

Abstract
Microsatellites are repeats of 1- to 6-bp units, and approximately 10 million microsatellites have been identified across the human genome. Microsatellites are vulnerable to DNA mismatch errors and have thus been used to detect cancers with mismatch repair deficiency. To reveal the mutational landscape of microsatellite repeat regions at the genome level, we analyzed approximately 20.1 billion microsatellites in 2717 whole genomes of pan-cancer samples across 21 tissue types. First, we developed a new insertion and deletion caller (MIMcall) that takes into consideration the error patterns of different types of microsatellites. Among the 2717 pan-cancer samples, our analysis identified 31 samples, including colorectal, uterus, and stomach cancers, with a higher proportion of mutated microsatellite (≥0.03), which we defined as microsatellite instability (MSI) cancers of genome-wide level. Next, we found 20 highly mutated microsatellites that can be used to detect MSI cancers with high sensitivity. Third, we found that replication timing and DNA shape were significantly associated with mutation rates of microsatellites. Last, analysis of mutations in mismatch repair genes showed that somatic SNVs and short indels had larger functional impacts than germline mutations and structural variations. Our analysis provides a comprehensive picture of mutations in the microsatellite regions and reveals possible causes of mutations, as well as provides a useful marker set for MSI detection.
Recent large-scale whole-genome sequencing (WGS) studies have revealed the complexity of the mutational landscape of the cancer genome (Fujimoto et al. 2016; Nik-Zainal et al. 2016; Hayward et al. 2017; Northcott et al. 2017). In cancer genomes, various types of mutations, such as single-nucleotide variants (SNVs), short indels (insertions and deletions [IDs]), genomic rearrangements, copy number alterations, insertion of retrotransposons, and virus genome integrations, have been identified, and their oncogenic roles have been characterized (Helman et al. 2014; Ewing et al. 2015; Fujimoto et al. 2016; Nik-Zainal et al. 2016; Hayward et al. 2017; Northcott et al. 2017). Additionally, genome sequencing studies have revealed the molecular basis of somatic mutations (Alexandrov et al. 2013, 2016; Chen and Zhang 2015; Tubbs and Nussenzweig 2017). However, somatic mutations in microsatellites or repeat sequences have not been well characterized in a large WGS cohort owing to difficulties in accurately detecting mutations using presently available short-read sequencing technologies.
A microsatellite is defined as a tract of repetitive DNA motif composed of short repeating units (Ellegren 2004). The mutation rate of microsatellites has been known to be higher than other genomic regions owing to DNA polymerase slippage during DNA replication and repair (Ellegren 2004). In cancer genetics studies, microsatellite instability (MSI) has been used for molecular diagnosis of Lynch syndrome and cancers with mismatch repair (MMR) deficiency (Boland and Goel 2010). Furthermore, MSI-positive tumors are generally burdened with higher numbers of somatic mutations and present many mutation-associated neoantigens, which might be recognized by the immune system. Currently, MSI can also be used as a marker to predict the effect of immune therapy (Le et al. 2017). The MSI phenotype is most common in colorectal (CR) cancers, stomach (ST) cancers, and uterine (UT) endometrial cancers (10%–15%), although it has also been observed across many tumor types at a few percentages (Boland and Goel 2010; Bailey et al. 2018). The MSI phenotype is defined by the presence of somatic indels of two to five microsatellite markers and immunohistochemistry (Shia 2008; Boland and Goel 2010; Geiersbach and Samowitz 2011).
Irrespective of the clinical importance of microsatellites, large-scale analysis of somatic changes in microsatellites across various type of cancers is limited for WGS data (Kim et al. 2013; Cortes-Ciriano et al. 2017). In the current study, we analyzed indels in microsatellites for 2913 International Cancer Genome Consortium (ICGC) pan-cancer samples from 21 tissues (The International Cancer Genome Consortium 2010; The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020) to reveal the whole-genome mutational landscape of microsatellite regions. We developed a method to detect somatic indels in microsatellite regions, selected appropriate parameters for our purpose, and identified indels in microsatellite regions. We identified MSI-positive samples and factors affecting the mutation rate of microsatellites, as well as highly mutated microsatellites. We also analyzed the association of mutation rate of microsatellites with somatic and germline mutations in DNA repair genes and compared mutational signatures between MSI and other samples. Our analysis provides a comprehensive picture of mutations in the microsatellite regions and reveals possible causes of mutations, as well as provides a useful marker set for MSI detection.
Results
Identification of microsatellite regions in the genome
We detected microsatellites using three methods (MsDetector, Tandem Repeat Finder, and MISA software) (Benson 1999; Girgis and Sheetlin 2013; Hause et al. 2016). To exclude microsatellites potentially arising from read mapping errors, we selected microsatellites based on the uniqueness of flanking sequences and pattern of repeats. A total of 9,292,677 microsatellites were used for subsequent analyses. Within these selected microsatellites, it was observed that the MISA software identified a larger number compared with other methods (Supplemental Fig. S1). Poly(A) tails of mobilized transposable elements are also included in this analysis as microsatellites, if the length is >5 bp.
Error rate estimation of microsatellites
During library preparation and sequencing processes, indel errors can be introduced by PCR in reads containing short repeats owing to replication slippage of DNA polymerases. Because the error rates should depend on the length and type of microsatellites, we first estimated the error rates of different types of microsatellites. The type of microsatellites was defined by length of the microsatellite region in the reference genome and repeat unit (see Methods). By using sequence data of Chr X from 32 normal tissues of male individuals, we estimated the error rate among different types and lengths of microsatellites. As the male Chr X is hemizygous, the error rate can be inferred without the influence of heterozygous polymorphisms (Supplemental Fig. S2; Fujimoto et al. 2010; Maruvka et al. 2017). As expected, error rates depended on the unit and length of the microsatellites, with longer microsatellites having higher error rates (Supplemental Fig. S3). In all types of microsatellites, deletion errors were more frequent than insertion errors, and smaller changes of unit number were predominant (Supplemental Fig. S3). These results suggest that PCR or sequencing processes are prone to induce short deletion errors. Error rates of microsatellites between 5 and 9 bp in length within the reference genome were very low (<0.2%), whereas those of longer microsatellites were higher (>5% error rate for 20–100 bp of microsatellites in the reference genome length); 2-bp repeats had higher error rates than other microsatellites (Supplemental Fig. S3). The A/T type of microsatellite was observed to have higher error rates compared with G/C microsatellites (Supplemental Fig. S3). Because the estimated error rates were quite different among the types and lengths of microsatellites, we used the difference between error rates to detect somatic indels in the microsatellites. We generated a table of error rates for analyzing mutations in microsatellite regions based on the estimated error rates (Supplemental Table S1).
Validation with simulation data sets and determination of thresholds
Mutations in microsatellite regions were identified based on likelihoods (see Methods). To estimate false-positive and false-negative rates and to select appropriate parameters, we generated simulation data sets by using sequence reads mapped on Chr X of male individuals. In this analysis, the false-positive rate was defined as heterozygous calls in homozygous loci (additional alleles are detected), and false negatives were homozygous calls in heterozygous loci (existing alleles are missed). First, we determined the genotype of each microsatellite on Chr X. Because Chr X is hemizygotic in males, we considered the major reads as the true genotype of each sample. We assumed that the major read type is the true genotype and others were errors. For example, when there were 10, three, and one reads containing (AT)8, (AT)7, and (AT)9 in a microsatellite locus, we considered (AT)8 the true genotype (Supplemental Fig. S2). We then mixed Chr X reads from two male individuals and identified the variation in the microsatellite regions with our algorithm (Supplemental Fig. S2). By comparing the true genotypes and identified variants from the mixed data, we estimated the false-positive and false-negative rates (Supplemental Fig. S2). The false-positive and -negative rates were varied according to the likelihood values (L), and higher Ls had higher false-negative and lower false-positive rates (Supplemental Fig. S4). To identify somatic mutations in microsatellites, we required reads that completely cover target microsatellites. The length of reads is ∼100 bp; therefore, longer microsatellites have fewer reads covering them compared with shorter microsatellites and thus have a lower sensitivity (Supplemental Fig. S4). Based on the analysis, L was set to −8 for cancer samples and −1 for matched normal samples.
