

Abstract
Swine based agro-industries throughout the world are in big threat of new PEDV infection due to lack of efficient prophylactic defenses as well as dependable curing agents. Bioactivity-guided fractionation of a methanol soluble extract from radix of Saposhnikovia divaricata led to the isolation of three new (1–3) together with 10 known coumarins (4–13). The structures of new isolates (1–3) were established by extensive spectroscopic analysis and their absolute configurations were assigned based on ECD spectra calculation and analysis. Among all isolates, compound 5 revealed strongest inhibitory effect on PEDV replication. Quantitative real-time PCR data showed inhibitory effect of 5 on genes responsible for synthesis of PEDV vital structural proteins (GP6 nucleocapsid, GP2 spike, and GP5 membrane) in a dose-dependent manner. Also, compound 5 demonstrated the inhibitory effect on PEDV GP6 nucleocapsid and GP2 spike protein synthesis as analyzed by western blotting. This study represents a new class of chemical entities for developing anti-PEDV agents.
Keywords: Saposhnikovia divaricata, Coumarin glycosides, Stereochemistry, PEDV inhibitory activity
Graphical abstract
1. Introduction
Porcine epidemic diarrhea virus (PEDV) is an enveloped single-stranded RNA virus belonging to the family Coronaviridae.1 It is the causative agent of porcine epidemic diarrhea, vomiting, dehydration, and high mortality in the piglets. Most of newborn piglets infected by PEDV would be dying and pigs of all ages are also affected and exhibit severe symptom like massive diarrhea and dehydration.2 Infection with this virus has been become a serious issue in the swine industry and outbreaks led to serious economic losses in many swine producing countries, notably in Europe and Asia.3 The outbreak in 2009 from Thailand reported that it spreads throughout the country's farrowing barns in 2007 with 100% piglet's mortality.4 An epidemiological survey of PEDV in China showed that severe diarrhea in piglets occurred with high mortality rate up to 100% in breeding piglets up to 10 days of age after birth.5 Recently, new PEDV was noticed and its outbreak affects 23 US states by the end of January of 2014. PEDV infection in US swine industry caused severe watery diarrhea, variable vomiting, dehydration, and high mortality in affected swine, especially neonatal piglets resulting severe economic losses.6, 7
In order to control over such infection, there had been many efforts to formulate a safe and efficient vaccines.8 Basically, live attenuated virus and the gene containing spike protein sequences are being tested as vaccines.9, 10 However, efficiency of such vaccines was reported to be just under 50% even at a very high dose.11 In Korea DR13 strain attenuated PEDV, licensed as vaccine against PEDV, is in use but the results are not satisfactory.12 Thus, new antiviral compounds from natural origin might be the possible candidates to develop effective therapeutic agents against such infection.
Saposhnikovia divaricata Schischk. (synonyms.: Ledebouriella seseloides WOLFF) belonging to the family Umbelliferae is an important Korean traditional medicine. The plant species, native to eastern Siberia and northern Asia, is a perennial herb listed as a high-grade drug in the old Chinese Materia Medica and is applied for generalized aching, vertigo, headaches, and arthralgia due to ‘wind, cold, and dampness’ in the traditional medical system.13
Biological activities of the root extract has been reported for suppressive nature on adjuvant arthritis, inhibitory effects on the CNS and peptic ulcers, and febrifugal analgesic, anti-convulsant, anti-oxidant, and anti-inflammatory activities.13 Furthermore, several chemical studies on this herb had been done, and many components such as chromones, coumarins, lignans, sterols, and polyacetylenes were isolated.14 Even though, it was established as a popular ingredient for preventing and treating people who are prone to catching cold as well as bacterial and viral swine disease in traditional Chinese medicine,15, 16, 17, 18 there are no any reports on the specific antiviral activity with chemical constituents.
2. Results and discussion
In a continuing effort to isolate antiviral compounds from natural sources,19, 20, 21 a methanol extract of the radix of S. divaricata showed inhibitory effect against the PEDV. The methanol soluble extract was suspended in water and partitioned successively with n-hexane, EtOAc, and n-BuOH. Bioactivity-guided fractionation of EtOAc-soluble fraction by successive chromatographic procedures (silica gel, RP-C18, and HPLC) yielded three new coumarin glycosides (1–3) together with 10 known coumarins (4–13) (Fig. 1 ).
Fig. 1.
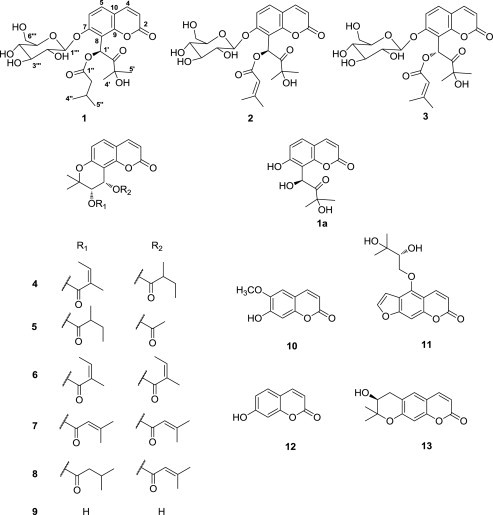
Chemical structures of coumarins 1–13 from Saposhnikovia divaricata and the structure 1a.
