

Abstract
A convenient and efficient synthesis of a novel class of acyclic nucleoside phosphonates derived from 2-(phosphonomethoxy)propanoic acid has been developed. The key step of the synthesis is the optimized oxidation of the 3-hydroxy-2-(phosphonomethoxy)propyl (HPMP) analogues to the corresponding 2′-carboxy-PME (CPME) derivatives using the TEMPO/NaClO2/NaClO oxidizing system. Although (S)-3-(adenin-9-yl)-2-(phosphonomethoxy)propanoic acid ((S)-CPMEA) has been designed as a compound with potential anti-HIV activity, none of the newly prepared CPME analogues exhibited any antiviral activity.
Keywords: Acyclic nucleoside phosphonates, CPMEA, HPMPA, PMEA, Oxidation, TEMPO, Microwave, Antiviral
Graphical abstract

1. Introduction
Acyclic nucleoside phosphonates (ANPs) constitute a distinguished class of antiviral agents.1 They also exhibit important cytostatic,2 antiparasitic3 and immunomodulatory activities.4 The main advantages of ANPs are their resistance towards degradation by enzymes in vivo, and the ability to by-pass the first phosphorylation step necessary for the activation of the bioactive nucleosides.5 Three ANPs have been approved worldwide for clinical use:5 HPMPC (cidofovir, Vistide®) for the treatment of CMV retinitis in AIDS patients,5, 6 PMEA (1, Fig. 1 ) (as its oral prodrug adefovir dipivoxil, Hepsera®) for the treatment of chronic HBV infections,5 and PMPA (2, Fig. 1) (as its oral prodrug tenofovir disoproxil fumarate, Viread®) for the treatment of HIV (itself or in combination with other antivirals) and chronic HBV infections.5 Many other ANPs were found to exhibit important antiviral properties, e.g., derivatives of 2,6-diaminopurine (HPMPDAP, PMEDAP, PMPDAP)1f and ‘open ring’ derivatives of 2,4-diaminopyrimidine (HPMPO–DAPy, PMEO–DAPy, PMPO–DAPy).(f), 7 Like nucleotides,8 due to their poor bioavailability, all ANPs have to be administered in the form of their prodrugs (or by injection in the case of cidofovir).9
Fig. 1.
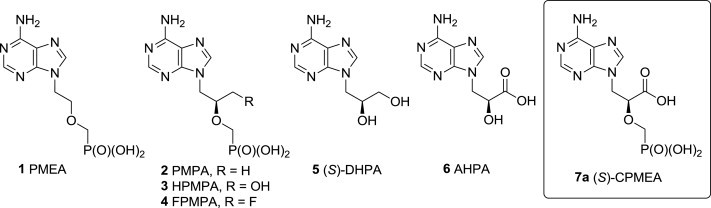
Examples of biologically important acyclic nucleoside and nucleotide analogues 1–6 and structure of the target compound 7a.
Analogues containing adenine play a special role among ANPs for their potent antiviral properties. HPMPA (3, Fig. 1),1a the prototype of ANPs derived from (S)-DHPA (5), possesses broad-spectrum activity against DNA viruses (including herpes-, adeno-, pox- and iridoviruses), as well as against retroviruses. While only the (S)-HPMP enantiomer 3 is active in the adenine series, its 3′-O-phosphonomethyl regioisomer (iso-HPMPA)10 exhibits no antiviral activity.1a The approved adenine analogues PMEA (1) and PMPA (2) are primarily active against hepadna- and retroviruses, while PMEA (1) also exhibits notable activity against herpesviruses.5 Finally, FPMPA (4), which proved to be less antivirally effective, but also less toxic than PMEA (1), was thoroughly studied for its antiretroviral activity but was not pursued further to the clinical use.11
A number of other adenine derivatives proved to have important antiviral activities.1f Acyclic nucleoside analogues like (S)-DHPA (5, Fig. 1), marketed in Czechoslovakia as Duvira® gel,12 or structurally related d-eritadenine13 and 3-(adenin-9-yl)-2-hydroxypropanoic acid (6, AHPA)14 constitute another class of broad-spectrum antivirals. These acyclic analogues of adenosine are targeted at S-adenosyl-l-homocysteine hydrolase,15 and thus at the capping process (methylation of 5′-end-guanine of viral mRNA).16
Herein we describe the synthesis and biological properties of the novel type of acyclic nucleoside phosphonates structurally related to all above-mentioned biologically important compounds. The novel type of ANPs is derived from 2-(phosphonomethoxy)propanoic acid, with the adenine derivative (S)-2′-carboxy-PMEA (7a, (S)-CPMEA, Fig. 1) being the most promising candidate with potential biological properties.
2. Results and discussion
The X-ray structure of HIV-1 RT–DNA complex after incorporation of tenofovir diphosphate is an ideal starting point for further rational and targeted drug design (Fig. 2a).17 In the complex, two amino acid residues (Arg 72 and Gln 151) in the binding site of the HIV-1 RT are oriented towards the C-2′ methyl group of tenofovir diphosphate (Fig. 2a). Apparent incapability of the C-2′ methyl group to substantially interact with the two polar amino acids of the HIV-1 RT led us to reason that replacement of the C-2′ methyl with a polar group able to interact with Arg 72 and/or Gln 151 would increase the binding affinity of such an inhibitor to HIV-1 RT. Our docking studies indicated that replacement of the C-2′ methyl group of tenofovir diphosphate (Fig. 2a) with a carboxyl group could, theoretically, lead to formation of up-to four new hydrogen bonds with arginine 72 and glutamine 151 (Fig. 2b).
Fig. 2.
