

Graphical abstract
Keywords: Viral macro domain, ADP-ribose, Conformational dynamics, NMR spectroscopy, 15N relaxation, Normal mode analysis
Highlights
-
•
ADPr’s binding triggers conformational changes to the whole VEEV macro domain.
-
•
High flexibility of the loops β5-α3 and α3-β6 assist the ADPr’s binding.
-
•
Loops around ADPr site undergo a transition pathway between apo and complex state.
Abstract
Venezuelan equine encephalitis virus (VEEV) is a new world alphavirus which can be involved in several central nervous system disorders such as encephalitis and meningitis. The VEEV genome codes for 4 non-structural proteins (nsP), of which nsP3 contains a Macro domain. Macro domains (MD) can be found as stand-alone proteins or embedded within larger proteins in viruses, bacteria and eukaryotes. Their most common feature is the binding of ADP-ribose (ADPr), while several macro domains act as ribosylation writers, erasers or readers. Alphavirus MD erase ribosylation but their precise contribution in viral replication is still under investigation. NMR-driven titration experiments of ADPr in solution with the VEEV macro domain (in apo- and complex state) show that it adopts a suitable conformation for ADPr binding. Specific experiments indicate that the flexibility of the loops β5-α3 and α3-β6 is critical for formation of the complex and assists a wrapping mechanism for ADPr binding. Furthermore, along with this sequence of events, the VEEV MD undergoes a conformational exchange process between the apo state and a low-populated “dark” conformational state.
1. Introduction
Venezuelan equine encephalitis virus (VEEV) has been recognized as a human pathogen since 1943 and causes flu-like symptoms and several central nervous system disorders like encephalitis and meningitis. VEEV is a new world alphavirus, i.e., specific to the Americas. It follows an enzootic life cycle between mosquito Culex sp. and wild rodents, and an epizootic cycle amongst humans and equines with mosquitos Aedes sp. and Psorophora sp. as epizootic vectors (Morrison et al., 2008). The virus is distributed over the Americas with main outbreaks located in Central and South America. Outbreaks of VEEV can lead to fatal cases in equines and VEEV is also considered as a high-risk pathogen for humans as well. Such outbreaks in Venezuela and Colombia in 1995, involving an estimated 100,000 human cases, indicate that VEEV poses a serious public health problem. It is worth mentioning that since 1995 there has not been any outbreak of the virus (Guillén et al., 2015, Morrison et al., 2008), however epidemics indicate that VEEV still represents a serious public health concern (Aguilar et al., 2011). So far, no efficient vaccines or drugs are licensed for the treatment of alphavirus infection (Chattopadhyay et al., 2013).
The 12 kb single-stranded positive sense RNA genome of the virus includes two large open reading frames (ORF). The first ORF encodes the four non-structural proteins (nsPs), which are necessary for virus replication, while the second is translated into the structural proteins: capsid, E1, E2 and E3 glycoproteins and 6K. In particular, nsP1 possesses guanine-7-methyltransferase and guanylyltransferase activity being responsible for the addition of a 5′ cap to viral RNA (Rupp et al., 2015), while nsP2 is a multifunctional protein consisting of 3 domains. The N-terminal domain acts as a helicase while it also exhibits nucleoside triphosphatase activity (NTPase). The C-terminus of nsP2 contains a protease domain and a non-functional methyltransferase-like domain. nsP3 is comprised of three domains: the conserved N-terminal domain, which is called macro domain (also known as “X domain”), a central alphavirus unique domain (AUD) and a hypervariable, phosphorylated C-terminus. Originally, an ADPr-1″-phosphatase was associated to the macro domain, as well as nucleic acid binding but recent studies showed its implication in de-ribosylation of proteins. The highly conserved AUD is characterized by a zinc-binding motif. Both length and sequence composition of the C-terminal domain are hypervariable among alphaviruses (Abu Bakar and Ng, 2018, Rupp et al., 2015). Last, nsP4 is the RNA-dependent RNA polymerase (Shin et al., 2012, Strauss and Strauss, 1994).
Macro domains are highly conserved proteins throughout evolution. They consist of 130–190 amino acids. They are named after the non-histone motif of the core histone variant macroH2A, in which they were initially characterized (Chakravarthy et al., 2005). High affinity binding to ADP-ribose (ADPr) is their most common feature and macro domains can be organized into three classes: ADP-ribosylation readers, erasers and writers. Many cellular processes are dependent on protein ADP-ribosylation which is a highly dynamic post-translational modification involved in DNA repair, apoptosis, protein degradation and chromatin remodeling (Fehr et al., 2017, Jankevicius et al., 2013). However, the biological role of the viral macro domains is still poorly understood. Several recent studies have demonstrated that they exhibit de-ADP-ribosylating activity (eraser) together with their ability to bind ADP-ribose (Fehr et al., 2017). Therefore, it was proposed that they participate in reversing the antiviral ADP-ribosylation of the host cells (Fehr et al., 2017, Li et al., 2016). Mutations of viral macro domains at residues critical for ADPr binding have been shown to lead to lower affinity towards ADPr resulting in disruption of loss of virulence and/or viral replication (Fehr et al., 2016, McPherson et al., 2017). Therefore, macro domains could serve as novel therapeutic targets for the design of antiviral agents in order to control Alphavirus, Hepevirus and Coronavirus-induced diseases. The crystal structure of the VEEV macro domain has shown that the protein forms a six-stranded β-sheet surrounded by 4 α-helices (Malet et al., 2009). The order of the β-strands within the β-sheet is β1, β6, β5, β2, β4, β3, all positioned in parallel except for β3. The N-terminus forms the β1-strand while the C- terminus forms the last α-helix (α4), which is located close to the N-terminus. This structural motif is quite conserved among the macro domains of different organisms (Cho et al., 2016, Malet et al., 2009, Peterson et al., 2011). The same study revealed the structural basis of the binding affinity of ADPr to the macro domain. The binding site of ADPr is located at the top of the β2, β4 and β5 strands surrounded by the loops connecting β2 to α1 and β5 to α3 defining a conserved binding pocket (Malet et al., 2009).