Compound 1 was isolated as a brownish powder with positive optical rotation ([α]D 25 +23.0 (c 0.2, MeOH)). Its molecular formula was assigned as C25H32O12 based on the [M+H]+ ion peak at m/z 525.1983 (calcd 525.1972) in the HRFABMS. In the 1H NMR spectrum (Table 1 ), a pair of protons at δ H 6.25 (d, J=9.5 Hz) and 7.90 (d, J=9.5 Hz) were assigned as H-3 and H-4, respectively, based on their HMBC correlations to characteristic C-2 at δ C 160.1 (Fig. 2 ). From the NOESY spectrum, H-4 showed a correlation to a proton at δ H 7.65 (d, J=8.7 Hz), indicating this proton being H-5, and the proton at δ H 7.22 (d, J=8.7 Hz) was determined as H-6. The absence of other coumarin protons suggested the presence of a 7,8-disubstituted coumarin skeleton. Acid hydrolysis of 1 yielded d-glucose, as analyzed by gas chromatography (GC) following treatment with l-cysteine methyl ester hydrochloride and TMS derivatization (Supplementary data). Besides, the HMBC correlation from the anomeric proton at δ H 5.03 (d, J=6.9 Hz) to C-7 (δ C 159.6) (Fig. 2) and the coupling constant (J=6.9 Hz) of the anomeric proton indicated the linkage of a β-d-glucopyranosyl moiety at C-7. The down-field singlet at δ H 7.23 in the 1H NMR spectrum suggested the presence of an O-benzylic esterified methine of compound 1.22 The detailed analysis of 1H and 13C NMR data (Table 1) suggested the presence of isovaleryl group, which was further confirmed by the HMBC correlations from H-2″ (δ H 2.23) to C-3″ (δ C 26.5), from H-2″ and H-3″ (δ H 2.08) to C-1″ (δ C 172.4), from H-4″/5″ (6H, δ H 0.93) to C-2″ (δ C 43.6) and C-3″ (Fig. 2). The HMBC correlation from H-1′ (δ H 7.23) to C-1″ (δ C 172.4) determined as the linkage of this isovaleryl group at C-1′ (Fig. 2). The 2-hydroxy-2-methylpropanol group was also elucidated at C-1′ from the HMBC correlations from H-1′ (δ H 7.23), H3-5′ (δ H 1.35), and H3-4′ (δ H 1.25) to C-2′ (δ C 209.4), and H3-5′ (δ H 1.35) and H3-4′ (δ H 1.25) to C-3′ (δ C 77.8) (Fig. 2). Finally, chemical structure of compound 1 was constructed by the HMBC correlations from H-1′ (δ H 7.23) to C-7 (δ C 159.6), C-8 (δ C 113.5), and C-9 (δ C 154.0), indicating the linkage of C-8 and C-1′, and was named as divaricoumarin A.
Table 1.
NMR spectroscopic data (500 MHz, acetone-d6) for compounds 1–3
| Position | 1 |
2 |
3 |
|||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| Aglycone | ||||||
| 2 | 160.1, C | 160.1, C | 160.1, C | |||
| 3 | 6.25, d (9.5) | 114.6, CH | 6.29, d (9.5) | 114.5, CH | 6.32, d (9.5) | 114.7, CH |
| 4 | 7.90, d (9.5) | 144.8, CH | 7.94, d (9.5) | 144.7, CH | 7.98, d (9.5) | 144.7, CH |
| 5 | 7.65, d (8.7) | 131.1, CH | 7.68, d (8.7) | 131.0, CH | 7.69, d (8.7) | 130.9, CH |
| 6 | 7.22, d (8.7) | 112.9, CH | 7.25, d (8.7) | 112.8, CH | 7.38, d (8.7) | 113.9, CH |
| 7 | 159.6, C | 159.5, C | 160.3, C | |||
| 8 | 113.5, C | 113.5, C | 114.1, C | |||
| 9 | 154.0, C | 154.0, C | 153.5, C | |||
| 10 | 115.1, C | 115.0, C | 115.5, C | |||
| 1′ | 7.23, s | 69.9, CH | 7.35, s | 69.1, CH | 7.33, s | 69.1, CH |
| 2′ | 209.4, C | 209.5, C | 211.2, C | |||
| 3′ | 77.8, C | 77.7, C | 77.8, C | |||
| 4′ | 1.25, s | 27.9, CH3 | 1.29, s | 27.9, CH3 | 1.28, s | 27.8, CH3 |
| 5′ | 1.35, s | 28.1, CH3 | 1.39, s | 27.9, CH3 | 1.39, s | 28.7, CH3 |
| 1″ | 172.4, C | 165.8, C | 165.3, C | |||
| 2″ | 2.23, m | 43.6, CH2 | 5.76, s | 116.1, CH | 5.74, s | 115.6, CH |
| 3″ | 2.08, m | 26.5, CH | 158.9, C | 159.4, C | ||
| 4″ | 0.93, d (6.7) | 22.7, CH3 | 2.16, s | 20.4, CH3 | 2.15, s | 20.2, CH3 |
| 5″ | 0.93, d (6.7) | 22.7, CH3 | 1.90, s | 27.3, CH3 | 1.90, s | 27.3, CH3 |
| Glucose | ||||||
| 1‴ | 5.03, d (6.9) | 102.4, CH | 5.06, d (6.0) | 102.4, CH | 4.88, d (7.7) | 104.3, CH |
| 2‴ | 3.50, t (6.9) | 74.6, CH | 3.54, t (6.0) | 74.5, CH | 3.39, t (7.7) | 74.7, CH |
| 3‴ | 3.52, overlap | 77.3, CH | 3.55, overlap | 77.1, CH | 3.51, t (7.7) | 76.8, CH |
| 4‴ | 3.44, t (7.2) | 71.1, CH | 3.47, d (8.0) | 71.0, CH | 3.43, t (7.7) | 71.2, CH |
| 5‴ | 3.54, overlap | 78.1, CH | 3.57, overlap | 78.0, CH | 3.55, m | 78.3, CH |
| 6‴ | 3.88, br d (11.8) 3.67, dd (11.8, 6.0) |
62.5, CH2 | 3.92, br d (11.6) 3.71, dd (11.6, 5.8) |
62.4, CH2 | 3.95, br d (12.0) 3.71, dd (12.0, 6.2) |
62.5, CH2 |
Fig. 2.
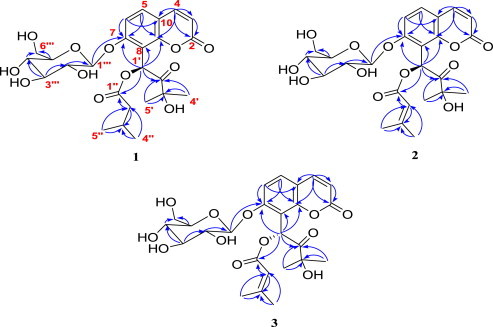
Key HMBC (H→C) correlations for compounds 1–3.