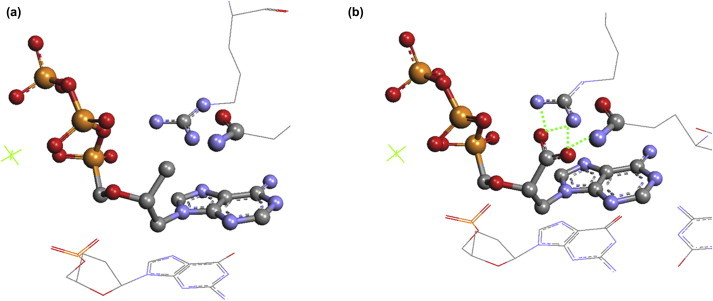
(a) Immediate binding site of the complex of HIV-1 RT–DNA and tenofovir diphosphate. (b) Docking of the complex of HIV-1 RT-DNA and (S)-CPMEA (7a) showing possible hydrogen bonding (dotted green lines) between the carboxyl group of (S)-CPMEA diphosphate and Arg 72 and Gln 151.
The initial docking studies indicated that (S)-CPMEA (7a, Fig. 1), an analogue of PMEA (1) bearing a carboxyl group at the C-2′ position of the aliphatic chain, potentially is a good candidate for strong binding affinity in the enzyme pocket of HIV-1 RT. Moreover, (S)-CPMEA (7a) can be considered to be a direct structural modification of PMPA (2) or HPMPA (3) with oxidized 2′-methyl or 2′-hydroxymethyl functions, respectively, or can also be looked upon as an O-phosphonomethyl derivative of AHPA (6, Fig. 1). Thus, compound 7a, as well as the other CPME analogues, was expected to display a wide range of biological properties.
Generally, two strategies could be utilized for the synthesis of the target analogue, (S)-CPMEA (7a, Fig. 1): (a) direct oxidation of the properly protected HPMPA followed by the removal of the protecting groups and (b) alkylation of adenine (or 6-chloropurine) with a suitable alkylating agent containing the protected carboxylic function, followed by deprotection (and ammonolysis in the case of 6-chloropurine).
The oxidation of the protected HPMPA derivative seems to be the more relevant and straightforward method for the synthesis of the compound 7a, while the ‘synthon’ approach seems to be too laborious and less effective. Thus, diisopropyl (S)-9-[3-hydroxy-2-(phosphonomethoxy)propyl]adenine (8a, Scheme 1 ) was chosen as the suitable starting material. This can be prepared in sufficient amounts according to the previously described procedures.18
Scheme 1.
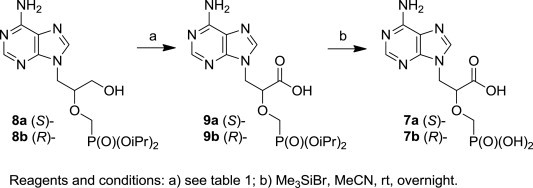
Synthesis of (S)-CPMEA (7a) and (R)-CPMEA (7b) through oxidation of the HPMPA derivatives 8a and 8b.
Nowadays, there are a considerable number of oxidizing agents, which can be used under relatively mild reaction conditions and which tolerate number of functional groups.19 Our goal was to develop a simple, mild and clean oxidation of the HPMPA derivative 8a, which would tolerate the presence of the unprotected amino group at the C-6 position of the purine moiety.
The conversion of the 5′-hydroxyl group of 2′,3′-protected purine nucleosides to the corresponding 5′-uronic acids is often accomplished by oxidation with an excess of KMnO4 in aqueous KOH solution under conditions of high dilution.20 An attempted oxidation of compound 8a under analogous reaction conditions only led to complex reaction mixtures (TLC).
A simple and efficient oxidative conversion of primary alcohols to the corresponding methyl esters using I2/K2CO3 in MeOH has been reported by Mori and Togo.21 Nevertheless, an attempt to prepare the methyl ester of derivative 9a by the direct oxidative esterification of compound 8a under the reported reaction conditions was not successful (Table 1 ).
Table 1.
Oxidation of diisopropyl HPMPA (8a) to diisopropyl (S)-CPMEA (9a)
| Oxidative agent; solvent | Desired product | Isolated yield |
|---|---|---|
| KMnO4/KOH; H2O | 9a | Complex mixture |
| I2/K2CO3; MeOH | Methyl ester of 9a | Complex mixture |
| RuO2/NaIO4; MeCN, CH2Cl2, H2O | 9a | 63% |
| TEMPO/BAIB; MeCN, H2O | 9a | 71% |
| TEMPO/NaClO2/NaClO; MeCN, H2O | 9a | 87% |
Ruthenium tetroxide (RuO4), another widely used oxidizing agent,19 is ideal when a very vigorous oxidizing agent is needed but mild reaction conditions must be maintained. Since RuO4 can decompose explosively, it is mostly prepared in situ by oxidation of RuCl3 or RuO2. A mild oxidation of nucleosides to the corresponding 5′-carboxylic acids by use of RuCl3/NaIO4 and RuCl3/K2S2O8/KOH systems has been reported.22 Since RuCl3 was found to be a very hygroscopic reagent, not easy to handle, RuO2 was our oxidizing agent of choice.
Oxidation of diisopropyl HPMPA (8a) with RuO2/NaIO4 afforded the derivative 9a in a satisfactory yield (63%, Table 1). The oxidation was carried out in a mixture of acetonitrile, chloroform, and water (1:1:2) according to the described procedure used for the preparation of adenosine 5′-uronamides,23 with an addition of concentrated HCl to pH 2.5. Despite the good isolated yield, purification was tedious and column chromatography had to be repeated several times to obtain pure (S)-CPMEA derivative 9a, probably owing to the fact that phosphonates strongly bind metal ions. Thus, a cleaner and less laborious procedure was still desirable.
Commercially available 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO) is a reagent of choice for oxidation of various organic substrates.24 TEMPO belongs to the class of stable organic nitroxyl radicals commonly used for the oxidation of primary and secondary alcohols.19, 24
Mild and efficient oxidation of alcohols to ketones and aldehydes using catalytic amounts of TEMPO and stoichiometric amounts of [bis(acetoxy)iodo]benzene (BAIB) has been developed.25 This method differs from most other TEMPO-mediated oxidations in that it generates acetic acid and iodobenzene as by-products and avoids inorganic contaminants.25 This oxidizing system was successfully employed for the oxidation of 2′,3′-protected nucleosides to give the corresponding 5′-carboxylic acids.26 In our case, oxidation of HPMPA derivative 8a with the TEMPO/BAIB oxidizing system afforded the expected product 9a in a 71% yield (Table 1).
