect eQTLs but not QTLs affecting organismal traits, which have smaller effects; because eQTLs need to be mapped in tissues relevant to the organismal trait; and because there are nonlinear (epistatic) relationships between QTLs for both transcripts and organismal phenotypes. The complex and highly connected cis-trans regulatory networks suggest that higher-order interactions need to be accommodated in systems genetic modeling, at least at the level of gene expression.
Methods
Drosophila lines
We used 200 inbred, sequenced DGRP lines (Mackay et al. 2012; Huang et al. 2014) established by 20 generations of full sib inbreeding from gravid females collected at the Raleigh, North Carolina, Farmer's Market. Genome sequences of the lines were obtained previously using the Illumina platform with an average of coverage of 27×. A total of 4,565,215 molecular variants (3,976,011 single/multiple-nucleotide polymorphisms [SNPs/MNPs], 169,053 polymorphic insertions [relative to the reference genome], 293,363 polymorphic deletions, and 125,788 polymorphic microsatellites) segregate in the DGRP.
Sample collection
All lines were reared on cornmeal-molasses-agar medium at 25°C, 60%–75% relative humidity, and a 12-h light–dark cycle at equal larval densities. We collected two replicates of 25 females and 30 males per line for a total of 800 samples. We used a strict randomized experimental design for sample collection. We collected mated 3- to 5-d-old flies between 1:00 and 3:00 pm. We transferred the flies into empty culture vials and froze them over ice supplemented with liquid nitrogen and then sexed the frozen flies. The samples were transferred to 2.0-mL nuclease-free microcentrifuge tubes (Ambion) and stored at −80°C until ready to process.
RNA sequencing
Total RNA was extracted with QIAzol lysis reagent (Qiagen) and the quick-RNA MiniPrep Zymo research kit (Zymo Research). Ribosomal RNA (rRNA) was depleted from 5 µg of total RNA using the Ribo-Zero gold kit (Illumina). Depleted mRNA was fragmented and converted to first-strand cDNA using SuperScript III reverse transcriptase (Invitrogen). During the synthesis of second-strand cDNA, dUTP instead of dTTP was incorporated to label the second-strand cDNA. cDNA from each RNA sample was used to produce barcoded cDNA libraries using NEXTflex DNA barcodes (Bioo Scientific) with an Illumina TruSeq compatible protocol. Libraries were size-selected for 250 bp (insert size ∼130 bp) using Agencourt AMPure XP beads (Beckman Coulter). Second-strand DNA was digested with uracil-DNA glycosylase before amplification to produce directional cDNA libraries. Libraries were quantified using Qubit dsDNA HS kits (Thermo Fisher Scientific) and Bioanalyzer (Agilent Technologies) to calculate molarity. Libraries were then diluted to equal molarity and requantified. A total of 50 pools of 16 libraries were made, again randomly assigning samples to each pool. Pooled library samples were quantified again to calculate final molarity and then denatured and diluted to 14 pM. Pooled library samples were clustered on an Illumina cBot; each pool was sequenced on one lane of Illumina HiSeq 2500 using 125-bp single-read v4 chemistry.
RNA sequence analysis
Barcoded sequence reads were demultiplexed using the Illumina pipeline v1.9. Adapter sequences were trimmed using cutadapt v1.6 (Martin 2011), and trimmed sequences <50 bp were discarded from further analysis. Trimmed sequences were then aligned to multiple target sequence databases in the following order, using BWA v0.7.10 (MEM algorithm with parameters “-v 2 –t 4”) (Li and Durbin 2010): (1) All trimmed sequences were aligned against a database containing the complete 5S, 18S-5p8S-2S-28S, mt:lrRNA, and mt:srRNA sequences to filter out residual rRNA that escaped depletion during library preparation; (2) remaining sequences were then aligned against a custom database of potential microbiome component species (see below) using BWA; and (3) sequences that did not align to either the rRNA or microbiome databases were aligned to all D. melanogaster sequences in Repbase (Jurka et al. 2005). The remaining sequences that did not align to any of the databases above were then aligned to the D. melanogaster genome (BDGP5) and known transcriptome (FlyBase v5.57) using STAR v2.4.0e (Dobin et al. 2013). Libraries with fewer than 5 million reads uniquely aligned to the D. melanogaster reference genome were resequenced to achieve sufficient read depth.
