

Abstract
Viruses are the most abundant biological entities on Earth and play key roles in host ecology, evolution, and horizontal gene transfer. Despite recent progress in viral metagenomics, the inherent genetic complexity of virus populations still poses technical difficulties for recovering complete virus genomes from natural assemblages. To address these challenges, we developed an assembly-free, single-molecule nanopore sequencing approach, enabling direct recovery of complete virus genome sequences from environmental samples. Our method yielded thousands of full-length, high-quality draft virus genome sequences that were not recovered using standard short-read assembly approaches. Additionally, our analyses discriminated between populations whose genomes had identical direct terminal repeats versus those with circularly permuted repeats at their termini, thus providing new insight into native virus reproduction and genome packaging. Novel DNA sequences were discovered, whose repeat structures, gene contents, and concatemer lengths suggest they are phage-inducible chromosomal islands, which are packaged as concatemers in phage particles, with lengths that match the size ranges of co-occurring phage genomes. Our new virus sequencing strategy can provide previously unavailable information about the genome structures, population biology, and ecology of naturally occurring viruses and viral parasites.
Viruses impact the ecology and evolution of virtually all cellular lifeforms on Earth. Because of their centrality in early molecular genetic studies and their small genome size, viruses were the first biological entities whose genomes were fully sequenced (Fiers et al. 1976; Sanger et al. 1977). More recently, microbial community shotgun genome sequencing (metagenomics) has advanced understanding of virus populations in the wild (Breitbart et al. 2002; Hurwitz et al. 2013; Sullivan 2015). Metagenomic studies to date have uncovered thousands of novel viral taxa, genes, and genomes from the oceans (Mizuno et al. 2013a; Brum et al. 2015; Roux et al. 2016b, 2019; Aylward et al. 2017; Luo et al. 2017).
However, the ecological richness, evenness, and genomic complexity of viral assemblages complicate the determination of full-length virus genome sequences from naturally occurring viral populations. Most previous viral metagenomic studies have relied primarily on one of three main sequencing strategies: (1) metagenomic short-read shotgun sequencing and assembly (Breitbart et al. 2002; Hurwitz et al. 2013; Sullivan 2015); (2) fosmid-based large DNA insert shotgun cloning followed by short-read sequencing and assembly (Mizuno et al. 2013a); and (3) amplification-based shotgun sequencing approaches, including techniques involving single-cell or single-virus multiple displacement amplification (Roux et al. 2014; Martinez-Hernandez et al. 2017), or PCR-based linker ligation amplification methods (Hurwitz et al. 2013; Warwick-Dugdale et al. 2019). Each approach has its own unique limitations. Challenges include amplification biases, ambiguities associated with De Bruijn graph short-read assemblies, and the limited DNA insert size range and cloning biases associated with fosmids. Because of these difficulties, obtaining whole-virus genome sequences from complex naturally occurring populations remains a challenge.
Given the typical size range of double-stranded DNA bacteriophages (∼3–300 kb), we reasoned that the determination of entire virus genome sequences from single reads should be possible using single-molecule sequencing approaches. Here, we describe the development of a method for obtaining high-quality, assembly-free virus genomes (AFVGs) from naturally occurring populations using single-molecule sequences spanning entire virus genomes. The method requires no amplification or de novo short-read assembly and so avoids the most common biases inherent in previous approaches. By using bioinformatic and experimental approaches, we validated the recovery of full-length, high-quality draft virus genome sequences from naturally occurring virus populations.
Results
Identification of marine virus genomes from metagenomic nanopore reads
An assembly-free phage discovery pipeline was developed to isolate and polish full-length phage genomes from nanopore reads (Fig. 1). Because direct terminal repeats (DTRs) that flank virus genome termini are characteristic of most dsDNA tailed phages (Casjens and Gilcrease 2009), we leveraged this feature to identify single reads comprising entire dsDNA tailed-phage genomes. Nanopore reads were first filtered so that only sequence reads containing DTRs were retained for downstream processing (Table 1). Briefly, the pipeline transformed each read to a vector of 5-mer counts and then used dimensionality reduction and clustering tools to embed these 5-mer count vectors into two dimensions. Read bins were then called and refined based on pairwise read alignments, and a single read from each refined cluster was polished using the other nanopore reads from the same cluster (see Methods). Finally, nanopore-polished genomes were polished using short reads sequenced from the same sample (Supplemental Methods).
Figure 1.
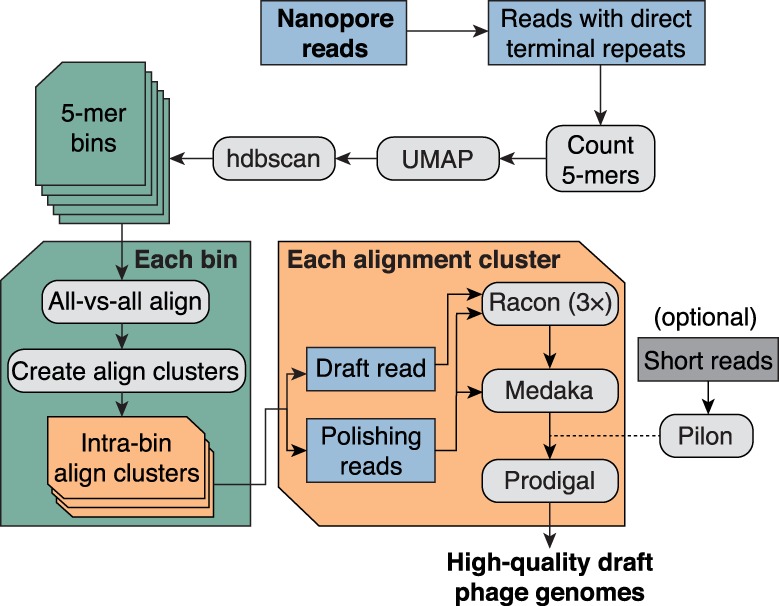
Bioinformatic pipeline for assembly-free discovery of marine phage genomes in nanopore sequences. Nanopore reads from each depth sample were first filtered to identify reads containing a direct terminal repeat (DTR). Read sequences were then decomposed into vectors containing their 5-mer counts, and all read vectors were embedded in a two-dimensional (2D) map using the dimensionality reduction tool UMAP (McInnes et al. 2018). Bins were called from the embedding using hdbscan (McInnes et al. 2017). Each 5-mer bin was refined by using minimap2 (Li 2018) to generate all-versus-all alignments of binned reads. Each pairwise alignment within each bin was assigned a score based on alignment length and sequence identity, and then these scores were hierarchically clustered to form one or more refined alignment clusters per 5-mer bin. Finally, a representative read was selected from each alignment cluster and polished by the remaining reads in the cluster using a combination of consensus polishing tools. Optionally, sample-matched short reads can be used to perform one last polishing round of the nanopore-only polished consensus sequence. Each polished draft genome was subsequently annotated for protein-coding sequences using Prodigal (Hyatt et al. 2010).