In this study we provide the solution structure and the dynamic properties of the VEEV macro domain both in the apo- and the ADPr-bound state. Using NMR spectroscopy, we demonstrate a local conformational transition on the µs-ms time scale between two states, involving residues in the crevice occupied by ADPr. The data presented here suggest that the flexibility of defined regions nearby the macro domain active site is crucial for recognition and binding of ADPr.
2. Material and methods
2.1. Protein expression and purification
The experimental protocol for the expression, purification and NMR sample preparation of the VEEV nsP3 macro domain (A1330-E1489) has been reported previously (Makrynitsa et al., 2015). The NMR samples, which were used for the structure determination of the macro domain and the macro domain-ADPr complex, contained 0.4 mM 15N-labeled protein or 0.8 mM 13C/15N-labeled protein and 0.18 mM 15N-labeled protein or 0.21 mM 13C/15N-labeled protein along with 0.26 mM ADPr, respectively.
2.2. NMR spectroscopy and structure determination
NMR measurements were performed at 298 K with an Avance III High-Definition four-channel 700 MHz spectrometer equipped with a cryogenically cooled 5 mm 1H/13C/15N/D Z-gradient probe (TCI) (Bruker). Proton chemical shifts were referenced to internal 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS). Sequence specific resonance assignments of the VEEV macro domain were obtained as reported previously (BMRB ID 25132) (Makrynitsa et al., 2015). For the structure determination of the complex the same set of NMR experiments (Makrynitsa et al., 2015) was used and the 1H, 15N and 13C resonance assignments were deposited in the BioMagResBank under the accession number 26753. However, in order to define the 3D structure of the VEEV macro domain-ADPr complex, a sufficient number of intermolecular distance restraints had to be obtained through 2D 13C-filtered/edited NOE experiments (Peterson et al., 2004).
NOE-derived distance constraints for the VEEV macro domain were obtained by the analysis of a 3D 15N-NOESY and two 3D 13C-NOESY spectra, which were recorded with the proton carrier frequency in the aliphatic and the aromatic region, respectively. DYANA’s routine CALIBA was used to generate distance restraints from the volume of each NOESY cross peak. Dihedral angles derived from TALOS+ (Makrynitsa et al., 2015, Shen et al., 2009) were incorporated in the structure calculations as well. The calculations were conducted with DYANA (Güntert et al., 1997) and the 20 conformers with the lowest target function values were subjected to energy minimization with AMBER (Case et al., 2005). The solution structure of the apoprotein was determined from 1648 unique, meaningful NOE constraints derived from a total number of 1486 NOEs from the 15N-NOESY and 1043 NOEs from the 13C-NOESY spectra (SI, Table S1). The coordinates of the 21 energy-refined conformers (including the average model) were deposited in the RCSB Protein Data Bank (PDB ID: 5ISN). For the solution structure of the biomolecular complex, the same procedure and software were used and it was defined by 1988 unique, meaningful NOE constraints: 1361 from the 15N-NOESY, 1991 from the 13C-NOESY spectra and 18 intermolecular NOE correlations from the 13C-filtered/edited NOESY. The coordinates of the 21 energy-refined models (20 DYANA models and the mean model) were deposited in the RCSB Protein Data Bank (PDB ID: 5MQX) as well.
2.3. NMR titration experiments
To monitor the behavior of the individual amino acids of the 15N-labeled VEEV macro domain in the presence of ADPr, we calculated the changes of their chemical shifts in 1H–15N HSQC spectra during the NMR titration experiment. The unlabeled ligand (ADPr stock concentration = 5 mM) was added in 16 steps in order to reach the following protein/ligand ratios: 1/0.125, 1/0.25, 1/0.4, 1/0.5, 1/0.75, 1./0.8, 1/0.9, 1/1, 1/1.125, 1/1.25, 1/1.4, 1/1.5, 1/1.75, 1/2, 1/3 and 1/4. Chemical shift perturbation values were calculated using the equation Δδppm = (Garrett et al., 1997). After the first addition (1/0.125) the ADPr concentration was 0.0225 mM; after the last addition (1:4) it was 0.64 mM.
