

Abstract
The coronavirus spike glycoprotein is a class I membrane fusion protein with two characteristic heptad repeat regions (HR1 and HR2) in its ectodomain. Here, we report the X-ray structure of a previously characterized HR1/HR2 complex of the severe acute respiratory syndrome coronavirus spike protein. As expected, the HR1 and HR2 segments are organized in antiparallel orientations within a rod-like molecule. The HR1 helices form an exceptionally long (120 Å) internal coiled coil stabilized by hydrophobic and polar interactions. A striking arrangement of conserved asparagine and glutamine residues of HR1 propagates from two central chloride ions, providing hydrogen-bonding “zippers” that strongly constrain the path of the HR2 main chain, forcing it to adopt an extended conformation at either end of a short HR2 α-helix.
Keywords: SARS, Membrane fusion, Heptad repeat, Spike protein, Structure, Class I fusion protein, Coiled coil, Asparagine zipper
Introduction
To initiate a productive infection, all viruses must translocate their genome across the cell membrane (Reviewed by Smith and Helenius, 2004). Enveloped viruses achieve this step by membrane fusion, a process mediated by specialized envelope proteins present at the virus surface. For coronaviruses such as the recently emerged SARS coronavirus (SARS-CoV), the spike (S) glycoprotein is responsible for both cell attachment and entry by triggering fusion of the viral and cellular membrane. This type I membrane protein can be divided into two domains of similar size, S1 and S2 (Fig. 1A). S1 forms the bulbous globular head and is responsible for cell attachment; its amino acid sequence is less conserved than S2, which forms the membrane-anchored stalk region and is responsible for membrane fusion. The coronavirus spike protein has all the essential features of a bona fide class I viral fusion protein (Smith and Helenius, 2004), including the occurrence of two heptad repeat regions in its S2 domain (de Groot et al., 1987). Most class I viral fusion proteins, with influenza virus hemagglutinin (HA) as a prototype (Skehel and Wiley, 2000), are expressed as precursor proteins, which are endoproteolytically cleaved by cellular proteases, giving rise to a metastable complex of a receptor-binding subunit and a membrane fusion subunit. Upon receptor binding at the cell membrane or as a result of protonation after endocytosis, the fusion proteins undergo a dramatic conformational transition that leads to the exposure of a hydrophobic fusion peptide and insertion into the target membrane. The free energy released upon subsequent refolding of the fusion protein to its most stable conformation through association of the two HR regions is believed to facilitate the close apposition of viral and cellular membranes and the subsequent lipid merger. The coronavirus spike protein has, however, some characteristics that set it apart from class I fusion proteins, such as the lack of a cleavage requirement and the presence of an internal fusion peptide. We and others have previously shown that, analogous to other class I fusion proteins, peptides corresponding to the HR regions of the mouse hepatitis coronavirus (MHV, Bosch et al., 2003) and SARS-CoV (Bosch et al., 2004, Ingallinella et al., 2004, Xu et al., 2004a, Tripet et al., 2004) can fold into a stable rod-like structure, consisting of three HR1 helices in association with three HR2 peptides in antiparallel orientation. This complex, supposedly representing the post-fusion conformation with the predicted fusion peptide upstream of HR1 and the transmembrane segment downstream of HR2 positioned at the same end, juxtaposes the cellular and viral membrane thereby facilitating membrane fusion and consequently virus entry. Here, we report our analysis of the structure of this HR1/HR2 complex of the SARS-CoV as determined by X-ray crystallography to 2.2 Å resolution. This analysis complements two recent reports (Supekar et al., 2004, Xu et al., 2004b) describing the structures of three related constructs of the core fragment of this protein. Indeed, the structure described here provides important additional insights for understanding the determinants of the stabilization of the HR1 central core and the constraints imposed to the HR2 main chain by the internal coiled coil.
Fig. 1.
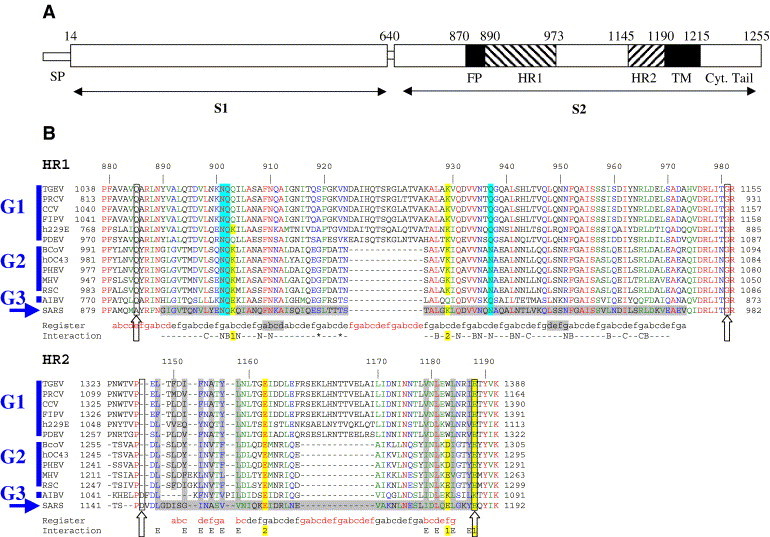
The HR1 and HR2 regions in the coronavirus S glycoprotein. (A) Linear diagram indicating the relative locations of the segments described in the text. The S1 and S2 regions are labeled. FP: putative fusion peptide region. TM: transmembrane region. SP: signal peptide. (B) Sequence alignments of the HR1 and HR2 regions of coronavirus spike proteins of the three different groups (G1 to G3) and the unclassified SARS coronavirus (blue arrow) using ClustalW (Thompson et al., 1994). See the Supplementary material for the complete names of the viruses used. The numbers at the top line correspond to the SARS amino acid sequence, the structure of which is described in the text. The alignment is color-coded according to sequence conservation: red, strictly conserved; green, highly conserved; blue, conserved; black, variable. The alignment in the HR2N region was manually modified to match the structural superposition with the corresponding region of the MHV