Table 1.
Sequencing, binning, and polished genome statistics for virus-enriched seawater samples obtained from three different depths

We first validated the phage discovery pipeline on a set of 192 known marine phage genomes from the uvMED collection (Mizuno et al. 2013a,b). Fifty simulated nanopore reads were generated for each uvMED genome and then combined to form a mock viral metagenome (see Methods). The phage discovery pipeline resulted in 183 5-mer bins (Supplemental Fig. S1) that were refined into 190 clusters based on all-versus-all read alignments within each 5-mer bin (Table 1). After polishing a representative read from each refined alignment cluster, 190/190 of polished draft genomes shared ≥99.67% accuracy and ≥99.86% coverage with their original uvMED reference genomes. Among these 190 polished sequences were two closely related genomes, AP013491 and AP013492, whose references share an overall 97.1% average nucleotide identity (ANI) and are distinguished by several small regions of sequence divergence and multiple small insertions and deletions totaling <1.5 kb. We successfully preserved these strain-level differences in the polished genomes produced from the pipeline (Supplemental Fig. S2).
The only two reference genomes that could not be recapitulated from the mock community were nearly identical to two of the polished genomes except for minor circular permutations (Supplemental Results). When only 10 nanopore reads were simulated from each of the 192 uvMED references, the overall sensitivity of the phage discovery pipeline decreased while specificity remained high: 115/117 of the polished genomes produced by the pipeline represented their original reference genome at ≥99.41% accuracy and ≥99.97% coverage.
We next applied the phage discovery pipeline to three virus-enriched seawater samples recovered from depths of 25 m, 117 m, and 250 m (Supplemental Methods). We identified 16,000–130,000 DTR-containing, putative full-length dsDNA tailed-phage reads in each sample, which were predominantly 20–90 kb in length (Supplemental Fig. S3). The assembly-free strategy facilitated identification and polishing of genomes containing complex repeat structures that can be problematic for short-read metagenomic assemblies. For example, a 4.2-kb complex repeat structure in genome AFVG_250M480 was easily resolved by selecting one 40.4-kb phage read from an alignment cluster and polishing it with the remaining reads from that cluster (Supplemental Fig. S4). Virus DTRs in the polished draft genomes ranged between 32 and 4829 bp in length, with average lengths of 452 bp, 449 bp, and 463 bp in the 25-m, 117-m and 250-m samples, respectively (Supplemental Table S1). Such repeats would not be readily resolved via short-read assembly approaches alone, because they would either collapse into a single copy or produce circular misassemblies.
The phage discovery pipeline also preserved levels of microdiversity known to produce fragmentation in short-read viral metagenomic assemblies (Roux et al. 2017). For example, genomes AFVG_250M1025 and AFVG_250M1026 were recovered from reads found in a single 5-mer bin in the 250-m sample (Fig. 2A). This bin was highly enriched for reads of ∼35 kb (Fig. 2B), suggesting that they derived from either the same phage genome or closely related genomes. Hierarchical clustering of pairwise alignment scores (see Methods) refined the 5-mer bin and separated the reads into two distinct alignment clusters, suggesting the existence of two phage populations differing at the strain level (Fig. 2C). Comparison of the two genomes polished from these clusters indicated they shared >95% sequence identity, but differed at a central multikilobase region of significant sequence divergence, as well as in several small insertions and deletions (Fig. 2D).
Figure 2.

Phage discovery steps for 250-m sample. (A) Nanopore reads that were found to contain DTRs were first represented by 5-mer count vectors and dimensionally reduced into a 2D embedding using UMAP (McInnes et al. 2018). Reads in the 2D embedding are colored based on their assignment to the 2386 5-mer bins called by hdbscan (McInnes et al. 2017). 5-mer bin colors are redundant owing to the large number of bins. Reads not assigned to a bin are colored gray. The 5-mer bin 75 is highlighted and contains 42 nanopore reads. (B) Read lengths from bin 75 were compared with read lengths from all bins, revealing an enrichment of ∼35-kb reads and a depletion of reads at other lengths. (C) Reads within bin 75 were aligned to each other to generate pairwise alignment scores, which were hierarchically clustered to reveal two main alignment clusters. A polished assembly-free virus genome (AFVG) was generated from each cluster: AFVG_250M1025 and AFVG_250M1026. (D) Comparison between the AFVG_250M1025 and AFVG_250M1026 sequences shows microdiversity between the two polished phage genomes. The genomes share large regions of varying degrees of sequence identity >95% with interspersed divergent sequence.
To further validate our methods, an environmental sample was spiked with 10 ng lambda phage DNA before sequencing and analysis (Methods), providing an internal standard reference sequence (Daniels et al. 1983). When 11 sequenced full-length lambda nanopore reads were included into the discovery pipeline, a 48,517-bp polished genome was recovered with 99.81% identity to the 48,502-bp lambda reference (Supplemental Fig. S5). When 23 lambda reads were instead included, a 48,510-bp polished genome was recovered sharing 99.92% identity with the lambda reference. These results were obtained using only nanopore reads for polishing and recapitulate the observed correlation between AFVG quality and the number of nanopore reads used for polishing (Supplemental Fig. S5).
In total, our phage discovery pipeline produced 1864 high-quality polished draft genomes, with the 25-m, 117-m, and 250-m samples generating 566, 93, and 1205 unique AFVGs, respectively (Supplemental Fig. S6). Numbers of viral genotypes from short-read Illumina sequencing versus nanopore sequencing in libraries prepared from the same DNA were compared for all three samples (Supplemental Fig. S7). In each of the three samples, the coverage of Illumina short reads on any given AFVG was generally comparable to the number of nanopore reads within the 5-mer bin that produced the AFVG. The nanopore sequencing approach described here recovered many more complete virus genome sequences than did short-read sequencing and assembly approaches alone. More specifically, all the AFVGs produced by the single-molecule nanopore sequencing method were longer and more complete compared with any homologous contigs recovered in short-read sequence assemblies from the identical DNA sample (Supplemental Fig. S8A). Conversely, all short-read contigs with homology with the nanopore AFVGs from the same DNA sample were fully covered by the nanopore AFVGs (Supplemental Fig. S8B).
As expected, further polishing of the nanopore polished AFVGs with Illumina short reads (Supplemental Methods) improved sequence quality. Both the mean annotated coding sequence (CDS) lengths and CDS coverage across the whole genome increased when AFVGs were further polished with short reads (Supplemental Table S1). CDS lengths of AFVGs after short-read polishing using Pilon (Walker et al. 2014) were 117% greater on average than those of nanopore-only polished sequences (Supplemental Fig. S9), suggesting that sequence quality can be further enhanced by leveraging both technologies.