For the determination of absolute configuration at C-1′ of divaricoumarin A (1), electronic circular dichroism (ECD) calculations based on time-dependent density functional theory were used. However, the calculated ECD profile for 1′S- and 1′R-divaricoumarin A showed the same profiles perhaps due to the hindrance by intramolecular hydrogen bond persisting between the sugar unit and isovaleryl substituent group. Thus, the ECD profile of a hydrolyzed 1a of compound 1 was determined. Interestingly, calculated ECD spectrum of 1a (S) isomer showed good agreement with the overall pattern of the experimental ECD spectrum of 1a,23 suggesting the assignment of 1′S configuration for 1a (Fig. 3 A). Furthermore, we found the similarity of experimental ECD profile between 1a and aglycone of compound 1 suggesting for the conservation of same configuration even though ester group was hydrolyzed from the structure by acid hydrolysis (Supplementary data).
Fig. 3.
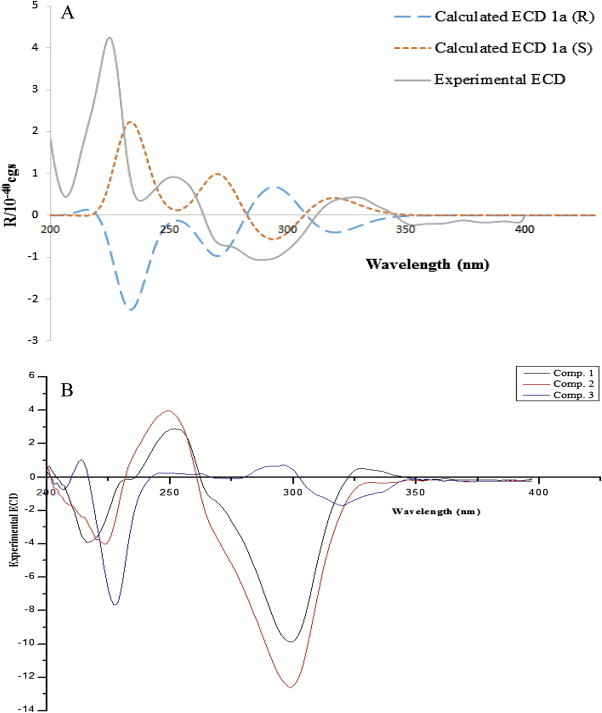
Comparative ECD profiles. (A) Experimental ECD (in MeOH) and calculated ECD (R,S) spectra of compound 1a. (B) Experimental ECD (in MeOH) spectra of compounds 1−3.
Compound 2 was isolated as a brownish powder with positive optical rotation ([α]D 25 +34.0 (c 0.2, MeOH)). Its molecular formula was assigned as C25H30O12 based on the [M+H]+ ion peak at m/z 523.1814 (calcd 523.1816) in the HRFABMS. As like compound 1, protons at δ H 6.29 (H-3, d, J=9.5 Hz), 7.94 (H-4, d, J=9.5 Hz), 7.68 (H-5, d, J=8.7 Hz), and 7.25 (H-6, d, J=8.7 Hz) in the 1H NMR spectrum suggested the presence of a 7,8-disubstituted coumarin nucleus. Compound 2 also possessed a β-d-glucopyranosyl moiety at C-7 as confirmed by GC analysis of the acid hydrolysis product of 2 (Supplementary data) and the HMBC experiment (Fig. 2), as well as the coupling constant (J=6.0 Hz) of the anomeric proton. The unusual singlet at δ H 7.35 revealed the presence of an O-benzylic esterified methine. Moreover, the detailed analysis of 1H and 13C NMR data (Table 1) suggested the presence of a senecioyl group instead of isovaleryl group of compound 1, which was confirmed by the HMBC correlations from H-2″ (δ H 5.76) to C-3″ (δ C 158.9) and C-1″ (δ C 165.9), from H-4″/5″ (6H, δ H 2.16, 1.90) to C-2″ (δ C 116.1) and C-3″ (Fig. 2). The HMBC correlation from H-1′ (δ H 7.35) to C-1″ showed the linkage of this senecioyl group at C-1′. The remaining part of the structure was also elucidated with HMBC correlations from H-1′ (δ H 7.35), H3-5′ (δ H 1.39), and H3-4′ (δ H 1.29) to C-2′ (δ C 209.6), and H3-5′ and H3-4′ to C-3′ (δ C 77.7) (Fig. 2). Finally, the structure of compound 2 was established and named as divaricoumarin B. Based on the similarity of experimental ECD profiles between divaricoumarin A (1) and 2, absolute configuration of C-1′ of 2 was also determined as S (Fig. 3B).
Compound 3 was isolated as a brownish powder with positive optical rotation ([α]D 25 +47.0 (c 0.2, MeOH)). Its molecular formula was assigned as C25H30O12 based on the [M+H]+ ion peak at m/z 523.1799 (calcd 523.1816) in the HRFABMS. A β-d-glucopyranosyl moiety was also attached at C-7 in 3, as analyzed by the same method for determining the sugar parts of 1 and 2. Moreover, the 1H and 13C NMR data showed that compound 3, apart from chemical shift of H-6 at δ H 7.38, had similar chemical shift values compared to compound 2 (Table 1). As the experimental ECD profile of compound 3 was an opposite to that of compound 2 (Fig. 3B), which suggested that compound 3 must be an epimer of 2 with 1′R configuration. Hence, the structure of compound 3 was established and named as divaricoumarin C.
All new compounds contained unusual down-fielded O-benzylic esterified methine at C-1′ position. Previously, few those types of coumarins were isolated from Murraya omphalocarpa 22 and Murraya paniculata,24 but the absolute configuration with respect to C-1′ position was not determined. Thus, this is the first report on the absolute configuration for 8-substituted coumarins with unusual down-fielded O-benzylic esterified methine.
The structures of known coumarins were identified as praeruptorin F (4) and cis-3′-isovaleryl-4′-acetylkhellactone (5),25 praeruptorin B (6) and cis-3′,4′-disenecioylkhellactone (7),26 cis-3′-isovaleryl-4′-senecioylkhellactone (8),27 (–)-cis-khellactone (9),28, 29 scopoletin (10),30 oxypeucedanin hydrate (11),31 decurcinol (12),32 and umbelliferone (13),33 respectively, by comparing their spectroscopic data and optical rotations with the literature values.