2.4. Protein dynamics
The backbone dynamics of the VEEV macro domain and the VEEV macro domain-ADPr complex on the ps-ns time scale were studied through the analysis of 15N R1 and R2 relaxation rates and heteronuclear {1HN}–15N NOEs. 1H–15N HSQC experiments were recorded with relaxation delays of 7, 18, 40, 85, 150, 230, 350, 500, 680, and 900 ms for obtaining the R1 relaxation rates and with delays of 18, 32, 50, 68, 86, 100, 120, 150, 190, and 240 ms for the R2 relaxation rates. All the relaxation data were analyzed with the Tensor2 (Dosset et al., 2000) program in order to define the dynamic properties of the apoprotein and the complex by obtaining the values of S2 order parameters for each amino acid.
The µs-ms dynamics of the amide backbone of VEEV macro domain was probed by SQ CPMG relaxation dispersion experiments. Dispersion profiles were obtained at 298 K and external magnetic field strengths of 14.1 T (600 MHz 1H) and 21.1 T (900 MHz 1H), respectively. 1H continuous wave decoupling was employed throughout both constant-time echo periods with a rf amplitude of 12.5 kHz (Hansen et al., 2008). The CPMG field νCPMG was varied from 25 to 1000 Hz, while T rel was set to 40 ms and the pre-scan delay to 2 s. 15N 180° refocusing pulses were applied at ∼5.6 kHz. The equilibration delay to achieve equilibrium populations of ground and excited states prior to the CPMG train was set to 5 ms. Amide resonance intensities I (νCPMG) were converted to transverse relaxation rates by , where I 0 is the intensity when omitting the CPMG train. The experimental error was set to two times the standard deviation of the spectral noise.
Dispersion curves were fitted employing the Carver-Richards equation for a system undergoing chemical exchange in two states, while fitting four independent parameters (k ex, p α, |Δω|, R 2,0) (Carver and Richards, 1972). For each residue curves at both available fields were fitted simultaneously. To quantitatively discriminate exchanging from non-exchanging residues, all curves were additionally fit by a linear function. The model, which represents the data the best, was selected based on the comparison of AICc values (d'Auvergne and Gooley, 2003). To improve the fitting performance, several residues localized on the same secondary structure element were grouped together, while employing the χ2 ratio for group over individual fitting (Mulder et al., 2001). Residues with ratios larger than two were discarded, assuming they undergo conformational fluctuations distinct from the global process.
2.5. Normal mode analysis
The free starting structure of the VEEV macro domain was obtained from the Protein Data Bank entry 5ISN and the ADPr-bound target from the calculated mean structure of the current study. In order to assess the effects of ligand binding, normal mode analysis (NMA) between the two states was performed using the iMOD program and specifically the imorph_gcc tool, which calculates potential transition pathways between two different conformations of a protein (Lopéz-Blanco et al., 2011). Elastic connections between all protein heavy atoms and their neighbors were considered, whilst sigmoid function was used, with a distance cut off of 10 Å. The step size used to control the iteration number was set at 10, the maximum stiffness constant at 1, the inflexion point at 3.8 Å and the cut-off for removing ineffective very weak springs from calculations at 10 Å. The RMSD to trigger NMA was set at 0.01 Å.
3. Results and discussion
3.1. Solution structure of the VEEV macro domain (A1330-E1489)
The NMR solution structure of the VEEV macro polypeptide shows that the domain falls into the α/β class of proteins and adopts the typical macro domain fold, consisting of a central six-stranded β sheet flanked by 4 α-helices namely α1 (V33-S44), α2 (E78-D98), α3 (L121-D133) and α4 (K147-R158). The β- sheet topology is β1β6β5β2β4β3 with the β3 strand being antiparallel to its neighbors (Fig. 1 A). The NMR structure of the VEEV macro domain in apo form superimposes well with the corresponding X-ray structure (Malet et al., 2009) with a rmsd of 2.1 Å for the backbone atoms (Fig. 1B). Although the two structures are very similar regarding the secondary structure elements and topology, loops Q28-A36, G116-R120 and the loop connecting the β6 strand with the α4 helix exhibit different conformations due to the lack of NOEs and unassigned residues in these regions (Makrynitsa et al., 2015). Taking into consideration that the NMR structure is of high resolution (target function: 1.49 Å, rmsd: 1.07 Å), the high value of the rmsd is the result of the behavior of the aforementioned loops. Amino acids without detectable spin patterns in the NMR spectra are located around the highly positively charged surface area, which defines the ADPr binding site (Malet et al., 2009). Similar behavior has been observed in the NMR study of the human orphan macro domain C6orf130 in which the amino acids of corresponding loops around the ADPr binding pocket were also missing in the NMR spectra (Peterson et al., 2011).
Fig. 1.

Solution structure of VEEV macro domain. (A) Representation of VEEV macro domain NMR solution structure with secondary structure elements labeled. (B) Superposition of ribbon representations of VEEV macro domain solved by NMR spectroscopy (purple) and X-ray diffraction (light blue). (C) Surface rendering of the calculated electrostatic potential of VEEV macro domain generated with the Adaptive Poisson-Boltzmann Solver (APBS) plug-in in PyMOL (Schrödinger, 2010) where blue and red indicate positively and negatively charged regions respectively (scale −3 kT/e to +3 kT/e).