Validating AFVG origins and preliminary characterization
The AFVGs were further validated using well-established approaches for characterizing viral metagenomic sequences (Supplemental Methods; Roux et al. 2015, 2019; Bolduc et al. 2017; Hurwitz et al. 2018). The primary 5-mer binning (Fig. 2A) generated nanopore sequence clusters with sequence length distributions that, unlike the bulk read size distributions, had single, well-defined peaks (Fig. 2B; Supplemental Table S1). We postulated that these read length peaks within bins, ranging between 28.0 and 87.3 kb across bins, represented full-length virus genomes contained in single nanopore sequence reads without assembly. The three samples from different depths varied with respect to AFVG size ranges (Supplemental Fig. S6; Supplemental Table S1). Specifically, AFVGs in the 25-m sample ranged from 28.0 to 65.2 kb (average 39.3 kb), in the 117-m sample from 29.8 to 87.4 kb (average 47.3 kb), and in the 250-m sample from 28.5 to 73.0 kb (average 41.6). These values are comparable to previously reported planktonic viral isolate and community genome size distributions (Steward et al. 2000; Holmfeldt et al. 2013). No AFVGs larger than ∼90 kb were detected in our current assembly-free nanopore sequencing method, because few sequence reads were >100 kb.
To further characterize the AFVGs, we screened them for virus sequence signatures (Roux et al. 2015; Hurwitz et al. 2018), viral gene content, and similarity to global viral gene sequence databases (Supplemental Methods; Hurwitz and Sullivan 2013; Mizuno et al. 2013b; Brum et al. 2015; Paez-Espino et al. 2016; Roux et al. 2016a). All of our 1864 AFVGs were flagged as viruses by the sequence classifier VirSorter (Roux et al. 2015). Because a final selection criterion in the pipeline included requiring DTRs be present in the putative AFVGs (a characteristic of many linear double stranded bacteriophage genomes) (Casjens and Gilcrease 2009), all of our AFVGs also contained this viral feature.
Although few of the protein-coding sequences in the AFVGs bore high similarity to those in NCBI's RefSeq database (Supplemental Fig. S10), a high proportion of AFVG annotated genes did share high sequence similarities to virus genes found in global metagenomic viral databases (Fig. 3; Roux et al. 2015; Bolduc et al. 2017; Hurwitz et al. 2018; Roux et al. 2019). The majority of AFVGs had >60% of their annotated genes matching at >60% average amino acid identity (AAI) to protein-coding virus genes in viral reference databases. AFVGs in the 250-m sample contained the largest proportion of novel genes (Fig. 3), in congruence with previous findings (Luo et al. 2017). The AFVGs also contained a high proportion of viral marker genes, including genes annotated as encoding terminase, tail, head, capsid, portal, and integrase proteins (Supplemental Fig. S11).
Figure 3.
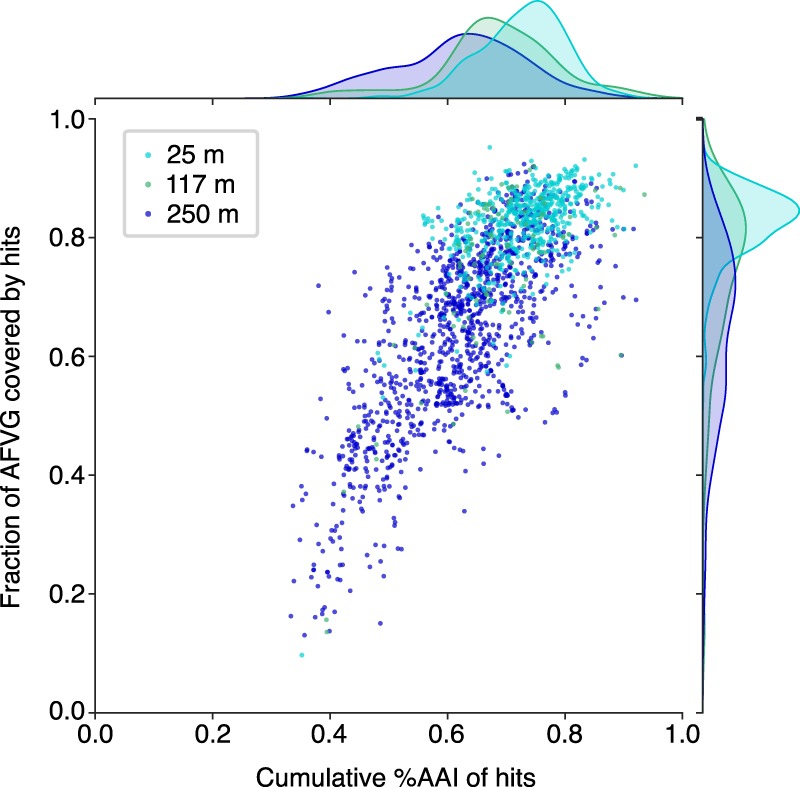
Similarity of AFVG annotated genes to known viral genes. Each point represents an AFVG colored by the depth at which it was found. The y-axis encodes the fraction of genes with matches to known viral genes, and the x-axis encodes the cumulative percentage of amino acid identity (%AAI) of those matches. The marginal histograms show the distribution of values for “cumulative %AAI” (top) and “fraction of genes” (right) grouped by sample depth.
The taxonomic affiliations of AFVGs were consistent with the known microbial community composition and depth distributions of their planktonic microbial hosts from the same oceanographic region sampled (Supplemental Fig. S12; DeLong et al. 2006; Pham et al. 2008; Aylward et al. 2017; Luo et al. 2017; Mende et al. 2017). A large proportion of AFVGs were most similar to viruses known to infect cyanobacteria or to common heterotrophic bacteria such as Pelagibacter (SAR11), Puniceispirillum (SAR116), Pseudomonas, and Vibrio species (Supplemental Fig. S13A–C).
Inference of DNA packaging mechanisms
Phage DNA packaging mechanisms are varied and diverse, but a “packaging by the headful” mechanism is a common strategy among many dsDNA bacteriophages (Casjens and Gilcrease 2009). To infer the likely phage DNA packaging strategy for each AFVG, the DTR sequence was extracted from each binned read and aligned to a single AFVG genome from its corresponding 5-mer bin. In the majority of cases (93.7%), the resulting alignments revealed that the DTRs were composed of the same genomic terminal sequence for all reads in the 5-mer bin (Fig. 4A). This observation suggests that these phage genomes were cleaved at specific sites during packaging, resulting in the observed exact DTRs similar to those found in T7 phage and other Podoviridae. However, in the remaining 6.3% of AFVGs, the DTR sequence alignments indicated that these DTRs were not fixed at specific sequences but rather shifted positions along the population reference AFVG sequence (Fig. 4B). This pattern is indicative of a strict “headful” DNA packaging mechanism, in which cleavage is instead determined by available volume within the phage head rather than the presence of specific cleavage sites (Casjens and Gilcrease 2009).