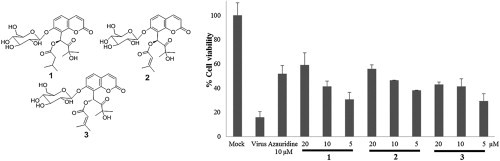
All isolates were evaluated for their inhibitory activity against PEDV through CPE analysis in Vero cells. After PEDV were infected to the Vero cells for 2 h, the cells were treated with test compounds at different concentrations. As shown in the S-Table 1 (Supplementary data), compounds 1–9 exhibited inhibitory effects on PEDV replication with high selective index (SI) values (higher than 4.07±0.25). Interestingly, the most potent compound, hyuganin C (5), showed stronger activity as compared to the positive control, azauridine, with EC50 value of 4.85±0.43. However, there was no obvious antiviral activity observed for compounds 10–13 in this assay. On the other hand, new coumarin glycosides 1–3 revealed moderate inhibitory activity against PEDV in a dose dependent manner without any cytotoxicity (Fig. 4 ).
Fig. 4.
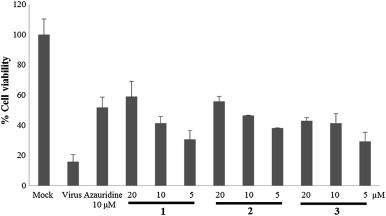
Dose-dependent inhibitory effect of new compounds 1–3 on PEDV-induced CPE.
To discuss the structure–activity relationships (SARs), coumarins 1–13 were divided into three groups based on the substitution patterns at C-7 and C-8: (1) 8-substituted coumarins 1–3 with O-sugar at C-7, (2) 8-substituted coumarins 4–9 with angular pyrano ring at C-8/C-1′/C-2′/C-3′/O/C-7, (3) non-8-substituted coumarins 10–13. Among these compounds, group 3 (10–13) had no anti-PEDV activity, indicating the key role of functionalization at C-8 in the antiviral property of coumarins (Supplementary data). Compared with group 1 (1–3), coumarins 4–8 showed stronger anti-PEDV affects based on EC50 values. However compound 9 in group 2 demonstrated similar EC50 values to those of coumarins 1–3. The above observations implied that the existence of an angular pyrano ring at C-8/C-1′/C-2′/C-3′/O/C-7 bearing functionalization at C-1′ and C-2′ positively influenced the antiviral property of coumarins.
The stages like viral entry, viral penetrations into the host cells, viral processing, viral replication, and viral release from the infected cells in PEDV life cycle are the potential targets for the possible antiviral compounds.34 During viral replication, genes encoding the viral structural proteins [nucleocapsid (N), membrane (M), and spike (S)] are key elements for viral integrity.5 Thus, to investigate the inhibitory effect of compound 5 on viral replication, quantitative real-time PCR was performed using specific primers for viral GP6 nucleocapsid, GP2 spike, and GP5 membrane encoding genes. As shown in Fig. 5 , compound 5 exhibited inhibitory effect on genes encoding PEDV GP6 nucleocapsid, GP2 spike, and GP5 membrane proteins in a dose-dependent manner at concentrations of 10, 5, and 2.5 μM. Compound 5 showed stronger inhibitory effects than positive control, azauridine, for all three structural proteins encoding genes at the same test concentration of 10 μM.
Fig. 5.
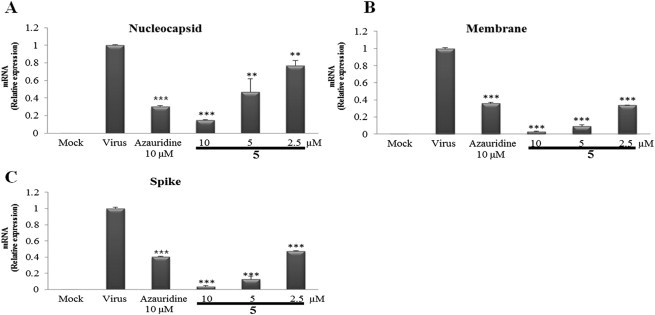
Quantitative real-time PCR analysis revealed the inhibitory effect of compound 5 on genes encoding viral structural proteins. Dose-dependent inhibitory effect of compound 5 on genes encoding GP6 nucleocapsid (A), GP5 membrane (B), and GP2 spike proteins (C), respectively.
Moreover, western blot analysis supported that compound 5 showed dose-dependent inhibitory effects on viral GP6 nucleocapsid and GP2 spike protein synthesis (Fig. 6 ). The GP6 nucleocapsid and GP2 spike proteins clearly indicated the stronger inhibitory tendency of compound 5 toward both structural protein syntheses than positive control at same concentration. Furthermore, PEDV replication analysis using an immunofluorescence assay revealed green fluorescence in virus-infected cells, but not in mock infected Vero cells. Treatment with different concentrations of compound 5 reduced the fluorescence-positive, PEDV infected cells in a dose-dependent manner at 20, 10, and 5 μM (Supplementary data).
Fig. 6.
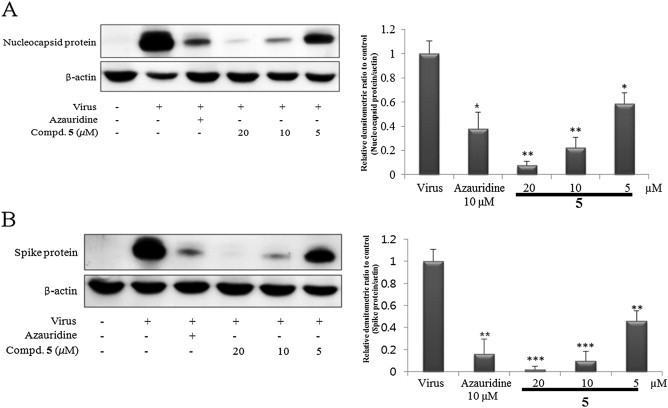
Western blot analysis showed the inhibitory effect of compound 5 on viral structural proteins synthesis. Dose-dependent GP6 nucleocapsid (A) and GP2 spike (B) proteins synthesis inhibitory effect of compound 5, respectively.
