

Abstract
Molecular clones of infectious bronchitis virus (IBV), derived from the Vero cell adapted Beaudette strain, were constructed, using an in vitro assembly method. In vitro transcribed RNA from a cDNA template that had been constructed from seven cDNA fragments, encompassing the entire genome of IBV, was electroporated into BHK-21 cells. The cells were overlaid onto the susceptible Vero cells and viable virus was recovered from the molecular clone. The molecularly cloned IBV (MIBV) demonstrated growth kinetics, and plaque size and morphology that resembled the parental Beaudette strain IBV. The recombinant virus was further manipulated to express enhanced green fluorescent protein (EGFP) by replacing an open reading frame (ORF) of the group-specific gene, ORF 5a, with the EGFP ORF. The rescued recombinant virus, expressing EGFP (GIBV), replicated to lower viral titers and formed smaller plaques compared to the parental virus and the MIBV. After six passages of GIBV, a minority of plaques were observed that had reverted to the larger plaque size and virus from these plaques no longer expressed EGFP. Direct sequencing of RT-PCR products derived from cells infected with the plaque-purified virus, which had lost expression of EGFP, confirmed loss of the EGFP ORF. The loss of EGFP expression (Δ5a IBV) was also accompanied by reversion to growth kinetics resembling the standard virus and intact recombinant virus. This study demonstrates that the 5a ORF is not essential for viral multiplication in Vero cells.
Keywords: Infectious bronchitis virus, Recombinant IBV, 5a ORF
Introduction
Infectious bronchitis virus (IBV), a coronavirus, continues to be one of the most economically important pathogens in the poultry industry. Coronaviruses are enveloped viruses with positive sense, 5′ capped and 3′ polyadenylated RNA genomes, that range from 27.6 to 32 kb (Lai and Cavanagh, 1997). Two thirds of the coronavirus genome encodes the replicase activity, including a viral RNA-dependent RNA polymerase (RdRp), helicase, and viral proteinases. The remaining one third of the genome encodes the structural proteins and small group-specific ORFs (Lai and Cavanagh, 1997).
IBV has four essential structural proteins, the three membrane proteins, the spike (S), integral membrane (M), and small envelope (E) proteins, and a phosphorylated, nucleocapsid (N) protein. The S protein interacts with cellular receptors and induces cell and viral membrane fusion (Bosch et al., 2003). N proteins, which interact with viral genomic RNA, forming ribonucleocapsid (RNP) complexes, have been associated with replication and transcription (Baric et al., 1988, Compton et al., 1987, Robbins et al., 1986). The E and M proteins are localized in ER-Golgi intermediate compartment and play critical roles in viral budding (de Haan et al., 1998, Fischer et al., 1998).
Coronaviruses are classified into three distinct groups by antigenic cross-reactivity and nucleotide sequence analysis (Fields, 1996). The group I coronaviruses include human coronavirus 229E, transmissible gastroenteritis virus (TGEV), feline coronavirus, and feline infectious peritonitis virus. The group II coronaviruses include human coronavirus OC43, murine hepatitis virus (MHV), and bovine coronavirus. IBV belongs to the group III coronaviruses, all of which are avian specific. Severe acute respiratory syndrome coronavirus (SARS CoV) respiratory disease in humans that resembles IBV in poultry may be distantly related to group II coronaviruses (Snijder et al., 2003, Stavrinides and Guttman, 2004). Coronavirus genomes also encode group-specific ORF that have been reported to express in infected cells (Lai and Cavanagh, 1997, Liu and Inglis, 1992, Liu et al., 1991). Functions of these genes are not well understood, however, deletion of combinations of several group-specific genes in MHV resulted in in vivo attenuation (de Haan et al., 2002). Although the functions of the four IBV group-specific ORF, 3a, 3b, 5a, and 5b are not known, a Vero cell passaged IBV mutant with a truncated 3b was viable in vitro and in vivo (Shen et al., 2003).
Reverse genetic systems for human coronavirus 229E, TGEV, MHV, SARS CoV, and IBV have been developed using three distinct methodologies. Bacterial artificial chromosomes (BAC) and vaccinia virus vectors, both of which can accommodate the large coronavirus genomes, have been utilized to carry a full-length cDNA clone of the genome (Almazan et al., 2000, Casais et al., 2001, Gonzalez et al., 2002, Thiel et al., 2001). A third system relied on the in vitro assembly of a modest number of cloned cDNAs. Instead of cloning the entire genome of TGEV into vectors, Yount et al. (2000) amplified the viral genome into several fragments by RT-PCR and the cloned amplicons were ligated through unique restriction sites for in vitro assembly of the entire genome. This strategy was further modified to construct an infectious cDNA clone of MHV with their “no see'm” technology, in which restriction endonuclease sequences were incorporated into amplicons, such that upon enzyme treatment, the endonuclease sites were eliminated prior to in vitro ligation. The assembled cDNA product was used as template for in vitro transcription to generate genomic RNA (Yount et al., 2002). More recently, this strategy was used to construct a reverse genetic system for the SARS CoV (Yount et al., 2003).
In the current study, molecular clones of IBV were generated using the in vitro assembly of cDNA fragments as a template for transcription of full-length genomic RNA. We further have demonstrated that the 5a ORF can be eliminated or replaced by the EGFP ORF while maintaining virus viability. Therefore, this gene is not essential for viral replication and its ORF may potentially be used as a site for heterologous gene delivery.