Figure 4.
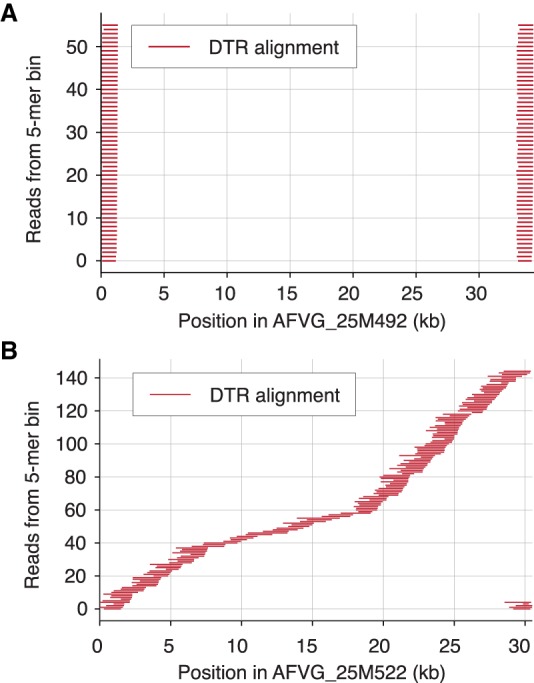
Aligned positions of DTR sequences in genomes. The DTR sequences can be either conserved (i.e., repeats all consist of the exact same genomic DNA subsequence) or circularly permuted (i.e., repeated termini sequences derive from different regions of the genome) depending on the mechanism used for phage DNA packaging. (A) DTR sequences flanking full-length genome reads in 5-mer bin 848 in the 25-m sample were aligned to a polished draft genome produced from that bin, AFVG_25M492, revealing that the DTR sequences were conserved across all reads and associated with fixed genomic positions. (B) DTR sequences from reads in 5-mer bin 903 in the same sample were aligned to a polished draft genome from that bin, AFVG_25M522. In this case, the DTRs were composed of sequences from throughout the genome instead of being associated with a single fixed subsequence. This observation reveals the circular permutation of the genome that is consistent with the “headful” mechanism of phage DNA packaging.
Phylogenetic analyses of terminases (proteins associated with phage DNA recognition and packaging) showed that AFVGs possessing circularly permuted DTRs were diverse and phylogenetically interspersed among other AFVGs and cultured bacteriophage reference sequences (Supplemental Methods; Supplemental Fig. S14). One cluster of AFVG terminases with circularly permuted DTRs was most similar (76% AAI) to homologs of cultured marine cyanophages known to infect the most abundant cyanobacterium in the ocean, Prochlorococcus. Other AFVG terminases with fixed DTRs were most closely related (88% AAI) to cultured bacteriophages known to infect members of a common oceanic bacterial genus, Puniceispirillum. Yet other AFVG terminases possessing fixed DTRs clustered with homologs of enterobacteriophages (T3, T5, and T7; also known to have fixed DTRs), as well as other marine bacteriophages (Supplemental Fig. S14).
Linear concatemer sequences isolated from seawater
Among the polished AFVGs, 16 were 33.1- to 66.2-kb sequences composed exclusively of concatenated repeats of 5.3- to 13.2-kb sequences (Supplemental Table S2). To further explore this phenomenon, we searched for other linear concatemers among all reads from each of the three samples. The 1546 linear concatemer reads found in the 25-m sample were mostly 20–40 kb in length, although there was a significant additional narrow peak in the length distribution near 60 kb (Fig. 5A), and a few were <10 kb. By defining the repeat count in the concatemeric reads as the read length divided by the length of the repeated sequence unit, we found that many of the reads contain integer repeat counts (between five and seven) with no partial repeat copies on either end (Fig. 5B). In the 117-m sample, 897 linear concatemer reads were mostly 20–40 kb, with a significant length enrichment between 60 and 65 kb (Fig. 5C). Many of these also contained integer repeat counts, most prominently at seven copies (Fig. 5D). Of the linear concatemer reads found in the 250-m sample, 1947 displayed a significant read length enrichment between 35 and 40 kb (Fig. 5E) but minimal enrichment near 60 kb. These concatemer read length distributions were generally consistent with those of the nonconcatemeric, virus-derived AFVGs. For example, 16 of 93 (17%) of the AFVGs produced from the 117-m sample were >60 kb compared with only 41 of 1205 (3%) for the 250-m sample, and 996 of 1205 (83%) of the AFVGs from 250 m were <45 kb (Supplemental Fig. S6). As in the 25-m and 117-m samples, we found that many concatemeric reads from the 250-m sample contained integer repeat counts (between four and seven whole repeat copies) with no partial repeat copies on either end (Fig. 5F).
Figure 5.

Read lengths and repeat counts in concatemeric nanopore reads. The length distribution of concatemeric nanopore reads is similar to the length distributions of polished AFVGs obtained from each sample (Supplemental Fig. S6). (A) Concatemeric reads obtained from the 25-m sample are predominately 20–40 kb with an additional enrichment for 60-kb reads. (B) In the 25-m sample, the length of the repeated unit in each concatemer and its associated repeat count. The repeat counts are enriched for whole integer values, primarily six and seven copies. Dashed lines indicate the sequence size associated with a given repeat unit count and length. (C) Concatemeric reads isolated from the 117-m sample show an enrichment in concatemer read lengths between 60 and 65 kb, which is similar in size to many of the complete phage genomes obtained from the 117-m sample. (D) The repeat unit counts are enriched at whole integer numbers, most prominently at five, six, or seven copies in the 117-m sample. Concatemers enriched for these counts in the 117-m sample are mostly associated with 60- to 65-kb concatemer reads. (E) Concatemer reads from the 250-m sample are predominantly between 35 and 40 kb in length. (F) The concatemer repeat unit counts in the 250-m sample are enriched at four, five, six, and seven whole copies. Given each specific combination of repeat unit count and length, the majority of concatemer reads remain just short of 40 kb.