protein (PDB accession numbers 1WDF and 1WDG, Xu et al., 2004c). Residues with ordered electron density have a grey background in the SARS line. The cloned fragment contained all residues between the boxed columns (further highlighted with a vertical empty arrow below the sequence). Two additional lines at the bottom summarize the following: the “Register” line provides the abcdefg heptad repeat assignment with letters in black for the residues actually observed in a helical conformation in the structure, with the two HR1 stutters in grey background. Note the insertion of exactly two heptad repeats, both in HR1 and HR2, in the S protein of group 1 coronaviruses. The “Interaction” line shows the two salt bridges (1 and 2, see Fig. 2B) in a yellow background. In the case of HR1, this line provides also the residues participating in the asparagine/glutamine zipper shown in Fig. 3 (labeled “N”), the knob-into-hole interactions with either partner within the trimer (labeled B or C), and the residues lining the central cavity shown in Fig. 2B (labeled with a star). Residues forming salt bridges 1 and 2 connecting HR1 with HR2 have a yellow background, with a number below to indicate the partner in each chain. Note that salt bridge 1 is conserved, but is sometimes made with HR1 residue 900 (from the previous helical turn) instead of 903. The structure shows that the side chain of residue 900 can contact the HR2 1188 side chain equally well. Blue background columns indicate HR1 residues interacting with the two central chloride ions. Vertical grey background columns identify HR2 residues in the extended segments (HR2N and HR2C) that pack their side chains into hydrophobic pockets in the HR1 interhelical grooves.
Results
Description of the molecule
The membrane fusion core fragment of the SARS-CoV S glycoprotein was crystallized in a rhombohedral space group and the crystals diffracted just beyond 2.2 Å resolution. The details of the construct used, the production and purification procedure, the crystallization and the X-ray structure determination are provided in Materials and methods, given as Supplementary material. The crystal asymmetric unit contains two HR1/HR2 heterodimers lying about two different crystallographic 3-fold axes. The packing environment of the two independent molecules is however very similar. The HR1/HR2 complex is an α-helical trimeric bundle (Figs. 1A and B) containing 378 amino acids in total, its maximal length and diameter being 123 Å and about 30 Å, respectively. The total molecular surface buried from solvent upon trimer formation is 3000 Å2 per HR1/HR2 heterodimeric subunit. In the intact protein, an intervening polypeptide segment of 173 amino acids – from residues 974 to 1146 – connects the two segments, HR1 and HR2, as diagrammed in Fig. 1A.
The HR1 segment folds as a 22-turns-long α-helix that is involved in parallel interactions with its 3-fold related symmetry mates throughout its whole length, forming a 120-Å-long central trimeric bundle which buries 2350 Å2 of accessible surface for each helix. The interhelical grooves of this bundle accommodate the HR2 segment running antiparallel to the HR1 helix in a pattern typical of the post-fusion conformation of class I fusion proteins. The accessible surface buried by each HR2 peptide is 1460 Å2. The overall fold and quaternary structure yields a very stable protein rod having the HR1 N-termini and the HR2 C-termini clustered together at one end. This organization of the post-fusion form implies that both of the membrane-interacting segments, the fusion peptide and the TM region, are brought into close proximity by the fusogenic conformational change of the S protein, similar to all the other membrane fusion proteins of known structure.
Determinants of the central coiled-coil interactions
As expected, there is essentially one side chain per turn of the HR1 α-helix participating to the central hydrophobic core of the molecule, resulting in 23 amino acids from each chain (labeled to the left of Fig. 2A) interacting with their symmetry mates at the central 3-fold axis. Positions “a” and “d” alternate every other turn according to the helical wheel diagram of Fig. 2D. The long HR1 α-helix does not display the typical “heptad repeat” parameters, in which the average periodicity is 3.5 residues per turn – that is, 7 residues every two turns (Crick, 1953) – but rather displays a mean periodicity of 3.64 residues per turn, which is very close to the canonical α-helix periodicity (Pauling et al., 1951). The “a” and “d” positions therefore drift away from the hydrophobic core (Fig. S1, Supplementary materials) and become out of register after several turns, effectively shifting the face of the helix that faces the hydrophobic core at the 3-fold axis. This results in the presence of “stutters” in which the 3–4–3–4 periodicity of the heptad repeat becomes 3–4–4–3–4 at two positions (indicated in Fig. 2A). The helical wheel of Fig. 2D has the residues labeled along their helical positions for the 22 turns, after correction for the stutters. This assignment is also indicated in the sequence alignment of Fig. 1B, which shows the “a” through “g” positions of the heptad repeats, and the “abcd” (first stutter) or “defg” (second stutter) insertions indicated under the HR1 sequence.
Fig. 2.