Results
cDNA was constructed that encompassed the entire IBV genome
The cloning strategy for a full-length IBV-Beaudette construct is illustrated in Fig. 1A. Five RT-PCR fragments were produced that cumulatively amplified the entire genome of the Vero cell adapted Beaudette IBV strain. Amplicons, overlapping by about 10 nucleotides, were designated A through E and the fragment lengths were as follows; A, 2269 nts; B, 6356 nts; C, 6895 nts; D, 5030 nts; and E, 7058 nts (Fig. 1B). However, consistent with the report that these regions were toxic or unstable in bacteria, difficulties arose in cloning the B and C fragments into bacterial plasmid based cloning vectors (Casais et al., 2001). Therefore, the B and C fragments were each cloned as two smaller fragments B1 and B2, and C1 and C2. Fragments A through C2 were cloned into the pSMART vector and fragments D through E2 were cloned into pCR-XL-TOPO cloning vectors. In order to determine the consensus sequence of the genome, two to five independent clones of each amplicon were sequenced using a panel of internal primers. Clones with consensus sequences were used for in vitro assembly of full-length cDNA template. There were several sequence differences, distributed throughout the entire genome of IBV, between our standard cell adapted IBV strain and the published Beaudette sequence (Table 1 ). The standard parental strain of IBV Beaudette used for these studies had a five nucleotide insertion at position 25,414 between the M and 5a ORFs, resulting in a genome size of 27,613, instead of 27,608 nt. The sequence differences were confirmed by sequencing three independent clones and direct sequencing of RT-PCR products. A nucleotide insertion was found in an intergenic sequence of our strain compared to the original published sequence. In addition, an E amplicon with a single unique nucleotide change of A to G at position 25,793 nt, resulting in amino acid change from Q to R, was used as a marker for the molecular clone. Full-length cDNA was in vitro assembled with amplicons conforming with our established consensus sequence. To increase full-length in vitro ligation efficiency, ordered ligation reactions were used. The appropriately sized ligation reaction products were purified from 1% agarose gels before further ligating with neighboring inserts (Fig. 1C).
Fig. 1.
SapI and BsmBI restriction enzyme sites were used for in vitro assembly of the cDNA for the full-length IBV genome. (A) The 5′ end of the A fragment was amplified with primers that incorporated the T7 RNA polymerase promoter and similarly, a 19 nucleotide polyadenylated tail was inserted at the 3′ end of E. Gray boxes represent ORF of the IBV genome. Open boxes represent genome fragments amplified by RT-PCR and black boxes represent 5′ and 3′ untranslated regions. (B) DNA amplicons used for in vitro ligation. Each amplicon was digested with the appropriate restriction enzyme and separated from the plasmid vectors using agarose gel electrophoresis. Lane M represents the 1 kb DNA ladder; lane 1, fragment A prepared by XhoI digestion, CIP treatment and then SapI restriction enzyme digestion; lane 2, fragment B1 prepared by BsmBI and SapI; lane 3, fragment B2 prepared by digestion with BsmBI and SapI restriction enzyme; lane 4, fragment C1 prepared by BsmBI and SapI digestion; lane 5, fragment C2 prepared by BsmBI digestion; lane 6, fragment D prepared by BsmBI digestion; lane 7, fragment E1 prepared by EcoRI digestion followed by BsmBI digestion, and lane 8, the E2 fragment. (C) In vitro orderly ligation of the generated cDNA fragments. The inserts were ligated stepwise as follows. Lane M represents the 1-kb DNA extension ladder; lane 1, ligation products of A and B1; lane 2, ligation products of B2 and C1; lane 3, purified ligation product of C2 and D; lane 4, purified ligation product of E1 and E2; lane 5, ligation products of lanes 1 and 2; lane 6, ligation products of lanes 3 and 4; lane 7, the final ligation product of lane 5 and 6 (shown by the arrow to the right).
Table 1.
Differences between the Vero cell-adapted IBV strain used in this study and the previously published IBV Beaudette strain
| Locationa | Nucleotide differenceb | Amino acid differencec | Location | Nucleotide difference | Amino acid difference | Location | Nucleotide difference | Amino acid difference |
|---|---|---|---|---|---|---|---|---|
| 32 | C-T | – | 9372 | A-G | – | 22,492 | A-G | – |
| 222 | C-T | – | 10,754 | T-C | V-A | 22,535 | T-C | F-L |
| 282 | G-A | – | 10,781 | T-C | I-T | 22,590 | C-T | – |
| 490 | T-C | – | 11,780 | T-A | V-D | 23,402 | G-T | S-I |
| 841 | C-T | S-P | 12,136 | A-G | S-G | 23,724 | T-A | – |
| 1374 | G-A | – | 13,106 | C-T | – | 24,338 | T-C | – |
| 1845 | T-C | – | 13,178 | A-C | Q-H | 24,508 | C-T | P-S |
| 2000 | C-T | A-V | 13,646 | A-G | – | 24,661 | A-G | I-V |
| 2015 | A-G | C-G | 15,302 | A-G | – | 24,716 | C-T | T-I |
| 2439 | A-T | G-E | 15,594 | C-A | – | 25,342 | G-T | – |
| 2651 | C-T | T-M | 16,286 | C-T | – | 25,426 | G-A | – |
| 3169 | A-T | – | 16,718 | A-G | – | 25,458 | T-C | – |
| 3210 | A-G | – | 17,546 | T-C | – | 25,465 | C-T | – |
| 3283 | A-G | K-E | 18,195 | T-C | Y-H | 25,793 | A-G | Q-R |
| 3322 | C-A | L-I | 20,731 | T-A | L-I | 25,901 | A-G | – |
| 3428 | C-A | A-D | 20,755 | C-T | L-F | 25,904 | A-G | – |
| 3926 | T-C | L-S | 21,149 | A-C | N-T | 25,928 | T-C | – |
| 4481 | C-A | T-N | 21,357 | A-T | K-N | 26,054 | C-T | – |
| 4791 | A-G | – | 21,457 | A-G | – | 26,208 | G-T | D-Y |
| 5456 | T-C | L-H | 21,459 | A-T | – | 26,403 | G-T | A-S |
| 5785 | C-T | L-F | 21,628 | A-C | N-H | 26,501 | G-T | Q-H |
| 5828 | C-T | P-L | 21,711 | G-A | – | 27,048 | T-G | Y-D |
| 6231 | G-A | – | 22,347 | T-C | – | 27,051 | G-T | D-Y |
| 7293 | A-C | – | 22,433 | A-G | K-R | 27,069 | A-G | N-D |
| 7755 | A-G | – | 22,415 | A-C | N-T | 27,466 | A-C | – |
| 9372 | A-G | – | 22,441 | C-G | L-V |
According to our Vero-adapted IBV derived sequence. The entire sequence has been submitted to Genbank.