An even better yield (87%) of the (S)-CPMEA derivative 9a was achieved when BAIB was replaced with NaClO2/NaClO (sodium hypochlorite is a readily available and inexpensive oxidant, household bleach). This oxidizing system (TEMPO/NaClO2/NaClO)27 offers a clean reaction and easy work-up based on the isolation of the product by flash chromatography on silica gel followed by crystallization. This efficient procedure offered compound 9a in very high purity.
Finally, the desired product (S)-CPMEA (7a) was obtained in a 76% yield by ester cleavage of compound 9a under the standard conditions (Me3SiBr in MeCN).28
Analogously, oxidation of enantiomeric diisopropyl (R)-HPMPA (8b) using the TEMPO/NaClO2/NaClO system afforded the derivative 9b in a 70% yield. Subsequent deprotection of compound 9b gave the free phosphonic acid 7b in a 50% yield.
Unfortunately, none of the oxidizing systems listed in Table 1 afforded (S)-CPMEA (7a) by direct oxidation of free HPMPA (3, Fig. 1), confirming the importance of the diester protection of the phosphonate group during the procedure, and synthetic transformations in general.
Eventually, we decided to verify the more general applicability of our optimized oxidation protocol, and it was utilized for the preparation of CPME derivatives bearing other nucleobases (Scheme 2 , Table 2 ). Thus, the expected diisopropyl CPME analogues 9 were obtained by the oxidation of the corresponding HPMP derivatives 8 with TEMPO/NaClO2/NaClO in high yields (76–87%).
Scheme 2.
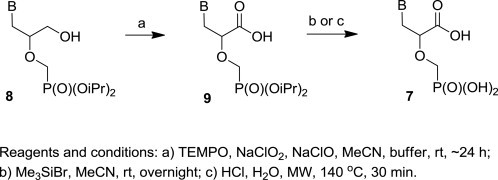
Synthesis of CPME compounds 7 starting from the HPMP analogues 8.
Table 2.
Oxidation of the HPMP analogues 8 with TEMPO/NaClO2/NaClO to give 9, followed by diester removal to yield 7
| Entry | Nucleobase B | Starting compound | Product (yield)a of oxidation | Product (yield)a of the phosphonate deprotection |
|---|---|---|---|---|
| 1 |  |
8a (S)- | 9a (87%) | 7a (68%),b (46%)c |
| 2 | 8b (R)- | 9b (76%) | 7b (50%)b | |
| 3 | 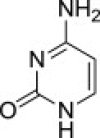 |
8c (S)- | 9c (83%) | 7c (62%),b (39%)c |
| 4 |  |
8d (S)- | 9d (82%) | 7dd (64%),b (44%)c |
| 5 |  |
8e (S)- | 9e (81%) | 7e (42%)c |
| 6 | 8f (R)- | 9f (80%) | 7f (48%)c | |
| 7 |  |
8g (S)- | Complex mixture (no 9g isolated)e | 7g (61%),b (37%)c |
| 8 | 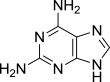 |
8h (S)- | Complex mixture (no 9h isolated) | 7h (38% from 9j)c,f |
| 9 | 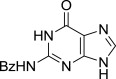 |
8i (S)- | 9i (78%) | 7i (52%)b |
| 10 |  |
8j (S)- | 9j (80%) | N.A.g |
The functional groups of the unprotected nucleobases are usually very well tolerated during the oxidation process. The only problem was observed with compounds bearing 2-aminopurine bases containing a guanidine motif (guanine and 2,6-diaminopurine). Treatment of the HPMPG derivative 8g 18b and HPMPDAP derivative 8h 18b with TEMPO/NaClO2/NaClO gave complex and colourful reaction mixtures and no desired products were isolated (entries 7 and 8, respectively, Table 2).
To overcome the undesirable reactivity of the 2-aminopurine bases, we decided to protect the amino groups of compounds 8g and 8h with the benzoyl group. Thus, the N 2-benzoylguanine derivative 8i 18b was prepared in 49% yield from compound 8g in pyridine by sequential treatment with Me3SiCl, BzCl, water and aqueous ammonia (Scheme 3 ). Subsequent oxidation of the benzoyl derivative 8i with TEMPO/NaClO2/NaClO afforded the carboxy analogue 9i in 78% yield (Table 2, entry 9). Direct deprotection of the phosphonate moiety of compound 9i using Me3SiBr in acetonitrile28 afforded the N 2-benzoylguanine derivative 7i in 52% yield (Table 2, entry 9, Scheme 3). In order to obtain CPMEG (7g), the benzoyl group of derivative 9i was removed first, using MeONa solution, followed by the removal of the isopropyl groups from the intermediate 9g (Scheme 3).
Scheme 3.
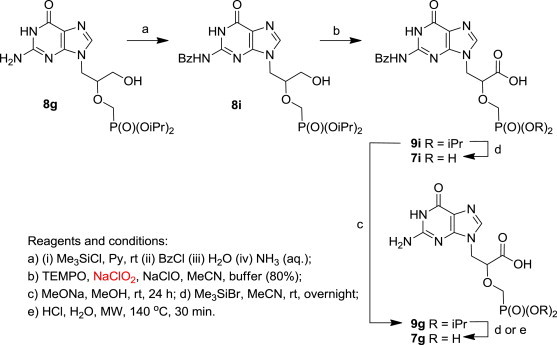
Synthesis of CPMEG (7g) using TEMPO/NaClO2/NaClO as a key step.