Overall structure: determinants of the hydrophobic core formation and central ions interactions. (A and B) Ribbon diagrams colored light and dark grey for the HR1 and HR2 polypeptides, respectively. Pink spheres on the central 3-fold axis indicate the chloride ions. In A, the axes of the HR1 helices in the trimer are drawn as green tubes, highlighting the two stutters in red. The coiled coil axis is dark red (vertical at the trimer center). The helical turns are numbered from N- to C-terminus for one of the subunits (black and white numbers are used for HR1 and HR2, respectively). The N- and C-terminal ends of the model are indicated for one HR1/HR2 heterodimer. The 2 columns between the vertical scale bar on the left and the ribbon diagram indicate the residues and the 3- and 4-residue repeat pattern of the side chains facing the 3-fold axis of the coiled coil. Black and red fonts indicate polar and non-polar side chains, respectively. Residues within green boxes are strictly conserved. Red boxes in the second column highlight the stutters. In B, the side chains of polar residues within the hydrophobic core are drawn in green and labeled. Water molecules are indicated as small red spheres. Inter chain salt bridges (1 and 2, labeled in blue, corresponding to Lys 903 to Glu 1188 and Lys 929 to Glu 1163, respectively) are also indicated (basic side chains are in blue, acidic in red). The central cavity is displayed as a gold surface. (C) Slab of the model viewed down the 3-fold axis to show the chloride ions. Pink arrows indicate the center of the slab in panel B: top and bottom panels display chloride ions 1 and 2, respectively. The hydrogen bonding network propagating from the central ions toward the outside—which highly constrains the HR2 main chain, is indicated. Several of the Asn and Gln residues labeled are part of the Asn/Gln zipper (see text). As a guide for orientation, the axes of the 3 HR1 α-helices are drawn and labeled in green. The top panel is a view from below the atom, and the bottom panel from above it, relative to panel 2B. (D) Helical wheel after correction for the stutters (as in the register line of Fig. 1B). HR1 left, HR2 middle: as in panel A, polar and non-polar side chains are black and red, respectively (notice the strong amphipathic character of the two helices). Positions a and d are highlighted within a circle with a red background. The right panel shows a diagram of the interactions in the 6-helix bundle.
Central ions on the 3-fold axis
Two strong peaks along the 3-fold molecular axis, with heights between 8 and 10 σ, are evident in the electron density maps calculated with the final phases, from a model refined in the absence of any atom at those sites (i.e., “omit maps”). Both of the independent molecules in the crystal display this feature. The oxygen atom of a water molecule does not have enough electrons to account for this extra density, and we have interpreted each of the peaks as corresponding to a bound chloride ion, which is the most common ion in all the buffers used to produce and purify the protein. Indeed, introducing a chloride ion in the model at these sites leads to refined thermal parameters (“B factors”) for the ion that are roughly the same as those of the surrounding amino acid side chains (about 20 Å2), whereas introducing a water molecule leads to abnormally low B factors.
The two chloride ions are labeled 1 and 2 according to their distance to the HR1 N-terminus, and are respectively chelated by Gln 902 and Asn 937 and their 3-fold symmetric counterparts. As shown in Figs. 2A and D, these are among the few polar buried residues of HR1. The Gln/Asn nature of residues at these two positions is strictly conserved among coronaviruses (see the alignment of Fig. 1B). Chloride ion 1 is directly liganded by the Nɛ atoms of the three equivalent Gln 902 side chains, with tetrahedral coordination geometry (Fig. 2C, top panel). The fourth ligand for such a sphere of coordination would have been on the 3-fold axis below the ion, but in this case, it is absent, the ion being held in place by Van der Waals contacts with hydrophobic side chains from the next helical turn (Ile 905 in the 4th turn) interacting at the 3-fold axis. Chloride ion 2 is liganded via water molecules because the Asn 937 side chain is also involved in interactions with the HR2 main chain (see below). This site contains 6 ordered water molecules in total, 2 per subunit, directly surrounding the 3-fold axis (Fig. 2C, bottom panel). Only one water molecule is in direct contact with the ion, the second is hydrogen bonded to the first one, to its own symmetry mate in the trimer and to the main chain carbonyl of Asn 937. The coordination geometry of chloride ion 2 is the same as that of the first ion but the tetrahedral “pyramid” is inverted, in this case, the fourth ligand would be on the 3-fold axis above the ion, which is held in place by the ring of hydrophobic side chains of Val 934 above it (with the molecule in the orientation of Fig. 2A).
The side chains of both Gln 902 (which is strictly conserved) and Asn 937 are engaged in a network of hydrogen bonds, both with main chain amide and carbonyl groups and with the side chains of adjacent residues (including Asn 901 which is also strictly conserved, see Fig. 1) and with the main chain of HR2 (at Gly 1182 for Gln 902, and Ser 1156 for Asn 937). Thus, a stabilizing hydrogen bonding network propagates from the central Cl− ion all the way to the periphery of the molecule, playing a role in constraining the HR2 main chain conformation (see below).
Internal cavities
In addition to Gln 902 and Asn 937, which chelate the central ions, the other polar side chains directed toward the 3-fold axis are Gln 895 (which is at the very first turn and so is not in the hydrophobic “core” of the molecule) and Thr 923, at the 9th turn (Fig. 2A). This region is where the HR2 helix inserts in the lateral groove, between turns 6 and 10 of HR1, resulting in the packing of 6 helices along 5 helical turns. Both Ser 919 (position “g”, 8th turn) and Thr 923 are found pointing toward the interior of the molecule here, where usually hydrophobic residues are present, leaving an internal hydrophilic cavity of about 21 Å3 at the 3-fold axis (depicted in gold in Fig. 2B). The sequence alignment (Fig. 1B) shows that position 923 is semi-conserved: it is threonine in about half of the sequences examined and valine in the others. Valine has a non-polar side chain of about the same volume as threonine and so is also unable to fill the cavity. Furthermore, the other residue lining the cavity, Ser 919, is often glycine or alanine in most coronaviruses, which have smaller side chains and so the corresponding proteins will have an even bigger cavity. In several group I coronaviruses, both valine and alanine are present at positions 923 and 919, in which case, the cavity will have only hydrophobic boundaries. The presence of this cavity will undoubtedly have a negative impact on the stability of the molecule, suggesting that the helical portion of HR2, which packs against the HR1 helix precisely in this region, strengthens the molecule by providing more rigidity in this weak point.