In this study, 3 new (1–3) and 10 known (4–13) coumarins were isolated from radix of S. divaricata. Absolute configuration of coumarins with 8-substituted down-fielded O-benzylic esterified methine was determined for the first time through ECD analysis. Compound 5 with angular pyrano coumarin skeleton showed the strongest inhibitory effect on viral replication. Human corona viruses like severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) from same coronaviridae family share some similar replication mechanism with PEDV. Further optimization of anti-PEDV coumarin molecules could be a worthy approach for finding new antiviral compounds against the fatal human corona viruses like SARS and MERS.
3. Experimental
3.1. General
Optical rotation values were determined on a Rudolph Autopol IV polarimeter in a 100 mm glass microcell. UV spectra were measured in MeOH on a JASCO V-550UV/vis spectrometer with a 0.5 nm resolution, and ECD spectra were recorded on a JASCO J-710 spectropolarimeter. IR spectra (KBr) were recorded on a Nicolet 6700 FT-IR (Thermo Electron Corp.). NMR spectra were acquired from a Varian Unity Inova 500 MHz spectrometer with TMS as the internal standard at the Seoul National University, Seoul, Korea. HRFABMS and ESIMS data were obtained with JEOL JMS-700 and Agilent 6130 Quadrupole mass spectrometer, respectively. Silica gel (Merck, 63–200 μm particle size), and RP-C18 (Merck, 40–63 μm particle size) were used for column chromatography. TLC was carried out with silica gel 60F254 and RP-C18 F254 plates. Preparative HPLC separation was performed by using a Gilson system with UV detection at 205 and 254 nm and Optima Pak C-18 column (10×250 mm, 10 μm particle size, RS Tech, Daejon, Korea). Analytical grade solvents were used for extraction and isolation.
3.2. Plant material
The dried radixes of S. divaricata were purchased from a market in Gwangju, South Korea, in February 2011 and were identified botanically by Prof. Won Keun Oh, College of Pharmacy, Seoul National University, Seoul, Korea. A voucher specimen (SNU2012–12) has been deposited at the Laboratory of Pharmacognosy, College of Pharmacy, Seoul National University.
3.3. Extraction and isolation
The dried radixes of S. divaricata (10 kg) were extracted with MeOH (3×15 L) at room temperature for 1 week. The combined MeOH extract was concentrated to yield a dry residue (458 g), which was suspended in H2O (3 L) and partitioned successively with n-hexane (3×2 L), EtOAc (3×2 L), and n-BuOH (3×2 L). The bioactive EtOAc-soluble part (72.0 g) was chromatographed over a silica gel open column (10×40 cm) by eluting with n-hexane/acetone gradient (20:1 to 0:1) to afford seven fractions (F1–F7). Fractions F1–F5 was found to be active against PEDV. Fraction F1 (7.8 g) was subjected to RP-C18 open column chromatography and eluted with MeOH/H2O (1:1 to 1:0) to yield eight subfractions (F1.1–F1.8). Further purification of F1.5 (200 mg) by preparative HPLC [mobile phase: MeOH in H2O containing 0.1% HCO2H (80:20, v/v); flow rate: 2 mL/min] resulted in the isolation of compound 4 (13 mg). Similarly, fraction F2 (9 g) was subjected to RP-C18 open column chromatography with mobile phase MeOH/H2O (1:1 to 1:0) and fractionated to seven subfractions (F2.1–F2.7). Then, part of subfraction F2.3 (1.4 g) was further purified using HPLC [mobile phase: MeOH in H2O containing 0.1% HCO2H (75:25, v/v); flow rate: 2 mL/min] leading to the isolation of compounds 5 (12 mg) and 6 (31 mg). Also, the subfraction F2.5 (550 mg) was purified using HPLC [mobile phase: MeOH in H2O containing 0.1% HCO2H (70:30, v/v); flow rate: 2 mL/min] to afford compounds 7 (12 mg) and 8 (22 mg). Fraction F3 (2 g) was subjected to RP-C18 open column chromatography with MeOH/H2O (1:2 to 1:0) and fractionated to five subfractions (F3.1–F3.5). After applying the fraction F3.1 to an RP-C18 open column (MeOH/H2O, 1:4 to 1:0), the subfraction F3.1.4 using HPLC with 50% MeOH in water at a flow rate of 2 mL/min yielded compounds 9, 10, and 11 (12, 6, and 7 mg, respectively). Fraction F4 yielded seven subfractions (F4.1–F4.7) after applying to an RP-C18 open column with MeOH/H2O (1:3 to 1:0). Compound 12 (50 mg) was purified via crystallization from subfraction F4.1 (1.5 g), and compound 13 (12 mg) was isolated from the subfraction F4.2 (650 mg) by using HPLC system with 40% MeOH in water as mobile phase at a flow rate of 2 mL/min. Fraction F5 (14 g) was purified by a silica gel column with a gradient of n-hexane/acetone (7:1 to 0:1). Subfraction F5.4 (4.7 g) was further purified by an RP-C18 column eluting with a stepwise gradient of MeOH/H2O (1:3 to 1:0) to afford six subfractions (F5.4.1–F5.4.6). Subfraction F5.4.2 (0.8 g) was subjected to preparative HPLC with 30% CH3CN in water as mobile phase at a flow rate of 2 mL/min to give compounds 1 (20.0 mg), 2 (3.5 mg), and 3 (2.5 mg).
3.3.1. Divaricoumarin A (1)
Brownish powder; [α]D 25 +23.0 (c 0.2, MeOH); UV (MeOH) λ max (log ɛ) 200 (3.19), 318 (2.19) nm; ECD (MeOH, Δɛ) 217 (−3.90), 252 (+2.89), 299 (−9.87); IR (KBr) ν max 3390, 2955, 1734, 1615, 1460, 1250, 1014 cm−1; 1H and 13C NMR data, Table 1; HRFABMS m/z 525.1983 [M+H]+ (calcd for C25H33O12, 525.1972).