3.2. Structural basis of ADP-r binding to VEEV macro domain
Macro domains are ADPr-binding molecules and contain a highly conserved binding-pocket (Karras et al., 2005, Li et al., 2016). To further investigate the binding of ADPr, we titrated 15N-labeled VEEV macro domain incrementally with ADPr and monitored backbone amide chemical shift changes in the 1H–15N HSQC spectra. During the titration experiments, 21 residues exhibited chemical shift perturbations (CSPs) above the threshold (calculated according to the standard deviation of all CSP values as determined by the equation Δδppm = (Garrett et al., 1997)) with the majority of these residues located around the binding groove (Fig. 2 A, B). Interestingly, 1H–15N HSQC spectra of the complex did not display any further changes regarding the chemical shifts of the perturbed amino acids after a 1:1 protein-ligand ratio had been reached. This strongly indicates that the VEEV macro domain binds specifically one ADPr molecule in its well-defined binding cleft. Cross peaks of residues affected by ADPr gradually vanished during the titration experiments and simultaneously appeared in a new region of the HSQC spectrum (Fig. 2C). This behavior is characteristic of a slow exchange process and is indicative for high affinity ligand binding (Kd of 3.9 ± 0.65 µM as measured by isothermal titration calorimetry (Malet et al., 2009)).
Fig. 2.

Interaction of VEEV macro domain with ADPr. (A) Overlay of 1H–15N HSQC spectra of the VEEV macro domain in the apo (purple) and ADPr bound state (ratio 1:4) (orange). The response of the NMR resonances of A22 (bottom) and N72 (top) to increasing ADPr concentrations. Peaks corresponding to the apo state decrease in intensity as the ones of the bound state increase (purple: free, red: 1:0.250, light blue: 1:0.5, grey: 1:0.750, green: 1:1, yellow: 1:2, orange: 1:4). (B) Chemical shift perturbation (CSP) in response to ADPr binding (ratio 1:4) with threshold value 0.26. Residues with CSP up to 0.2 from the threshold value are colored yellow, residues with CSP from 0.2 to 0.5 above the threshold are colored green and residues with CSP higher than 0.5 from the threshold are colored red. (C) Surface representation of the VEEV macro domain in complex with ADPr. The residues with CSPs above the threshold are mapped onto the surface using the same color code as in B. (D) Superposition of ribbon representations of VEEV macro domain in the apo (purple) and ADPr bound states (grey) as determined by NMR spectroscopy. (E) Superposition of ribbon representations of the VEEV macro domain-ADPr complex as determined by X-ray diffraction (orange) and NMR spectroscopy (grey).
Analysis of 13C-filtered/edited NOESY spectra along with the three-dimensional structure of the VEEV-ADPr complex gave insights into the interactions between the residues in the binding pocket and the ADPr moieties (SI, Fig. S1). In particular, the adenine moiety is accommodated in a hydrophobic cavity (P1 pocket) and shows NOE contacts with I11, V33, Y142, and R144. Concerning the proximal ribose (the adenosine ribose), we identified NOE contacts with V33, L108, T111, and R144 (P2 pocket), while the distal ribose of ADPr is hydrogen bonded with the backbone NH of G32 and shows NOEs with A22 and I113, fitting into a pocket (P3 pocket) comprising the N-terminal part of the α1 helix and the loop preceding the α3 helix (residues L108-R120). The phosphate binding site is stabilized through hydrogen bonds with the main chain NH groups of V33 and I113. Analysis of the VEEV/ADPr complex by molecular dynamics also revealed that the above residues participate strongly in the ADPr binding with the distal ribose playing a key role in the complex formation (Rungrotmongkol et al., 2010). Due to exchange with the bulk solvent the assignment of the ADPr OH groups was not feasible. The residues that map the binding sites for each ADPr moiety are not random. A sequence alignment of macro domains from different organisms reveals the high degree of conservation for the amino acids residing in the binding pocket (Fehr et al., 2017, McPherson et al., 2017) (SI, Fig. S2).
Mapping amino acids with CSP values above threshold to the surface of the complex revealed that the majority of these residues form the ADPr cavity. However, despite their high CSPs residues K26, L37, K40, H64, A68, V69, F70, N72, and G116 do not participate directly in the binding of ADPr (Fig. 2D). Comparison of the structures in the free and complex state revealed conformational differences in loop S25-G31 and the C-terminal helix α4 although ADPr binding to the VEEV macro domain does not dramatically alter the overall structure of the protein (Fig. 2E). Likewise, the solution structure of the VEEV macro domain-ADPr complex is very similar to the corresponding crystal structure except for the aforementioned regions that exhibit conformational differences (Malet et al., 2009) (Fig. 2F). These differences are coupled with the binding of the ADPr molecule to the macro domain cavity. The loops comprising S25-G31 and L109-R120 undergo conformational changes resulting in a slight widening of the crevice where the distal ribose is located. Helices α1 and α4 helices seem to exhibit a displacement towards the binding pocket as well, but in this case the conformational changes cause a closure of the pocket around the ligand (Fig. 3 A, B).
Fig. 3.