Left nucleotide according to the previously published sequences and the right nucleotide from our Vero cell-adapted derived strain.
Left amino acid from the previously published sequences and the right amino acid from our Vero cell-adapted derived strain. – indicates that there was no amino acid difference.
IBV generated by in vitro assembly of cDNAs was indistinguishable from the wild type virus
A full-length genomic transcript of IBV was produced in vitro using T7 RNA polymerase with the T7 RNA promoter, incorporated at the 5′ end of fragment A. The final transcript product was verified by Northern blot analysis using a probe against 3′ UTR (data not shown). Any transcript of a size comparable to viral genomic RNA with a detectable 3′ UTR presumably contained the entire genome of IBV because only the A fragment had a T7 RNA promoter and thus could initiate transcription.
The transcripts from the full-length cDNA (along with the N transcript alone), total cellular RNA from IBV infected Vero cells (positive control), and PBS (negative control) were electroporated into non-permissive BHK-21 cells, which were then cultured with Vero cells. Three days after transfection, typical cytopathic effects (CPE), including syncytia formation, were seen in cells transfected with the positive control. Supernatant from the cells, which were overgrown by 3 days after transfection, were harvested and passaged into fresh Vero cells. One day after passage, the positive control showed CPE. Two days after passage, cells transfected with the transcript of the full-length ligated cDNAs showed CPE. CPE was never observed in the negative control cells. We were unable to rescue virus from full-length IBV RNA transcript without co-transfection of N transcript, confirming the possible essential role of N transcript as described by previous reports (Casais et al., 2001, Yount et al., 2000, Yount et al., 2002). The virus generated by the molecular cDNA clone was referred to as MIBV. The derivation of MIBV from the ligated cDNA was confirmed by RT-PCR sequence identification of the marker mutation of a G at nt 25,793.
MIBV was further characterized after three rounds of plaque purification. MIBV formed plaques which were indistinguishable in size and morphology from standard Beaudette cell adapted viral plaques (Figs. 2A and B). The growth kinetics of MIBV and the standard virus were also compared and found to be similar (Fig. 3 ).
Fig. 2.
MIBV generated similar sized plaques as the wild type IBV. Vero cells were infected with each virus and then overlaid with media containing 0.8% agarose. Three days after infection, agarose was removed and cells were stained with crystal violet. The MIBV plaques are shown in panel A and standard cell adapted Beaudette IBV, in panel B.
Fig. 3.
Comparison of the replication kinetics of standard cell-adapted and recombinant IBV. Standard IBV and recombinant IBV, MIBV, GIBV, and Δ5a IBV were used to infect Vero cells and triplicate samples of the infected cells were harvested every 4 h for 24 h. Total virus was harvested by freezing and thawing cells three times and the viral titer was measured by counting plaque forming units/ml at each time point.
The 5a gene is not essential for IBV replication in Vero cells
The reverse genetic strategy we adapted for IBV was used to evaluate whether a small group-specific gene was necessary for the survival of IBV in cell culture. Furthermore, the replacement with a foreign ORF would be a first step in developing constructs of IBV as expression vectors. Because it has been shown that the expression level of coronavirus genes can be modified by their transcription regulatory sequences (TRS), rather than add an additional TRS, a reporter gene was used to replace most of the 5a ORF, thus maintaining the 5a TRS sequence. Based on results with MHV, we hypothesized that IBV group-specific genes would not be necessary for viral replication in cell culture. The 5a ORF was used rather than the 3a, 3b region which contains an internal ribosomal entry site (IRES) needed for expression of the essential structural, E protein (Le et al., 1994). The EGFP ORF was used to replace the 5a ORF, beginning at the start codon, while the 3′ end of 5a ORF, that included the stop codon, was kept intact because this region overlaps with the 5b ORF.
PCR mutagenesis and the “no see'm” methodology were used to replace ORF5a with EGFP. The cloning strategy used to replace most of the 5a ORF with EGFP (Δ5a/EGFP) is illustrated in Fig. 4A. Three regions, including the EGFP ORF (Fig. 4B, lane 1), and sequences between a region upstream of 5a, including ORF M (Fig. 4B, lane 2), a region downstream of 5a through the 3′ UTR (Fig. 4B, lane 3), were PCR amplified using primers that incorporated the BsmBI restriction enzyme recognition sequence (Table 2). These three PCR products were cloned into the pSTBlue cloning vector and the inserts were excised from the vectors with the BsmBI restriction enzyme.