Determinants of the HR2/HR1 interaction
The HR2 polypeptide contains a central helix of 5 complete turns flanked by two extended regions, HR2N and HR2C, at the amino- and carboxy-terminal ends of the central helix, respectively. In Fig. 1B, the heptad repeat positions are labeled such that the first hydrophobic position “d” is right before the first helical turn (turn 0), and the last is at the end of the 5th turn, amounting to a total of 6 hydrophobic side chains from positions “a” and “d” – mostly leucines and isoleucines – which interact with the hydrophobic core of HR1. The corresponding helical wheel is shown in Fig. 2D, with the helix oriented such that the “a” and “d” positions of the helix face the HR1 interhelical groove. The extended segments of HR2 also contribute hydrophobic side chains to the central hydrophobic cluster, 10 in total (6 from HR2N and 4 from HR2C, shown in Fig. 3 ). Thus, in total, 16 side chains from HR2 contribute to the stability of the core. The N-terminus of HR2 lies in between turns 16 and 17 of the HR1 helix, and the C-terminus reaches turn 1, so that HR2 contacts most of the HR1 helix, except for its 5 C-terminal turns, as illustrated in Fig. 2, Fig. 3.
Fig. 3.

HR1/HR2 interactions: Asn/Gln zippers and hydrophobic pockets along the HR1 grooves. Left panel. Ribbon representation, colored and oriented as in Figs. 2A and B, in which all the main chain atoms of the HR2N and HR2C extended segments of one of the subunits are represented as ball and stick, as well as the side chains of the Asn and Gln residues participating in the zipper. The atoms are colored according to atom type: grey are carbon atoms (light and dark for atoms of HR1 and HR2, respectively), red and blue indicate oxygen and nitrogen atoms, respectively. Hydrogen bonds between Gln and Asn side chains and main chain atoms are displayed as hatched cyan tubes. At the top, the arrows indicate the segments that connect to the fusion peptide (FP) and trans-membrane (TM) region. The boxes indicate the regions blown up in the right panels. The α-helical 5 turns of HR2 are numbered. Right panels. The top and bottom panel zoom into the Asn/Gln zippers that constrain the HR2C and HR2N segments, respectively. The model was slightly rotated in each of the two panels, with respect to the view in the left panel, for clarity. Hydrophobic HR2 side chains fitting into pockets in the HR1 grooves are shown in green. Asn and Gln side chains are colored as in the left panel. All residues indicated are labeled, red boxes highlighting highly conserved residues. The interactions displayed in this figure, together with the interactions that form an N- and C-cap to the HR2 helix as described in the text, highly constrain the HR2N and HR2C main chain.
Constraints to the HR2 main chain
The main-chain amide groups of α-helices are – by definition – engaged in hydrogen bonding with the main chain carbonyl of a residue located four residues downstream (Pauling et al., 1951) in an N to N + 4 pattern – where N denotes the number of an amino acid – in contrast to 3/10 helices in which the pattern is N to N + 3. The ends of the helix are special because the amide groups at the N-terminus and the carbonyl groups at the C-terminus do not have a hydrogen bonding partner from within the helical main-chain; in general these interactions have to be satisfied by alternative hydrogen bond acceptors (constituting an N-cap) or donors (C-cap), except when the ends of the helix are directly exposed to solvent (Presta and Rose, 1988, Richardson and Richardson, 1988). In the HR1/HR2 complex, the long HR1 α-helix has both its termini exposed to solvent, with a few non-helical residues at its N-terminus in which the sequence 890-Gly-Ile-Gly-892 breaks the helix. In contrast, the short HR2 helix is strongly capped at both ends.
HR2 N-cap
The three exposed amide groups at the N-terminus of HR2 are residues 1161 to 1163. They are capped by Asn 1159—a position in which there is either Asn or Asp in the sequence alignment of Fig. 1. The Oδ atom of Asn 1159 accepts a hydrogen bond from the amide group of residue 1161, and at the same time interacts with the amide group 1162 via ordered water molecules. These waters are also stabilized by lateral hydrogen bonding to the salt bridge Glu-1163 to Lys-929 (from HR2, 10th turn—Fig. 2B). Thus, all three residues (1159, 1163 and 929—two from HR2 and one from HR1, and which are all nearly strictly conserved, see Fig. 1) participate indirectly in the capping of this amide group. Finally, the amide 1163 donates a hydrogen bond to the main chain carbonyl of residue 1160, in a 3/10 helix interaction. The latter carbonyl also accepts a hydrogen bond from the amide 1164, within a standard N to N + 4 α-helix pattern. Thus, the N-cap is provided almost entirely from within the HR2 sequence.