Dynamic properties of VEEV macro domain. (A) Surface representations of the VEEV macro domain in the apo state (purple) and the ADPr-bound state (grey). Regions G30-V33, I113-R120 and helix α4 are colored cyan. (B) Close-up view of the ADPr binding pocket – left: VEEV macro domain apo state, right: VEEV macro domain-ADPr complex. Panels A and B display the conformational changes upon ADPr binding and some of the affected residues. (C) S2 values plotted as a function of residue number of VEEV macro domain in the apo state (top row) and complex (bottom row).
3.3. Dynamic and exchange properties of the VEEV macro domain (apo state and complex with ADPr)
So far, a remarkable wealth of studies has focused on the structure and function of macro domains (Götte et al., 2018). However, information about their dynamics and the conformational changes triggered by substrate binding is limited.
Model-free analysis of 15N NMR relaxation data as implemented in the Tensor2 program (Dosset et al., 2000) (Fig. 3C, SI, Fig. S3) shows that the VEEV macro domain in solution exhibits a rather rigid structure on the ps-ns time scale. However, a lot of residues in the sequential stretches G30-Y38 and G112- R120 could not be assigned (Makrynitsa et al., 2015) suggesting that these residues undergo an exchange between different conformational states on an intermediate μs-ms time scale, where the signals are broadened.
To quantify the protein dynamics on this timescale, we performed 15N CPMG relaxation dispersion experiments (SI, Fig. S4). There are essentially two regions at the N- and C-terminus that undergo µs-ms motion. We simultaneously fit both clusters with residues 1–44 and 103–168, respectively, and found exchange rates kex of ∼3500 and ∼2580 s−1 and populations of the excited state pB of around 0.35–0.5%. A23 as well as loops β5-α3 (L108-R120) and α3-β6 (T134-D138) exhibit the largest exchange contribution to R2, being responsible for the flexibility of the nearby residues (Fig. 4 A, B). Remarkably, after ADPr binding, the flexibility of loop β5-α3 was reduced as all residues of this loop were assigned in the 1H–15N HSQC spectrum and showed relatively high order parameters S 2.
Fig. 4.

µs-ms dynamics of VEEV macro domain without ADPr as probed by SQ 15N CPMG relaxation dispersion experiments. (A) The residues undergoing significant µs-ms conformational exchange were mapped to the structure. The tube radius corresponds to the exchange contribution to the R2 rate (Rex) as plotted in panel B. Inset (C) Linear correlation of absolute experimental 15N chemical shift differences between 1) apo and ADPr-bound macro domain (x-axis) and 2) ground and excited state of apoprotein from relaxation dispersion (y-axis).
Besides populations and exchange rates, relaxation dispersion curves are sensitive to the absolute chemical shift difference between ground and excited state, with the chemical shift being a valuable structural probe reporting on secondary and tertiary structure. We determined absolute 15N chemical shift differences from HSQC spectra in the apo and bound state and plotted them against CPMG derived values (Fig. 4C). Residues in the ADPr binding crevice (L108-G116) showed a linear correlation suggesting that the ADPr-bound pose can be nearly adopted by the excited state of the apo protein, however, most likely with a more open conformation compared to the ground apo and ADPr-bound state to allow entering of the ligand. We note that deviations are in part also attributed to chemical shift contributions imposed by the ligand, which are not present in the CPMG experiment.
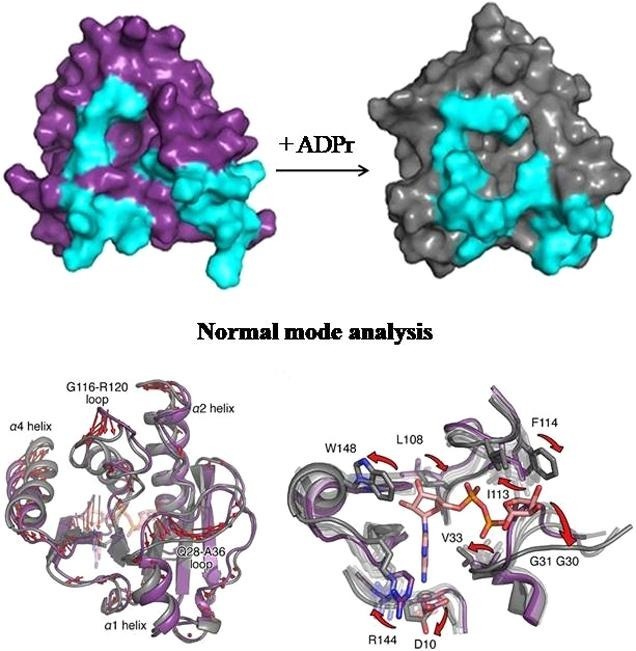
To study a potential transition pathway from the apo to the bound form of the domain during binding of ADPr, we performed normal mode analysis (NMA) calculations. The most pronounced topological differences observed between the two NMR resolved states of the domain are in the two loops L108-R120 and Q28-A36 and in helices α1, α2, and α4 (Fig. 5 A), which relates well to the experimental CPMG data (Fig. 4). Using NMA to address these conformational changes, the structure converged smoothly to the target ADPr-bound conformation with final heavy atom RMSD of 0.55 Å (initial RMSD: 1.8 Å), indicating a pathway of backbone and side chain transitions to reshape the binding cavity of the domain and accommodate the ligand. Specifically, the Q28-A36 loop undergoes a significant translational movement of ∼3 Å with respect to the L108-R120 loop, increasing the distance between the Cα atoms of G31 and I113 from ∼7 to ∼10 Å in order to admit the distal ribose of ADPr (Fig. 5B). Consequently, F114 also moves to interact with the same ribose. Mutational insertions in the Q28-A36 loop can conditionally alter virus replication and, at the protein scale, destabilize the macro domain and partly affect its ADPr binding property (Beitzel et al., 2010, Guillén et al., 2015), highlighting the role this loop in the transition mechanism required for the accommodation of ADPr.