Analogously, the HPMPDAP analogue 8h was protected as its dibenzoyl derivative 8j (in 47% yield), which was then efficiently oxidized using the TEMPO/NaClO2/NaClO system to compound 9j in 80% yield (Table 2, entry 10, Scheme 4 ). Finally, CPMEDAP (7h) was obtained in 38% yield from compound 9j by simultaneous hydrolysis of both benzoyl groups and diisopropyl esters using 1 M aqueous HCl under microwave irradiation (Scheme 4), a general method for the convenient deprotection of phosphonate diesters recently developed in our laboratory.29
Scheme 4.

Synthesis of CPMEDAP (7h) using TEMPO/NaClO2/NaClO as a key step.
All of the prepared diisopropyl CPME phosphonates 9 were deprotected to the final free phosphonic acids 7 (Table 2, Scheme 2). As already mentioned above, two general methods for the removal of alkyl esters from the phosphonate group can be employed: (a) commonly used deprotection with Me3SiBr in acetonitrile followed by hydrolysis (method A),28 (b) recently described microwave-assisted hydrolysis using aqueous HCl (method B).29 Method A gave better yields (50–68%) of the free phosphonic acids compared to the method B (37–46%). Diisopropyl phosphonate 9d, with the methoxy group in position C-4 of the pyrimidine ring, afforded under the deprotection conditions (by both methods A and B) the corresponding thymine derivative 7d (Table 2, entry 4).
All newly prepared CPME analogues 7 were tested for their antiviral properties. Although completely nontoxic in all assays, none of the compounds (including the most promising (S)-CPMEA (7a)) exhibited any promising activity against the HIV-1 virus or any other viruses tested (HSV-1 (KOS), HSV-1 (KOS TK−), HSV-2 (G), RSV, Vaccinia virus, Vesicular stomatitis virus, Parainfluenza-3 virus, Reovirus-1, Sindbis virus, Coxsackie B4 virus, Punta Toro virus, Feline Corona virus and Feline Herpes virus). One of the obvious reasons for the lack of the biological properties of the CPME derivatives 7 may be their poor bioavailability caused by their high polarity (CPME derivatives are even more polar that the corresponding PME, PMP and HPMP analogues). Since the majority of the therapeutically successful ANPs have to be administered in the form of their prodrugs, the preparation of various prodrugs of the lead (S)-CPMEA (7a) is currently in progress and will be reported elsewhere.
3. Conclusion
A novel acyclic nucleoside analogue, (S)-3-(adenin-9-yl)-2-(phosphonomethoxy)propanoic acid (7a, (S)-CPMEA), was designed as an inhibitor of HIV-1 RT and its synthesis was developed and optimized. The key step of the (S)-CPMEA (7a) synthesis consists of oxidation of the (S)-HPMPA derivative 8a with TEMPO/NaClO2/NaClO to give compound 9a in a high yield (87%). Subsequently, a whole series of CPME derivatives 9 was prepared in high yields from the corresponding HPMP analogues 8 by the optimized oxidative methodology using TEMPO. The oxidation process tolerates the functional groups present on the common nucleobases (A, C, T, U), with the exception of compounds containing the amino group in position C-2 of the purine ring (i.e., 8g and 8h). Compounds 8g and 8h had to be oxidized as their benzoyl derivatives 8i and 8j, respectively. Finally, two methods were employed for the deprotection of the phosphonate diesters 9: (a) treatment with Me3SiBr in acetonitrile followed by hydrolysis; (b) microwave-assisted hydrolysis with aqueous HCl. Unfortunately, none of the newly synthesized CPME compounds (including the most promising (S)-CPMEA) showed any interesting activity against the viruses tested.
4. Experimental section
4.1. General
Analytical TLC was performed on silica gel pre-coated aluminium plates with fluorescent indicator (Merck 5554, 60 F254). Spots were visualized with UV light (254 nm) or by spraying with 4-nitrobenzylpyridine (2% solution in ethanol) followed by short heating to 600 °C and spraying with concentrated ammonia (26% solution in water). Column chromatography was carried out on silica gel (Sigma S-0507, 40–63 μm). Mass spectra were measured on a Q-Tof micro (Waters) and HRMS were taken on an LTQ Orbitrap XL (Thermo Fisher Scientific) spectrometer. 1H and 13C NMR spectra were recorded on a Bruker Avance 400 (1H at 400 MHz and 13C 100.6 MHz), Bruker Avance 500 (1H at 500 MHz and 13C 125.7 MHz) and Bruker Avance 600 (1H at 600.1 MHz and 13C 150.9 MHz) in DMSO-d 6 (referenced to the solvent signal δ=2.50 and 39.70 ppm, respectively), or in D2O (referenced to dioxane as an internal standard δ=ppm). Complete assignment is based on heteronuclear correlation experiments HSQC and H,C-HMBC. Chemical shifts (δ) are in parts per million and coupling constants (J) in hertz. UV spectra were taken on a Beckman DU-65 spectrophotometer in methanol or water solution. IR spectra were obtained on a FT IR Bruker Equinox IFS 55 spectrometer in KBr pellets.
The microwave-assisted reactions were carried out in CEM Discover (Explorer) microwave apparatus, 24-position system for 10-mL vessels sealed with a Teflon septum. It was operated at a frequency of 2.45 GHz with continuous irradiation power from 0 to 300 W. The solutions were steadily stirred during the reaction. The temperature was measured with an IR sensor on the outer of the process vessel. The vials were cooled to ambient temperature with gas jet cooling system. The pressure was measured with an inboard CEM Explorer pressure control system (0–21 bar).