Analysis of indels in the microsatellite regions in pan-cancer samples
We analyzed the whole-genome sequence data of 2917 pan-cancer samples (The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020) with our method and compared them against somatic and germline variants detected by the Pan-Cancer Analysis of Whole Genomes (PCAWG) project (The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020). To compare our results with somatic consensus indels from the four PCAWG indel callings, we gathered indels located ±5 bp in the microsatellite regions in the PCAWG calls. On average, 1826.5 indels were detected by our indel caller (MIMcall) in the microsatellite regions (Supplemental Code 1). Of these, 1185.1 were found only by MIMcall (Supplemental Fig. S5), suggesting a higher sensitivity of our indel calls compared with the other PCAWG callers for microsatellite regions. PCAWG calls removed repetitive regions to achieve highly accurate mutation calling; therefore, our result can complement the PCAWG calls. We then compared the number of indels in the microsatellite regions between our indel caller and PCAWG callers. In the microsatellite regions, the number of indels uniquely identified by our indel caller was significantly correlated with that of commonly identified indels (identified by two or more PCAWG callers; Pearson product-moment correlation coefficient; r = 0.90, P-value < 10−16) (Supplemental Fig. S5). We further performed experimental validation with Japanese liver (LI) cancer samples for the mutation candidates in longer microsatellites by capillary electrophoresis (Supplemental Fig. S6). The false-discovery rate of our method was estimated to be 7% (2/29) (Supplemental Fig. S6). These results indicate that MIMcall can effectively identify indels in the microsatellite region.
Microsatellites covered by 15 or more reads in 2500 or more samples (7,650,128 microsatellites) were subjected to further analysis, and samples with 6 million or more testable microsatellites were used for the analysis (2717 samples) (Supplemental Table S2). On average, 7,407,000 microsatellites were analyzed in each sample. We compared the proportion of mutated samples for each microsatellite. Most of the microsatellites in whole genomes were not mutated in the pan-cancer samples (Fig. 1A), we therefore selected 198,578 microsatellites with proportions of mutated samples 0.001 or more (more than two to three mutated samples in the pan-cancer samples) and considered them as informative microsatellites. The proportions of mutated samples were different among the types and lengths of microsatellite, with A/T microsatellites more frequently mutated than other types (Fig. 1B). We then calculated the proportion of mutated microsatellite by (total number of mutated microsatellite)/(total number of microsatellite) and compared the proportions among annotations. The proportions of mutated microsatellites were significantly lower in exonic and intronic regions but were higher in nongenic regions (Supplemental Fig. S7). Microsatellites in coding sequence (CDS) regions would evolve to be more stable to avoid mutations, and this would cause lower mutation rates in the CDS regions. Lower mutation rates in intronic regions suggests the influence of transcription-coupled repair (Gonzalez-Perez et al. 2019).
Figure 1.
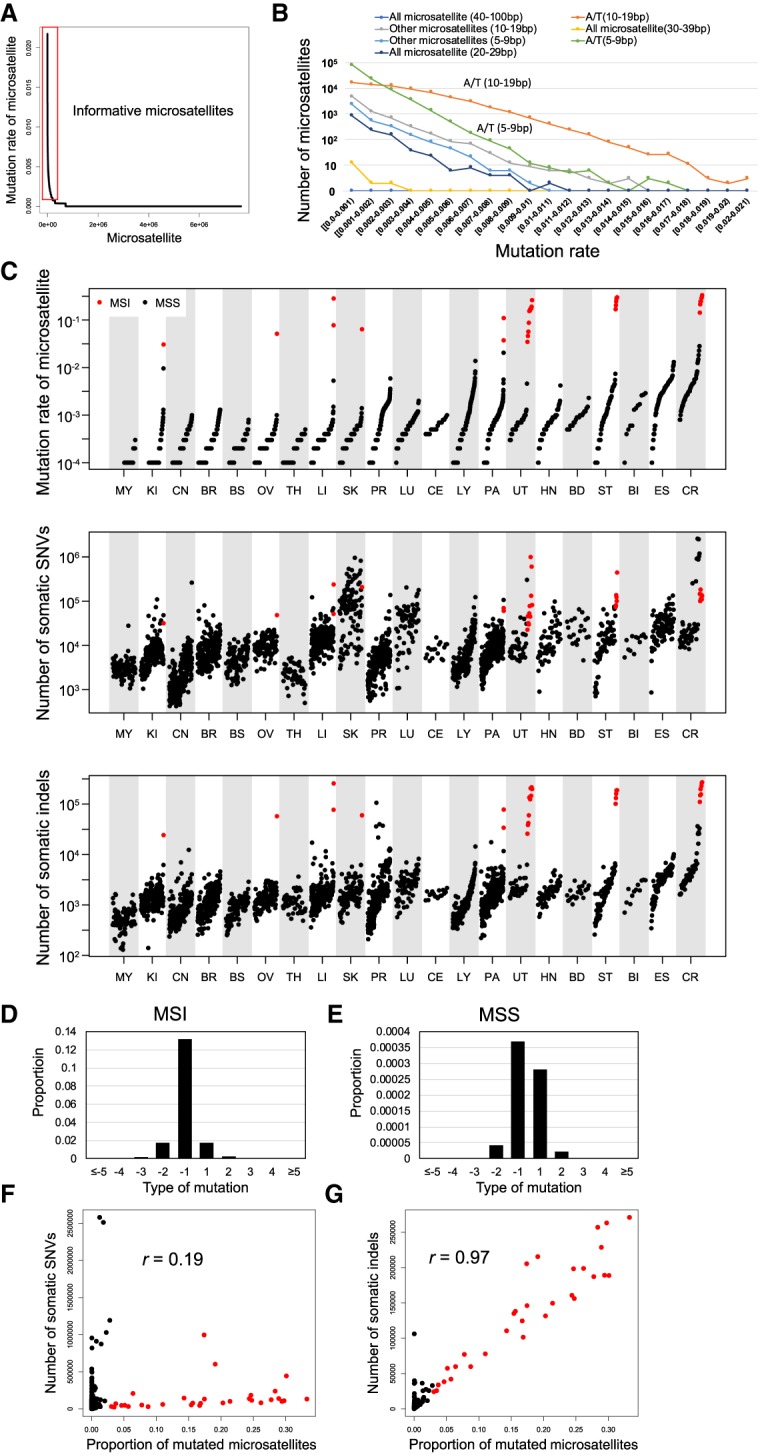
Pattern of somatic indels in microsatellite regions. (A) Mutation rate of each microsatellite; 7,650,128 microsatellites were sorted by the proportion of mutated samples. The red box indicates informative microsatellites defined in this study (proportion of mutated samples 0.001 or more). (B) Mutation rate among different microsatellites. Microsatellites are classified based on sequence of unit and length of each microsatellite in the reference genome. (C) Comparison of mutation rate of microsatellites and number of somatic SNVs and indels in different types of cancer. MSI samples are shown in red. (D,E) Pattern of insertions (positive change in repeat length in x-axis) and deletions (negative change in repeat length in x-axis) in microsatellites of the MSI (D) and MSS (E) samples. Correlation between the mutation rate of microsatellites and the number of somatic SNVs. Pearson product-moment correlation; r = 0.19, P-value = 1.6 × 10−23. MSI samples are shown in red. (F) Correlation between the mutation rate of microsatellites and the number of somatic SNVs. Pearson product-moment correlation; r = 0.19, P-value = 1.6 × 10−23. MSI samples are shown in red. (G) Correlation between the mutation rate of microsatellites and the number of somatic indels. Pearson product-moment correlation; r = 0.97, P-value < 1.0 × 10−200. MSI samples are shown in red.
We next defined samples with proportions of mutated microsatellites 0.03 or more as MSI samples (n = 31) at the genome-wide level (Fig. 1C; Supplemental Fig. S8A), and others as microsatellite stable (MSS) samples. As expected, CR, UT, and ST cancers had a larger number of MSI samples, but MSI was also observed in a minority of samples for liver, pancreas (PA), ovary (OV), kidney (KI), and skin (SK) cancers (Fig. 1C). The proportions of MSI samples were 11.9% for CR (7/59, 95% C.I. 4.9%–22.9%), 7.7% for ST (6/78, 95% C.I. 2.8%–16.0%), and 22.0% for UT (11/50, 95% C.I. 11.5%–36.0%) cancers, which are consistent with previous studies (Hampel et al. 2005, 2006; Arai et al. 2013; Bailey et al. 2018). We additionally analyzed Bethesda markers, which are a conventional marker set for MSI definition. Although the number of reads that mapped to these regions was quite small and we could not analyze their mutations in most of the samples, the pattern of Bethesda markers was consistent with that of all microsatellites (Supplemental Fig. S8B–D).