HR2 C-cap
The HR2 helix makes four α-helical turns followed by a 3/10 helical turn (labeled in Fig. 2B), that is, there is a disruption of the normal helical pattern, which is obvious in the ribbon diagrams. The exposed carbonyls of the 4th turn belong to residues 1171 to 1173. The first two carbonyls are capped by Asn 1175 (strictly conserved) and Gln 917, respectively, the side chains of which also hydrogen bond to each other. The Asn 1175 side chain, a residue within the HR2 helix, thus perturbs the geometry of the helix by introducing a side chain to main chain hydrogen bond. However, Asn 1175 is part of a strictly conserved N-glycosylation site, and it is very likely that, once glycosylated, its Nδ atom would not be available for hydrogen bonding to the main chain. Thus, the observed perturbation of the HR2 helix, in which the chain switches from an α to a 3/10 helix at the 4th turn, may simply be an artifact resulting from producing the protein in Escherichia coli. The third carbonyl, 1173, makes a 3/10 interaction with the amide group of residue 1176. The 3/10 5th helical turn of HR2 also exposes its carbonyl groups (1175 and 1176), the second one being capped by Gln 908 and Lys 911. Therefore, the HR2 C-cap is set in place, to an important extent, by interactions with HR1.
“Asparagine/Glutamine zippers”
The polypeptide chain at either end of the HR2 helix is maintained in an extended conformation by a string of asparagine and glutamine side chains from the HR1 coiled coil, which hydrogen bond to the HR2 main chain as illustrated in Fig. 3. The asparagine Nδ atoms (or the glutamine Nɛ atoms) donate a hydrogen bond to the main chain carbonyls, and the Oδ (or Oɛ) accept one from the main-chain amide groups, resulting in a pattern that partially mimics the one seen by a β-strand. This extensive hydrogen-bonding network zips the HR2 main chain along the HR1 interhelical grooves.
Stabilizing external salt-bridges
Two important peripheral ionic HR1/HR2 interactions are also observed at either end of the HR2 helix. The first one is between E1163 and K929 and the second one between K903 and E1188, as illustrated in Fig. 2B. The residues participating in these salt bridges – especially the first one – are also highly conserved among coronaviruses.
Discussion
The most important discovery revealed by the structure of this particular construct of the fusion core of the SARS CoV S glycoprotein is the extent of the constraints imposed on the HR2 main chain by the central HR1 coiled coil. The striking arrangement of conserved Asn and Gln residues, which provides a hydrogen bonding network propagating from two central ions at the 3-fold axis of the coiled coil, stretches the main chain of HR2 and results in confinement of its helical portion to only 5 turns in total at the center, capped at either side. From the amino acid sequences of many coronaviruses S proteins, it had been predicted that HR2 by itself would display at least four to five heptad repeats, which would lead to a helical region of at least 8 turns (see Fig. S2 in the Supplementary material). This prediction has been further substantiated experimentally by the high degree of α-helicity (81% for SARS-CoV and 89% for MHV) observed by circular dichroism with the HR2 peptide in the absence of HR1 (Bosch et al., 2003, Bosch et al., 2004).
One of the structures recently reported by Supekar et al. (2004), displayed in Fig. 4 (third panel, PDB code 1BEQ) shows that HR2 segments not interacting with HR1 do adopt an α-helical conformation. This led the authors to propose that the C-terminal segment of HR2 would continue as a helix as it approaches the N-terminus of HR1. Fig. 4 (compare the 1st and the 3rd panels) shows that this is not the case, as the internal HR1 coiled coil actually maintains this HR2 segment in an extended conformation all the way up to the HR1 first helical turn. The structure drawn in the second panel (1BEZ) lacked the first turns of HR1 and the C-terminal end of HR2 to provide this information, while the resolution – and thus the quality of the resulting atomic model – of the 1WNC structure (Xu et al., 2004b) was not enough for a detailed analysis of the hydrogen-bonding interactions.
Fig. 4.
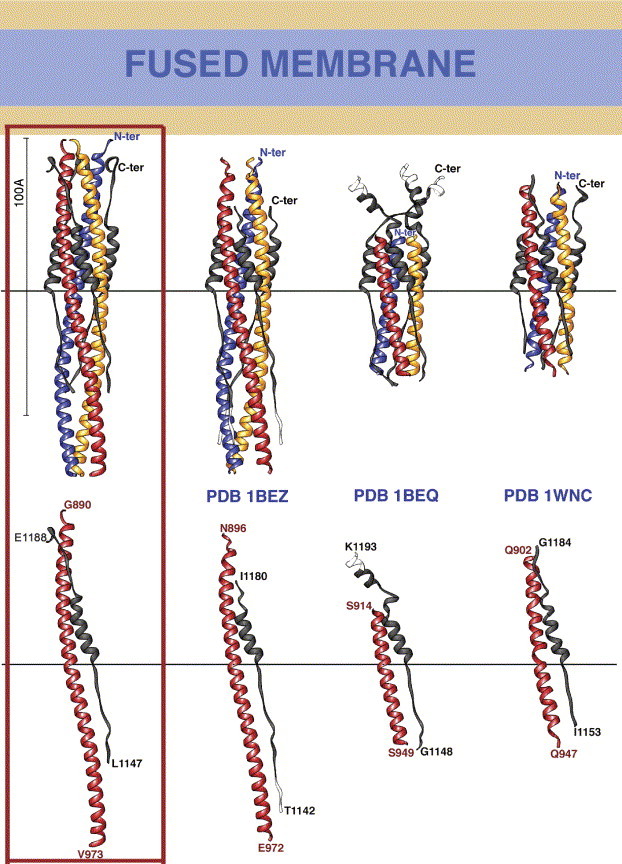
Recapitulation of the current structural data on the fusion core of the SARS CoV glycoprotein S. The left panel (framed in red) displays the structure described in this report in a ribbons representation in which the three HR1 segments are in primary colors and HR2 in grey. The other three panels are labeled with the corresponding PDB accession code of the structure depicted (pdb codes 1BEQ and 1BEZ from Supekar et al., 2004, and 1WNC from Xu et al., 2004b). In all panels, both the trimeric molecule (top) and one subunit (bottom) are displayed. All panels are colored identically, with segments containing amino acids that are not present in our current model (in the left panel) colored white. The images are all at the same scale, with the horizontal bars providing a means to align them so that the N-terminus of the HR2 helix is at the same height in each panel. The bottom panel indicates the number of the N- and C-terminal ends of the constructs represented. At the top, a roughly-to-scale diagram of a putative “fused membrane”, with its aliphatic portion in blue and the hydrophilic lipid heads in orange, is drawn at about the expected distance from the structures, as deduced from the amino acids that are missing between the N- and C-termini and the membrane interacting segments of the protein, the N-terminal fusion peptide and the C-terminal trans-membrane region.