Household bleach SAVO purchased from BOCHEMIE a.s. (Lidická 326, 735 95 Bohumín, Czech Republic) and the concentration of sodium hypochlorite was determined by iodometric titration (40.7 mg/mL). Starting HPMP analogues were prepared according to the published procedures.18
4.2. Computational details
The docking studies (Fig. 2) were performed with the ArgusLab30 program with the use of the ‘ArgusDock’ algorithm. An exhaustive search was performed by enabling the ‘High precision’ option in the Docking precision menu, ‘Dock’ was chosen as the calculation type, and ‘flexible’ for the ligand. Spacing of 0.2 Å between the grid points was used. The atomic co-ordinates for the X-ray structures of the HIV-1 RT–DNA complexes after incorporation of the anti-HIV drug tenofovir were downloaded from the Protein Data Bank (PDB code: 1T05).17 The final inhibitor–protein complex was visualized using ArgusLab.30
4.3. Synthesis of the N-benzoyl derivatives 8i and 8j
Starting HPMP derivative 8g or 8h (1.5 mmol) was dissolved in dry pyridine (20 mL) in a round-bottomed flask with a drying tube, and the reaction mixture was cooled in an ice bath. Me3SiBr (7.5 mmol) was added to the reaction mixture and after 30 min benzoyl chloride (7.5 mmol) was added. The reaction flask was removed from the ice bath and the mixture was stirred for 2 h at room temperature. Then the reaction mixture was cooled in ice bath and cold water (4 mL) was added, followed after 15 min of stirring by concentrated aqueous ammonia (4 mL). The mixture was stirred at room temperature for another 30 min and solvents were removed in vacuo to give crude oil. The chromatography on silica gel column (0–3% methanol in chloroform) afforded the product 8i or 8j as thick yellowish oil.
4.3.1. Diisopropyl (S)-N2-benzoyl-9-[3-hydroxy-2-(phosphonomethoxy)propyl]guanine (8i)18b
Yield 49%, yellowish solid (recryst EtOAc/hexane), mp 171–173 °C (mp 173–174 °C);18b MS-ESI+ m/z (%) 508 (15, M+H+), 530 (100, M+Na+); HRMS-ESI+: m/z calcd for C22H31O7N5P (M+H+) 508.1956, found 508.1954. 1H NMR (DMSO-d 6) spectrum is identical with the original spectroscopic data.18b
4.3.2. Diisopropyl (S)-N2,N6-dibenzoyl-9-[3-hydroxy-2-(phosphonomethoxy)propyl]-2,6-diaminopurine (8j)
Yield 47%, thick yellowish oil; 1H NMR (DMSO-d 6) δ: 11.16 (s, 1H, NH), 11.01 (s, 1H, NH), 8.31 (s, 1H, H-8), 8.06 (m, 2H, H-2″), 7.99 (m, 2H, H-2″), 7.50–7.66 (m, 6H, H-3″, 4″), 5.04 (t, J OH–CH2=5.7 Hz, 1H, OH), 4.44–4.56 (m, 3H, CHipr, H-1′a), 4.35 (dd, 1H, Jgem=14.7 Hz, J 1′b–2′=6.4 Hz, H-1′b), 3.89–3.94 (m, 3H, H-2′, CH2P), 3.50 (m, 1H, H-3′), 3.43 (m, 1H, H-3′), 1.20 (d, J CH3–CH=6.2 Hz, 3H, CH3ipr), 1.17 (d, J CH3–CH=6.2 Hz, 3H, CH3ipr), 1.16 (d, J CH3–CH=6.2 Hz, 3H, CH3ipr), 1.13 (d, J CH3–CH=6.2 Hz, 3H, CH3ipr); 13C NMR (DMSO-d 6) δ: 165.86 (CON), 165.74 (CON), 153.90 (C-4), 152.30 (C-2), 150.68 (C-6), 145.28 (C-8), 134.46 (C-1″), 133.56 (C-1″), 132.64 (C-4″), 132.20 (C-4″), 128.70 (C-2″, 3″), 128.63 (C-2″, 3″), 128.59 (C-2″, 3″), 128.28 (C-2″, 3″), 123.08 (C-5), 79.91 (d, J 2′-P=11.5 Hz, C-2′), 70.48 (d, J C–O–P=6.3 Hz, CHipr), 63.57 (d, J C–P=165.1 Hz, CH2P), 59.97 (C-3′), 43.36 (C-1′), 23.78–24.00 (m, CH3ipr); MS-ESI+ m/z (%) 611 (5, M+H+), 633 (100, M+Na+), 655 (12, M+2Na+); HRMS-ESI+: m/z calcd for C29H36O7N6P (M+H+) 611.2378, found 611.2376.
4.4. Oxidation of diisopropyl (S)-HPMPA (8a) with ruthenium tetroxide
Compound 8a (1.0 mmol, 387 mg), RuO2 (0.15 mmol, 20 mg) and NaIO4 (5.0 mmol, 1.1 g) were vigorously stirred in a mixture of CH3CN (15 mL), CHCl3 (15 mL) and H2O (30 mL). Concentrated HCl (eight drops) was added to ∼pH 2.5. The heterogenous mixture was vigorously stirred at room temperature for 72 h. The solid was removed by filtration of the reaction mixture through a silica gel layer and washed with methanol. The solvents were evaporated and the residue was purified by column chromatography on silica gel (CHCl3/MeOH/AcOH, 88:6:6). The purification was repeated two more times to remove colourful by-products and inorganic salts. The desired product 9a (253 mg, 63%) was obtained as a yellowish oil, which upon standing in EtOAc/MeOH mixture crystallized.
4.5. Oxidation of diisopropyl (S)-HPMPA (8a) with TEMPO/BAIB
Compound 8a (1.0 mmol, 387 mg), TEMPO (0.2 mmol, 32 mg) and BAIB (2.2 mmol, 644 mg) in a mixture of CH3CN (2 mL) and H2O (2 mL) were stirred at room temperature for 24 h and the solvents were evaporated. Flash chromatography on silica gel (CHCl3/MeOH, 4:1) afforded the product 9a (285 mg, 71%) as a yellowish oil, which upon standing in EtOAc/MeOH mixture crystallized.