Fig. 5.

Normal mode analysis of VEEV macor domain. (A) Differences between the NMR solution structures of VEEV macro domain in the apo state (purple) and the ADPr-bound state (grey) are indicated by red arrows (minimum length of the displayed vectors is 2 Å). (B) Normal mode analysis from the apo (purple) to the bound state (grey) shows a transition pathway in the binding cavity. Transition states are shown in transparent white to dark grey colors as they progress to the bound state. Red arrows indicate significant reorientations of amino acids participating in binding of ADPr.
4. Conclusions
Our study demonstrates that ADPr binding to the VEEV macro domain results in a conformational exchange process of loops S25-G31 and L109-R120 in order to adopt an open conformation. Similar rearrangements have been found in human C6orf130, an orphan macro domain protein (Peterson et al., 2011), and in the macro domain from SARS coronavirus (Götte et al., 2018). The data reveals that the VEEV macro domain exhibits high adaptability to bind ADPr. The ligand does not simply bind to a well-defined, highly conserved binding pocket of the protein, but several regions of the protein contribute to the formation of the biomolecular complex. A number of residues undergo both backbone and side-chain conformational changes to adopt a suitable geometry that allows ADPr to enter the cavity and bind strongly to the protein.
Regarding the mobility analysis, the movement of helix α4 results in a reorientation of the W148 and L108 side chains which upon binding interact with the proximal ribose of ADPr. V33, as part of the moving helix α1, plays a crucial role in binding, positioning itself to a σ-π interacting arrangement with the aromatic group of adenine. The charged residues R144 and D10 also undergo certain rearrangements, the first to widen the binding pocket and the second to form a crucial hydrogen bond with the adenine amine. R144 in particular showed minor but significant conformational exchange as determined by the CPMG experiment (Fig. 4, SI, Fig. S4). The conformational changes not only contribute to the ADPr binding but also to the efficient de-ribosylation activity by properly positioning the reactive molecule, likely a water molecule, close to the substrate. Indeed, it was evidenced that a mutation in the corresponding Q28-A36 loop of Hepatitis E virus macro domain or O. iheyensis macro domains greatly impaired the catalysis without affecting significantly the substrate recognition (Li et al., 2016, Zapata-Pérez et al., 2017). These conformational changes along the transition path thus provide a framework to understand the dynamic nature of the macro domains and to dissect the events during ADPr binding and de-ribosylation.
Taken together, our structural and mobility data strongly suggest that in solution the VEEV macro domain shows dynamics on the μs-ms timescale in the absence of ADPr, while the VEEV macro-ADPr complex seems to adopt a compact structure. The analysis of the 3D structures allows the identification of key conformational changes around the binding cleft that are critical for ADPr binding and depend on the plasticity of the loops β5-α3 and α3-β6. The sensitivity of loop β5-α3 to ADPr binding in combination with the mobility of loop α3-β6 indicates that they serve as gates of the binding cleft and assist the ADPr entrance. ADPr binding drives the protein molecule to switch from an open conformation with flexible segments to a rigid closed one.
We identified a conformational exchange process between the solution structure of the apoprotein presented herein and another, low-populated conformational state (∼0.5%). A comparison of the structures of the VEEV macro domain in the apo and ADPr bound state reveals that flexibility of the loop β5-α3 is crucial for the key conformational changes around the active site favoring ADPr binding. The high exchange rates of loops β5-α3 and α3-β6 contribute to the flexibility of these segments, which may serve as micro-switches that allow ADPr to enter the ligand-binding cleft. The correlation of chemical shifts of the excited apo to the bound ground state suggests that ADPr binding occurs through a conformational selection mechanism. It is well established that polypeptide segments with high adaptability play a key role in partner or ligand identification, selectivity and binding (Peterson et al., 2011, Zapata-Pérez et al., 2017). This may also be the function of these regions in many macro domains, including the one from VEEV. It is worth mentioning, that so far most of the studies concerning the structure of macro domains have focused on the characterization of the ADPr binding pocket before and after binding of the ligand (Egloff et al., 2006). In the present study, for the first time, it is demonstrated by experimental data that an exchange process between two states of certain loop regions in the apo form of the macro domain is crucial for ADPr binding.
