

Graphical abstract

Keywords: Heterocycles, NNRTI, Heteroaromatic, Pyrimidines
Abstract
A series of heterocyclic compounds were designed as potential nonnucleoside HIV reverse transcriptase inhibitors. Although the compounds ultimately proved inactive against HIV, during the course of the synthesis, a new and highly facile method to realize N-phenylacetamides was developed. Notably, the new route avoids the intractable workups and byproducts previously reported procedures have been associated with, thereby making this approach highly attractive to adaptation with other heterocyclics.
As a part of our ongoing efforts to synthesize aryl and heteroaromatic nonnucleoside HIV reverse transcriptase inhibitors (NNRTIs), we sought to construct a new series of N-3 phenylacetamides for use in alkylating various uracils. The amide unit is one of the most important and widely occurring functional groups; it is present in many natural products and is a key feature of many important drugs. Among the more notable examples of amide-containing compounds, those found in conjunction with N-heterocycles exhibit diverse and marked pharmacological activities. In particular, the HIV-1 NNRTIs,1 dihydrofolate reductase (DHFR) inhibitors,2 and histone deacetylase (HDAC) inhibitors stand out.3 It is not surprising therefore that there are numerous papers in which amide bond formation has been described.4 Despite the plethora of available routes, most are plagued with problems, in particular, low yields and difficult workups. As a result, a new and more facile route was sought.
A preliminary search of the literature revealed several known methods for the formation of 2-chloroacetanilides. The most commonly used methodology involves condensation of chloroacetyl chloride with various anilines in aprotic solvents (e.g. ether, dioxane, chloroform, dichloromethane, ethyl acetate, or DMF) in the presence of anhydrous K2CO3,5 triethylamine6 or pyridine7 as the base. Another variant utilizes methylethylketone and CaCO3 8 while other references describe condensation promoted by triethylamine in acetic acid.9 Examples of aniline acylation in acetate buffer10 and aqueous K2CO3 11 are also known, while another approach proposes the synthesis of 2-chloroacetanilides in base-free conditions. In the latter case, the reaction takes place in nonpolar solvent12 or in a mixed solvent system.13 Although there are many options available, it is important to note that most of these methods suffer from numerous side reactions arising from either N-quaternization of the organic base or hydrolysis, which lead to low yields and tedious work-up procedures.
It has however, been shown that amide bond formation could be achieved by the cleavage of the silicon–nitrogen bonds in TMS-protected alkylamines and anilines by acid halides.14 The absence of a base renders the silyl method very attractive for the synthesis of 2-chloroacetanilides, as this serves to bypass the problems with side reactions such as quaternization and hydrolysis that are common with the aforementioned routes. Surprisingly, there was no information in the literature for the preparation of 2-chloroacetanilides that utilized the silyl methodology. As a result, we have now employed this approach to synthesize a series of 2-chloroacetanilides, as shown in Scheme 1 , which were then used for the synthesis of the desired NNRTIs.
Scheme 1.
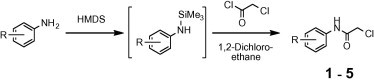
Proposed pathway to 2-chloroacetanilides.
As shown in Scheme 1, the starting anilines were converted into N-trimethylsilyl derivatives by refluxing in excess HMDS to form the N-silylated intermediates, followed by treatment with chloroacetyl chloride in anhydrous 1,2-dichloroethane to produce target amides 1–5 in 82–98% yield (Table 1 ).15 Notably, this occurred without any of the tar-like by-products typically observed with these reactions.
Table 1.

| Compd | R | Rfa | Yield (%) | Mp (°C) |
|---|---|---|---|---|
| 1 | H | 0.70 | 82 | 135–136.5 |
| 2 | 2-Me | 0.60 | 86 | 111.5–113 |
| 3 | 4-Ме | 0.67 | 87 | 163–165 |
| 4 | 3,4-Me2 | 0.71 | 97 | 110–112 |
| 5 | 3,5-Me2 | 0.73 | 98 | 143–145 |
Ethyl acetate/hexane 1:1.
The target 2-(1-benzyluracil-3-yl)-N-phenylacetamides 6–13 were then obtained in excellent to almost quantitative yields (78–96%, see Table 2 ) by alkylation of 1-benzyluracils16 14 with the appropriate 2-chloroacetanilides 1–5 in the presence of a 1.5-fold molar excess of K2CO3 in DMF as shown in Scheme 2 .17
Table 2.