4.6. Oxidation of diisopropyl HPMP derivatives 8 with TEMPO/NaClO2/NaClO. General procedure
The corresponding HPMP derivative 8 (1.0 mmol), TEMPO (0.3 mmol, 47 mg) and NaClO2 (2.0 mmol, 226 mg) in a mixture of CH3CN (10 mL) and 0.67 M aqueous sodium phosphate buffer (10 mL, ∼pH 6.7) were vigorously stirred at room temperature. An aqueous solution of sodium hypochlorite (40.7 mg/ml), prepared by dilution of household bleach SAVO (1.0 mL) with H2O (1.0 mL) was added. The mixture was stirred at room temperature until the reaction was complete (∼24 h) and the solvents were evaporated. Flash chromatography on silica gel column (CHCl3/MeOH, 4:1) afforded the desired CPME products 9 in good yields (76–87%) as yellowish oils, which upon standing in EtOAc/MeOH mixture gave yellowish crystals.
4.6.1. (S)-3-(6-Amino-9H-purin-9-yl)-2-[(diisopropoxyphosphono)methoxy]propanoic acid (9a)
Yellowish crystals, dec >150 °C, yield 87%; 1H NMR (DMSO-d 6) δ: 8.15 (s, 1H, H-2), 8.03 (s, 1H, H-8), 7.28 (br s, 2H, NH2), 4.37–4.55 (m, 5H, H-1′, H-2′, CHipr), 3.95 (dd, Jgem=13.7 Hz, J H–C–P=8.7 Hz, 1H, CH2Pb), 3.73 (dd, Jgem=13.7 Hz, J H–C–P=9.4 Hz, 1H, CH2Pa), 1.18 (d, J=6.2 Hz, 3H, CH3ipr), 1.14 (d, J=6.2 Hz, 3H, CH3ipr), 1.13 (d, J=6.2 Hz, 3H, CH3ipr), 1.07 (d, J=6.2 Hz, 3H, CH3ipr); 13C NMR (DMSO-d 6) δ: 170.61 (C-3′), 155.93 (C-6), 152.37 (C-2), 149.75 (C-4), 141.47 (C-8), 118.55 (C-5), 77.99 (d, J 2′-P=12.7 Hz, C-2′), 70.58 (d, J C–O–P=6.5 Hz, CHipr), 70.53 (d, J C–O–P=6.5 Hz, CHipr), 64.08 (d, J C–P=163.9 Hz, CH2P), 44.36 (C-1′), 23.92 (d, J C–C–O–P=4.0 Hz, CH3ipr), 23.88 (d, J C–C–O–P=3.9 Hz, CH3ipr), 23.78 (d, J C–C–O–P=4.5 Hz, CH3ipr), 23.67 (d, J C–C–O–P=4.5 Hz, CH3ipr); MS-ESI+ m/z (%) 402 (60, M+H+), 825 (100, 2M+Na+); HRMS-ESI+: m/z calcd for C15H25O6N5P (M+H+) 402.1537, found 402.1536; FTIR (KBr, cm−1) ν: 3396, 3181, 2981, 1688, 1651, 1599, 1233, 996. [α]D 20 −26.2 (c 0.302, MeOH).
4.6.2. (R)-3-(6-Amino-9H-purin-9-yl)-2-[(diisopropoxyphosphono)methoxy]propanoic acid (9b)
Yellowish crystals, dec >150 °C, yield 76%; 1H NMR (DMSO-d 6) δ: 8.15 (s, 1H, H-2), 8.03 (s, 1H, H-8), 7.28 (br s, 2H, NH2), 4.37–4.55 (m, 5H, H-1′, H-2′, CHipr), 3.95 (dd, Jgem=13.7 Hz, J H–C–P=8.7 Hz, 1H, CH2Pb), 3.73 (dd, Jgem=13.7 Hz, J H–C–P=9.4 Hz, 1H, CH2Pa), 1.18 (d, J=6.2 Hz, 3H, CH3ipr), 1.14 (d, J=6.2 Hz, 3H, CH3ipr), 1.13 (d, J=6.2 Hz, 3H, CH3ipr), 1.07 (d, J=6.2 Hz, 3H, CH3ipr); 13C NMR (DMSO-d 6) δ: 170.61 (C-3′), 155.93 (C-6), 152.37 (C-2), 149.75 (C-4), 141.47 (C-8), 118.55 (C-5), 77.99 (d, J 2′-P=12.7 Hz, C-2′), 70.58 (d, J C–O–P=6.5 Hz, CHipr), 70.53 (d, J C–O–P=6.5 Hz, CHipr), 64.08 (d, J C–P=163.9 Hz, CH2P), 44.36 (C-1′), 23.92 (d, J C–C–O–P=4.0 Hz, CH3ipr), 23.88 (d, J C–C–O–P=3.9 Hz, CH3ipr), 23.78 (d, J C–C–O–P=4.5 Hz, CH3ipr), 23.67 (d, J C–C–O–P=4.5 Hz, CH3ipr); MS-ESI+ m/z (%) 402 (60, M+H+), 825 (100, 2M+Na+); HRMS-ESI+: m/z calcd for C15H25O6N5P (M+H+) 402.1537, found 402.1536; FTIR (KBr, cm−1) ν: 3396, 3181, 2981, 1688, 1651, 1599, 1233, 996. [α]D 20 +28.0 (c 0.613, MeOH).