The 16 nanopore-polished concatemer sequences were further polished using short reads to remove residual sequence errors (Supplemental Methods). Subsequent gene annotation revealed the presence of integrase genes in all repeat copies, as well as DNA primases in several concatemers (Fig. 6). A similar arrangement of genes, repeats, and gene contents is known to occur in small mobile elements that can “hijack” helper-phage packaging machinery, called phage-inducible chromosomal islands (PICIs) (Penades and Christie 2015). In particular, the integrases and DNA primases we found in the concatemers are hallmark genes commonly found in PICIs. Furthermore, the concatemeric whole number repeat copies and read lengths we observed are consistent with predicted PICI-like DNA synthesis and packaging strategies. One of the assembly-free putative PICIs (AFPPs) we found (AFPP_117M2) (Fig. 6) appears to have originated from one of the most common heterotrophic bacterial groups in the ocean, Pelagibacter. This mobile element presumably co-opts Pelagibacter phage packaging machinery intracellularly and is consistent with recent reports that pelagiphages in the family Podoviridae integrate into Pelagibacter host genomes (Zhao et al. 2019). Although these mobile phage parasites were originally discovered in cultures of Gram-positive bacteria (Ruzin et al. 2001; Chen and Novick 2009; Novick et al. 2010), more recent studies indicate that PICIs may be much more widespread (Martinez-Rubio et al. 2017; Fillol-Salom et al. 2018; Dokland 2019). To the best of our knowledge, the results reported here are among the first to show that PICI-like genome concatemers are likely packaged in “wild” phage particles, with concatemer repeat sizes reflecting the genome size of phages they parasitize.
Figure 6.

Structure of putative phage-packaged, PICI-like concatemers. Protein annotations from four representative concatemeric sequences recovered from the 250-m sample are shown. Background shading represents repetitive regions in the PICI-like concatemers. Protein-coding sequences are color-coded based on functional annotations on Pfam (bit score >30) (El-Gebali et al. 2019). Taxonomic annotations are shown with top hit to organism of RefSeq release 92 (O'Leary et al. 2016), amino acid identity (AAI), and bit score. Functional annotations are shown with top hits and bit scores to protein domains in Pfam and eggNOG (Huerta-Cepas et al. 2016).
Discussion
Generation of dsDNA virus genome fragments from natural populations via short-read metagenomic assembly has greatly advanced our understanding of wild dsDNA virus populations. For example, these approaches have led to a greater appreciation of the ecological roles (Roux et al. 2016a), taxonomic diversity (Paez-Espino et al. 2016), gene content (Hurwitz and Sullivan 2013), and genomic variability of native virus populations (Warwick-Dugdale et al. 2019). Despite these advances, short-read assembly approaches do have limitations (Roux et al. 2019). These include generation of fragmented assemblies, chimeras, or false positive circular contigs (Roux et al. 2017, 2019; Hurwitz et al. 2018); an inability to accurately identify virion linear genome termini (Casjens and Gilcrease 2009); and difficulties in differentiating highly repetitive sequence regions, rare island regions, or genetically similar population variants (Warwick-Dugdale et al. 2019). Consequently, long repeat elements like DTRs, or repetitive concatemer sequences (produced, e.g., via rolling circle replication), are not recovered in short-read assemblies. Although metagenomic virion genomes assembled from short reads are typically reported as complete if they are “circular” (Roux et al. 2015, 2017), this apparent circularity can in fact be artefactually produced, because short-read assemblies do not fully recover DTRs from linear phage genome termini (Casjens and Gilcrease 2009). Consequently, considering that virtually all tailed-phage virion genomes are linear and not circular, complete virion genome sequences that include their termini cannot be recovered by short-read assembly methods alone (Casjens and Gilcrease 2009).
In contrast, the single-molecule nanopore sequencing strategy reported here fully captured complete virion genomes, including their DTRs and termini. Furthermore, the method makes it possible to reveal more detailed population microheterogeneity, as well as highly variable, rare genomic island regions, that would be difficult to detect using standard methods (Thompson et al. 2005; Warwick-Dugdale et al. 2019). The approach also facilitates inference of phage packaging strategy, based on the nature of a given phage population's DTRs. Additionally, the discovery, reported here, of PICI-like concatemers that appear to be packaged in phage particles, shows the utility of the approach for revealing novel repeat sequences derived from phage-mobilized genetic elements that would not be detectable using short-read sequence assemblies.
The assembly-free virus sequencing strategy reported here has room for improvement with respect to its efficiency as well as the quantity, diversity, and size range of virus genomes recovered from complex communities. Our initial approach required a starting input of ∼1 μg of virus-enriched high-molecular-weight DNA, but lowering the DNA input requirement should be achievable with future methodological refinement. Additionally, our current phage discovery pipeline discarded many reads by stringently requiring the presence of DTRs to be considered as a virus genome. Virus genome sequences lacking a DTR on their termini, virus genome fragments of unequal lengths, or viral genotypes that simply were rare in the sample (so their bin sizes were lower than our cutoff threshold) would be omitted in our current method. Improving upstream virus purification methods, using more inclusive strategies to identify viral sequences, incorporating long-read assembly strategies, and polishing with short reads from other sequencing platforms all have potential to enhance the yield and sequence accuracy, as well as shed light on virus genomes beyond the <90-kb dsDNA tailed-phage genomes described here.
The detection limit of our method for any given virus population is difficult to precisely quantify given that recovery will depend on several variables inherent within complex environmental samples. These include the concentration factor of the original virus-enriched preparation (determined here by the amount of seawater filtered), the relative number, evenness, and diversity of coexisting phage populations in the sample, and the fraction of phage genomic DNA that has remained fully intact during extraction and library preparation. In the case of the lambda spike-in experiment reported here, the added lambda phage genomes comprised ∼0.5% of the total phage genomes present in the mixture and were readily detected using our reported methodology (Supplemental Fig. S5).
Further application of the assembly-free, single-molecule nanopore approach described should help advance a more detailed understanding of dsDNA virus genome structure and variability, their fine-scale population biology, and the nature and prevalence of viral parasites like PICIs in complex naturally occurring assemblages.
Methods
Virus particle collection and DNA extraction
To concentrate and enrich viral particles, seawater was collected, prefiltered to remove bacterioplankton cells, and concentrated via tangential flow filtration (TFF) over a 30-kDa filter (Biomax 30-kDa membrane, Millipore P3B030D01). DNA extractions were performed using a Qiagen Genomic-tip 20/G (Qiagen) purification kit following the manufacturer's recommendations. For more detail, see Supplemental Methods.
Nanopore sequencing
A total of 1–1.5 μg of purified DNA from each sample was used to prepare sequencing libraries using the standard ligation sequencing kit LSK109 (Oxford Nanopore Technologies), modifying the DNA repair and end-prep incubation step to 20 min. Sequencing was conducted on GridION X5 with FLO-MIN106 (R 9.4.1) flowcells (Oxford Nanopore Technologies).
Identification of reads containing DTRs
Subsequences representing the first and last 20% of each nanopore read were aligned using minimap2 v2.17 (Li 2018) with the flag -x map-ont. A read is considered to have a DTR if an alignment is produced and (1) the aligned positions in the starting subsequence are within the first 200 bp and (2) the aligned positions in the ending subsequence are with last 200 bp.