The mutation pattern of microsatellites was different between the MSI and MSS samples. In the MSI samples, deletions were more predominant compared with insertions (Fig. 1D,E). We compared the proportion of mutated microsatellites and the number of somatic SNVs and somatic indels (Fig. 1F,G). The number of somatic SNVs and indels showed significant strong correlation (r > 0.7) in three and 11 cancers, respectively (Supplemental Figs. S9, S10). In the overall samples, the numbers of indels (r = 0.97) showed stronger correlation with the proportion of mutated microsatellites than that of somatic SNVs (r = 0.19) (Fig. 1F,G). One may think that the high correlation between the indels and the proportion of mutated microsatellites is strongly influenced by specific regions. To examine whether SNVs and indels in specific regions were correlated with SNV and indel numbers at the whole-genome level, we divided the genome into 1-Mbp bins and tested the correlation between the number of all SNVs and indels and these within bins. All bins showed significantly high correlation (Supplemental Table S3), suggesting that the correlations between the proportion of mutated microsatellites and the number of SNVs/indels are a genome-wide pattern and are not influenced by specific genome regions. These results suggest that microsatellite and nonmicrosatellite indels are affected by common mechanisms of mutation and repair.
Mutability of microsatellites
Recent studies have suggested that epigenetic factors, such as DNA structures, and sequence motif influence the mutation rate (Fungtammasan et al. 2012; Woo and Li 2012; Chen and Zhang 2015; Lemmens et al. 2015; Tubbs and Nussenzweig 2017). However, little is known about factors that influence the mutability of microsatellites. We first analyzed the replication timings and microsatellite mutation rates (proportion of mutated samples for a microsatellite) (Fig. 2A–D; The ENCODE Project Consortium 2012). The late-replicating regions had lower mutation rates for IDs in MSS samples and insertions in MSI samples (Fig. 2A–C). However, an inverse pattern was observed for deletions of the MSI samples, with early-replicating regions having higher mutation rates (Fig. 2D).
Figure 2.

Analysis of mutation rate of each microsatellite. The mutation rates of 198,578 informative microsatellites were analyzed. (A–D) Association of replication timing with insertion and deletion rates. The edges of the boxes represent the 25th and 75th percentile values. The whiskers represent the most extreme data points, which are no more than 1.5 times the interquartile range from the boxes. (A) Insertions in MSS samples (χ2 test; P-value < 1 × 10−200); (B) insertions in MSI samples (χ2 test; P-value < 1 × 10−200); (C) deletions in MSS samples (χ2 test; P-value < 1 × 10−200); (D) deletions in MSI samples (χ2 test; P-value < 1 × 10−200). (E–H) Association of DNA shape with mutation rate. The top 1000 A/T microsatellites with 10- to 30-bp length were used for the analysis. The microsatellites were divided into the three categories based on the mutation rate. (E) Helix twist (HelT) of insertions in MSS samples; (F) the ·OH Radical Cleavage Intensity (ORChID2) of insertions in MSS samples; (G) Propeller Twist (ProT) of deletions in MSI samples; (H) slide of deletions in MSI samples. In this figure, we divided the microsatellites with mutation rates (0–0.001, 0.001–0.003, and greater than 0.003 for MSS; 0–0.4, 0.4–0.8, and greater than 0.8 for MSI) and showed the DNA shape values. The arrows show base positions with significant association between the DNA shape values and mutation rates (Supplemental Table S4).
For a more detailed analysis, we performed multiple regression. In our analysis, the majority of mutated microsatellites were A/T mononucleotide repeat as previously reported, suggesting that the fragility is primarily determined by the base composition (Fig. 1B; Maruvka et al. 2017). Therefore, to find other factors that associate with the mutation rate of microsatellites, we selected 1000 highly mutated A/T microsatellites of 10- to 30-bp length in the reference genome and analyzed them for IDs in the MSS and MSI samples. We considered replication timing, nuclear lamina binding region, G-quadruplexes, and predicted DNA shapes. Nuclear lamina binding regions are known to be associated with genomic fragile sites (Fungtammasan et al. 2012), whereas G-quadruplexes can cause replication errors (Lemmens et al. 2015). The impact of DNA shapes is not well known, but one DNA shape parameter ·OH Radical Cleavage Intensity [ORChID2]) has been reported to be associated with mutation rate of somatic indels (Chen and Zhang 2015). Multiple regression analyses for these factors showed that the length of microsatellite, replication timing, and several DNA shapes were significantly associated with the mutation rate of microsatellites (Supplemental Table S4).
The predicted DNA shapes of the flanking sequences were significantly associated with the proportion of mutated samples (Fig. 2E—H; Supplemental Table S4; Chiu et al. 2015). Several DNA shape features—such as ORChID2, Helix Twist (HelT), Opening, Minor Groove Width (MGW), Rise, Propeller Twist (ProT), Roll, and Slide—were significantly associated with the prevalence of IDs in microsatellite regions (Fig. 2E–H; Supplemental Fig. S11; Supplemental Table S4). The nuclear lamina binding region and G-quadruplexes were not significantly associated (Supplemental Table S4). The adjusted R2-values of the multiple regression analysis were 0.25 in the deletions of MSI, 0.14 in the insertions of MSI, 0.28 in the deletions of MSS, and 0.29 in the insertions of MSS (Supplemental Table S4).
Microsatellites are highly polymorphic and have also been used as genetic markers for population genetics studies (Ellegren 2004). To evaluate the genetic polymorphism, we detected germline polymorphism with MIVcall method (Supplemental Code 2) and estimated the heterozygosity of each microsatellite locus in normal tissues. The proportion of mutated samples in cancers and the heterozygosity in normal tissues was significantly correlated (Pearson product-moment correlation coefficient; r = 0.31, P-value < 10−16) (Supplemental Fig. S12), suggesting that genetic variations and somatic mutations are influenced by the same factors.
Highly mutated microsatellites
We compared mutability of each microsatellite between the MSI and MSS samples and selected the top 20 highly mutated microsatellites with the highest mutation rates (proportion of mutated samples for a microsatellite) (Supplemental Table S5). We performed a clustering analysis with these microsatellite markers and confirmed that they perfectly distinguished the MSI and MSS samples in CR, UT, and ST cancers (Fig. 3A–C; Supplemental Fig. S13).
Figure 3.

Highly mutated microsatellite markers. Result of clustering analysis with the top 20 microsatellites. (A) Colon/rectum (CR) cancer; (B) stomach (ST) cancer; (C) uterus (UT) cancer. Mutation status for the 20 microsatellites in each sample are shown. Colors indicates mutation status of each microsatellite (red, mutated; blue, unmutated; and white, unanalyzed owing to low depth). (D) Result of validation study in the independent CR and UT cancer cohort (n = 48). MSI status was defined by BAT25, BAT26, NR21, NR24, MONO27, D5S346, D17S250, and D2S123. Standard microsatellite markers, 18 highly mutated microsatellites, and homopolymers in coding regions were analyzed by MiSeq (Supplemental Fig. S19). Number of mutated samples in the MSI samples are shown in the bar plot (right). Colors indicates mutation status of each microsatellite (orange, mutated; white, unmutated; and gray, not tested). (MS) Microsatellite.
For validation in an independent cohort, we analyzed DNA from 36 CR and 12 UT cancer tissues, which were evaluated for MSI using the standard microsatellite markers (BAT25, BAT26, NR21, NR24, MONO27, D5S346, D17S250, and D2S123) at Saitama Cancer Center. These standard MSI markers identified 36 MSI-positive (24 CRs and 12 UTs) and 12 negative CRs. For these samples, we analyzed microsatellite mutations of 18 new microsatellite markers among the 20 highly mutated microsatellites and 11 recurrently mutated coding microsatellites (see below). Figure 3D summarizes the presence/absence of the indels in each of these highly mutated microsatellites. Three microsatellites were mutated in >90% of the validation samples (MS05, MS11, and MS20), which was a comparable sensitivity to conventional markers (BAT26, NR21, and NR24). Although the efficiency of these markers should be evaluated by a larger cohort, we consider that they have a technical advantage over known MSI markers in availability in WGS and combinations of the markers can be used as a new marker set.