4.6.3. (S)-3-(4-Amino-2-oxopyrimidin-1(2H)-yl)-2-[(diisopropoxyphosphono)methoxy]propanoic acid (9c)
Yellowish crystals, dec >150 °C, yield 83%; 1H NMR (DMSO-d 6) δ: 7.43 (d, J 6–5=7.3 Hz, 1H, H-6), 7.34 (br s, 1H, NH2), 7.19 (br s, 1H, NH2), 5.66 (d, J 5–6=7.3 Hz, 1H, H-5), 4.49–4.58 (m, 2H, CHipr), 4.26 (dd, J CH–CH2=8.7 and 3.7 Hz, 1H, 1-CH2-CH), 4.15 (dd, Jgem=13.8 Hz, J CH2–CH=3.7 Hz, 1H, 1-CH2a), 3.91 (dd, Jgem=13.8 Hz, J H–C–P=8.6 Hz, 1H, CH2Pa), 3.64–3.71 (m, 2H, CH2Pb, 1-CH2b), 1.17–1.23 (m, 12H, CH3ipr); 13C NMR (DMSO-d 6) δ: 171.04 (COOH), 165.84 (C-4), 155.22 (C-2), 147.29 (C-6), 93.24 (C-5), 77.77 (d, J C–O–C–P=12.8 Hz, 1-CH2–CH), 70.50 (d, J C–O–P=6.1 Hz, CHipr), 64.15 (d, J C–P=163.9 Hz, CH2P), 50.39 (N–CH2), 23.77–23.98 (m, CH3ipr); MS-ESI+ m/z (%) 378 (20, M+H+), 400 (85, M+Na+), 422 (100, M+2Na+); HRMS-ESI−: m/z calcd for C14H23O7N3P (M−H+) 376.1279, found 376.1280; FTIR (KBr, cm−1) ν: 3357, 3183, 2979, 2901, 1723, 1652, 1376, 1238, 1002. [α]D 20 −75.7 (c 0.264, MeOH).
4.6.4. (S)-2-[(Diisopropoxyphosphono)methoxy]-3-(4-methoxy-5-methyl-2-oxopyrimidin-1(2H)-yl)propanoic acid (9d)
Yellowish crystals, mp 82–90 °C, dec at 130 °C, yield 82%; 1H NMR (DMSO-d 6) δ: 7.63 (q, J 6-CH3=1.0 Hz, 1H, H-6), 4.43–4.54 (m, 2H, CHipr), 4.27 (dd, Jgem=13.7 Hz, J 1′a–2′=3.3 Hz, 1H, H-1′a), 4.03 (dd, Jgem=13.8 Hz, J H–C–P=7.8 Hz, 1H, CH2Pa), 3.96 (dd, J 2′–1′=9.8 and 3.3 Hz, 1H, H-2′), 3.82 (s, 3H, OCH3), 3.62 (dd, Jgem=13.7 Hz, J 1′b–2′=9.7 Hz, 1H, H-1′b), 3.54 (dd, J H–C–P=9.1 Hz, Jgem=13.8 Hz, 1H, CH2Pb), 1.84 (d, J CH3-6=1.0 Hz, 3H, 5-CH3), 1.20 (d, J CH3–CH=6.2 Hz, 3H, CH3ipr), 1.17 (d, J CH3–CH=6.2 Hz, 3H, CH3ipr), 1.15 (d, J CH3–CH=6.2 Hz, 3H, CH3ipr), 1.12 (d, J CH3–CH=6.2 Hz, 3H, CH3ipr); 13C NMR (DMSO-d 6) δ: 171.38 (C-3′), 169.99 (C-4), 155.45 (C-2), 147.59 (C-6), 101.43 (C-5), 79.65 (d, J 2′-P=12.5 Hz, C-2′), 70.16 (m, CHipr), 63.45 (d, J C–P=163.6 Hz, CH2P), 53.95 (OCH3), 51.64 (C-1′), 23.66–23.90 (m, CH3ipr), 11.81 (5-CH3); MS-ESI+ m/z (%) 407 (5, M+H+), 429 (15, M+Na+), 451 (100, M+2Na+); HRMS-ESI+: m/z calcd for C16H27O8N2NaP (M+H+) 429.1397, found 429.1397; FTIR (KBr, cm−1) ν: 3440, 2981, 2935, 1670, 1541, 1480, 1374, 1244, 1106, 994. [α]D 20 −84.4 (c 0.399, MeOH).
4.6.5. (S)-2-[(Diisopropoxyphosphono)methoxy]-3-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)propanoic acid (9e)
Yellowish crystals, dec >150 °C, yield 81%; 1H NMR (DMSO-d 6) δ: 11.23 (br s, 1H, H-3), 7.49 (d, J 6–5=7.9 Hz, 1H, H-6), 5.48 (dd, J 5–6=7.9 Hz, J 5–3=2.1 Hz, 1H, H-5), 4.49–4.59 (m, 2H, CHipr), 4.14 (dd, Jgem=14.0 Hz, J CH2–CH=3.5 Hz, 1H, 1-CH2a), 4.08 (dd, J CH–CH2=9.1 and 3.5 Hz, 1H, 1-CH2–CH), 3.97 (dd, Jgem=13.9 Hz, J H–C–P=8.3 Hz, 1H, CH2Pa), 3.69 (dd, Jgem=14.0 Hz, J CH2–CH=9.2 Hz, 1H, 1-CH2b), 3.65 (dd, Jgem=13.9 Hz, J H–C–P=8.5 Hz, 1H, CH2Pb), 1.17–1.22 (m, 12H, CH3ipr); 13C NMR (DMSO-d 6) δ: 171.22 (COOH), 164.01 (C-4), 151.06 (C-2), 146.76 (C-6), 100.42 (C-5), 78.92 (d, J C–O–C–P=11.9 Hz, 1-CH2–CH), 70.39 (m, CHipr), 63.72 (d, J C–P=163.7 Hz, CH2P), 49.63 (1-CH2), 23.79–23.99 (m, CH3ipr); MS-ESI+ m/z (%) 379 (5, M+H+), 401 (60, M+Na+), 423 (100, M+2Na+); HRMS-ESI−: m/z calcd for C14H22O8N2P (M−H+): 377.1119, found: 377.1119; FTIR (KBr, cm−1) ν: 3417, 2981, 1691, 1630, 1464, 1388, 1245, 1105, 1001. [α]D 20 −53.0 (c 0.322, MeOH).
4.6.6. (R)-2-[(Diisopropoxyphosphono)methoxy]-3-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)propanoic acid (9f)
Yellowish crystals, dec >150 °C, yield 80%; 1H NMR (DMSO-d 6), 13C NMR (DMSO-d 6), MS and FTIR spectra are identical with compound 9e. [α]D 20 +68.1 (c 0.584, MeOH).