Fig. 4.
Construction of EGFP/Δ5a amplicon. (A) The 5a ORF was replaced with the EGFP ORF. Open boxes represent ORF of the IBV genome. Black lines represent PCR products. Arrows indicate the restriction enzyme sites inserted into the PCR products. (B) PCR products containing EGFP in place of ORF 5a. M represents the 1-kb DNA molecular size marker; lane 1, PCR product of EGFP, denoted as F in (A); lane 2, PCR product of M ORF and junction of 5′ of ORF 5a, denoted as G in (A); lane 3; PCR product encompassing 3′ end of 5a ORF and downstream of the ORF through 3′ UTR of IBV, denoted as H in (A). (C) Underlined are IBV sequences and bold are EGFP sequences.
Table 2.
Primer pairs used for cloning and mutagenesis of the IBV and GIBV amplicons
| Designation | Amplicona | Polarity | Genome locationb | Nucleotide sequencec |
|---|---|---|---|---|
| T7P1F | A | + | 1–22 | 5′-TAATACGACTCACTATAGGACTTAAGATAGATATTAATATA-3′ |
| P1R | A | − | 2254–2276 | 5′-CCTTTCCAGAAGAGCAAATCTCC-3′ |
| P1F | B-1 | + | 2254–2276 | 5′-GGAGATTTGCTCTTCTGGAAAGG-3 |
| NHER | B-1 | − | 5731–5752 | 5′-CGTCTCGCGACAACACTCTTAACACTAGC-3′ |
| NHEF | B-2 | + | 5748–5771 | 5′-CGTCTCTGTCGCTAGCTATAAGACCGTGTT-3′ |
| P2R | B-2 | − | 8611–8635 | 5′-CAAAAGTGCTCTTCGCAGCAAGATC-3′ |
| P2F | C-1 | + | 8611–8635 | 5′-GATCTTGCTGCGAAGAGCACTTTTG-3′ |
| SACR | C-1 | − | 11,926–11,949 | 5′-CGTCTCGGGATCTACTGCAAATGAACATAG-3′ |
| SACF | C-2 | + | 11,944–11,972 | 5′-CGTCTCGATCCCCGCGGACACATATTGTAAATATG-3′ |
| P3R | C-2 | − | 15,508–15,531 | 5′-CAAAACGTCTCAATGAATCACTAC-3′ |
| P3F | D | + | 15,508–15,531 | 5′-GTAGTGATTCATTGAGACGTTTTG-3′ |
| BEAUSSR | D | − | 20,543–20,566 | 5′-CGTCTCCAACATCTCTTACCAGTAACTTAC-3′ |
| BEAUSSF | E | + | 20,543–20,566 | 5′-CGTCTCATGTTGGTAACACCTCTTTTACTAG-3′ |
| GNRT | E/H | − | 27,593–27,613 | 5′-TTTTTTTTTTTTTTTTTTTGCTCTAACTCTATACTAGCC-3′ |
| EGFPSF | F | + | 5′-CGTCTCATGGTGAGCAAGGGCGAGGAG-3′ | |
| EGFPER | F | − | 5′-CGTCTCCGCTTACTTGTACAGCTCGTCC-3′ | |
| BMF | G | + | 24,509–24,535 | 5′-ATGGCGGAAAATTGCACACTTGATTC-3′ |
| 5ASR | G | − | 25,574–25,592 | 5′-CGTCTCCACCATCGTCCGTATTTGTTAAGT-3′ |
| 5AEF | H | + | 25,658–25,678 | 5′-CGTCTCTAAGCGTATACGCCCACCCA-3′ |
As described in Fig. 1.
As derived from the sequence of our Vero cell-adapted, laboratory Beaudette strain.
The T7 RNA polymerase recognition sequences shown in bold. Underlined are BsmBI restriction enzyme recognition sequences.
The E amplicon was replaced with the Δ5a/EGFP amplicon and the full-length infectious cDNA template was assembled and transcribed as described previously. Three days after electroporation into BHK cells and co-culturing with Vero cells, CPE was observed in the positive control and in the cells transfected with the GIBV RNA transcript (Fig. 5A). The cells that showed CPE by GIBV infection also expressed EGFP which was easily detected under a UV microscope (Fig. 5B). Interestingly, some cells expressed EGFP before even showing virus infection induced morphological changes. Following nucleotide sequencing analysis, it was confirmed that no unintentional mutations were incorporated into the amplicons used to construct the recombinant GIBV. Therefore, not only is 5a expression not essential for viral replication but also it is possible to replace the 5a with a functional foreign ORF.
Fig. 5.
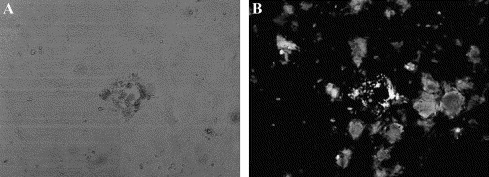
Expression of EGFP in Vero cells infected with GIBV. (A) GIBV infection produced syncytia typical of the CPE from Vero cells infected with the standard IBV. (B) Expression of EGFP was observed by UV microscopy. Expression of EGFP could be observed prior to the typical IBV-induced CPE.