Generation of microbiome database
We first performed a preliminary alignment of RNA-seq reads by filtering only rRNA sequences and then aligning directly to the D. melanogaster genome using the tools and parameters described above. Sequences that did not align to the rRNA database or D. melanogaster reference genome were then analyzed with Trinity v2.1.1 (Garbherr et al. 2011) to perform de novo assembly of longer sequences from the short reads. Assembled sequences >1 kb in length were then searched against the RefSeq_genomic database (downloaded from NCBI on 1/27/16) using BLAST. We then compiled a list of all RefSeq genomes that were found as a top BLAST hit for at least two assembled sequences. We compiled all FASTA files for each of these RefSeq genomes into a single database for alignment with BWA.
Genotype validation
To validate the DGRP line assigned to each RNA-seq sample, we identified SNPs from the RNA-seq reads that aligned to the D. melanogaster reference genome using STAR as described above. We retained only those SNP calls covered by at least three reads and at least 75% of all reads supporting the major genotype (note that DGRP lines are inbred, and therefore, the majority of SNPs are homozygous). This filtering process produced >400,000 usable SNPs per sample, primarily located in transcribed regions of the genome. We then performed two validation tests of the DGRP line assigned to each sample X by comparing to the previously published genotype calls for each DGRP line (Huang et al. 2014; http://dgrp2.gnets.ncsu.edu/data/website/dgrp2.tgeno). First, we computed the “line mean error” (LME) for each line as follows: given the set of homozygous SNPs from line Y that have sufficient coverage (described above) in sample X, LME(X,Y) = # of mismatching SNPs/total # of comparable SNPs. We confirmed that for each sample X, the DGRP line Ylab labeled for that samples produced the minimum value of LME(X,Y) compared with all other possible line assignments Yalt, and further confirmed that LME(X,Ylab) was <1%. Second, we performed competitive tests between the labeled line Ylab and each possible alternate line Yalt. Under this test, we considered only the SNPs that are homozygous for different genotypes in Ylab and Yalt (i.e., only the segregating SNPs for the two lines) and that have sufficient coverage in sample X. We then computed the “line error ratio” (LER) = # of SNPs matching Ylab/# of SNPs matching Yalt. We confirmed that for each sample X, the lowest LER for any Yalt was more than one (i.e., the majority of SNP calls always supported the labeled line compared with any alternative line).
Inference of novel transcripts
We constructed a de novo transcriptome for each individual sample by inputting the RNA-seq reads aligned to the D. melanogaster reference genome into Cufflinks v2.2.1 (Trapnell et al. 2012). We also considered the NTRs identified in a previous study based on unstranded pooled RNA sequencing of the DGRP lines (Huang et al. 2015). However, the previously published data do not provide strand-specific signal, whereas our current RNA-seq data use a strand-specific library preparation. Therefore, we reassigned the strand for each of the previously published NTRs that was supported by the greater number of total aligned reads across all samples. We then merged all de novo sample transcriptomes and the previously published NTRs using the cuffmerge tool included with Cufflinks v2.2.1 and then removed all merged transcript models with any exon overlapping on the same strand any exon in the known D. melanogaster transcriptome. We defined the known transcriptome here as all gene models in FlyBase v5.57 plus all subsequently added gene models in FlyBase v6.11 to account for recently discovered lncRNA sequences. Thus, the final output of this analysis was a set of NTRs constructed from both our current RNA-seq data and previously published pooled RNA-seq data that do not overlap any known gene exons on the same strand.