| Compd | R1 | R | Rfa | Yield (%) | Mp (°C) |
|---|---|---|---|---|---|
| 6 | H | H | 0.57 | 96 | 212–213 |
| 7 | 2,5-Me2 | H | 0.65 | 90 | 175–176.5 |
| 8 | 3,5-Ме2 | H | 0.61 | 84 | 248–250 |
| 9 | 4-tBu | Н | 0.63 | 90 | 219–220 |
| 10 | H | 2-Me | 0.48 | 84 | 201–202 |
| 11 | H | 4-Ме | 0.59 | 83 | 221–222 |
| 12 | H | 3,4-Me2 | 0.56 | 78 | 222.5–224 |
| 13 | H | 3,5-Me2 | 0.64 | 87 | 214–215 |
| 15 | — | — | 0.71 | 90 | 197.5–198.5 |
| 16 | — | — | 0.78 | 96 | 125–127 |
Ethyl acetate/1,2-dichloroethane 1:1.
Scheme 2.

Synthesis of target uracil derivatives.
In an analogous fashion, N-phenylacetamides 15 and 16 were synthesized starting with 1-benzylthymine18 and 1-(benzhydryl)uracil,19 respectively. It should be noted that all the compounds were obtained without suffering the intractable workups or side products the typical procedures have been associated with, thereby making this approach highly attractive to adaptation with other heterocyclic compounds.
Compounds 6–13, 15, and 16 were evaluated as inhibitors against a large set of DNA and RNA viruses. Disappointingly, none of them proved to be active against any of the strains tested including HIV-1, HIV-2, HSV-1, HSV-2, HCMV, VZV, Vaccinia virus, Para-influenza-3 virus, Reovirus-1, Sindbis virus, Vesicular stomatitis virus, Respiratory syncytial virus, Coxsackie virus B4 or Feline Corona Virus.20, 21
In summary, while the target NNRTIs synthesized proved inactive, a facile and high-yielding route to N-phenylacetamides was developed, which should prove useful for many synthetic applications employing heterocyclic amides.
Acknowledgments
This work was supported by the Russian Foundation for Basic Research (Grant 10-04-0056a). The authors would like to thank Professor Jan Balzarini of the Rega Institute for antiviral evaluation of synthesized compounds.
Footnotes
Supplementary data (specific experimental details for each of the target compounds including 1H and 13C NMR data) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.tetlet.2012.11.090.
Supplementary data
Specific experimental details for each of the target compounds including 1H and 13C NMR data.
References and notes
- 1.(a) De La Rosa M., Kim H.W., Gunic E., Jenket C., Boyle U., Koh Y.-H., Korboukh I., Allan M., Zhang W., Chen H., Xu W., Nilar S., Yao N., Hamatake R., Lang S.A., Hong Z., Zhang Z., Girardet J.-L. Bioorg. Med. Chem. Lett. 2006;16:4444–4449. doi: 10.1016/j.bmcl.2006.06.048. [DOI] [PubMed] [Google Scholar]; (b) Gagnon A., Amad M.H., Bonneau P.R., Coulombe R., DeRoy P.L., Doyon L., Duan J., Garneau M., Guse I., Jakalian A., Jolicoeur E., Landry S., Malenfant E., Simoneau B., Yoakim C. Bioorg. Med. Chem. Lett. 2007;17:4437–4441. doi: 10.1016/j.bmcl.2007.06.012. [DOI] [PubMed] [Google Scholar]; (c) Yu M., Liu X., Li Z., Liu S., Pannecouque C., Clercq E.D. Bioorg. Med. Chem. Lett. 2009;17:7749–7754. doi: 10.1016/j.bmc.2009.09.035. [DOI] [PubMed] [Google Scholar]
- 2.Al-Omary F.A.M., Abou-Zeid L.A., Nagi M.N., Habib E.-S.E., Abdel-Aziz A.A.-M., El-Azab A.S., Abdel-Hamide S.G., Al-Omar M.A., Al-Obaid A.M., El-Subbagh H.I. Bioorg. Med. Chem. 2010;18:2849–2863. doi: 10.1016/j.bmc.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 3.(a) Zhang Y., Fang H., Feng J., Jia Y., Wang X., Xu W. J. Med. Chem. 2011;54:5532–5539. doi: 10.1021/jm200577a. [DOI] [PubMed] [Google Scholar]; (b) Wang H., Yu N., Chen D., Lee K.C.L., Lye P.L., Chang J.W.W., Deng W., Ng M.C.Y., Lu T., Khoo M.L., Poulsen A., Sangthongpitag K., Wu X., Hu C., Goh K.C., Wang X., Fang L., Goh K.L., Khng H.H., Goh S.K., Yeo P., Liu X., Bonday Z., Wood J.M., Dymock B.W., Kantharaj E., Sun E.T. J. Med. Chem. 2011;54:4694–4720. doi: 10.1021/jm2003552. [DOI] [PubMed] [Google Scholar]