Given the different lengths of the structures reported (Fig. 4), a question arises concerning the actual N-terminus of the HR1 coiled-coil in the post-fusion conformation of the intact S protein. The hydrophobic cluster analysis (HCA) provided in Fig. S2 (Supplementary material) suggests that the two glycines at positions 890 and 892 are indeed likely to break the helix also in the intact molecule, as they do in the structure of the fragment reported here. Accordingly, the α-helical HCA pattern switches at this position to one of alternating polar/non-polar side-chains along the sequence, which is more typical of β-strands or extended conformations. This extended segment of the HR1 chain, of about 10 amino acids, would connect to a glycine/alanine-rich region (residues 855 to 880) that has all the characteristics of typical viral fusion peptides. This is reminiscent of the influenza virus HA2 protein in its post-fusion conformation, in which the fusion peptide – which is comprised between HA2 residues 1 to 22 – is connected to the N-terminus of the central coiled coil (at residue 38) by a segment of polypeptide for which there is visible electron density in the crystals between amino acids 33 and 38. The visible portion of the connector is in an extended conformation and provides an N-cap to the neighboring α-helix in the trimer (Chen et al., 1999). In the present case, we see ordered electron density for residues 890 to 892 in an extended conformation (although in this case, they do not provide an N-cap to the HR1 helix). The 5 N-terminal amino acids of the construct (885 to 889) are disordered. In contrast, at the C-terminus of HR2, there is clear density in the crystals all the way to the last amino acid of the construct, Tyr 1188, with the chain ending with a single turn of α-helix between residues 1184 and 1188. Interestingly, the structure 1BEQ shows that these residues are indeed part of a longer helix (these amino acids are actually in turn 7 of the 1BEQ-HR2 helix, labeled in Fig. 4, third panel, top) ending after turn 8 at residue 1193. It is clear from Fig. S2 that the TM region begins around amino acid 1194. The presence of one α-helical turn in our structure suggests that the HR2 chain may connect in α-helical conformation to the lipid bilayer, bringing the fused membrane to about the location drawn in Fig. 4. It is likely that in the 1BEQ structure, the presence of the shorter HR1 segment causes the observed disruption of the HR2 helix at turns 4 and 5 (see Fig. 4, 3rd panel, bottom) because all the elements for capping the helix at that position are present, but the downstream zipper is missing. Indeed, the residues corresponding to turn 6 in 1BEQ are seen in extended conformation in our structure, owing to the strong constrains imposed to the HR2 main chain by the central coiled coil. In the absence of HR1, HR2 could well adopt a straight α-helical conformation since there are no glycine nor proline residues in this region, which are known to be α-helix breakers. Our separate observations that in the absence of HR1, the HR2 peptide has a very high α-helical content as observed by EM and circular dichroism (Bosch et al., 2003, Bosch et al., 2004) argue in favor of this interpretation.
The HR1 coiled coil – or at least its N-terminal portion – is believed to form only when the fusogenic conformational change of the S protein takes place, triggered by receptor binding at the target cell surface, so that it is not present in the fusion-active conformation of the molecule at the surface of infectious virions. In the absence of the N-terminal portion of the HR1 coiled coil, the HR2 regions are likely to be part of the stalk of the S protein pre-fusion conformation, since they are connecting to the TM regions and have a strong propensity to adopt an α-helical conformation as discussed above. The pre-fusion conformation of S is trimeric (Delmas and Laude, 1990), and it is thus likely that the α-helical HR2 segments are also part of its trimerization interface, stabilized by additional segments of this very large glycoprotein. Indeed, several (iso)leucine to alanine mutations in HR2 were shown to strongly impair oligomerization of the MHV S protein (Luo et al., 1999). The results from the structural studies on the fusion core would therefore suggest that the prefusion arrangement of this region of the S protein is strongly disturbed and refolds after formation of the HR1 coiled coil. The rearrangement is such that presumably the pre-fusion trimer has to dissociate and then reassociate around the HR1 coiled coil. A putative transient dissociation of the pre-fusion trimer may help explain the topological problems encountered when all three subunits of a trimer are simultaneously connected to two separate membranes to then fuse them into a single lipid bilayer. It is, therefore, possible that trimer dissociation and re-association during the fusogenic conformational transition is indeed part of a more general membrane fusion mechanism, which would be valid for all class I fusion proteins.
An additional unnoticed feature in the previously reported structures is the presence of ions at the 3-fold axis, liganded by polar residues, a feature that has been observed in the structures of similar coiled coils from class I fusion proteins of other viruses (Baker et al., 1999, Fass et al., 1996, Malashkevich et al., 1999, Weissenhorn et al., 1998). The presence of these polar residues within the hydrophobic cores has been proposed to provide a register to the interactions (Akey et al., 2001), resulting in a single pattern of hydrophobic contacts, and so avoid possible miss-folding by packing of helices shifted by one or two turns along the axis of interaction. In the particular case reported here, it is obvious that the presence of the two ions provides clear anchors.