The phenotypes of the recombinant IBV expressing EGFP and the standard cell adapted Beaudette IBV differed
To confirm the stability of the recombinant GIBV, the virus was subsequently passaged in Vero cells. EGFP expression was detected in each of seven sequential passages. GIBV was further characterized by comparing plaque morphology and one step growth curves. Compared to our standard virus and the virus from the cloned MIBV, GIBV-generated plaques were much smaller in size (Fig. 6A). The growth of GIBV was similar to the standard IBV with maximum growth at 14 h post-infection. However, GIBV grew to a 10-fold lower titer compared with the standard Beaudette IBV or MIBV (Fig. 3).
Fig. 6.
EGFP deletion revertant of GIBV showed comparable plaques to standard IBV or MIBV. After six serial passages of GIBV, heterogeneous-sized plaques were observed. The arrow indicates typical GIBV plaques with the smaller size compared to MIBV and the arrow head indicates a larger plaque from the GIBV stock that was comparable to the standard cell adapted IBV, and plaques purified from the larger plaques in A are shown in panel B.
Beyond passage five in Vero cells, the plaque assays of the GIBV stocks began to demonstrate plaques similar in size to those formed by our standard virus. Viruses derived from these plaques had lost the ability to express EGFP. To explain the phenomena, the region of EGFP insertion/5a replacement from several plaque purified viruses was RT-PCR amplified and compared with RT-PCR products of MIBV and the GIBV expressing EGFP (forming smaller plaques). The RT-PCR product of the latter was smaller than the GIBV and MIBV (Fig. 7A). In addition, the region across the insertion site of EGFP ORF for ten GIBV derived strains that had lost EGFP expression was amplified by RT-PCR. Sequencing of the RT-PCR products confirmed that these stains had either partially or completely lost the EGFP ORF. The 10 strains examined could be categorized into four distinct groups, depending on the deletion pattern of the EGFP ORF (Fig. 7B). The 5b ORF of all was maintained and completely intact while the start codon at the insertion site of the EGFP was deleted in all of these phenotypic revertants. Interestingly, the size of plaques with the 5a deletions, including a revertant that had completely lost the EGFP ORF, was comparable to plaques generated by our standard Beaudette cell-adapted Beaudette virus (Fig. 6B). When compared with the standard virus, growth kinetics and maximum titer of the revertants with the 5a deletion was similar to the standard cell adapted Beaudette virus (Fig. 3).
Fig. 7.
GIBV phenotypic revertant had lost the EGFP ORF. The absence of the EGFP ORF was confirmed by RT-PCR and nucleotide sequencing of the region corresponding to nucleotides 25,162 to 25,613 of the standard cell adapted IBV. Compared to GIBV, the GIBV revertant produced a smaller RT-PCR product, smaller than the E amplicon, suggesting that the revertant had lost the EGFP and 5a ORFs. (A) RT-PCR and PCR products of the region corresponding to nucleotides 25,162 to 25,613 of GIBV, the phenotypic 5aGFP deletion revertant and the control, E amplicon. The M lanes represent the DNA molecular size marker; lane 1, RT-PCR product of GIBV expressing EGFP; lane 2, the RT-PCR product of the phenotypic revertant that had lost expression of EGFP, and 3, as a control, the PCR product from the E fragment. (B) Alignment of the nucleotide sequences of the region spanning the 5a/EGFP ORF of Δ5aGFP viruses identified four distinct types of EGFP deletions. Italicized nucleotides and the positions starred below the sequence alignment represent conserved sequences. Every mutant lost the EGFP start codon but maintained the 5b start codon (underlined).
Discussion
Molecular clones of several coronaviruses have been constructed using three different methodologies, vaccinia virus vectors and BAC as cloning systems, and the in vitro assembly strategy. In this study, we constructed an infectious cDNA clone of IBV and recombinant IBV expressing EGFP instead of 5a protein, using in vitro assembly which is less labor intensive and allows for easy manipulation, such as the introduction of targeted mutations. We chose to use highly attenuated strain of IBV, the Vero cell-adapted Beaudette US strain as the cDNA backbone because we hope to eventually use IBV as a gene transfer vector, as well as to elucidate mechanisms of pathogenesis and generate new appropriate vaccines. Contrary to the difficulties stated in a previous report (Casais et al., 2001), our study has demonstrated that the in vitro assembly method used to generate infectious RNA from in vitro assembled cloned cDNAs of TGEV, MHV, and SARS coronavirus also can be applied to IBV.
This is also the first report that the group III coronavirus group-specific 5a gene is not essential for viral replication. Based on studies with MHV and TGEV, in which deletion of multiple group-specific genes resulted in attenuation of virulence, group-specific genes of IBV are reasonable candidates for viral attenuation and foreign gene replacement (de Haan et al., 2002, Sola et al., 2003). However, it has been shown that these small ORF, including 5a, are conserved among the various strains of IBV (Brooks et al., 2004). Therefore, it is likely that 5a is important, if not critical, for in vivo replication. Further studies will determine the in vivo role of 5a in pathogenesis and whether its deletion results in attenuation.
GIBV produced smaller sized plaques compared to our standard parental virus. Although many recombinant viruses have been constructed to express EGFP or GFP without known reports that EGFP is toxic to virus replication, EGFP was shown in at least one study to induce cellular apoptosis (Liu et al., 1999). In the current study, it is possible that infected cells died by apoptosis because of high expression of EGFP prior to producing maximum amounts of virus viral replication. This is consistent with the finding that we could detect expression of EGFP before cells showed CPE. Alternatively, manipulation of ORF 5a could affect ORF 5b expression. It has been assumed that gene 5b is expressed by leaky scanning mechanism (Liu and Inglis, 1992). The EGFP ORF contained an appropriate Kozak sequence and may have reduced the expression of 5b. Therefore, reduction in 5b expression could potentially have been associated with the observed change in viral replication.