- 4.Bailey P.D., Mills T.J., Pettecrew R., Price R.A. Elsevier; Vol. null: 2005. In Comprehensive Organic Functional Group Transformations II. pp 201–294. [Google Scholar]
- 5.(a) Sauter F., Stanetty P. Monatsh. Chem. 1975;106:1111–1116. [Google Scholar]; (b) Vejdělek Z., Holubek J., Bartošová M., Protiva M. Collect. Czech. Chem. Commun. 1982;47:3297–3305. [Google Scholar]; (c) Vejdělek Z., Bartošová M., Protiva M. Collect. Czech. Chem. Commun. 1983;48:156–162. [Google Scholar]; (d) Effland R.C., Helsley G.C., Tegeler J.J. J. Heterocycl. Chem. 1982;19:537–539. [Google Scholar]
- 6.(a) Rudkevich D.M., Verboom W., Reinhoudt D.N. J. Org. Chem. 1994;59:3683–3686. [Google Scholar]; (b) Szmuszkovicz J., Chidester C.G., Laurian L.G., Scahill T.A. J. Org. Chem. 1986;51:5001–5002. [Google Scholar]; (c) Kizuka H., Hanson R.N. J. Med. Chem. 1987;30:722–726. doi: 10.1021/jm00387a025. [DOI] [PubMed] [Google Scholar]; (d) Wolfbeis O.S., Marhold H. Monatsh. Chem. 1983;114:599–604. [Google Scholar]
- 7.Reddy P.S.N., Nagaraju C. Synth. Commun. 1991;21:173–181. [Google Scholar]
- 8.Perez M., Ayerbe N., Fourrier C., Sigogneau I., Pauwels P., Palmier C., John G., Valentin J., Halazy S. Eur. J. Med. Chem. 1997;32:129–134. doi: 10.1021/jm00018a020. [DOI] [PubMed] [Google Scholar]
- 9.(a) Deak G., Doda M., Gyorgy L., Hazai L., Sterk L. J. Med. Chem. 1977;20:1384–1388. doi: 10.1021/jm00221a006. [DOI] [PubMed] [Google Scholar]; (b) Gall-Istok K., Sterk L., Toth G., Deak G. J. Heterocycl. Chem. 1984;21:1045–1048. [Google Scholar]
- 10.(a) Löfgren N., Tegnér C., Bonnichsen R., Virtanen A.I. Acta Chem. Scand. 1955;9:493–496. [Google Scholar]; (b) Byrnes E.W., McMaster P.D., Smith E.R., Blair M.R., Boyes R.N., Duce B.R., Feldman H.S., Kronberg G.H., Takman B.H., Tenthorey P.A. J. Med. Chem. 1979;22:1171–1176. doi: 10.1021/jm00196a005. [DOI] [PubMed] [Google Scholar]
- 11.Kim S.-C., Kwon B.-M. Synthesis. 1982;1982:795–796. [Google Scholar]
- 12.(a) Meth-Cohn O., Rhouati S., Tarnowski B., Robinson A. Perkin Trans. 1. 1981:1537–1543. [Google Scholar]; (b) Neumeyer J.L., Perianayagam C., Ruchirawat S., Feldman H.S., Takman B.H., Tenthorey P.A. J. Med. Chem. 1977;20:894–898. doi: 10.1021/jm00217a005. [DOI] [PubMed] [Google Scholar]; (c) Hromatka O., Knollmüller M., Foroutan-Rad M. Monatsh. Chem. 1974;105:1057–1066. [Google Scholar]; (d) Lin W.O., Souza Coutinho E. Monatsh. Chem. 1983;114:1231–1235. [Google Scholar]
- 13.Martelli S., Dzieduszycka M., Stefanska B., Bontemps-Gracz M., Borowski E. J. Med. Chem. 1988;31:1956–1959. doi: 10.1021/jm00118a015. [DOI] [PubMed] [Google Scholar]
- 14.(a) Bowser J.R., Williams P.J., Kurz K. J. Org. Chem. 1983;48:4111–4113. [Google Scholar]; (b) Taddei M., Tempesti F. Synth. Commun. 1985;15:1019–1024. [Google Scholar]; (c) Oishi Y., Kakimoto M., Imai Y. Macromolecules. 1987;20:703–704. [Google Scholar]; (d) Lozano A.E., de Abajo J., de la Campa J.G. Macromolecules. 1997;30:2507–2508. [Google Scholar]; (e) Rajeswari S., Jones R.J., Cava M.P. Tetrahedron Lett. 1987;28:5099–5102. [Google Scholar]
- 15.General method for synthesis of 2-chloro-N-phenylacetamides 1–5. A mixture of 16.50 mmol of aniline, 20 mL of HMDS and NH4Cl (250 mg) was refluxed for 10 h until a homogeneous solution was obtained. Excess of HMDS was removed under reduced pressure, and the residual clear oil dissolved in 40 mL of anhydrous 1,2-dichloroethane. A solution of chloroacetyl chloride (1.4 ml, 17.60 mmol) in 1,2-dichloroethane (10 mL) was added dropwise with stirring at 0 °С. The reaction mixture was stirred overnight at rt and then evaporated to 1/3 volume. Hexane (30 mL) was added and the mixture was kept overnight in a refrigerator. The resulting precipitate was filtered and recrystallized from EtOAc/hexane.