Genes with large number of mutated microsatellites
To find genes with high mutation rates (proportion of mutated samples), we tested the total number of indels in microsatellites for each gene across the 21 tumor types. For the analysis, microsatellites within each gene (located between the transcription start and end sites) were used. We counted the number of mutated microsatellites and the total number of analyzed microsatellites for each gene and identified genes with larger numbers of mutated microsatellites compared with others. After adjusting for multiple testing, 1134 genes had significantly larger numbers of mutated microsatellites for at least one tissue (q-value < 0.01) (Supplemental Table S6). Of these genes, ALB, which is known to be highly expressed in LI, showed the largest number of mutated microsatellite (Fisher's exact test; q-value = 6.5 × 10−15, odds ratio = 65.1) in LI cancer (Supplemental Fig. S14). A previous study suggested that some cell lineage–specific highly expressed genes, including ALB in LI, had recurrent short indels (Imielinski et al. 2017). This result is consistent with the previous study, and strong DNA damage in cell lineage–specific highly expressed genes would influence mutation rate of microsatellites (Supplemental Table S6).
Recurrently mutated microsatellites in the coding regions
To compare mutation rates in the same microsatellite in the coding regions between MSS and MSI samples, we calculated the proportion of mutated samples for each microsatellite in coding regions (Fig. 4; Supplemental Table S7). Most of all mutations were frameshift indels (Supplemental Table S7). Microsatellites or repeat sequences in ACVR2A and TGFBR2, which have been reported to be frequently mutated in MSI tumors (Kim et al. 2013; Cortes-Ciriano et al. 2017; Maruvka et al. 2017), were recurrently mutated in 60% and 47% of the MSI samples, respectively. In addition, microsatellites in ASTE1, USF3, LIN1, and CDH26 were also mutated in >50% of the MSI samples. Mutations in microsatellites in COSMIC Cancer Genes (MSH6, JAK1, BLM, IL7R, and CSF3R) were identified as MSI-specific mutations. Of these, indels in MSH6, which is a MMR gene, are likely to cause the MSI phenotype (Boland and Goel 2010). Mutations in JAK1 in MSI cancers were reported to associate with tumor immune evasion (Albacker et al. 2017). In the MSS samples, microsatellites or repeat sequences in APC and TCF12 were mutated only in MSS samples, suggesting that these mutations cause cancer without MSI.
Figure 4.
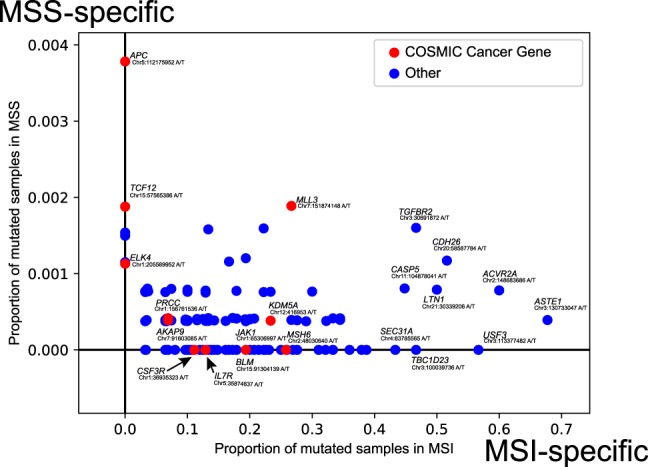
Proportion of mutated samples in coding microsatellites. (x-axis) Proportion of mutated samples in the MSI samples; (y-axis) proportion of mutated samples in the MSS samples. COSMIC cancer genes are shown in red.
Although many of the recurrently mutated coding microsatellites have been reported by whole-genome or exome sequencing studies (Supplemental Table S7; Kim et al. 2013; Cortes-Ciriano et al. 2017; Kondelin et al. 2017; Maruvka et al. 2017), our analysis identified new genes with recurrently mutated microsatellites. Of these, the GINS1 gene encodes a subunit of DNA replication complex (Ueno et al. 2005). MBD4 has been reported to contribute to tumorigenesis and work as a modifier of mutation in MMR-deficient cancer (Tricarico et al. 2015). BLM is included in the COSMIC Cancer Gene database and has functions in DNA replication and DNA double-strand break repair (Patel et al. 2017). These results suggest that mutations in microsatellites can work as driver events.
Association between microsatellite mutations and gene expression
To further examine the functional impact of mutations in microsatellites, we tested the association between the mutations and gene expression levels. We analyzed CR, ST, and UT, which had highly mutated microsatellites, and focused on microsatellites in the promoter and UTR regions. We compared gene expression levels between samples with and without mutations in promoter and UTR microsatellites. However, after adjusting for multiple testing, no significant association was observed (Supplemental Tables S8–S10).
MMR and proofreading genes
We analyzed the association between the proportion of mutated microsatellites, and somatic and germline variants of eight DNA repair genes (MLH1, MLH3, MSH2, MSH3, MSH6, PMS2, POLE and POLD1). First, we focused on stop gain, splice site, nonsense mutations, and gene-disrupting structural variations (SVs) in tumor and matched normal samples (Fig. 5A–D). Two samples in CR and UT had loss-of-function germline mutations in the MSH2 or PSM2 gene, suggesting that they cause Lynch syndrome in these patients (Fig. 5A,C; Boland and Goel 2010). The number of samples with somatic SNVs and indels in these genes were significantly enriched in the MSI samples (Fisher's exact test; CR: P-value = 2.0 × 10−8, odds ratio = 10.6; ST: P-value = 1.2 × 10−4, odds ratio = 49.0; UT: P-value = 1.4 × 10−5, odds ratio = 40.6), whereas those with germline variants and SVs were not significantly enriched. These results suggest that most of the MSI phenotypes in cancer were mainly caused by somatic short indels or somatic SNVs. Germline SVs of PMS2 were frequently observed in MSS tumors, indicating that PMS2 could have a lower impact on DNA MMR deficiency or MSI (Senter et al. 2008).
Figure 5.

Mutation in mismatch repair (MMR) and proofreading genes. (A) Colon/rectum (CR) cancer; (B) stomach (ST) cancer; (C) uterus (UT) cancer; (D) other MSI cancers. LI, liver; SK, skin; OV, ovary; PA, pancreas; and KI, kidney. Mutated genes and mutation rate of microsatellites are shown.
Most MSI samples had larger numbers of somatic SNVs (Fig. 1C) owing to functional deficiency of MMR genes. However, 40 MSS samples had larger numbers of somatic SNVs than the average number of SNVs in the MSI samples (151,816.6 SNVs). Of these, eight had somatic missense mutation in the exonuclease domain of POLE (residues 268–471) (Supplemental Fig. S15; Supplemental Table S11; Church et al. 2013; Shinbrot et al. 2014), suggesting that exonuclease domain mutations of POLE were associated with a large number of SNVs in MSS, not MSI.
Association with somatic mutational signatures in PCAWG
We compared mutational signatures found in single base substitution (SBS), doublet base substitution (DBS), as well as IDs between the MSI and MSS samples (Fig. 6A–C; Supplemental Fig. S16; Supplemental Table S12). The PACWG signature analysis detected 49 SBS, 11 DBS, and 17 ID signatures (Alexandrov et al. 2020). We compared the fraction of each mutational signature between MSI and MSS samples in CR, ST, and UT and found that six SBS signatures (SBS5, SBS15, SBS20, SBS21, SBS26, and SBS44), one ID signature (ID2), and four DBS signatures (DBS3, DBS7, DBS8, and DBS10) were significantly different among the MSI and MSS samples in at least one cancer type (Wilcoxon signed-rank test, q-value < 0.05). Except for DBS3 and DBS8, most of these mutational signatures have been reported to be associated with tumors having defective DNA MMR (Alexandrov et al. 2020). We found DBS3 and DBS8 to be associated with MSI. DBS3 was also associated with the mutations in exonuclease domain of POLE in the current study (Wilcoxon signed-rank test, q-value < 0.05) (Supplemental Table S13), and no etiology has been proposed for DBS8, which was observed in esophagus (ES) adenocarcinoma and CR (Alexandrov et al. 2020). In the ID signatures, the fraction of ID2 (A/T deletion) was significantly different between MSI and MSS in CR, ST, and UT, which is consistent with an excess of A/T indels in the microsatellite regions (Fig. 1B).