GIBV phenotypic revertants, which lost the EGFP ORF, maintained an intact 5b ORF, and regained standard Beaudette cell adapted virus plaque size, growth kinetics and titer. Based on sequence analysis, four distinct deletion events were indicated. It has been shown that the GFP gene has homology with the MHV TRS and hinders expression of GFP because of abortive transcription (Fischer et al., 1997). However, we do not favor this explanation because, contrary to Fisher et al., we had sufficient expression of EGFP, with recombinant virus during the first several passages, to be easily detectable and no sequence similarity could be identified in or around the deleted region between the IBV TRS and the EGFP gene. Non-homologous recombination events during IBV replication likely accounts for the deletion of a gene, such as EGFP, that is not necessary for the viral replication. It is not clear whether the foreign gene itself or its specific location in the viral genome causes this spontaneous deletion event. Furthermore, we cannot conclude whether deletion of the EGFP gene, under the control of the 5a TRS, is a general phenomenon of recombinant IBV or a result of EGFP toxicity.
Coronaviruses have several attractive traits as gene delivery systems, including their unusually high packaging capacity, exclusive replication in cytoplasm, excluding any chance of viral genome incorporation into host chromosomes, and the ability to express multiple genes which has been shown by Thiel et al. (2003). The stability of a foreign ORF replacing the 5a ORF should be further examined with genes that encode non-toxic proteins, in order to determine the replication status and pathogenesis of these recombinant viruses in vivo. Furthermore, this methodology provides with a convenient reverse genetic tool for correlating the pathogenesis with the group-specific genes of IBV.
Materials and methods
Cultured cells and virus
A V5ero cell-adapted strain of IBV Beaudette US, from American Type Culture Collection (Manassas, VA) was plaque purified three times before use. The virus was propagated in an African green monkey kidney Vero cell line, obtained from ViroMed Laboratory (Minnetonka, MN) and maintained in Dulbecco's modified eagle medium (DMEM) containing 5% fetal bovine serum (FBS) supplemented with penicillin G (100 units/ml) and streptomycin (100 μg/ml). The baby hamster kidney cell line, BHK-21, was also obtained from ViroMed and maintained in DMEM containing 10% FBS supplemented with antibiotics as described for Vero cells.
RT-PCR cloning and sequencing of the IBV-Beaudette genome
Total cellular RNA was extracted from IBV-Beaudette infected Vero cells with Trizol reagent (Invitrogen, Carlsbad, CA), according to the manufacturer's directions. Reverse transcription was performed with Superscript II (Invitrogen) and reverse direction primers P1R, P2R, P3R, BEAUSSR, and GNRT (Table 2 ), containing BsmBI and SapI restriction endonuclease recognition sequences. The IBV sequence, as reported in GenBank (accession number M95169), was used for primer design and nucleotide sequencing. Each DNA fragment was amplified from cDNA templates by PCR using Expand Long polymerase (Boehringer Mannheim Biochemical). In order to transcribe RNA using T7 RNA polymerase, the T7 RNA promoter sequence was incorporated into the T7P1F primer. PCR primer pairs used to amplify genomic regions are listed in Table 2. PCR amplification of cDNA fragments was performed using the following conditions; denaturation at 94 °C for 2 min, 10 cycles at 94 °C for 30 s, 55 °C for 30 s, and 68 °C for 2–6 min depending on the size of the fragment and 18 cycles of 94 °C for 30 s, 55 °C for 30 s, and 68 °C for 2–6 min (with an additional 20 s for each subsequent cycle). The PCR products were isolated from agarose gels and cloned into pCR-XL-TOPO (Invitrogen) or pSMART (Lucigen, Middleton, WI) vectors, according to the manufacturer's directions.
Two to four independent clones of each amplicon were isolated and sequenced by using specific primers and the ABI dye termination sequencing method. A consensus sequence was determined by comparison of direct PCR product sequencing from each independent clone and amplicons, encoding the consensus sequence of each region of IBV, were cloned using standard recombinant DNA techniques (Sambrook et al., 1989).
The entire IBV N ORF, including the 3′ UTR, was amplified by RT-PCR from total cellular RNA extracted from IBV infected Vero cells, and the RT-PCR product was cloned into a transcription vector, pGEM-3Zf(+) (Promega, Madison, WI).
Construction of the Δ5a/EGFP amplicon
The EGFP ORF (F fragment) was PCR amplified from pLEGFP-N1 (Clontech Laboratories, Inc., Palo Alto, CA), using the GFPSF and GFPER primer pair, and cloned into pSTBlue (Table 2). The region upstream of the 5a ORF (G fragment) was PCR amplified using the E amplicon as a template with the 5ASR and BMF primer pair and the product cloned into pSTBlue (Novagen, Darmstadt, Germany), referred to as 5aS. The region downstream of the 5a ORF (fragment H which included the 3′UTR and polyadenylated tail) was PCR amplified using the E amplicon and the 5AEF and GNRT primer pair before cloning into the pCR-XL-TOPO vector (referred to as 5aE). The G fragment was prepared by digesting 5aS with XhoI and BsmBI restriction enzymes. The F fragment was digested from the vector with BsmBI. The G and F fragments were ligated into the 5aE amplicon which was digested with XhoI and BsmBI restriction enzymes. Replacement of 5a sequences with EGFP ORF was confirmed by sequencing of the Δ5a/EGFP amplicon.