Gene expression estimation
Read counts for individual microbial species were computed as all reads aligning to any sequence in any genome for any strain of that species. Reads aligning to multiple species were ignored for individual species read counts. We also aligned microbiome-aligning reads to the D. melanogaster genome and removed all reads that aligned to both microbial and D. melanogaster sequences before computing read counts to account for several domains that are highly conserved between microbial and metazoan species. Read counts were computed for transposon sequences by computing the number of reads uniquely aligned to each transposon sequence in Repbase. Highly homologous sequences were grouped together for computing transposon read counts. Read counts were computed for known and novel gene models using HTSeq-count (Anders et al. 2015) with the “intersection-nonempty” assignment method for exonic reads only. Tabulated read counts for each expression feature type (microbiome, transposon, endogenous genes) were then normalized across all samples using edgeR (Robinson et al. 2010) as follows. First, genes with low expression overall (fewer than 10 aligned reads in >75% of the libraries) were excluded from the analysis. Library sizes were recomputed as the sum of reads assigned to the remaining genes and further normalized using the trimmed mean of M-values (TMM) method (Robinson and Oshlack 2010). At this point, we retained only genes (known or novel) whose expression in both biological replicates was above an empirical threshold in more than 200 line-sex combinations (400 samples total). This criterion retains genes expressed in only one sex. The threshold was determined by fitting all log2 transformed FPKM expression data points using a two-component Gaussian mixture model and by finding the expression value (FPKM = 0.280263) where the posterior probability of being in the lower expression component is 0.95. Genes on Chr U and Chr Uextra were also removed. We further adjusted transposon expression estimates to account for differences in transposon copy number across lines by fitting a linear model: RNA ∼ DNA + ε, where RNA = the normalized log2 (RNA-seq read count), and DNA = normalized log2 (DNA read count) derived from the previously published DNA-seq data for each DGRP line (Huang et al. 2014). After fitting the linear model for each transposon sequence, ε estimates the relative transcription rate in each line independent of copy number and was used as the adjusted transposon expression for all subsequent analysis. We further adjusted endogenous gene expression values by estimating and removing the effect of alignment bias resulting from higher rates of nonreference variants clustering in some lines. We computed the alignment bias score A(g,L) defined as the number of nonreference nucleotides per kilobase in all exons of gene g in DGRP line L, based on the previous map of genomic variation in the DGRP (Huang et al. 2014). We then fit a linear model for each endogenous gene: Y = A + ε, where Y is the normalized expression profile for gene g after the read counting and edgeR normalization described above. After fitting these linear models, ε represents the alignment bias–corrected expression and was used as the normalized gene expression in all subsequent analysis. Read mapping ambiguity could affect the confidence in defining NTRs. We assessed this by using BLAT (https://genome.ucsc.edu/FAQ/FAQblat.html) to map all RNA transcripts to the fly genome and identified all possible alignments. We used a metric (ΔBitscore) to characterize the mapping ambiguity of the full-length RNA transcripts for known genes, NTRs filtering for low expression across the DGRP, and NTRs retained after filtering. ΔBitscore is defined as the difference between the bit score for the best alignment and the second best one. The greater the ΔBitscore, the less ambiguous is the alignment. In addition, we assessed whether the NTRs identified in the DGRP were present in an independent data set of 41 Drosophila cell lines that were either untreated or treated with the hormone ecdysone (Stoiber et al. 2016). We computed the median and maximum expression across all cell lines for each transcript using kallisto (Bray et al. 2016), an alignment-free abundance estimator, and calculated the median and maximum expression RNA transcripts for known genes, NTRs filtered for low expression across the DGRP, and NTRs retained after filtering.
Genetics of gene expression
For each class of expression features (endogenous genes, transposons, microbiome), we fit mixed-effect models to the gene expression data corresponding to Y = S + W + W × S + L + L × S + ε, where Y is the observed log2 (normalized read count), S is sex, W is Wolbachia infection status, W × S is Wolbachia by sex interaction, L is DGRP line, L × S is the line by sex interaction, and ε is the residual error. We also performed reduced analyses (Y = W + L + ε) independently for males and females. We identified genetically variable transcripts as those that passed a 5% FDR threshold (based on Benjamini and Hochberg [1995] corrected P-values) for the L and/or L × S terms. We computed the broad sense heritabilities (H2) for each gene expression trait separately for males and females as , where and are, respectively, the among line and within line variance components.