Acknowledgements
CPMG experiments were performed at the Bavarian NMR center (BNMRZ, http://www.bnmrz.org). EU FP7 REGPOT CT-2011-285950 – “SEE-DRUG” project is acknowledged for financial support (DB, GAS) as well as for the purchase of UPAT’s 700 MHz NMR equipment. This work was also supported by the European program H2020 under the EVAg Research Infrastructure (grant agreement No. 653316)
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jsb.2019.02.008.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- Abu Bakar F., Ng L. Nonstructural proteins of alphavirus—potential targets for drug development. Viruses. 2018;10:71. doi: 10.3390/v10020071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar P.V., Estrada-Franco J.G., Navarro-Lopez R., Ferro C., Haddow A.D., Weaver S.C. Endemic Venezuelan equine encephalitis in the Americas: hidden under the dengue umbrella. Fut. Virol. 2011;6:721–740. doi: 10.2217/FVL.11.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitzel B.F., Bakken R.R., Smith J.M., Schmaljohn C.S. High-resolution functional mapping of the venezuelan equine encephalitis virus genome by insertional mutagenesis and massively parallel sequencing. PLoS Pathogens. 2010;6 doi: 10.1371/journal.ppat.1001146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver J., Richards R. A general two-site solution for the chemical exchange produced dependence of T2 upon the Carr-Purcell pulse separation. J. Magn. Reson. 1969;6:89–105. [Google Scholar]
- Case D.A., Cheatham T.E., Darden T., Gohlke H., Luo R., Merz K.M., Onufriev A., Simmerling C., Wang B., Woods R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy S., Gundimella S.K.Y., Caron C., Perche P.-Y., Pehrson J.R., Khochbin S., Luger K. Structural characterization of the histone variant macroH2A. Mol. Cell. Biol. 2005;25:7616–7624. doi: 10.1128/MCB.25.17.7616-7624.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay A., Wang E., Seymour R., Weaver S.C., Rose J.K. A chimeric vesiculo/alphavirus is an effective alphavirus vaccine. J. Virol. 2013;87:395–402. doi: 10.1128/JVI.01860-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C.-C., Lin M.-H., Chuang C.-Y., Hsu C.-H. Macro domain from middle east respiratory syndrome coronavirus (MERS-CoV) is an efficient ADP-ribose binding module: crystal structure and biochemical studies. J. Biol. Chem. 2016;291:4894–4902. doi: 10.1074/jbc.M115.700542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Auvergne E.J., Gooley P.R. The use of model selection in the model-free analysis of protein dynamics. J. Biomol. NMR. 2003;25:25–39. doi: 10.1023/a:1021902006114. [DOI] [PubMed] [Google Scholar]
- Dosset P., Hus J.-C., Blackledge M., Marion D. Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. J. Biomol. NMR. 2000;16:23–28. doi: 10.1023/a:1008305808620. [DOI] [PubMed] [Google Scholar]
- Egloff M.-P., Malet H., Putics Á., Heinonen M., Dutartre H., Frangeul A., Gruez A., Campanacci V., Cambillau C., Ziebuhr J. Structural and functional basis for ADP-ribose and poly (ADP-ribose) binding by viral macro domains. J. Virol. 2006;80:8493–8502. doi: 10.1128/JVI.00713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr A.R., Jankevicius G., Ahel I., Perlman S. Viral macrodomains: unique mediators of viral replication and pathogenesis. Trends Microbiol. 2017;26:598–610. doi: 10.1016/j.tim.2017.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr A.R., Channappanavar R., Jankevicius G., Fett C., Zhao J., Athmer J., Meyerholz D.K., Ahel I., Perlman S. The conserved coronavirus macrodomain promotes virulence and suppresses the innate immune response during severe acute respiratory syndrome coronavirus infection. MBio. 2016;7:e01721–01716. doi: 10.1128/mBio.01721-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güntert P., Mumenthaler C., Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program Dyana1. J. Mol. Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- Garrett D.S., Seok Y.-J., Peterkofsky A., Clore G.M., Gronenborn A.M. Identification by NMR of the binding surface for the histidine-containing phosphocarrier protein HPr on the N-terminal domain of enzyme I of the Escherichia coli phosphotransferase system. Biochemistry. 1997;36:4393–4398. doi: 10.1021/bi970221q. [DOI] [PubMed] [Google Scholar]
- Götte B., Liu L., McInerney G. The enigmatic alphavirus non-structural protein 3 (nsP3) revealing its secrets at last. Viruses. 2018;10:105. doi: 10.3390/v10030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillén J., Lichière J., Rabah N., Beitzel B.F., Canard B., Coutard B. Structural and biophysical analysis of sequence insertions in the Venezuelan Equine Encephalitis Virus macro domain. Virus Res. 2015;201:94–100. doi: 10.1016/j.virusres.2015.02.018. [DOI] [PubMed] [Google Scholar]
- Hansen D.F., Vallurupalli P., Kay L.E. An improved 15N relaxation dispersion experiment for the measurement of millisecond time-scale dynamics in proteins. J. Phys. Chem. B. 2008;112:5898–5904. doi: 10.1021/jp074793o. [DOI] [PubMed] [Google Scholar]