3.3.2. Divaricoumarin B (2)
Brownish powder; [α]D 25 +34.0 (c 0.2, MeOH); UV (MeOH) λ max (log ɛ) 202 (3.22), 222 (3.09), 318 (2.83) nm; ECD (MeOH, Δɛ) 223 (−4.0), 249 (+3.97), 299 (−12.61); IR (KBr) ν max 3387, 2945, 1721, 1609, 1448, 1246, 1026 cm−1; 1H and 13C NMR data, Table 1; HRFABMS m/z 523.1814 [M+H]+ (calcd for C25H31O12, 523.1816).
3.3.3. Divaricoumarin C (3)
Brownish powder; [α]D 25 +47.0 (c 0.2, MeOH); UV (MeOH) λ max (log ɛ) 202 (2.97), 222 (2.79), 318 (2.49) nm; ECD (MeOH, Δɛ) 214 (+1.05), 228 (−7.66), 296 (+0.73), 320 (−1.70); IR (KBr) ν max 3383, 2941, 1724, 1603, 1452, 1242, 1023 cm−1; 1H and 13C NMR data, Table 1; HRFABMS m/z 523.1799 [M+H]+ (calcd for C25H31O12, 523.1816).
3.4. Acid hydrolysis of compound 1
A solution of compound 1 (9.0 mg) in water was treated with 5% HCl and heated on a water bath maintained at 50 °C for 24 h. After the sample solution (monitored by TLC) was partitioned with EtOAc, its residue was analyzed by LC–MS system (Agilent Technologies, 6130) with one major and one minor compounds. The mixture was further purified by HPLC to give aglycone of compound 1 (0.2 mg) and 1a (0.7 mg), respectively, to determine absolute configuration. Aglycone of compound 1: white powder; ECD (MeOH, Δɛ) 223 (+1.09), 263 (+0.05), 296 (−0.15), 332 (+0.13); 1H NMR (600 MHz, acetone-d 6) δ H 7.89 (1H, d, J=9.7 Hz, H-4), 7.59 (1H, d, J=7.8 Hz, H-5), 6.89 (1H, s, H-1′), 6.78 (1H, d, J=7.8 Hz, H-6), 6.18 (1H, d, J=9.7 Hz, H-3), 2.15 (2H, m, H-2″), 1.36 (3H, s, H-5′), 1.30 (3H, s, H-4′), 0.98 (6H, d, J=6.9 Hz, H-4″/5″); ESIMS (negative) m/z 361.1 [M−H]−. Compound 1a: white powder; ECD (MeOH, Δɛ) 225 (+4.25), 252 (+0.91), 289 (−1.06), 329 (+0.43); 1H NMR (600 MHz, acetone-d 6) δ H 7.89 (1H, d, J=9.6 Hz, H-4), 7.53 (1H, d, J=8.2 Hz, H-5), 6.73 (1H, d, J=8.2 Hz, H-6), 6.17 (1H, d, J=9.6 Hz, H-3), 5.89 (1H, s, H-1′), 1.34 (3H, s, H-5′), 1.32 (3H, s, H-4′); ESIMS (positive) m/z 279.1 [M+H]+.
3.5. Computational chemistry for ECD calculation
Turbomole at the basis set def-SV(P) for all atoms and the functional B3LYP was used for the ground-state geometries optimization with density functional theory (DFT) calculations. Harmonic frequency calculation was used for the further confirmation of the ground states. The calculated ECD data corresponding to the optimized structures were obtained with TDDFT at the B3LYP functional. The ECD spectra were simulated by overlapping Gaussian functions for each transition (σ is the width of the band at 1/e height). ΔE i and R i are the excitation energies and rotatory strengths for transition i, respectively. For this report, the value of σ was 0.10 eV.
3.6. Cell culture and virus stock
Vero cells (African green monkey kidney cell line; ATCC CCR-81) were supported by American Type Culture Collection (ATCC, Manassas, VA, U.S.A.) and maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 100 μg/mL streptomycin, 100 U/mL penicillin, and 10% fetal bovine serum (FBS). PEDV (porcine epidemic diarrhea virus) was obtained from Choong Ang Vaccine Laboratory, Korea. Virus stock was stored at −80 °C until use.
3.7. Cytotoxicity assay
The cell viability was assessed using an MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) based cytotoxicity assay. Vero cells were grown in 96-well plates at 1×105 cells per well and allowed to adhere for 24 h prior to treatment. The cells in 96-well plates were treated with various concentrations of compounds and incubated for 48 h. The final concentration of DMSO in the culture medium was maintained at 0.05% (v/v) to avoid solvent toxicity. Subsequently, 20 μL of the 2 mg/mL MTT solution was added to each well of the plate and incubated for 4 h. After removal of the supernatant, 100 μL DMSO was added for solubilization of formazan crystals. Then, the absorbance was measured at 550 nm. The percentage cell viability was expressed as toxicities of the compounds, and defined as the absorbance in the experiment compared to that in the control wells. The regression analysis was used to determine the 50% cytotoxic concentration (CC50).
3.8. Cytopathic effect (CPE) inhibition assay
Vero cells were seeded onto 96-well culture plates at 1×105 cells per well. Next day, after removal of the medium and washing with phosphate buffered saline (PBS), PEDV at 0.01 MOI was inoculated onto near confluent Vero cell monolayers for 2 h. Then, the media was removed and replaced by DMEM with different concentrations of test compounds. Each concentration of compounds was determined in triplicate. The cultures were incubated for 3 days at 37 °C under 5% CO2 atmosphere. Then, cells were replaced with DMEM and 20 μL of the 2 mg/mL MTT to each well and incubated at 37 °C for 4 h. After that, cytotoxicity assay and the 50% effective concentration (EC50) were calculated by regression analysis. A selective index (SI) was determined using the formula SI=CC50/EC50.