Clustering by genetic correlation
For each feature type (microbiome, transposons, endogenous genes), we clustered line means using the WGCNA R package v1.51 (Langfelder and Horvath 2008) as follows. Only genes with sufficient average expression (log2 FPKM > 0) and genetic variance (line mean variance >0.05) were considered in these analyses. First, the Pearson correlation coefficient for every pair of lines means that the soft-power threshold was computed using the pickSoftThreshold function and used to convert the correlation matrix to an adjacency matrix with approximately scale-free connectivity. The adjacency matrix was then converted to a dissimilarity matrix based on the topological overlap map (Langfelder and Horvath 2008). Expression features were then clustered using hierarchical clustering (hclust function) based on the dissimilarity matrix and split into distinct modules using the cutreeDynamic with deepSplit = 4 and minClusterSize = 20 (for endogenous gene expression, minClusterSize = 4 was used for microbiome and transposon clustering). Module eigengenes were computed for each cluster, and highly similar clusters were combined using the mergeCloseModules function with cutHeight = 0.25. Expression features assigned to module 0 (insufficient similarity) were discarded. Modules consisting of more than 1000 features were also discarded as insufficiently split into distinct modules. For each expression feature, the degree was computed as the overage topological overlap with all other features assigned to the same module. The average degree of each module was computed as the mean degree across all features in the module. Modules were sorted by average degree, such that module 1 has the highest average degree in each analysis.
Gene set enrichment analyses
Lists of known gene IDs (FlyBase FBgn accessions) were uploaded to FlyMine (Lyne et al. 2007) or Panther (Mi et al. 2017) for functional enrichment. For analysis of gene lists from WGCNA clusters, the list of known genes input to WGCNA was used as the background set to correct for any biases inherent to highly heritable expression patterns in general.
eQTL mapping
For each gene expression feature, we performed eQTL analysis as previously described (Huang et al. 2015). Briefly, we adjusted mean expression values in each sex for fixed effects of Wolbachia infection status, five major polymorphic inversions (In2L(t), In2R(NS), In3R(P), In3R(K), In3R(Mo)), and the first 10 principal components of the genetic relatedness matrix of all DGRP lines using a linear model. We mapped QTLs for the adjusted line means using PLINK (Purcell et al. 2007) against 1,932,427 SNPs with major allele frequency >0.05 and missing genotypes in <25% of the 200 DGRP lines profiled by RNA-seq. Instead of controlling for experiment-wise type I error rate, which can be overly conservative, we controlled for the FDR (Benjamani and Hochberg 1995). We computed FDR of eQTL calls by comparing observed eQTL P-value distributions to those obtained from running PLINK on 100 permutations of the observed line means for each expression feature. At any given P-value cut-off X, the estimated false-positive rate of eQTLs for a specific gene expression feature is the average number of eQTLs with P-value < X across all permutations. The FDR at the same P-value is then computed as the estimated false-positive rate divided by the number of eQTLs with P-value < X in the observed data. By using this formulation of FDR, we identified the unadjusted P-value cut-off corresponding to 5% FDR for each gene expression feature. No further model selection was performed; however, we classified eQTLs as being either cis-eQTLs (within 1 kb of the gene body for the associated expression feature) or trans-eQTLs (>1 kb of the gene body). To eliminate eQTLs whose genotypes are correlated with each other and cannot be genetically distinguished, we used forward model selection to iteratively add eQTLs to the model in the order of their conditional association (Huang et al. 2015). The model selection was stopped when none of the remaining putative eQTLs can enter the model with P < 0.00001. When two putative eQTLs had equal P-values, the one closer to the transcription start site was added.
Construction of eQTL networks
We then constructed regulatory eQTL networks based on individual SNPs that were called as both cis- and trans-eQTLs for multiple expression features. Specifically, we assign a directed edge X→Y if there is at least one variant that is both a cis-eQTL for gene X (defined as within 1 kb of gene X) and a trans-eQTL for gene Y at 5% FDR. We then broke all loops in the regulatory network for each sex by dropping the edge in each loop with the highest minimum P-value from all associated SNPs to create a directed, acyclic network.