Figure 6.

Comparison of mutational signatures between the MSI and MSS samples. (A) CR cancer; (B) ST cancer; (C) UT cancer. Signatures showing significant difference between the MSI and MSS samples are shown in rectangles in the legends (Wilcoxon signed-rank test, q-value < 0.05).
Neoantigen load from the microsatellite or repeat coding regions
MSI cancers are known to show specific immune reactions such as Crohn-like reaction and diffused infiltration of lymphocytes in pathology (Umar et al. 2004), and PD-1 inhibiting immune therapy is a highly effective treatment for all types of tumors showing MSI (Le et al. 2017). Its specific immune reaction should be related to neoantigen burden, and we thus calculated the neoantigen burdens of MSI and MSS tumors by using somatic mutations and HLA genotypes, taking into account neopeptides generated from indel or frameshift mutations of the coding microsatellites. The number of predicted neoantigens in MSI tumors (median 393) was significantly higher than MSS tumors (median 11; P-value = 5.9 × 10−20) (Supplemental Fig. S17), which is consistent with the mutational burden. Although 95% of neoantigens were derived from SNVs in MSS tumors, in MSI tumors, 51% of the predicted neoantigens were derived from short indels and 5% were derived from indels of the microsatellites. To examine their relevance to tumor immunology, we analyzed RNA-seq of 967 tumor samples in PCAWG. The number of neoantigens generated from microsatellite indels was zero for 938 tumors, one to 10 for 15 tumors, and greater than 10 for 14 tumors. mRNA expression levels of antitumor immunity genes GZMA and IFNG were positively correlated with microsatellite neoantigens (P-values < 0.05, Jonckheere-Terpstra trend test) (Supplemental Fig. S17). This suggests that microsatellite neoantigens may be involved in antitumor attack by immune cells.
Discussion
Because of the clinical importance of MSI phenotypes, exome sequencing and small-scale WGS studies were performed for the MSI samples (Kim et al. 2013; Cortes-Ciriano et al. 2017; Kondelin et al. 2017; Maruvka et al. 2017). These studies identified recurrently mutated microsatellites and driver genes mainly located in coding regions, as well as created an algorithm to find MSI with smaller number of microsatellite sets. However, detailed analysis for factors that influence mutation rate and validation of the selected microsatellite marker sets in independent cohorts was limited. Here, we performed an analysis of microsatellite mutations in the largest WGS cohort with the largest number of microsatellites to date, so as to characterize microsatellite mutations and MSI tumors at the genome-wide level.
To identify microsatellite regions in the human genome, we used results from three types of software (Tandem Repeat Finder, MS Detector, and MISA) (Benson 1999; Girgis and Sheetlin 2013; Hause et al. 2016). After filtering, we obtained 9,292,677 microsatellites in the genome for analysis. Among the methods for detecting microsatellites, the MISA software identified the largest number of microsatellites (Hause et al. 2016). Although most of them were short and may not be considered as microsatellites by the other two methods, they contained highly mutated repeat regions. It has been reported that the rate of mutation of longer microsatellites is higher than that of shorter microsatellites (Sun et al. 2012). However, current short-read sequencing technologies are unable to analyze longer microsatellites. Indeed, we could not obtain sufficient number of reads for Bethesda markers (Supplemental Fig. S8). We therefore decided to prioritize shorter repeats as the main targets for the current WGS study. Alternatively, in this study, we detected highly mutated short microsatellites (Fig. 3; Supplemental Table S5; Supplemental Fig. S13), and they could be useful for clinical diagnosis of MSI with current short-read technologies.
The analysis of WGS and the validation study in an independent cohort found 20 novel microsatellite markers (Fig. 3), which can be used to predict tumors with MSI. In addition to the MSI samples, our analysis found samples with larger number of SNVs in MSS (Supplemental Fig. S15). As the analysis of neoantigens showed that SNVs can also produce a larger number of neoantigens (Supplemental Fig. S17), identification of these samples is also important for diagnosis. The analysis of MSS samples with a larger number of SNVs showed that mutations in the exonuclease domain of POLE can partly explain the high mutation rate of SNVs, instead of indels and microsatellites (Supplemental Fig. S15). Therefore, analysis of mutations in the POLE gene in MSS samples can identify more tumors with high mutational burdens (Mehnert et al. 2016).
Our WGS analysis found high rates of short deletions in the microsatellites of both the MSI and MSS samples (Fig. 1D,E). The excess of deletion events was also observed in previous studies (Maruvka et al. 2017). Therefore, the excess of short deletions should not be owing to a bias of our mutation calling method and can be considered as a common feature of cancers. A microsatellite mutation model suggests that deletions are generated by a misalignment loop in the template strand, and insertions are subsequently generated in the nascent strand (Ellegren 2004). During the DNA replication of cancer cells, template strands could exist as single strands for a longer period compared with nascent strands, resulting in a higher chance to generate misaligned loop structures, which would induce larger numbers of deletions.
The analysis of replication timing showed a different pattern between deletions in MSI and others (Fig. 2A–D; Supplemental Table S4). As observed in the SNVs, replication timing and mutation rate were positively correlated with the IDs of MSS samples, as well as insertions of MSI samples (Fig. 2A–D). It is suggested that early-replicating regions are more accessible for DNA repair machineries, resulting in more chances for repair (Tubbs and Nussenzweig 2017). However, deletions in microsatellites were enriched in the early-replicating regions of MSI samples (Fig. 2D). A recent exome sequencing study also reported the inverse correlation between the microsatellite indels and replication timing in MSI tumors (Maruvka et al. 2017). Because MSI tumors should have defects in their DNA MMR machinery, this result would reflect the pattern of mutation without DNA MMR. In early replication, template strands may exist as single strands for a longer period, facilitating the occurrence of deletions.
In addition to replication timing, DNA shape parameters were also associated with the mutation rate of microsatellites. Microsatellites with lower HelT, higher ProT, higher Roll, and higher Slide had higher insertion rates (Fig. 2E–H; Supplemental Fig. S11). Microsatellites with higher HelT, higher Opening, higher ProT, higher Roll, and lower Slide had higher deletion rates (Fig. 2E–H; Supplemental Fig. S11). Because DNA shapes were associated in both the MSI and the MSS tumors, they would affect the fragility of DNA strand and mainly influence the mutation generation instead of the repair process.
In the present study, we considered that the mutation rate of microsatellites is mainly influenced by the following: fragility of DNA sequences (length and unit type of microsatellite, DNA shape) (Figs. 1B, 2E–H), activity of DNA repair machinery (mutations or activities in MMR machinery genes) (Fig. 5), DNA damage against cell lineage–specific highly expressed genes (Supplemental Fig. S14; Supplemental Table S6), and accessibility of DNA repair machinery (DNA replication timing) (Fig. 2A–D). Furthermore, because the mutation rate in cancer was correlated with the heterozygosity of germline variations, these factors may also affect the mutation rate in germline variations (Supplemental Fig. S12). Of these factors, activity of DNA repair machinery affects mutation rate among samples, and other factors influence the difference of mutation rate among microsatellite loci. We consider that the strongest factors are length and unit type of microsatellites. In addition, local replication timing, which reflects accessibility of DNA repair machinery, and DNA shape also influence the mutation rate. The effect of DNA damage against cell lineage–specific highly expressed genes is limited to microsatellites within specific genes in specific tissues (such as ALB gene in LI).
The current study analyzed somatic indels in microsatellite regions in the largest WGS cohort to date. We found a microsatellite marker set to detect MSI, factors that influence the mutation rate of microsatellite, genes with recurrently mutated microsatellites, and the influence somatic mutations in MMR and proofreading genes have on MSI. Our analysis provides a mutational landscape of microsatellites in cancer samples for future clinical applications.