One interesting observation is the likely conservation of the central cavity with its destabilizing effect on the molecule. This suggests that the residues lining the cavity in the post-fusion form are important for an alternative conformation of the protein, before the fusogenic conformational change. It thus appears from the structure that the protein has been forced to evolve alternative ways to compensate for the destabilizing effect of the cavity, strengthening the molecule from the outside with the presence of the HR2 helical segment and further stabilizing the arrangement by the salt bridges and the zipping of the asparagines at either end.
Taken together, all of these features of the structure account for the observed relatively high stability of the (HR1/HR2)3 complex, as indicated by its resistance to proteolytic degradation and the fact that the thermal dissociation in SDS gels is 60 °C and 80 °C for SARS-CoV and MHV, respectively (Bosch et al., 2004). These values suggest that the melting point in the absence of SDS would be much higher, although calorimetry measurements with the proteins resulting from these constructs have not been made. In contrast to most of the class I fusion proteins that have been studied structurally until now, the coronavirus S protein is special because is does not undergo an activating cleavage near the N terminus of the fusion peptide. In influenza virus, the structural studies have shown that the conformational transition of the hemagglutinin projects the fusion peptide by a distance of 100 Å away from its original location in the metastable form of the protein. This is possible because this peptide is located at the very N-terminus of HA2, and therefore it does not carry along any upstream polypeptide segment. The situation is completely different in the case of the coronaviruses, and it is likely that the polypeptide segment preceding the fusion peptide will have to act like a rope that can follow the projection of the fusion peptide. This would imply that the segment preceding the fusion peptide does not have a very rigid and stable structure in the pre-fusion form so that it can be unwound during the conformational change. However, such flexibility is not obvious from the amino acid sequence (see the HCA pattern in Fig. S2), and only a structure of the pre-fusion form of the S-protein can clarify this issue. These features of the S protein suggest, however, that in order for the fusogenic conformational transition to take place, a relatively high energy barrier has to be overcome, and this process could be facilitated also by having a very low energy minimum for the final stable conformation.
Finally, the structure described in this manuscript can provide a rational basis for developing potent inhibitors of entry of the SARS-CoV, by blocking the formation of the membrane-fusion core of the molecule. As shown in Fig. 3, there are indeed a number of pockets identified along the interhelical grooves of HR1 into which putative inhibitors can be designed to bind and disturb the correct association of HR2.
Acknowledgments
We thank Stephane Bressanelli and Enrico Stura for their participation in parts of this project. We gratefully acknowledge Matthijs Raaben for his technical assistance in the construction of the HR1-HR2 linker plasmid and the expression and purification of the HR1-HR2 protein complex; C. Schulze-Briese and T. Tomikazi for help during diffraction data collection; J. Navaza and J. Lepault for discussions; G. Aumont and C. Branlant for support. Diffraction data were collected at synchrotrons: SLS, Paul Scherrer Institut, Villigen, Switzerland and ESRF, Grenoble, France. FAR acknowledges support from the CNRS and INRA, the SESAME Program of the “Région Ile-de-France”, the CNRS programs PCV and “Dynamique et réactivité des assemblages biologiques”.
Footnotes
The coordinates have been deposited in the Protein Data Bank and the accession code is 1WYY.
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.virol.2005.02.022.
Appendix A. Supplementary data
Supplementary Fig. 1.
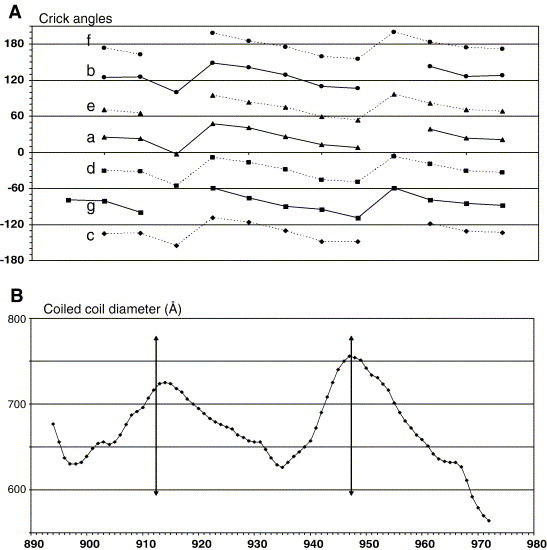
Mechanism of stutter formation. Examination of crick's angles along the helix (A) shows a progressive decrease of the values at each position. This reflects the regular 3.64 residues per turn observed in the helix and constant phase yield between successive residues of 98.9° rather than 3.5 and 102.8° expected for a perfect coiled coil. Thus, to keep in phase hydrophobic residues, a stutter must be inserted which cause a shift of around 50° in all values. At each stutter position (arrow), an increase of the local coiled coil radius is observed (B). Crick's angles and coiled coil radius calculation were made with Twister program (Strelkov and Burkhard, 2002).
Supplementary Fig. 2.
Hydrophobic Cluster Analysis. Hydrophobic Cluster Analysis (Gaboriaud, 1987) Plot for HR1 and TM regions (A) and HR2, TM and cytoplasmic tail (B).