Quantitative traits
We retrieved phenotypic data documented from previous publications on the same fly lines for male aggression (Shorter et al. 2015), chill coma recovery time and startle response (Mackay et al. 2012), food consumption (Garlapow et al. 2015), phototaxis (Carbone et al. 2016), sleep traits (day and night bout number, day and night total sleep duration, total waking activity) (Harbison et al. 2013), and starvation resistance (Huang et al. 2014).
To measure body weight and size, we collected 10 replicates of 10 flies per line and sex into preweighed 1.7-mL tubes and weighed and flash-froze them for downstream analyses. Virgin flies were used to avoid body weight variation owing to variation in egg production. In addition, we measured thorax length and thorax width as metrics for body size.
Frozen flies were homogenized in 250 μL Dulbecco's Phosphate Buffered Saline, and after gentle centrifugation, supernatants were collected for measurements of free glucose, glycogen, free glycerol, triglyceride, and total protein (further diluted 10-fold). For free glucose and glycogen, samples were denatured for 25 min at 95°C to prevent glycogenolysis. Measurements were performed following protocols provided by the glycogen colorimetric/fluorometric assay kit (BioVision). For free glycerol and triglyceride, we used the serum triglyceride determination kit (Sigma-Aldrich) and incubated samples with the triglyceride reagent for 1 h at 37°C. For total protein measurement, we used the Qubit protein assay kit (Thermo Fisher Scientific).
Quantitative trait genetic parameters
We used mixed-model, factorial ANOVAs (Y = S + L + L × S + Rep(L) + S × Rep(L) + ε) to partition variation of the quantitative traits between sexes (S), DGRP lines (L), and replicate vials within lines (Rep). Broad sense heritabilities were estimated as , where , and are, respectively, the among line, sex by line, and within line variance components.
eQTL-GWA enrichment analysis
We performed GWA analyses for all quantitative traits separately for females and males. All phenotypes (line means) were first adjusted for the effect of Wolbachia infection and major polymorphic inversions using a linear model. The residuals (plus the intercept) from this analysis were then used as phenotype in a linear mixed model to test for the effect of each common variant individually, while adjusting for sample structure using a genomic relationship matrix (GRM), as implemented in GCTA-MLMA (Yang et al. 2011). The GRM was built as WW′/p, where W is a matrix of centered and scaled genotypes for the 200 lines, and p is the total number of genetic variants. Similarly, we have also mapped trait QTLs using the same procedure as the eQTL mapping described above by deriving empirical FDR based on 100 permutations of phenotypes.
For each trait and sex, variants with P < 10−5 were retained for downstream analysis. We then combined the lists of variants associated with each trait, separately for females and males, to obtain a single list of unique variants (i.e., no duplicates) associated with any of the traits of interest. The enrichment analysis proceeded as described by Nicolae et al. (2010) within each sex. Briefly, GWAS hits were divided into minor allele frequency bins of width equal to 0.05. Then, an equal number of common variants (which may or may not have included actual GWAS hits) per bin were sampled at random, and the overlap with eQTLs was calculated. This procedure was repeated 10,000 times, and an empirical P-value for the enrichment was calculated as the number of replicates where the overlap between randomly sampled variants and eQTLs was greater than or equal to the observed overlap between GWAS hits and eQTLs over the total number of replicates.
Association of expression and quantitative traits
A TWAS, namely, regressing the phenotype on each gene's expression level, was performed for each sex separately for each quantitative trait. We developed a method that accounts for structure present in the transcriptome due correlations between transcripts. This was achieved by fitting a linear mixed model of the type: y = 1μ + wβ + t + e, where y = n-vector of mean phenotypic values for n lines, μ = fixed population mean effect, w= n-vector of the tested gene's centered and scaled expression level, β = fixed effect of the gene, t= n-vector of random transcriptomic line effect, and e= n-vector of random error.