Methods
Samples and data
WGS data were obtained by the ICGC pan-cancer project (The International Cancer Genome Consortium 2010; The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020). The list of analyzed samples is shown in the Supplemental Tables S2 and S14. Data sets of somatic point mutations, short indels, SVs, and copy number alterations were generated as part of the PCAWG project (The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020). Overall, WGS data from 2834 donors (2913 tumor samples) are represented in the PCAWG data sets, spanning a range of cancer types (bladder, sarcoma, breast, LI-biliary, cervix, leukemia, CR, lymphoma, prostate, ES, ST, central nervous system, head/neck, KI, lung, melanoma, OV, PA, thyroid, and UT). Mapping reads to GRCh37 was performed by PCAWG, and we used the generated BAM files (The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020). Realigning the reads to GRCh38 would not significantly affect the conclusions because we used microsatellites in nonrepetitive regions (see below). The consensus somatic SNVs and short indels in PCAWG samples were determined using different algorithms; calls made by at least two algorithms were used in downstream analyses (The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020).
Definition of microsatellite regions for the analysis
We determined microsatellite regions using MsDetector, Tandem Repeat Finder, and MISA software (Benson 1999; Girgis and Sheetlin 2013; Hause et al. 2016). Microsatellite regions defined by the Tandem Repeat Finder were obtained from the UCSC Genome Browser (Girgis and Sheetlin 2013). Identification of microsatellites with MISA was performed by (unit size) = 1–5, (minimum number of repeats) = 5 and (max difference between two microsatellites) = 10. Because these three methods used different algorithms to define microsatellites, we first defined the repeat unit of each microsatellite. We divided each region by different lengths (1–6 bp) and calculated the entropy of the character string. The length with the lowest entropy was selected as the unit length of each microsatellite region. For the analysis of the microsatellite, we filtered microsatellite regions according to the following criteria: (1) the proportion of the most frequent unit ≥0.8, and (2) distance between closest neighboring microsatellite ≥30 bp. (3) If the microsatellite regions were detected by two or more methods, we selected the longest one and discarded others; (4) upstream and downstream flanking sequences (100 bp) of each microsatellite were mapped against human reference genome (GRCh37) by BLAT software (Kent 2002) with the options of -stepSize = 5 and -repMatch = 2253, and microsatellites that had ≥90 bp of flanking sequences mapped to different positions were removed; and (5) the length of microsatellite region was <80 bp. As a result of this selection procedure, 9,292,677 microsatellites, in which 8,817,054 were autosomal, remained and were used for the subsequent analyses (Supplemental Figs. S1, S18).
Error rate estimation of each repeat unit
To identify somatic indels in the microsatellite regions, we first estimated the error rate of the different types of repeat units. The type of microsatellites was defined by length of microsatellite region in the reference genome and repeat unit. Microsatellites were categorized by length (6–9, 10–19, 20–29, 30–39, and 40–100 bp), and repeat unit and error rates were estimated for each category (see Supplemental Table S1). For this purpose, we used data from Chr X of 32 male normal samples because Chr X is hemizygous, and error rates can be estimated without the influence of heterozygous polymorphisms (Fujimoto et al. 2010; Maruvka et al. 2017). We assumed that the major read type is the true genotype and that others were errors. For example, when 10, three, and one reads contain (AT)8, (AT)7, and (AT)9 in a microsatellite locus, we considered (AT)8 the true genotype and three deletion error reads ((AT)8 to (AT)7) and one insertion error read ((AT)8 to (AT)9) observed. The estimated error rates are shown in Supplemental Figure S3.
Identification of change of repeat unit from whole-genome sequence (algorithm in MIMcall)
Microsatellites are repeat sequences and mapping errors can influence the accuracy of detection. To remove possible mapping errors, we removed improper pairs and reads with low mapping quality (less than 30), as well as reads with large (>550-bp) or small (<100-bp) read pair distance.
We counted the number of repeat units in each microsatellite region. We then determined the genotype of the matched normal tissues and detected somatic indels by comparing the genotype of the normal and cancer samples. To distinguish the mutation or variation from sequencing errors, we incorporated the binomial distribution with the estimated error rates (Supplemental Fig. S3) and calculated a likelihood for each variant candidate.
For normal samples, we calculated the likelihood for the second most frequent number of repeat:
where n is the total number of reads that cover the microsatellite, r is the number of reads containing ith repeat, and pi is the estimated error rate of the ith repeat. If the likelihood is lower than a threshold value, the genotype was assumed to be heterozygous for the major repeat and second major repeat. We next calculated the likelihood for the number of repeats in the cancer. If the likelihood of the nongermline repeat was lower than a threshold value, we defined the repeat as a somatic indel candidate.
To find the appropriate likelihood threshold values, we applied this algorithm on data from other male Chr X. Based on the estimated false-positive and false-negative rates, we set −1 and −8 for germline genotyping and somatic mutation calling (Supplemental Fig. S4), respectively. Based on the comparison, we set L = −8 for tumor and L = −1 for matched normal samples. We selected microsatellites that were covered by five or more reads in both the cancer and matched normal samples. Additionally, we selected somatic indels with variant allele frequencies in cancer or 0.15 or more and number of support reads in cancer of two or more and one or less in normal samples.
Estimation of false discovery rate
We randomly selected 29 somatic MS mutations detected in LI cancer samples RK001, RK249, and RK308 and performed validation with a previously reported method (Schuelke 2000). Amplicons were analyzed using the ABI PRISM 3100 genetic analyzer (Applied Biosystems), and GeneMapper software (Applied Biosystems). Validation for the selected microsatellites was also performed using the Sanger sequencing method (Supplemental Fig. S6).
Selection of highly mutated microsatellites
To select microsatellites, we compared the number of mutated samples in MSI and MSS samples for each microsatellite using a Fisher's exact test. Nine microsatellites with odds ratio [(number of mutated samples in the MSI)/(number of unmutated samples in the MSI)/(number of mutated samples in the MSS)/(number of unmutated samples in the MSS)] ≥ 500 and proportion of the mutated samples in the MSI samples of 0.8 or more were selected and genotyped in the additional samples. Three microsatellites were selected from highly mutated microsatellites in the MSI samples. Ten microsatellites were selected from recurrently mutated coding microsatellites.
Validation study of the CR and UT cancer cohort
For new microsatellite markers or mutated regions, we designed primers to amplify them (primer information in Supplemental Table S15) and performed multiplex-PCR on DNA from 48 frozen cancer tissues and their corresponding normal tissues, which were collected at the Saitama Cancer Center, Japan. Ethical committees at RIKEN, the Saitama Cancer Center, and all groups participating in this study approved this work. Standardized MSI analysis was performed using fluorescence-based PCR, as described previously (Ishikubo et al. 2004) at the Saitama Cancer Center. MSI status was determined using five Bethesda markers (BAT25, BAT26, D5S346, D2S123, and D17S250) or Promega panel (BAT25, BAT26, NR21, NR24, and MONO27) and classified as MSI-H (two or more markers shown to be unstable), MSI-low (MSI-L; only one marker unstable), and MSS (no markers unstable). MSI-positive markers were re-examined at least twice to confirm the results. MSI-L was included with MSS in this study. We selected 24 MSI-positive CR cancers, 12 MSI-positive UT cancers, and 12 MSI-negative CR cancers for the validation study. Multiplex-PCR of these new MSI markers was performed, and pools of the amplicons were sequenced by MiSeq after Illumina adaptor ligation. We then determined indel mutations by read number distributions mapped to the target microsatellite regions (Supplemental Fig. S19).
Threshold determination for MSI
Because the mutation status of conventional MSI markers could not be obtained (see Supplemental Fig. S8), we needed to determine the threshold value to select MSI. We first excluded CR, ST, and UT cancers (n = 186) from all samples (n = 2717). We assumed that the other cancers contained negligible number of MSI samples. We then calculated the average and standard deviation of the mutation rates. We also assumed that the distribution of the mutation rates follows a normal distribution with the obtained average and standard deviation. The 99.99th percentile of the normal distribution was 0.0254. Therefore, we adapted 0.03 (slightly conservative value from the 99.99th percentile) for the criteria for MSI in genome-wide level, and 31 samples were defined as MSI in this study. No CR, ST, or UT cancers were used to determine the threshold value; however, the value still gave a reasonable grouping for CR, ST, and UT cancers (please see Fig. 1B; Supplemental Fig. S8).