- 16.Kundu N.G., Sikdar S., Hertzberg R.P., Schmitz S.A., Khatri S.G. Perkin Trans. 1. 1985:1295–1300. [Google Scholar]
- 17.General method for the synthesis of 2-(3-benzyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)-N-phenylacetamides 6–13, 15 and 16. A suspension of 7.72 mmol of 1-benzyluracil1614, 1-benzylthymine18 or 1-(diphenylmethyl)uracil19 and K2CO3 (1.6 g, 11.58 mmol) in 15 mL of DMF was stirred at 80 °С for 1 h then cooled to rt. After addition of 2-chloroacetanilide 1–5 (7.66 mmol), the reaction mixture was stirred for 2 h, chilled overnight, filtered and the solvent removed under reduced pressure. The residue was treated with 50 mL of water, and the resulting precipitate filtered and recrystallized from a mixture of acetone-iPrOH-DMF.
- 18.Lopez F.J., Arias L., Chan R., Clarke D.E., Elworthy T.R., Ford A.P.D.W., Guzman A., Jaime-Figueroa S., Jasper J.R., Morgans D.J., Padilla F., Perez-Medrano A., Quintero C., Romero M., Sandoval L., Smith S.A., Williams T.J., Blue D.R. Bioorg. Med. Chem. Lett. 2003;13:1873–1878. doi: 10.1016/s0960-894x(03)00305-6. [DOI] [PubMed] [Google Scholar]
- 19.Wu F., Buhendwa M.G., Weaver D.F. J. Org. Chem. 2004;69:9307–9309. doi: 10.1021/jo0485076. [DOI] [PubMed] [Google Scholar]
- 20.The methodology of the anti-HIV assays in MT-4 cell cultures was as follows: Virus stocks were titrated in MT-4 cells and expressed as the 50% cell culture infective dose (CCID50). MT-4 cells were suspended in culture medium at 1 × 105 cells/ml and infected with HIV at a multiplicity of infection of 0.02. Immediately after viral infection, 100 μl of the cell suspension was placed in each well of a flat-bottomed microtiter tray containing various concentrations of the test compounds. After 4 days of incubation at 37°C, the number of viable cells was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. The selection and characterization of mutant virus strains have been performed previously.
- 21.The compounds were evaluated against the following viruses: Herpes simplex virus type 1 (HSV-1) strain KOS, thymidine kinase-deficient (TK−) HSV-1 KOS strain resistant to ACV (ACVr), herpes simplex virus type 2 (HSV-2) strains Lyons and G, varicella-zoster virus (VZV) strain Oka, TK− VZV strain 07−1, human cytomegalovirus (HCMV) strains AD-169 and Davis, vaccinia virus Lederle strain, respiratory syncytial virus (RSV) strain Long, vesicular stomatitis virus (VSV), Coxsackie B4, Parainfluenza 3, Influenza virus A (subtypes H1N1, H3N2), influenza virus B, Reovirus-1, Sindbis and Punta Toro. The antiviral assays were based on the inhibition of virus-induced cytopathicity or plaque formation in human embryonic lung (HEL) fibroblasts, African green monkey cells (Vero), human epithelial cells (HeLa) or Madin-Darby canine kidney cells (MDCK). Confluent cell cultures in microtiter 96-well plates were inoculated with 100 CCID50 of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures) or with 20 plaque forming units (PFU) (VZV) in the presence of varying concentrations of the test compounds. Viral cytopathicity or plaque formation was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. Antiviral activity was expressed as the EC50 or compound concentration required to reduce virus-induced cytopathogenicity or viral plaque formation by 50%.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Specific experimental details for each of the target compounds including 1H and 13C NMR data.