The key term in the model that accounts for sample structure is T, the transcriptomic relationship matrix (TRM). The TRM was computed as , where W− is a matrix of centered and scaled gene expression levels for the 200 lines, excluding the gene tested to maximize the power to find an association (Yang et al. 2014), and p is the total number of genes. The TRM in TWAS has similar role to the GRM in GWAS.
The effect of each gene's expression level on the phenotype was tested using a Wald test of the form . Raw P-values and Benjamini and Hochberg (1995) FDR-corrected P-values were computed.
The phenotypes were adjusted for the effects of Wolbachia and major polymorphic inversions as described in the previous section. Because the phenotypes were adjusted, we did not adjust gene expression in this analysis to avoid spurious associations owing to adjustment on both sides of the equation.
We also performed similar associations of quantitative traits with TEs and microbial gene expression, using the same models as for TWAS but substituting TE and microbial expression for gene expression levels. Quantitative trait phenotypes were adjusted for the effects of Wolbachia and major polymorphic inversions but the TE and microbial expression data were not. The TE analysis was performed for males and females separately, whereas sex-pooled microbe expression data were used with female or male quantitative trait phenotypes because microbial gene expression was not sex specific.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE117850. The DGRP lines are available from the Bloomington Drosophila Stock Center (Bloomington, IN). All analysis codes are available in Supplemental Codes and on GitHub (https://github.com/qgg-lab/dgrp-rna-seq/).
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants R01 AA016560, R01 AG043490, and U01 DA041613 to T.F.C.M. and R.R.H.A. and Genomic Selection in Animals and Plants (GenSAP) funded by The Danish Council for Strategic Research to T.F.C.M. and F.M.
Author contributions: L.J.E., W.H., S.Z., F.M., R.R.H.A., and T.F.C.M. wrote the manuscript. L.J.E., W.H., S.Z., F.M., and T.F.C.M. analyzed the data. M.A.C., R.F.L., G.H.A., M.S.G., J.M., G.S.A., and L.T. performed the research.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.257592.119.
Freely available online through the Genome Research Open Access option.
References
- Albert FW, Kruglyak L. 2015. The role of regulatory variation in complex traits and disease. Nat Rev Genet 16: 197–212. 10.1038/nrg3891 [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayroles JF, Carbone MA, Stone EA, Jordan KW, Lyman RF, Magwire MM, Rollmann SM, Duncan LH, Lawrence F, Anholt RR, et al. 2009. Systems genetics of complex traits in Drosophila melanogaster. Nat Genet 41: 299–307. 10.1038/ng.332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B (Methodological) 57: 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Boyle EA, Li YI, Pritchard JK. 2017. An expanded view of complex traits: from polygenic to omnigenic. Cell 169: 1177–1186. 10.1016/j.cell.2017.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34: 525–527. 10.1038/nbt.3519 [DOI] [PubMed] [Google Scholar]
- Bryois J, Buil A, Evans DM, Kemp JP, Montgomery SB, Conrad DF, Ho KM, Ring S, Hurles M, Deloukas P, et al. 2014. Cis and trans effects of human genomic variants on gene expression. PLoS Genet 10: e1004461 10.1371/journal.pgen.1004461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone MA, Yamamoto A, Huang W, Lyman RA, Meadors TB, Yamamoto R, Anholt RR, Mackay TFC. 2016. Genetic architecture of natural variation in visual senescence in Drosophila. Proc Natl Acad Sci 113: E6620–E6629. 10.1073/pnas.1613833113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhu J, Lum PY, Yang X, Pinto S, MacNeil DJ, Zhang C, Lamb J, Edwards S, Sieberts SK, et al. 2008. Variations in DNA elucidate molecular networks that cause disease. Nature 452: 429–435. 10.1038/nature06757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelek M, Lusis AJ. 2014. Systems genetics approaches to understand complex traits. Nat Rev Genet 15: 34–48. 10.1038/nrg3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. 2009. Mapping complex disease traits with global gene expression. Nat Rev Genet 10: 184–194. 10.1038/nrg2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, et al. 2008. Genetics of gene expression and its effect on disease. Nature 452: 423–428. 10.1038/nature06758 [DOI] [PubMed] [Google Scholar]