Read simulation for mock viral community
Nanopore reads were simulated using NanoSim v2.2.0 (Yang et al. 2017). The model was trained on existing nanopore sequencing data from Escherichia coli K12 using the NanoSim script read_analysis.py with default parameters. Reads were generated for each reference genome from uvMED using the NanoSim script simulator.py using the parameters “linear -n 50” with the parameter --min_len set for each reference to represent 98% of the reference length. Because the reference sequences lack large DTRs, the DTR-filtering step of the phage discovery pipeline was bypassed for the simulated reads.
5-mer binning of nanopore reads
Read binning was performed by projecting all DTR-containing reads into a two-dimensional embedding of their k-mer count vectors (k = 5). In contrast with the procedure used for conventional k-mer binning (Alneberg et al. 2014), the 5-mer counts for each read in this study were not normalized by read length. For genome-spanning reads, read length can serve as a useful feature for binning. By omitting a normalization step, we retain the read length information in the 5-mer vectors. Specifically, all 5-mers in a read were counted, and the counts of reverse-complement 5-mers were combined, resulting in a vector, Z, for each read, i, denoted as Zi = (Zi,j, …, Zi,V), where V = 512 possible combined 5-mers. The resulting N × V matrix of 5-mer counts, where N is the number of reads, was subjected to dimensionality reduction with UMAP v0.3.2 (McInnes et al. 2018) using the options n_neighbors = 15 and min_dist = 0.1. Bins were automatically called in the resulting two-dimensional embedding with hdbscan v0.8.18 (McInnes et al. 2017) using the option min_cluster_size = 10. All reads not assigned to a bin were omitted from further analysis.
5-mer bin refinement through alignment clustering
Each 5-mer bin was refined using an all-versus-all alignment approach for the binned reads. Alignment was performed using minimap2 (Li 2018) with the parameters “-x ava-ont --no-long-join -r100.” Each pairwise alignment between read i and read j was assigned an alignment score Sij:
where Lij is the alignment length, and Dij is the sequence divergence as reported by the minimap2 tag “dv.” Alignment scores were hierarchically clustered with the cluster.hierachy.linkage function from the Python package SciPy v1.1.0 using the Ward variance minimization algorithm. Alignment clusters were assigned using cluster.hierachy.fcluster by cutting the dendrogram at three times the median distance in the linkage matrix (Fig. 2C). An alignment cluster was retained for genome polishing if the cluster had (1) 11 or more reads (one draft genome read and 10 reads for polishing), and (2) a mean alignment score, S, among clustered reads that was ≥80% of the theoretically maximum alignment score S* for the cluster mean read length (S* = M2, where M is the mean length of clustered reads). These criteria, along with the strict alignment parameters used by minimap2, helped to ensure that the reads contained in each cluster fully aligned to one another with a high degree of sequence similarity. If these criteria were met, the read in the cluster with the highest mean alignment score was designated to serve as the draft genome sequence and the remainders were designated for use in polishing the draft genome.
Draft genome polishing and coding sequence annotation
Polishing was conducted using both Racon (Vaser et al. 2017) and Medaka (https://github.com/nanoporetech/medaka). The first polishing step, which was iteratively performed three times, used minimap2 v2.15 (Li 2018) with “-ax map-ont” and Racon v1.3.1 with the options “--include-unpolished --quality-threshold=9.” The polished output of this step was further polished with Medaka v1.4.3 using the supplied model file r941_flip_model.hdf5. Any residual adapter sequences were then pruned from the polished draft genomes with Porechop v0.2.3 (https://github.com/rrwick/Porechop) using the option “--no_split.” All polished sequences <20 kb were discarded owing to a lack of evidence for DTR-containing phage genomes (shortest observed AFVG was 28.0 kb). Finally, the CDSs of the polished and deduplicated draft genomes (Supplemental Methods) were annotated with Prodigal v2.6.3 (Hyatt et al. 2010) using the option “-p meta.”
Lambda phage DNA spike-in analyses
Lambda reads were identified among the sequencing output by aligning them to the appropriate reference (Daniels et al. 1983). Because the lambda phage genome contains only short DTRs, we selected for full-length reads based on alignment to the reference sequence. Reads between 45 and 50 kb in length and whose alignments spanned from the first 1% to the last 1% of the reference sequence were considered full-length reads. These were included among the DTR-containing environmental phage reads for processing by the phage discovery pipeline.
Identification of linear concatemer reads
Subsequences of 3 kb were taken from the beginning of all reads >15 kb and aligned to their full-length reads using minimap2 v2.15 (Li 2018) with the option “-x ava-ont.” All reads in which >90% of the 3 kb starting subsequence aligned at least twice on the full-length read were considered concatemeric. The concatemer repeat size was determined by taking the median of the differences between the consecutive alignment start positions in the full-length read. Concatemer repeat counts were calculated by dividing the full read length by the concatemer repeat size.
Phage genome validation: taxonomic and functional annotations
Predicted proteins were taxonomically annotated using LAST v756 (Kielbasa et al. 2011) against the RefSeq release 84 database (O'Leary et al. 2016). Phage genomes were annotated at the genus level if they contained one, three, or five or more proteins with top hits to phages infecting the same bacterioplankton genus (Supplemental Fig. S12). Predicted proteins were functionally annotated using hmmsearch (Finn et al. 2011) against the Pfam-A v30 database (El-Gebali et al. 2019), and top hits at >30 bit score were retained (Supplemental Fig. S11). Predicted proteins from four concatemeric sequences were also functionally annotated with the eggNOG database (Huerta-Cepas et al. 2016), and all functional top hits with any bit score were retained (Fig. 6).
Characterization and comparative analyses of AFVGs
Known viral genes were placed in a single database from multiple sources: RefSeq release 84 (Brister et al. 2015), the Earth Virome (Paez-Espino et al. 2016), the Global Ocean Virome (Roux et al. 2016a), and three Mediterranean metagenomic viromes (Mizuno et al. 2013a, 2016; López-Pérez et al. 2017). Nucleotide gene sequences predicted from AFVGs were compared with this database and also with all RefSeq genes using lastal v828 (Kielbasa et al. 2011). The highest-scoring match was taken for each gene, and these matches were grouped by AFVG. For each AFVG, the total fraction of genes with matches and the cumulative AAI of matches were calculated (Fig. 4; Supplemental Fig. S4).
Data access
All sequencing reads, AFVGs, and AFPP concatemers generated in this study have been submitted to the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA529454. All custom scripts used to perform bioinformatic analyses are available at GitHub (https://github.com/nanoporetech/DTR-phage-pipeline) and as Supplemental Code.