3.9. Quantitative real-time PCR
Vero cells were grown to about 90% confluence in 6-well plates, then infected with PEDV at 0.01 MOI and incubated for 2 h. After media was removed, cells were cultured with DMEM in the presence of various concentrations of compounds. Total RNA was isolated from the cells after 24 h, based on the TRIzol method. The total RNA was reverse transcribed using random primer (iNtRON Biotechnology, INC, Korea) according to manufacturer's instruction. Real-time PCR was carried out by using selective primers for PEDV (Supplementary data) and conducted by using 2 μL of cDNA and Maxima SYBR Green qPCR master mix 2X (Thermo sci., Rockford, IL, U. S. A.). Regarding cycling conditions for real-time PCR, the following procedure was used: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 1 min. The Step one Plus Real-time PCR system (Applied Biosystems) was used for real-time PCR experiments and the StepOne software v2.3 was for the data analysis.
3.10. Western blot analysis
According to the similar procedure to quantitative Real-time PCR experiment, the cultures for western blot analysis were prepared. The cells were first washed with cold PBS and then stored at −80 °C. To prepare the whole cell lysate, cells were lysed on ice by using 100 μL lysis buffer [50 mM Tris–HCl (pH 7.6), 120 mM NaCl, 1 mM EDTA, 0.5% NP-40, 50 mM NaF] and centrifuged for 20 min at 12,000 rpm. Supernatants were collected from the lysates and protein concentrations were determined using protein assay kit (Bio-Rad Laboratories, Inc., CA, U.S.A.). Aliquots of lysates were boiled for 5 min and electrophoresed on 10% or 12% SDS-polyacrylamide gels, and the protein in the gels were electrotransferred to PVDF membranes (PVDF 0.45 μm, Immobilon-P, U. S. A.). Before incubation with secondary antibodies, the membranes were first incubated with primary antibodies, like spike (S) protein, nucleocapsid (N) (Abfrontier Co. Ltd., Seoul, Korea) or mouse monoclonal actin antibody. These were detected by enhanced chemiluminescence western blotting detection kit (Thermo sci., Rockford, IL, U.S.A.).
3.11. Immunofluorescence assay
Vero cells were grown on 8-well chamber slides (LAB-TEK, NUNC, U. S. A.) and the cell monolayers were injected with PEDV at 0.01 MOI for 2 h. The solution was removed and replaced by DMEM, and treated with test compounds at the corresponding concentration. The incubation of the cultures continued for 24 h at 37 °C under 5% CO2 atmosphere. After washing with PBS (pH 7.4) three times, the cells were fixed with a 4% paraformaldehyde solution at room temperature for 30 min. After the cells were blocking with 1% BSA for 1 h, and then incubated overnight with monoclonal antibody against N protein of PEDV (Abfrontier Co. Ltd., Seoul, Korea) diluted 1:50 in PBS (pH 7.4). Then, the cells were further incubated with FITC-conjugated goat anti-mouse lgG antibody (Jackson, ImmunoResearch, Inc., PA, U.S.A.) for 1 h. After the cells were stained with 500 nM DAPI solution at room temperature for 10 min, slides were mounted with mounting reagent for fluorescence (Vectashield, Vector Lab, Inc., CA, U.S.A.) and observed by fluorescence microscopy (Olympus ix70 Fluorescence Microscope, U.S.A.).
3.12. Statistical analysis
The results are expressed as the means±SD of three independent experiments. Statistical analysis was performed using Sigma Plot Statistical Analysis software. Differences between group mean values were determined by one-way analysis of variance followed by a two-tailed Student's t-test for unpaired samples, assuming equal variances. Statistical significance was accepted at p<0.05.
Acknowledgements
This research was supported in part by grants from the Marine Biotechnology Program of the Ministry of Oceans and Fisheries (PJT200669) and from the Procurement and Development of Foreign Biological Resources (2012-K1A1A3307871) funded by the Korean Government.
Footnotes
Analysis of the absolute configuration of sugar moieties in compounds 1–3, 1D (1H and 13C) and 2D (HSQC and HMBC) NMR spectra for compounds 1–3, ESIMS spectra of the aglycone of compound 1 and 1a, experimental ECD spectra of the aglycone of compound 1 and 1a, immunofluorescence image revealing the inhibition of PEDV replication by compound 5 in a dose dependent manner, inhibitory effect of compounds 1–13 on PEDV induced CPE, and primers used for Real-time PCR. These materials are available free of charge via the internet at http://www.elsevier.com. Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.tet.2015.04.092.
Supplementary data
Analysis of the absolute configuration of sugar moieties in compounds 1–3, 1D (1H and 13C) and 2D (HSQC and HMBC) NMR spectra for compounds 1–3, ESIMS spectra of the aglycone of compound 1 and 1a, experimental ECD spectra of the aglycone of compound 1 and 1a, immunofluorescence image revealing the inhibition of PEDV replication by compound 5 in a dose dependent manner, inhibitory effect of compounds 1–13 on PEDV induced CPE, and primers used for Real-time PCR. These materials are available free of charge via the internet at http://www.elsevier.com. The following is the supplementary data related to this article:
References and notes