Comparison of mutational signatures between the MSS and MSI
Mutational signatures and their proportions were obtained from the result of the PCAWAG signature working group (Alexandrov et al. 2020). The proportion of each signature was compared between the MSI and MSS samples in CR, UT, and ST using the Wilcoxon signed-rank test. Multiple testing adjustment was performed using the Benjamini and Hochberg's FDR method (Benjamini and Hochberg 1995).
Correlation between the total number of SNVs/indels and the number of SNVs/indels in 1-Mbp bins
To examine whether SNVs and indels in specific regions were correlated with total number of SNV and indel, we divided the genome into 1-Mbp bins and tested the correlation between the number of all SNVs and indels and these within bins. The Pearson's correlation coefficient was calculated and tested by R (R Core Team 2017).
Identification of genes with recurrently mutated microsatellites
To identify highly mutated genes, we compared the mutation rate of microsatellites in each gene. We compared the total number of analyzed microsatellites and total number of mutated microsatellites in introns and exons in each gene. We also counted the total number of analyzed microsatellites and total number of mutated microsatellites in the entire genome in MSS samples for each cancer type; (total number of mutated microsatellites in gene i in all MSS samples in cancer j)/(total number of unmutated microsatellite in gene i in all MSS samples in cancer j) and (total number of mutated microsatellite in entire genome in all MSS samples in cancer j)/(total number of unmutated microsatellite in entire genome i in all MSS samples in cancer j), and these were compared with a Fisher's exact test. Multiple testing adjustment was performed using the Benjamini and Hochberg's FDR method (Benjamini and Hochberg 1995), and from this analysis, we could obtain genes with larger numbers of mutated microsatellites compared to the entire genome.
Driver genes shown in Figure 4 were based on COSMIC database (https://cancer.sanger.ac.uk/cosmic).
Association between microsatellite mutations and gene expression
To further examine the functional impact of mutations of microsatellites, we tested the association between the mutations and gene expression levels. First, we selected microsatellite in promoter (1000 bp from transcription start sites) and UTR regions. Then, we compared gene expression levels between samples with and without mutations in promoter and UTR microsatellites (syn5553985) (The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020). A statistical test was performed using a Mann–Whitney U test with Python 3 (https://docs.python.org/3.8/). We applied this analysis for CR, ST, and UT cancers, which have highly mutated microsatellites. In the promoter and UTR regions, 799, 260, and 784 genes had mutated microsatellites in CT, ST, and UT, and adjustment of the multiple testing was performed by the number of tested genes.
Analysis of epigenetic factors on mutability of microsatellites
To find the factors that influence the mutability of microsatellites, we considered replication timing, nuclear lamina binding region, G-quadruplexes, and predicted DNA shapes (replication timing: https://genome.ucsc.edu/cgi-bin/hgFileUi?db=hg19&g=wgEncodeUwRepliSeq; DNA shapes: http://rohsdb.cmb.usc.edu/GBshape/; and nuclear lamina binding region: https://static-content.springer.com/esm/art%3A10.1038%2Fnature06947/MediaObjects/41586_2008_BFnature06947_MOESM252_ESM.txt). G-quadruplex was estimated using software (Lemmens et al. 2015; https://github.com/dariober/bioinformatics-cafe/blob/master/fastaRegexFinder.py). For the replication timing, we downloaded data of HepG2, K562, MCF-7, SK-N-SH, and GM12878 cells. We averaged the replication timing within 1-Mbp bins for each cell line, and bins with standard deviation of 15 or less were used for the analysis. The presence or absence of a nuclear lamina binding region and G-quadruplexes within ±1000 bp from the start and end of microsatellites was examined. The predicted DNA shapes (Buckle, HelT, MGW, ORChID2, Opening, ProT, Rise, Roll, Shear, Shift, Slide, Stagger, Stretch, and Tilt) of ±5 bp from the start and end of each microsatellite were used for the analysis. We performed a multiple regression analysis of the parameters with the lm function of the R software, and parameter selection was performed with the step() function (R Core Team 2017). We tested 144 parameters for IDs of MSI and MSS samples. Therefore, we adjusted P-values by 144.
Prediction of neoantigens
HLA genotyping from WGS data was generated as part of the PCAWG project. Somatic point mutations, non-MS indels detected by the PCAWG project, and MS indels detected by our method were combined and annotated using ANNOVAR. Mutant peptides of the length of eight to 11 residues were assessed for their binding affinity (IC50) to the HLA class I of matched patients using NetMHCpan-3.0 (Nielsen and Andreatta 2016). Mutant peptides of IC50 < 50 nM were predicted as neoantigens.
Data access
The result of MSI call was released from ICGC (https://dcc.icgc.org/releases/PCAWG/msi). Source code for microsatellite analysis can be found at GitHub (https://github.com/afujimoto/MIMcall) and as Supplemental Code (MIMcall, Supplemental Code 1; MIVcall, Supplemental Code 2). Genotype table of all identified mutations is provided as supplemental material (Supplemental Table S16).
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
This work was supported partially by grant-in-aid for RIKEN CGM and IMS, grant-in-aid for scientific research on innovative areas from Japan Society for the Promotion of Science London grants (25134717, 25670375, 23114001, 15H04814), Princess Takamatsu Cancer Research Fund, Project for Cancer Research and Therapeutic Evolution (P-CREATE) (grant number 16cm0106519h0001, to H.N.), and Platform Program for Promotion of Genome Medicine (grant number 18km0405207h0003, to A.F.) in the Japan Agency for Medical Research and Development (AMED). The super-computing resource SHIROKANE was provided by the Human Genome Center, The University of Tokyo (http://sc.hgc.jp/shirokane.html). We thank Prof. Michael R. Stratton, Steven G. Rosen, who led PCAWG Signature working group, PCAWG steering committee members, and ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Network for their assistance and coordination. We also thank Prof. Shu Narumiya for his constructive comments and encouragement.
Author contributions: Study design was by A.F. and H.N. Data analysis was by A.F., M.F., T.H., Y.S., S.M., S.I., and H.N. Molecular analysis was by K.M., A.O.-S., K.N., G.Y., K.A., and H.N. Manuscript writing was by A.F., M.F., J.H.W., and H.N.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.255026.119.
Freely available online through the Genome Research Open Access option.