Competing interest statement
J.B., X.D., D.J.T., M.P., S.J., and E.H. are employed by Oxford Nanopore Technologies.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the Gordon and Betty Moore Foundation (GBMF 3777 to E.F.D.) and the Simons Foundation (329108 to E.F.D.).
Author contributions: E.F.D. and M.P. conceived of the project. E.F.D. led the project. P.D.U., A.B., E.L., and E.F.D. collected the seawater samples, prepared the virus-enriched fractions, and extracted and purified the DNA. D.J.T., M.P., E.H., S.J. and X.D., E.F.D., and A.B. coordinated and conducted ONT sequence generation and its preliminary quality checks and analyses. J.B. engineered, developed, and tested the bioinformatic pipelines. J.B., E.L., and J.M.E. performed analysis of all sequence data sets. E.F.D., J.B., J.M.E., and E.L. wrote the manuscript. E.F.D., J.B., J.M.E., E.L., E.H., S.J., and D.J.T. contributed to the figures or to editing of the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.251686.119.
Freely available online through the Genome Research Open Access option.
References
- Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, Lahti L, Loman NJ, Andersson AF, Quince C. 2014. Binning metagenomic contigs by coverage and composition. Nat Methods 11: 1144–1146. 10.1038/nmeth.3103 [DOI] [PubMed] [Google Scholar]
- Aylward FO, Boeuf D, Mende DR, Wood-Charlson EM, Vislova A, Eppley JM, Romano AE, DeLong EF. 2017. Diel cycling and long-term persistence of viruses in the ocean's euphotic zone. Proc Natl Acad Sci 114: 11446–11451. 10.1073/pnas.1714821114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc B, Youens-Clark K, Roux S, Hurwitz BL, Sullivan MB. 2017. iVirus: facilitating new insights in viral ecology with software and community data sets imbedded in a cyberinfrastructure. ISME Journal 11: 7–14. 10.1038/ismej.2016.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M, Salamon P, Andresen B, Mahaffy JM, Segall AM, Mead D, Azam F, Rohwer F. 2002. Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci 99: 14250–14255. 10.1073/pnas.202488399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brister JR, Ako-Adjei D, Bao Y, Blinkova O. 2015. NCBI Viral Genomes Resource. Nucleic Acids Res 43: D571–D577. 10.1093/nar/gku1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum JR, Ignacio-Espinoza JC, Roux S, Doulcier G, Acinas SG, Alberti A, Chaffron S, Cruaud C, de Vargas C, Gasol JM, et al. 2015. Ocean plankton. Patterns and ecological drivers of ocean viral communities. Science 348: 1261498 10.1126/science.1261498 [DOI] [PubMed] [Google Scholar]
- Casjens SR, Gilcrease EB. 2009. Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. Methods Mol Biol 502: 91–111. 10.1007/978-1-60327-565-1_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Novick RP. 2009. Phage-mediated intergeneric transfer of toxin genes. Science 323: 139–141. 10.1126/science.1164783 [DOI] [PubMed] [Google Scholar]
- Daniels, DL, Schroeder JL, Szybalski W, Sanger F, Coulson AR, Hong GF, Hill DF, Petersen GB, Blattner FR. 1983. Appendix II: complete annotated λ sequence. In Lambda-II (ed. Hendrix RW, et al. ), pp. 519–676. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York. [Google Scholar]
- DeLong EF, Preston CM, Mincer T, Rich V, Hallam SJ, Frigaard N-u, Martinez A, Sullivan MB, Edwards R, Brito BR, et al. 2006. Community genomics among stratified microbial assemblages in the ocean's interior. Science 311: 496–503. 10.1126/science.1120250 [DOI] [PubMed] [Google Scholar]
- Dokland T. 2019. Molecular piracy: redirection of bacteriophage capsid assembly by mobile genetic elements. Viruses 11: pii: E1003 10.3390/v11111003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, et al. 2019. The Pfam protein families database in 2019. Nucleic Acids Res 47: D427–D432. 10.1093/nar/gky995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiers W, Contreras R, Duerinck F, Haegeman G, Iserentant D, Merregaert J, Min Jou W, Molemans F, Raeymaekers A, Van den Berghe A, et al. 1976. Complete nucleotide sequence of bacteriophage MS2 RNA: primary and secondary structure of the replicase gene. Nature 260: 500–507. 10.1038/260500a0 [DOI] [PubMed] [Google Scholar]
- Fillol-Salom A, Martínez-Rubio R, Abdulrahman RF, Chen J, Davies R, Penadés JR. 2018. Phage-inducible chromosomal islands are ubiquitous within the bacterial universe. ISME Journal 12: 2114–2128. 10.1038/s41396-018-0156-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39: W29–W37. 10.1093/nar/gkr367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmfeldt K, Solonenko N, Shah M, Corrier K, Riemann L, Verberkmoes NC, Sullivan MB. 2013. Twelve previously unknown phage genera are ubiquitous in global oceans. Proc Natl Acad Sci 110: 12798–12803. 10.1073/pnas.1305956110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, Rattei T, Mende DR, Sunagawa S, Kuhn M, et al. 2016. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 44: D286–D293. 10.1093/nar/gkv1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz BL, Sullivan MB. 2013. The Pacific Ocean Virome (POV): a marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS One 8: e57355 10.1371/journal.pone.0057355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz BL, Deng L, Poulos BT, Sullivan MB. 2013. Evaluation of methods to concentrate and purify ocean virus communities through comparative, replicated metagenomics. Environ Microbiol 15: 1428–1440. 10.1111/j.1462-2920.2012.02836.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz BL, Ponsero A, Thornton J, U'Ren JM. 2018. Phage hunters: computational strategies for finding phages in large-scale ‘omics datasets. Virus Res 244: 110–115. 10.1016/j.virusres.2017.10.019 [DOI] [PubMed] [Google Scholar]
- Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11: 119 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielbasa SM, Wan R, Sato K, Horton P, Frith MC. 2011. Adaptive seeds tame genomic sequence comparison. Genome Res 21: 487–493. 10.1101/gr.113985.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Pérez M, Haro-Moreno JM, Gonzalez-Serrano R, Parras-Moltó M, Rodriguez-Valera F. 2017. Genome diversity of marine phages recovered from Mediterranean metagenomes: Size matters. PLoS Genet 13: e1007018 10.1371/journal.pgen.1007018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo E, Aylward FO, Mende DR, DeLong EF. 2017. Bacteriophage distributions and temporal variability in the ocean's interior. mBio 8: e01903-17 10.1128/mBio.01903-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Hernandez F, Fornas O, Lluesma Gomez M, Bolduc B, de la Cruz Peña MJ, Martínez JM, Anton J, Gasol JM, Rosselli R, Rodriguez-Valera F, et al. 2017. Single-virus genomics reveals hidden cosmopolitan and abundant viruses. Nat Commun 8: 15892 10.1038/ncomms15892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Rubio R, Quiles-Puchalt N, Martí M, Humphrey S, Ram G, Smyth D, Chen J, Novick RP, Penadés JR. 2017. Phage-inducible islands in the Gram-positive cocci. ISME J 11: 1029–1042. 10.1038/ismej.2016.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, Astels S. 2017. hdbscan: hierarchical density based clustering. J Open Source Softw 2: 205 10.21105/joss.00205 [DOI] [Google Scholar]
- McInnes L, Healy J, Melville J. 2018. UMAP: Uniform Manifold Approximation and Projection for dimension reduction. arXiv:1802.03426 [stat.ML].