- Fagny M, Paulson JN, Kuijjer ML, Sonawane AR, Chen CY, Lopes-Ramos CM, Glass K, Quackenbush J, Platig J. 2017. Exploring regulation in tissues with eQTL networks. Proc Natl Acad Sci 114: E7841–E7850. 10.1073/pnas.1707375114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiston-Lavier AS, Singh ND, Lipatov M, Petrov DA. 2010. Drosophila melanogaster recombination rate calculator. Gene 463: 18–20. 10.1016/j.gene.2010.04.015 [DOI] [PubMed] [Google Scholar]
- Garbherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng D, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29: 644–652. 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlapow ME, Huang W, Yarboro MT, Peterson KR, Mackay TFC. 2015. Quantitative genetics of food intake in Drosophila melanogaster. PLoS One 10: e0138129 10.1371/journal.pone.0138129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G, Powell JE, Marigorta UM. 2015. Expression quantitative trait locus analysis for translational medicine. Genome Med 7: 60 10.1186/s13073-015-0186-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramates LS, Marygold SJ, Santos GD, Urbano JM, Antonazzo G, Matthews BB, Rey AJ, Tabone CJ, Crosby MA, Emmert DB, et al. 2017. FlyBase at 25: looking to the future. Nucleic Acids Res 45: D663–D671. 10.1093/nar/gkw1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacisuleyman E, Goff LA, Trapnell C, Williams A, Henao-Mejia J, Sun L, McClanahan P, Hendrickson DG, Sauvageau M, Kelley DR, et al. 2014. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat Struct Mol Biol 21: 198–206. 10.1038/nsmb.2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison ST, McCoy LJ, Mackay TFC. 2013. Genome-wide association study of sleep in Drosophila melanogaster. BMC Genomics 14: 281 10.1186/1471-2164-14-281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Massouras A, Inoue Y, Peiffer J, Ràmia M, Tarone AM, Turlapati L, Zichner T, Zhu D, Lyman RF, et al. 2014. Natural variation in genome architecture among 205 Drosophila melanogaster Genetic Reference Panel lines. Genome Res 24: 1193–1208. 10.1101/gr.171546.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Carbone MA, Magwire MM, Peiffer JA, Lyman RF, Stone EA, Anholt RR, Mackay TFC. 2015. Genetic basis of transcriptome diversity in Drosophila melanogaster. Proc Natl Acad Sci 112: E6010–E6019. 10.1073/pnas.1519159112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. 2005. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110: 462–467. 10.1159/000084979 [DOI] [PubMed] [Google Scholar]
- Kang YJ, Yang DC, Kong L, Hou M, Meng YQ, Wei L, Gao G. 2017. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res 3: 45 10.1093/nar/gkx428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, et al. 2009. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci 106: 11667–11672. 10.1073/pnas.0904715106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9: 559 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YC, Langley CH. 2010. Transposable elements in natural populations of Drosophila melanogaster. Phil Trans Roy Soc B 365: 1219–1228. 10.1098/rstb.2009.0318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26: 589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, de la Fuente A, Hoeschele I. 2008. Gene network inference via structural equation modeling in genetical genomics experiments. Genetics 178: 1763–1776. 10.1534/genetics.107.080069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyne R, Smith R, Rutherford K, Wakeling M, Varley A, Guillier F, Janssens H, Ji W, Mclaren P, North P, et al. 2007. FlyMine: an integrated database for Drosophila and Anopheles genomics. Genome Biol 8: R129 10.1186/gb-2007-8-7-r129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TFC, Huang W. 2018. Charting the genotype-phenotype map: lessons from the Drosophila melanogaster Genetic Reference Panel. Wiley Interdiscip Rev Dev Bio 7 10.1002/wdev.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TFC, Stone EA, Ayroles JF. 2009. The genetics of quantitative traits: challenges and prospects. Nat Rev Genet 10: 565–577. 10.1038/nrg2612 [DOI] [PubMed] [Google Scholar]