References
- Akey D.L., Malashkevich V.N., Kim P.S. Buried polar residues in coiled-coil interfaces. Biochemistry. 2001;40(21):6352–6360. doi: 10.1021/bi002829w. [DOI] [PubMed] [Google Scholar]
- Baker K.A., Dutch R.E., Lamb R.A., Jardetzky T.S. Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell. 1999;3(3):309–319. doi: 10.1016/s1097-2765(00)80458-x. [DOI] [PubMed] [Google Scholar]
- Bosch B.J., Van Der Zee R., De Haan C.A., Rottier P.J. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 2003;77(16):8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch B.J., Martina B.E., Van Der Zee R., Lepault J., Haijema B.J., Versluis C., Heck A.J., De Groot R., Osterhaus A.D., Rottier P.J. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. U. S. A. 2004;101(22):8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Skehel J.J., Wiley D.C. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA2 subunit to form an N cap that terminates the triple-stranded coiled coil. Proc. Natl. Acad. Sci. U. S. A. 1999;96:8967–8972. doi: 10.1073/pnas.96.16.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F.H.C. The Fourier transform of a coiled-coil. Acta Crystallogr. 1953;6:685–689. [Google Scholar]
- de Groot R.J., Luytjes W., Horzinek M.C., van der Zeijst B.A., Spaan W.J., Lenstra J.A. Evidence for a coiled-coil structure in the spike proteins of coronaviruses. J. Mol. Biol. 1987;196(4):963–966. doi: 10.1016/0022-2836(87)90422-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas B., Laude H. Assembly of coronavirus spike protein into trimers and its role in epitope expression. J. Virol. 1990;64(11):5367–5375. doi: 10.1128/jvi.64.11.5367-5375.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fass D., Harrison S.C., Kim P.S. Retrovirus envelope domain at 1.7 angstrom resolution. Nat. Struct. Biol. 1996;3(5):465–469. doi: 10.1038/nsb0596-465. [DOI] [PubMed] [Google Scholar]
- Ingallinella P., Bianchi E., Finotto M., Cantoni G., Eckert D.M., Supekar V.M., Bruckmann C., Carfi A., Pessi A. Structural characterization of the fusion-active complex of severe acute respiratory syndrome (SARS) coronavirus. Proc. Natl. Acad. Sci. U. S. A. 2004;101(23):8709–8714. doi: 10.1073/pnas.0402753101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z., Matthews A.M., Weiss S.R. Amino acid substitutions within the leucine zipper domain of the murine coronavirus spike protein cause defects in oligomerization and the ability to induce cell-to-cell fusion. J. Virol. 1999;73(10):8152–8159. doi: 10.1128/jvi.73.10.8152-8159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malashkevich V.N., Schneider B.J., McNally M.L., Milhollen M.A., Pang J.X., Kim P.S. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9—A resolution. Proc. Natl. Acad. Sci. U. S. A. 1999;96(6):2662–2667. doi: 10.1073/pnas.96.6.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L., Corey R.B., Branson H.R. The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. U. S. A. 1951;37(4):205–211. doi: 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presta L.G., Rose G.D. Helix signals in proteins. Science. 1988;240:1632–1641. doi: 10.1126/science.2837824. [DOI] [PubMed] [Google Scholar]
- Richardson J.S., Richardson D.C. Amino acid preferences for specific locations at the ends of a helices. Science. 1988;240:1648–1652. doi: 10.1126/science.3381086. [DOI] [PubMed] [Google Scholar]
- Skehel J.J., Wiley D.C. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- Smith A.E., Helenius A. How viruses enter animal cells. Science. 2004;304:237–242. doi: 10.1126/science.1094823. [DOI] [PubMed] [Google Scholar]
- Strelkov S.V., Burkhard P. Analysis of alpha-helical coiled coils with the program TWISTER reveals a structural mechanism for stutter compensation. J. Struct. Biol. 2002;137(1–2):54–64. doi: 10.1006/jsbi.2002.4454. [DOI] [PubMed] [Google Scholar]
- Supekar V.M., Bruckmann C., Ingallinella P., Bianchi E., Pessi A., Carfi A. Structure of a proteolytically resistant core from the severe acute respiratory syndrome coronavirus S2 fusion protein. Proc. Natl. Acad. Sci. U. S. A. 2004;101(52):17958–17963. doi: 10.1073/pnas.0406128102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripet B., Howard M.W., Jobling M., Holmes R.K., Holmes K.V., Hodges R.S. Structural characterization of the SARS-coronavirus spike S fusion protein core. J. Biol. Chem. 2004;279(20):20836–20849. doi: 10.1074/jbc.M400759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenhorn W., Carfi A., Lee K.H., Skehel J.J., Wiley D.C. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol. Cell. 1998;2(5):605–616. doi: 10.1016/s1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- Xu Y., Zhu J., Liu Y., Lou Z., Yuan F., Liu Y., Cole D.K., Ni L., Su N., Qin L., Li X., Bai Z., Bell J.I., Pang H., Tien P., Gao G.F., Rao Z. Characterization of the heptad repeat regions, HR1 and HR2, and design of a fusion core structure model of the spike protein from severe acute respiratory syndrome (SARS) coronavirus. Biochemistry. 2004;43(44):14064–14071. doi: 10.1021/bi049101q. [DOI] [PubMed] [Google Scholar]
- Xu Y., Lou Z., Liu Y., Pang H., Tien P., Gao G.F., Rao Z. Crystal structure of severe acute respiratory syndrome coronavirus spike protein fusion core. J. Biol. Chem. 2004;279(47):49414–49419. doi: 10.1074/jbc.M408782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Liu Y., Lou Z., Qin L., Li X., Bai Z., Pang H., Tien P., Gao G.F., Rao Z. Structural basis for coronavirus-mediated membrane fusion. Crystal structure of mouse hepatitis virus spike protein fusion core. J. Biol. Chem. 2004;279(29):30514–30522. doi: 10.1074/jbc.M403760200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.