- Mende DR, Bryant JA, Aylward FO, Eppley JM, Nielsen T, Karl DM, Delong EF. 2017. Environmental drivers of a microbial genomic transition zone in the ocean's interior. Nature Microbiology 2: 1367–1373. 10.1038/s41564-017-0008-3 [DOI] [PubMed] [Google Scholar]
- Mizuno CM, Rodriguez-Valera F, Garcia-Heredia I, Martin-Cuadrado AB, Ghai R. 2013a. Reconstruction of novel cyanobacterial siphovirus genomes from Mediterranean metagenomic fosmids. Appl Environ Microbiol 79: 688–695. 10.1128/AEM.02742-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno CM, Rodriguez-Valera F, Kimes NE, Ghai R. 2013b. Expanding the marine virosphere using metagenomics. PLoS Genet 9: e1003987 10.1371/journal.pgen.1003987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno CM, Ghai R, Saghaï A, López-García P, Rodriguez-Valera F. 2016. Genomes of abundant and widespread viruses from the deep ocean. mBio 7: e00805-16 10.1128/mBio.00805-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick RP, Christie GE, Penadés JR. 2010. The phage-related chromosomal islands of Gram-positive bacteria. Nat Rev Microbiol 8: 541–551. 10.1038/nrmicro2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. 2016. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44: D733–D745. 10.1093/nar/gkv1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Espino D, Eloe-Fadrosh EA, Pavlopoulos GA, Thomas AD, Huntemann M, Mikhailova N, Rubin E, Ivanova NN, Kyrpides NC. 2016. Uncovering Earth's virome. Nature 536: 425–430. 10.1038/nature19094 [DOI] [PubMed] [Google Scholar]
- Penadés JR, Christie GE. 2015. The phage-inducible chromosomal islands: a family of highly evolved molecular parasites. Annu Rev Virol 2: 181–201. 10.1146/annurev-virology-031413-085446 [DOI] [PubMed] [Google Scholar]
- Pham VD, Konstantinidis KT, Palden T, DeLong EF. 2008. Phylogenetic analyses of ribosomal DNA-containing bacterioplankton genome fragments from a 4000 m vertical profile in the North Pacific Subtropical Gyre. Environ Microbiol 10: 2313–2330. 10.1111/j.1462-2920.2008.01657.x [DOI] [PubMed] [Google Scholar]
- Roux S, Hawley AK, Beltran MT, Scofield M, Schwientek P, Stepanauskas R, Woyke T, Hallam SJ, Sullivan MB. 2014. Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta-genomics. eLife 3: e03125 10.7554/eLife.03125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Enault F, Hurwitz BL, Sullivan MB. 2015. VirSorter: mining viral signal from microbial genomic data. PeerJ 3: e985 10.7717/peerj.985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Brum JR, Dutilh BE, Sunagawa S, Duhaime MB, Loy A, Poulos BT, Solonenko N, Lara E, Poulain J, et al. 2016a. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 537: 689–693. 10.1038/nature19366 [DOI] [PubMed] [Google Scholar]
- Roux S, Solonenko NE, Dang VT, Poulos BT, Schwenck SM, Goldsmith DB, Coleman ML, Breitbart M, Sullivan MB. 2016b. Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 4: e2777 10.7717/peerj.2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Emerson JB, Eloe-Fadrosh EA, Sullivan MB. 2017. Benchmarking viromics: an in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 5: e3817 10.7717/peerj.3817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Adriaenssens E, Dutilh B, Koonin E, Kropinski A, Krupovic M, Kuhn J, Lavigne R, Brister J, Varsani A, et al. 2019. Minimum information about an uncultivated virus genome (MIUViG). Nat Biotechnol 37: 29–37. 10.1038/nbt.4306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzin A, Lindsay J, Novick RP. 2001. Molecular genetics of SaPI1—a mobile pathogenicity island in Staphylococcus aureus. Mol Microbiol 41: 365–377. 10.1046/j.1365-2958.2001.02488.x [DOI] [PubMed] [Google Scholar]
- Sanger F, Air GM, Barrell BG, Brown NL, Coulson AR, Fiddes CA, Hutchison CA, Slocombe PM, Smith M. 1977. Nucleotide sequence of bacteriophage φ X174 DNA. Nature 265: 687–695. 10.1038/265687a0 [DOI] [PubMed] [Google Scholar]
- Steward GF, Montiel JL, Azam F. 2000. Genome size distributions indicate variability and similarities among marine viral assemblages from diverse environments. Limnol Oceanogr 45: 1697–1706. 10.4319/lo.2000.45.8.1697 [DOI] [Google Scholar]
- Sullivan MB. 2015. Viromes, not gene markers, for studying double-stranded DNA virus communities. J Virol 89: 2459–2461. 10.1128/JVI.03289-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JR, Pacocha S, Pharino C, Klepac-Ceraj V, Hunt DE, Benoit J, Sarma-Rupavtarm R, Distel DL, Polz MF. 2005. Genotypic diversity within a natural coastal bacterioplankton population. Science 307: 1311–1313. 10.1126/science.1106028 [DOI] [PubMed] [Google Scholar]
- Vaser R, Sović I, Nagarajan N, Šikić M. 2017. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27: 737–746. 10.1101/gr.214270.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9: e112963 10.1371/journal.pone.0112963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warwick-Dugdale J, Solonenko N, Moore K, Chittick L, Gregory AC, Allen MJ, Sullivan MB, Temperton B. 2019. Long-read viral metagenomics captures abundant and microdiverse viral populations and their niche-defining genomic islands. PeerJ 7: e6800 10.7717/peerj.6800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Chu J, Warren RL, Birol I. 2017. NanoSim: nanopore sequence read simulator based on statistical characterization. Gigascience 6: 1–6. 10.1093/gigascience/gix010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Qin F, Zhang R, Giovannoni SJ, Zhang Z, Sun J, Du S, Rensing C. 2019. Pelagiphages in the Podoviridae family integrate into host genomes. Environ Microbiol 21: 1989–2001. 10.1111/1462-2920.14487 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.