- Jankevicius G., Hassler M., Golia B., Rybin V., Zacharias M., Timinszky G., Ladurner A.G. A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat. Struct. Mol. Biol. 2013;20:508. doi: 10.1038/nsmb.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karras G.I., Kustatscher G., Buhecha H.R., Allen M.D., Pugieux C., Sait F., Bycroft M., Ladurner A.G. The macro domain is an ADP-ribose binding module. EMBO J. 2005;24:1911–1920. doi: 10.1038/sj.emboj.7600664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Debing Y., Jankevicius G., Neyts J., Ahel I., Coutard B., Canard B. Viral macro domains reverse protein ADP-ribosylation. J. Virol. 2016;90:8478–8486. doi: 10.1128/JVI.00705-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopéz-Blanco J.R., Garzón J.I., Chacón P. iMod: multipurpose normal mode analysis in internal coordinates. Bioinformatics. 2011;27:2843–2850. doi: 10.1093/bioinformatics/btr497. [DOI] [PubMed] [Google Scholar]
- Makrynitsa G.I., Ntonti D., Marousis K.D., Tsika A.C., Lichière J., Papageorgiou N., Coutard B., Bentrop D., Spyroulias G.A. NMR study of non-structural proteins—part II: 1 H, 13 C, 15 N backbone and side-chain resonance assignment of macro domain from Venezuelan equine encephalitis virus (VEEV) Biomol. NMR Assignments. 2015;9:247–251. doi: 10.1007/s12104-014-9584-9. [DOI] [PubMed] [Google Scholar]
- Malet H., Coutard B., Jamal S., Dutartre H., Papageorgiou N., Neuvonen M., Ahola T., Forrester N., Gould E.A., Lafitte D. The crystal structures of Chikungunya and Venezuelan equine encephalitis virus nsP3 macro domains define a conserved adenosine binding pocket. J. Virol. 2009;83:6534–6545. doi: 10.1128/JVI.00189-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson R.L., Abraham R., Sreekumar E., Ong S.-E., Cheng S.-J., Baxter V.K., Kistemaker H.A., Filippov D.V., Griffin D.E., Leung A.K. ADP-ribosylhydrolase activity of Chikungunya virus macrodomain is critical for virus replication and virulence. Proc. Natl. Acad. Sci. U.S.A. 2017;114:1666–1671. doi: 10.1073/pnas.1621485114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison A.C., Forshey B.M., Notyce D., Astete H., Lopez V., Rocha C., Carrion R., Carey C., Eza D., Montgomery J.M. Venezuelan equine encephalitis virus in Iquitos, Peru: urban transmission of a sylvatic strain. PLoS Neglect. Trop. Dis. 2008;2 doi: 10.1371/journal.pntd.0000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder F.A., Mittermaier A., Hon B., Dahlquist F.W., Kay L.E. Studying excited states of proteins by NMR spectroscopy. Nat. Struct. Mol. Biol. 2001;8:932. doi: 10.1038/nsb1101-932. [DOI] [PubMed] [Google Scholar]
- Peterson F.C., Chen D., Lytle B.L., Rossi M.N., Ahel I., Denu J.M., Volkman B.F. Orphan macrodomain (human C6ORF130) is an o-acyl-ADP-ribose deacylase: solution structure and catalytic properties. J. Biol. Chem. 2011;286:35955–35965. doi: 10.1074/jbc.M111.276238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson R.D., Theimer C.A., Wu H., Feigon J. New applications of 2D filtered/edited NOESY for assignment and structure elucidation of RNA and RNA-protein complexes. J. Biomol. NMR. 2004;28:59–67. doi: 10.1023/B:JNMR.0000012861.95939.05. [DOI] [PubMed] [Google Scholar]
- Rungrotmongkol T., Nunthaboot N., Malaisree M., Kaiyawet N., Yotmanee P., Meeprasert A., Hannongbua S. Molecular insight into the specific binding of ADP-ribose to the nsP3 macro domains of chikungunya and Venezuelan equine encephalitis viruses: molecular dynamics simulations and free energy calculations. J. Mol. Graph. Modell. 2010;29:347–353. doi: 10.1016/j.jmgm.2010.09.010. [DOI] [PubMed] [Google Scholar]
- Rupp J.C., Sokoloski K.J., Gebhart N.N., Hardy R.W. Alphavirus RNA synthesis and non-structural protein functions. J. General Virol. 2015;96:2483–2500. doi: 10.1099/jgv.0.000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger L. 2010. Schrödinger, LLC2010The PyMOL Molecular Graphics System, Version 1.4. The PyMOL Molecular Graphics System Version 1. [Google Scholar]
- Shen Y., Delaglio F., Cornilescu G., Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin G., Yost S.A., Miller M.T., Elrod E.J., Grakoui A., Marcotrigiano J. Structural and functional insights into alphavirus polyprotein processing and pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 2012;109:16534–16539. doi: 10.1073/pnas.1210418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss J.H., Strauss E.G. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 1994;58:491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapata-Pérez R., Gil-Ortiz F., Martínez-Moñino A.B., García-Saura A.G., Juanhuix J., Sánchez-Ferrer Á. Structural and functional analysis of Oceanobacillus iheyensis macrodomain reveals a network of waters involved in substrate binding and catalysis. Open Biol. 2017;7:160327. doi: 10.1098/rsob.160327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.