- Mackay TFC, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, Casillas S, Han Y, Magwire MM, Cridland JM, et al. 2012. The Drosophila melanogaster Genetic Reference Panel. Nature 482: 173–178. 10.1038/nature10811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. 2009. Finding the missing heritability of complex diseases. Nature 461: 747–753. 10.1038/nature08494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17: 10–12. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Massouras A, Waszak SM, Albarca-Aguilera M, Hens K, Holcombe W, Ayroles JF, Dermitzakis ET, Stone EA, Jensen JD, Mackay TFC, et al. 2012. Genomic variation and its impact on gene expression in Drosophila melanogaster. PLoS Genet 8: e1003055 10.1371/journal.pgen.1003055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. 2017. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res 45: D183–D189. 10.1093/nar/gkw1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. 2010. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 6: e1000888 10.1371/journal.pgen.1000888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura T, Busch W. 2016. Genotypes, networks, phenotypes: moving toward plant systems genetics. Annu Rev Cell Dev Biol 32: 103–126. 10.1146/annurev-cellbio-111315-124922 [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575. 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff JD, Wei Y, Khavari PA. 2018. The functions and unique features of long intergenic non-coding RNA. Nat Rev Mol Cell Biol 19: 143–157. 10.1038/nrm.2017.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle NC, Minoda A, Kharchenko PV, Alekseyenko AA, Schwartz YB, Tolstorukov MY, Gorchakov AA, Jaffe JD, Kennedy C, Linder-Basso D, et al. 2011. Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin. Genome Res 21: 147–163. 10.1101/gr.110098.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A. 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11: R25 10.1186/gb-2010-11-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV. 2008. Reverse engineering the genotype–phenotype map with natural genetic variation. Nature 456: 738–744. 10.1038/nature07633 [DOI] [PubMed] [Google Scholar]
- Rogoyski OM, Pueyo JI, Couso JP, Newbury SF. 2017. Functions of long non-coding RNAs in human disease and their conservation in Drosophila development. Biochem Soc Trans 45: 895–904. 10.1042/BST20160428 [DOI] [PubMed] [Google Scholar]
- Schughart K, Williams RW. 2017. Systems genetics methods and protocols. Humana Press, New York. [Google Scholar]
- Shorter J, Couch C, Huang W, Carbone MA, Peiffer J, Anholt RR, Mackay TFC. 2015. Genetic architecture of natural variation in Drosophila melanogaster aggressive behavior. Proc Natl Acad Sci 112: E3555–E3563. 10.1073/pnas.1510104112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieberts SK, Schadt EE. 2007. Moving toward a system genetics view of disease. Mamm Genome 18: 389–401. 10.1007/s00335-007-9040-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC, Rubin GM. 1981. Drosophila genome organization: conserved and dynamic aspects. Annu Rev Genet 15: 219–264. 10.1146/annurev.ge.15.120181.001251 [DOI] [PubMed] [Google Scholar]
- Stoiber M, Celniker S, Cherbas L, Brown B, Cherbas P. 2016. Diverse hormone response networks in 41 independent Drosophila cell lines. G3 (Bethesda) 6: 683–694. 10.1534/g3.115.023366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher PM, Brown MA, McCarthy MI, Yang J. 2012. Five years of GWAS discovery. Am J Hum Genet 90: 7–24. 10.1016/j.ajhg.2011.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, et al. 2011. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472: 120–124. 10.1038/nature09819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Samuels DC, Zhao S, Xiang YY, Zhao Y-Y, Guo Y. 2017. Current research on non-coding ribonucleic acid (RNA). Genes (Basel) 8: 366 10.3390/genes8120366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM. 2011. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88: 76–82. 10.1016/j.ajhg.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zaitlen NA, Goddard ME, Visscher PM, Price AL. 2014. Advantages and pitfalls in the application of mixed-model association methods. Nat Genet 46: 100–106. 10.1038/ng.2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Morgante F, Geisz MS, Ma J, Anholt RRA, Mackay TFC. 2020. Systems genetics of the Drosophila metabolome. Genome Res (this issue). 10.1101/gr.243030.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.