In vitro assembly of full-length IBV infectious constructs
Each amplicon, having the consensus sequence, was prepared from an overnight bacterial culture. Plasmid was isolated and digested with the indicated restriction endonucleases according to the manufacturer's directions. Briefly, the IBV A amplicon was digested with XhoI and treated with calf intestine alkaline phosphatase (CIP), before digesting with the SapI enzyme. The IBV E amplicon was digested with EcoRI, CIP treated and digested with BsmBI. The B1, B2 and C1 amplicons were digested with BsmBI at 55 °C and then digested with SapI at 37 °C. The C2 and D amplicons were digested with BsmBI. Inserts of each amplicon were resolved by electrophoresis on 1% agarose gels containing crystal violet and isolated with QIAquick gel extraction kit (QIAGEN Inc., Valencia, CA) according to the manufacturer's directions. One picomole of each cDNA insert was ligated in an ordered reaction (A + B1, B2 + C1, C2 + D, E1 + E2 or EΔ5α/EGFP + E2). The appropriately sized ligation reaction products were purified from 1% agarose gels and further ligated with neighboring inserts for 4 h, pooled and ligated overnight (Fig. 1C). The final ligation product was purified by extracting with a phenol/chloroform/isoamyl alcohol (25:24:1) mixture and precipitated with ethanol before using as a template for in vitro transcription reactions.
In vitro transcription and electroporation
Full-length transcripts of the IBV cDNA constructs were generated in vitro using the mMESSAGE mMACHINE T7 Ultra kit (Ambion, Austin, TX) according to the manufacturer's direction with certain modifications. The in vitro transcription reaction was performed at 37 °C for 2 h in 20 μl reaction mixtures, supplemented with 3 μl of 30 mM GTP, resulting in a 1:1 ratio of GTP to capping analog. A similar reaction was performed for the N transcript using a 1:3 ratio of GTP and capping analog. Before electroporation, the transcripts were treated with DNase I and analyzed by 1% denaturing agarose gel (containing 2.2M formaldehyde) electrophoresis. The identity of the full-length in vitro transcript was confirmed by northern blot analysis using a probe against 3′ UTR of IBV.
After BHK-21 cells were grown to subconfluence (80%), treated with trypsin, and washed with cold DEPC treated PBS twice, they were resuspended in DEPC-treated PBS at a concentration of 107 cells/ml. RNA transcripts were added to 400 μl of the BHK-21 cell suspension in microfuge tubes on ice, gently pipetted and transferred to electroporation cuvettes. Three consecutive electrical pulses of 850 V at 25 μF were given, using the electro-cell manipulator 600, BTX (Genetronics, Inc., San Diego, CA). The transfected BHK-21 cells were diluted 1 to 20 with complete DMEM in 100 mm cell culture Petri dishes and co-cultured with 2 × 106 Vero cells/dish.
One-step growth curve
Triplicate wells of Vero cells in six-well plates were infected with virus at a multiplicity of infection (m.o.i.) of 2 to 3. The cells were harvested every 4 h for 24 h and stored at −80 °C until they were used for quantification by titration. The cells were three times frozen and thawed, and 10-fold serially diluted in DMEM without serum. Two hundred microliters of each dilution were inoculated into six-well plates for an hour, washed with PBS and overlaid with DMEM, containing 2% FBS, 0.8% agarose, before incubating at 37 °C for 3 days. The plaques were counted after removing the agarose overlay from the cells and staining the monolayer with crystal violet.
Acknowledgments
We thank Dr. Ralph Baric for providing valuable advice in developing the in vitro assembly procedure for IBV. This work was supported in part by grants from the U.S. Poultry and Egg Association (297), USDA Formula Animal Health Funds (1433), and NIH grant AI 51493 and the Institute of Food Safety and Engineering at Texas A & M University.