References
- Albacker LA, Wu J, Smith P, Warmuth M, Stephens PJ, Zhu P, Yu L, Chmielecki J. 2017. Loss of function JAK1 mutations occur at high frequency in cancers with microsatellite instability and are suggestive of immune evasion. PLoS One 12: e0176181 10.1371/journal.pone.0176181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, et al. 2016. Mutational signatures associated with tobacco smoking in human cancer. Science 354: 618–622. 10.1126/science.aag0299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom E, et al. 2020. The repertoire of mutational signatures in human cancer. Nature 578: 94–101. 10.1038/s41586-020-1943-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Sakurai U, Sawabe M, Honma N, Aida J, Ushio Y, Kanazawa N, Kuroiwa K, Takubo K. 2013. Frequent microsatellite instability in papillary and solid-type, poorly differentiated adenocarcinomas of the stomach. Gastric Cancer 16: 505–512. 10.1007/s10120-012-0226-6 [DOI] [PubMed] [Google Scholar]
- Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. 2018. Comprehensive characterization of cancer driver genes and mutations. Cell 174: 1034–1035. 10.1016/j.cell.2018.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57: 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27: 573–580. 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland CR, Goel A. 2010. Microsatellite instability in colorectal cancer. Gastroenterology 138: 2073–2087.e3. 10.1053/j.gastro.2009.12.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Zhang L. 2015. The pattern of DNA cleavage intensity around indels. Sci Rep 5: 8333 10.1038/srep08333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu TP, Yang L, Zhou T, Main BJ, Parker SC, Nuzhdin SV, Tullius TD, Rohs R. 2015. GBshape: a genome browser database for DNA shape annotations. Nucleic Acids Res 43: D103–D109. 10.1093/nar/gku977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church DN, Briggs SE, Palles C, Domingo E, Kearsey SJ, Grimes JM, Gorman M, Martin L, Howarth KM, Hodgson SV, et al. 2013. DNA polymerase ε and δ exonuclease domain mutations in endometrial cancer. Hum Mol Genet 22: 2820–2828. 10.1093/hmg/ddt131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Ciriano I, Lee S, Park WY, Kim TM, Park PJ. 2017. A molecular portrait of microsatellite instability across multiple cancers. Nat Commun 8: 15180 10.1038/ncomms15180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H. 2004. Microsatellites: simple sequences with complex evolution. Nat Rev Genet 5: 435–445. 10.1038/nrg1348 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AD, Gacita A, Wood LD, Ma F, Xing D, Kim MS, Manda SS, Abril G, Pereira G, Makohon-Moore A, et al. 2015. Widespread somatic L1 retrotransposition occurs early during gastrointestinal cancer evolution. Genome Res 25: 1536–1545. 10.1101/gr.196238.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto A, Nakagawa H, Hosono N, Nakano K, Abe T, Boroevich KA, Nagasaki M, Yamaguchi R, Shibuya T, Kubo M, et al. 2010. Whole-genome sequencing and comprehensive variant analysis of a Japanese individual using massively parallel sequencing. Nat Genet 42: 931–936. 10.1038/ng.691 [DOI] [PubMed] [Google Scholar]
- Fujimoto A, Furuta M, Totoki Y, Tsunoda T, Kato M, Shiraishi Y, Tanaka H, Taniguchi H, Kawakami Y, Ueno M, et al. 2016. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet 48: 500–509. 10.1038/ng.3547 [DOI] [PubMed] [Google Scholar]
- Fungtammasan A, Walsh E, Chiaromonte F, Eckert KA, Makova KD. 2012. A genome-wide analysis of common fragile sites: What features determine chromosomal instability in the human genome? Genome Res 22: 993–1005. 10.1101/gr.134395.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiersbach KB, Samowitz WS. 2011. Microsatellite instability and colorectal cancer. Arch Pathol Lab Med 135: 1269–1277. 10.5858/arpa.2011-0035-RA [DOI] [PubMed] [Google Scholar]
- Girgis HZ, Sheetlin SL. 2013. MsDetector: toward a standard computational tool for DNA microsatellites detection. Nucleic Acids Res 41: e22 10.1093/nar/gks881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A, Sabarinathan R, Lopez-Bigas N. 2019. Local determinants of the mutational landscape of the human genome. Cell 177: 101–114. 10.1016/j.cell.2019.02.051 [DOI] [PubMed] [Google Scholar]
- Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, et al. 2005. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 352: 1851–1860. 10.1056/NEJMoa043146 [DOI] [PubMed] [Google Scholar]
- Hampel H, Frankel W, Panescu J, Lockman J, Sotamaa K, Fix D, Comeras I, La Jeunesse J, Nakagawa H, Westman JA, et al. 2006. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res 66: 7810–7817. 10.1158/0008-5472.CAN-06-1114 [DOI] [PubMed] [Google Scholar]
- Hause RJ, Pritchard CC, Shendure J, Salipante SJ. 2016. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med 22: 1342–1350. 10.1038/nm.4191 [DOI] [PubMed] [Google Scholar]
- Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, Patch AM, Kakavand H, Alexandrov LB, Burke H, et al. 2017. Whole-genome landscapes of major melanoma subtypes. Nature 545: 175–180. 10.1038/nature22071 [DOI] [PubMed] [Google Scholar]
- Helman E, Lawrence MS, Stewart C, Sougnez C, Getz G, Meyerson M. 2014. Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res 24: 1053–1063. 10.1101/gr.163659.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. 2020. Pan-cancer analysis of whole genomes. Nature 578: 82–93. 10.1038/s41586-020-1969-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Guo G, Meyerson M. 2017. Insertions and deletions target lineage-defining genes in human cancers. Cell 168: 460–472.e14. 10.1016/j.cell.2016.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Cancer Genome Consortium. 2010. International network of cancer genome projects. Nature 464: 993–998. 10.1038/nature08987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikubo T, Nishimura Y, Yamaguchi K, Khansuwan U, Arai Y, Kobayashi T, Ohkura Y, Hashiguchi Y, Tanaka Y, Akagi K. 2004. The clinical features of rectal cancers with high-frequency microsatellite instability (MSI-H) in Japanese males. Cancer Lett 216: 55–62. 10.1016/j.canlet.2004.07.017 [DOI] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664. 10.1101/gr.229202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TM, Laird PW, Park PJ. 2013. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell 155: 858–868. 10.1016/j.cell.2013.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondelin J, Gylfe AE, Lundgren S, Tanskanen T, Hamberg J, Aavikko M, Palin K, Ristolainen H, Katainen R, Kaasinen E, et al. 2017. Comprehensive evaluation of protein coding mononucleotide microsatellites in microsatellite-unstable colorectal cancer. Cancer Res 77: 4078–4088. 10.1158/0008-5472.CAN-17-0682 [DOI] [PubMed] [Google Scholar]
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. 2017. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357: 409–413. 10.1126/science.aan6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmens B, van Schendel R, Tijsterman M. 2015. Mutagenic consequences of a single G-quadruplex demonstrate mitotic inheritance of DNA replication fork barriers. Nat Commun 6: 8909 10.1038/ncomms9909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruvka YE, Mouw KW, Karlic R, Parasuraman P, Kamburov A, Polak P, Haradhvala NJ, Hess JM, Rheinbay E, Brody Y, et al. 2017. Analysis of somatic microsatellite indels identifies driver events in human tumors. Nat Biotechnol 35: 951–959. 10.1038/nbt.3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehnert JM, Panda A, Zhong H, Hirshfield K, Damare S, Lane K, Sokol L, Stein MN, Rodriguez-Rodriquez L, Kaufman HL, et al. 2016. Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J Clin Invest 126: 2334–2340. 10.1172/JCI84940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Andreatta M. 2016. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med 8: 33 10.1186/s13073-016-0288-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, et al. 2016. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534: 47–54. 10.1038/nature17676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, Gröbner S, Segura-Wang M, Zichner T, Rudneva VA, et al. 2017. The whole-genome landscape of medulloblastoma subtypes. Nature 547: 311–317. 10.1038/nature22973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel DS, Misenko SM, Her J, Bunting SF. 2017. BLM helicase regulates DNA repair by counteracting RAD51 loading at DNA double-strand break sites. J Cell Biol 216: 3521–3534. 10.1083/jcb.201703144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Schuelke M. 2000. An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18: 233–234. 10.1038/72708 [DOI] [PubMed] [Google Scholar]
- Senter L, Clendenning M, Sotamaa K, Hampel H, Green J, Potter JD, Lindblom A, Lagerstedt K, Thibodeau SN, Lindor NM, et al. 2008. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 135: 419–428.e1. 10.1053/j.gastro.2008.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shia J. 2008. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome, part I: the utility of immunohistochemistry. J Mol Diagn 10: 293–300. 10.2353/jmoldx.2008.080031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinbrot E, Henninger EE, Weinhold N, Covington KR, Göksenin AY, Schultz N, Chao H, Doddapaneni H, Muzny DM, Gibbs RA, et al. 2014. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res 24: 1740–1750. 10.1101/gr.174789.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JX, Helgason A, Masson G, Ebenesersdóttir SS, Li H, Mallick S, Gnerre S, Patterson N, Kong A, Reich D, et al. 2012. A direct characterization of human mutation based on microsatellites. Nat Genet 44: 1161–1165. 10.1038/ng.2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricarico R, Cortellino S, Riccio A, Jagmohan-Changur S, Van der Klift H, Wijnen J, Turner D, Ventura A, Rovella V, Percesepe A, et al. 2015. Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis. Oncotarget 6: 42892–42904. 10.18632/oncotarget.5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs A, Nussenzweig A. 2017. Endogenous DNA damage as a source of genomic instability in cancer. Cell 168: 644–656. 10.1016/j.cell.2017.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Itoh M, Kong L, Sugihara K, Asano M, Takakura N. 2005. PSF1 is essential for early embryogenesis in mice. Mol Cell Biol 25: 10528–10532. 10.1128/MCB.25.23.10528-10532.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, et al. 2004. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 96: 261–268. 10.1093/jnci/djh034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo YH, Li WH. 2012. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat Commun 3: 1004 10.1038/ncomms1982 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.