- 1.Zhao M., Sun Z., Zhang Y., Wang G., Wang H., Yang F., Tian F., Jiang S. J. Virol. 2012;86:13858–13859. doi: 10.1128/JVI.02674-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho W.K., Kim H., Choi Y.J., Yim N.H., Yang H.J., Ma J.Y. Evid. Based Complement. Altern. Med. 2012:1–10. doi: 10.1155/2012/985151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi H.J., Kim J.H., Lee C.H., Ahn Y.J., Song J.H., Baek S.H., Kwon D.H. Antiviral Res. 2009;81:77–81. doi: 10.1016/j.antiviral.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Puranaveja S., Poolperm P., Lertwatcharasarakul P., Kesdaengsakonwut S., Boonsoongnern A., Urairong K., Kitikoon P., Choojai P., Kedkovid R., Teankum K. Emerg. Infect. Dis. 2009;15:1112–1115. doi: 10.3201/eid1507.081256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ge F.F., Yang D.Q., Ju H.B., Wang J., Liu J., Liu P.H., Zhou J.P. Arch. Virol. 2013;158:2227–2231. doi: 10.1007/s00705-013-1722-7. [DOI] [PubMed] [Google Scholar]
- 6.Chen Q., Li G., Stasko J., Thomas J.T., Stensland W.R., Pillatzki A.E., Gauger P.C., Schwartz K.J., Madson D., Yoon K.J. J. Clin. Microbiol. 2014;52:234–243. doi: 10.1128/JCM.02820-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L., Byrum B., Zhang Y. Emerg. Infect. Dis. 2014;20:917–919. doi: 10.3201/eid2005.140195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kweon C.H., Kwon B.J., Lee J.G., Kwon G.O., Kang Y.B. Vaccine. 1999;17:2546–2553. doi: 10.1016/S0264-410X(99)00059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song D., Oh J., Kang B., Yang J.S., Moon H., Yoo H.S., Jang Y., Park B. Res. Vet. Sci. 2007;82:134–140. doi: 10.1016/j.rvsc.2006.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suo S., Ren Y., Li G., Zarlenga D., Bu R.E., Su D., Li X., Li P., Meng F., Wang C. Virus Res. 2012;167:259–266. doi: 10.1016/j.virusres.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pospischil A., Stuedli A., Kiupel M. J. Swine Health Prod. 2002;10:81–85. [Google Scholar]
- 12.Song D., Park B. Virus Genes. 2012;44:167–175. doi: 10.1007/s11262-012-0713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okuyama E., Hasegawa T., Matsushita T., Fujimoto H., Ishibashi M., Yamazaki M. Chem. Pharm. Bull. 2001;49:154–160. doi: 10.1248/cpb.49.154. [DOI] [PubMed] [Google Scholar]
- 14.Dai J., Chen X., Cheng W., Liu X., Fan X., Shen Z., Bi K. J. Chromatogr., B. 2008;868:13–19. doi: 10.1016/j.jchromb.2008.03.031. [DOI] [PubMed] [Google Scholar]
- 15.Hiromi K., Takeshi D., Junko K., Kaisuke Y. Shoyakugaku Zasshi. 1983;37:276–280. [Google Scholar]
- 16.Sasaki H., Taguchi H., Endo T., Yoshioka I. Chem. Pharm. Bull. 1982;30:3555–3562. [Google Scholar]
- 17.Wang C.C., Chen L.G., Yang L.L. Cancer Lett. 1999;145:151–157. doi: 10.1016/s0304-3835(99)00248-7. [DOI] [PubMed] [Google Scholar]
- 18.Xue B., Li W., Li L., Xiao Y. China J. Chin. Mater. Med. 2000;25:297–299. [PubMed] [Google Scholar]
- 19.Dao T.T., Tung B.T., Nguyen P.H., Thuong P.T., Yoo S.S., Kim E.H., Kim S.K., Oh W.K. J. Nat. Prod. 2010;73:1636–1642. doi: 10.1021/np1002753. [DOI] [PubMed] [Google Scholar]
- 20.Dao T.T., Nguyen P.H., Lee H.S., Kim E., Park J., Lim S.I., Oh W.K. Bioorg. Med. Chem. Lett. 2011;21:294–298. doi: 10.1016/j.bmcl.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 21.Dao T.T., Dang T.T., Nguyen P.H., Kim E., Thuong P.T., Oh W.K. Bioorg. Med. Chem. Lett. 2012;22:3688–3692. doi: 10.1016/j.bmcl.2012.04.028. [DOI] [PubMed] [Google Scholar]
- 22.Chia Y.C., Chang F.R., Wang J.C., Wu C.C., Chiang M.Y.N., Lan Y.H., Chen K.S., Wu Y.C. Molecules. 2008;13:122–128. doi: 10.3390/molecules13010122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schramm A., Ebrahimi S.N., Raith M., Zaugg J., Rueda D.C., Hering S., Hamburger M. Phytochemistry. 2013;96:318–329. doi: 10.1016/j.phytochem.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 24.Steck W. Can. J. Chem. 1972;50:443–445. [Google Scholar]
- 25.Song Y.L., Zhang Q.W., Li Y.P., Yan R., Wang Y.T. Molecules. 2012;17:4236–4251. doi: 10.3390/molecules17044236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu R., Feng L., Sun A., Kong L. J. Chromatogr., A. 2004;1057:89–94. doi: 10.1016/j.chroma.2004.09.047. [DOI] [PubMed] [Google Scholar]
- 27.Jong T.T., Hwang H.C., Jean M.Y., Wu T.S., Teng C.M. J. Nat. Prod. 1992;55:1396–1401. doi: 10.1021/np50088a002. [DOI] [PubMed] [Google Scholar]
- 28.Chen I.S., Chang C.T., Sheen W.S., Teng C.M., Tsai I.L., Duh C.Y., Ko F.N. Phytochemistry. 1996;41:525–530. doi: 10.1016/0031-9422(95)00625-7. [DOI] [PubMed] [Google Scholar]
- 29.Macias F., Massanet G., Rodríguez-Luis F., Salvá J., Fronczek F. Magn. Reson. Chem. 1989;27:653–658. doi: 10.1002/mrc.1260270913. [DOI] [PubMed] [Google Scholar]
- 30.Adfa M., Yoshimura T., Komura K., Koketsu M. J. Chem. Ecol. 2010;36:720–726. doi: 10.1007/s10886-010-9807-1. [DOI] [PubMed] [Google Scholar]
- 31.Fujioka T., Furumi K., Fujii H., Okabe H., Mihashi K., Nakano Y., Matsunaga H., Katano M., Mori M. Chem. Pharm. Bull. 1999;47:96–100. doi: 10.1248/cpb.47.96. [DOI] [PubMed] [Google Scholar]
- 32.Kim S., Ko H., Son S., Shin K.J., Kim D.J. Tetrahedron Lett. 2001;42:7641–7643. [Google Scholar]
- 33.Singh R., Singh B., Singh S., Kumar N., Kumar S., Arora S. Food Chem. 2010;120:825–830. [Google Scholar]
- 34.Kwon H.J., Ryu Y.B., Kim Y.M., Song N., Kim C.Y., Rho M.C., Jeong J.H., Cho K.O., Lee W.S., Park S.J. Bioorg. Med. Chem. 2013;21:4706–4713. doi: 10.1016/j.bmc.2013.04.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.