References
- Almazan F., Gonzalez J.M., Penzes Z., Izeta A., Calvo E., Plana-Duran J., Enjuanes L. Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. U.S.A. 2000;97(10):5516–5521. doi: 10.1073/pnas.97.10.5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baric R.S., Nelson G.W., Fleming J.O., Deans R.J., Keck J.G., Casteel N., Stohlman S.A. Interactions between coronavirus nucleocapsid protein and viral RNAs: implications for viral transcription. J. Virol. 1988;62(11):4280–4287. doi: 10.1128/jvi.62.11.4280-4287.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch B.J., Van Der Zee R., De Haan C.A., Rottier P.J. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 2003;77(16):8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks J.E., Rainer A.C., Parr R.L., Woolcock P., Hoerr F., Collisson E.W. Comparisons of envelope through 5B sequences of infectious bronchitis coronaviruses indicates recombination occurs in the envelope and membrane genes. Virus Res. 2004;100(2):191–198. doi: 10.1016/j.virusres.2003.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casais R., Thiel V., Siddell S.G., Cavanagh D., Britton P. Reverse genetics system for the avian coronavirus infectious bronchitis virus. J. Virol. 2001;75(24):12359–12369. doi: 10.1128/JVI.75.24.12359-12369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton S.R., Rogers D.B., Holmes K.V., Fertsch D., Remenick J., McGowan J.J. In vitro replication of mouse hepatitis virus strain A59. J. Virol. 1987;61(6):1814–1820. doi: 10.1128/jvi.61.6.1814-1820.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A., Kuo L., Masters P.S., Vennema H., Rottier P.J. Coronavirus particle assembly: primary structure requirements of the membrane protein. J. Virol. 1998;72(8):6838–6850. doi: 10.1128/jvi.72.8.6838-6850.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan C.A., Masters P.S., Shen X., Weiss S., Rottier P.J. The group-specific murine coronavirus genes are not essential, but their deletion, by reverse genetics, is attenuating in the natural host. Virology. 2002;296(1):177–189. doi: 10.1006/viro.2002.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields B.N. Coronaviridae: the viruses and their replication. In: Peter D.E.G., Howley M., Lamb R.A., Martin M.A., Roizman B., Straus S.E., Knipe D.M., editors. Fields-Virology. Lippincott Williams and Wilkins Publishers; 1996. pp. 1163–1220. [Google Scholar]
- Fischer F., Stegen C.F., Koetzner C.A., Masters P.S. Analysis of a recombinant mouse hepatitis virus expressing a foreign gene reveals a novel aspect of coronavirus transcription. J. Virol. 1997;71(7):5148–5160. doi: 10.1128/jvi.71.7.5148-5160.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F., Stegen C.F., Masters P.S., Samsonoff W.A. Analysis of constructed E gene mutants of mouse hepatitis virus confirms a pivotal role for E protein in coronavirus assembly. J. Virol. 1998;72(10):7885–7894. doi: 10.1128/jvi.72.10.7885-7894.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez J.M., Penzes Z., Almazan F., Calvo E., Enjuanes L. Stabilization of a full-length infectious cDNA clone of transmissible gastroenteritis coronavirus by insertion of an intron. J. Virol. 2002;76(9):4655–4661. doi: 10.1128/JVI.76.9.4655-4661.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M., Cavanagh D. The molecular biology of coronaviruses. Adv. Virus Res. 1997;48:1–100. doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le S.Y., Sonenberg N., Maizel J.V., Jr. Distinct structural elements and internal entry of ribosomes in mRNA3 encoded by infectious bronchitis virus. Virology. 1994;198(1):405–411. doi: 10.1006/viro.1994.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.X., Inglis S.C. Identification of two new polypeptides encoded by mRNA5 of the coronavirus infectious bronchitis virus. Virology. 1992;186(1):342–347. doi: 10.1016/0042-6822(92)90094-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.X., Cavanagh D., Green P., Inglis S.C. A polycistronic mRNA specified by the coronavirus infectious bronchitis virus. Virology. 1991;184(2):531–544. doi: 10.1016/0042-6822(91)90423-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.S., Jan M.S., Chou C.K., Chen P.H., Ke N.J. Is green fluorescent protein toxic to the living cells? Biochem. Biophys. Res. Commun. 1999;260(3):712–717. doi: 10.1006/bbrc.1999.0954. [DOI] [PubMed] [Google Scholar]
- Robbins S.G., Frana M.F., McGowan J.J., Boyle J.F., Holmes K.V. RNA-binding proteins of coronavirus MHV: detection of monomeric and multimeric N protein with an RNA overlay-protein blot assay. Virology. 1986;150(2):402–410. doi: 10.1016/0042-6822(86)90305-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. 2nd ed. Cold Spring Harbor Laboratory Press; Plainview, NY: 1989. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- Shen S., Wen Z.L., Liu D.X. Emergence of a coronavirus infectious bronchitis virus mutant with a truncated 3b gene: functional characterization of the 3b protein in pathogenesis and replication. Virology. 2003;311(1):16–27. doi: 10.1016/S0042-6822(03)00117-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331(5):991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola I., Alonso S., Zuniga S., Balasch M., Plana-Duran J., Enjuanes L. Engineering the transmissible gastroenteritis virus genome as an expression vector inducing lactogenic immunity. J. Virol. 2003;77(7):4357–4369. doi: 10.1128/JVI.77.7.4357-4369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavrinides J., Guttman D.S. Mosaic evolution of the severe acute respiratory syndrome coronavirus. J. Virol. 2004;78(1):76–82. doi: 10.1128/JVI.78.1.76-82.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V., Herold J., Schelle B., Siddell S.G. Infectious RNA transcribed in vitro from a cDNA copy of the human coronavirus genome cloned in vaccinia virus. J. Gen. Virol. 2001;82(Pt. 6):1273–1281. doi: 10.1099/0022-1317-82-6-1273. [DOI] [PubMed] [Google Scholar]
- Thiel V., Karl N., Schelle B., Disterer P., Klagge I., Siddell S.G. Multigene RNA vector based on coronavirus transcription. J. Virol. 2003;77(18):9790–9798. doi: 10.1128/JVI.77.18.9790-9798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount B., Curtis K.M., Baric R.S. Strategy for systematic assembly of large RNA and DNA genomes: transmissible gastroenteritis virus model. J. Virol. 2000;74(22):10600–10611. doi: 10.1128/jvi.74.22.10600-10611.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount B., Denison M.R., Weiss S.R., Baric R.S. Systematic assembly of a full-length infectious cDNA of mouse hepatitis virus strain A59. J. Virol. 2002;76(21):11065–11078. doi: 10.1128/JVI.76.21.11065-11078.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount B., Curtis K.M., Fritz E.A., Hensley L.E., Jahrling P.B., Prentice E., Denison M.R., Geisbert T.W., Baric R.S. Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. U.S.A. 2003;100(22):12995–13000. doi: 10.1073/pnas.1735582100. [DOI] [PMC free article] [PubMed] [Google Scholar]