4.6.7. (S)-3-(2-Benzamido-6-oxo-1H-purin-9(6H)-yl)-2-[(diisopropoxyphosphono)methoxy]propanoic acid (9i)
Colourless oil, yield 78%; 1H NMR (DMSO-d 6) δ: 10.59 (br s, 1H, NH), 8.01 (s, 1H, H-8), 7.95 (m, 2H, H-2″), 7.58 (m, 1H, H-4″), 7.49 (m, 2H, H-3″), 7.43 (br s, 2H, NH2), 4.35–4.54 (m, 5H, H-2′, CHipr, H-1′), 3.97 (dd, Jgem=13.7 Hz, J H–C–P=8.9 Hz, 1H, CH2Pa), 3.77 (dd, Jgem=13.7 Hz, J H–C–P=9.7 Hz, 1H, CH2Pb), 1.09–1.20 (m, 12H, CH3ipr); 13C NMR (DMSO-d 6) δ: 170.59 (C-3′), 165.88 (CON), 152.42 (C-2 or C-6), 150.61 (C-4), 141.37 (C-8), 134.65 (C-1″), 131.99 (C-4″), 128.50 (C-3″), 128.18 (C-2″), 116.23 (C-5), 77.98 (d, J 2′-P=13.5 Hz, C-2′), 70.54–70.63 (m, CHipr), 64.16 (d, J C–P=164.4 Hz, CH2P), 44.25 (C-1′), 23.68–23.95 (m, CH3ipr); MS-ESI+ m/z (%) 522 (75, M+H+), 544 (100, M+Na+); HRMS-ESI+: m/z calcd for C22H29O8N5P (M+H+): 522.1748, found: 522.1747; FTIR (KBr, cm−1) ν: 3318, 3143, 2981, 2500, 1674, 1602, 1529, 1453, 1443, 1387, 1255, 955. [α]D 20 −40.7 (c 0.194, MeOH).
4.6.8. (S)-3-(N2,N6-Dibenzoyl-2,6-diamino-9H-purin-9-yl)-2-[(diisopropoxyphosphono)methoxy]propanoic acid (9j)
Colourless oil, yield 80%; MS-ESI+ m/z (%) 625 (7, M+H+), 647 (15, M+Na+) 669 (100, M+2Na+); HRMS-ESI+: m/z calcd for C29H34O8N6P (M+H+): 625.2170, found: 625.2171.
4.7. (S)-3-(2-Amino-6-oxo-1H-purin-9(6H)-yl)-2-((diisopropoxyphosphoryl)methoxy)propanoic acid (9g)
To a solution of compound 9i (1 mmol, 521 mg) in MeOH (10 mL) was added 1.0 M MeONa (2 mmol, 2 mL). After stirring at room temperature for 24 h, the reaction mixture was neutralized with glacial AcOH. The solvents were evaporated and the residue was codistilled with toluene (2×10 mL) and ethanol (2×10 mL). Chromatography on silica gel column (gradient 5–20% MeOH in chloroform) followed by crystallization from EtOAc afforded 296 mg (71%) of compound 9g as a yellowish oil: 1H NMR (DMSO-d 6) δ: 12.32 (br s, 1H, H-1), 7.56 (s, 1H, H-8), 7.22 (br s, 2H, NH2), 4.41–4.53 (m, 2H, CHipr), 4.33 (dd, Jgem=13.5 Hz, J 1′a–2′=1.9 Hz, 1H, H-1′a), 4.10 (dd, Jgem=13.7 Hz, J H–C–P=8.1 Hz, 1H, CH2Pa), 3.88–3.97 (m, 2H, H-1′b, H-2′), 3.56 (dd, Jgem=13.7 Hz, J H–C–P=9.0 Hz, 1H, CH2Pb), 1.19 (d, J CH3–CH=6.2 Hz, 3H, CH3ipr), 1.15 (d, J CH3–CH=6.2 Hz, 3H, CH3ipr), 1.14 (d, J CH3–CH=6.2 Hz, 3H, CH3ipr), 1.11 (d, J CH3–CH=6.2 Hz, 3H, CH3ipr); 13C NMR (DMSO-d 6) δ: 171.53 (C-3′), 157.95 (C-6), 154.61 (C-2), 151.56 (C-4), 137.78 (C-8), 116.26 (C-5), 81.36 (d, J 2′-P=13.0 Hz, C-2′), 70.24–70.38 (m, CHipr), 63.28 (d, J C–P=163.7 Hz, CH2P), 45.54 (C-1′), 23.74–23.98 (m, CH3ipr); MS-ESI− m/z (%) 416 (100, M−H+), 438 (10, M+Na+); HRMS-ESI−: m/z calcd for C15H23O7N5P (M−H+) 416.1340, found 416.1339; FTIR (KBr, cm−1) ν: 3423, 3163, 1686, 1630, 1579, 1485, 1410, 1242, 1178, 1105, 997. [α]D 20 −35.6 (c 0.407, MeOH).
4.8. Deprotection of diisopropyl esters 9 to free phosphonic acids 7
4.8.1. Method A.28General procedure
Diisopropyl esters 9 (0.5 mmol) in CH3CN (10 mL) were treated with Me3SiBr (2 mL) at room temperature overnight. Volatiles were evaporated, and the reaction mixture codistilled with dry acetonitrile (3×10 mL) and water (1×10 mL). Crystallization from MeOH gave the products 7 (50–68%) as white or yellowish crystals.
4.8.2. Method B.29General procedure
A solution of diisopropyl esters 9 (0.5 mmol) in 0.5 M aqueous HCl (2 mL) was heated in a microwave at 140 °C for 30 min. The volatiles were evaporated, the residue was dissolved in H2O (0.5–1.0 mL) and the product was isolated by HPLC (gradient: 2% aqueous MeOH to 80% aqueous MeOH). Crystallization from a water/ethanol mixture afforded the products 7 (37–46%) as white or yellowish crystals.
4.8.3. (S)-3-(6-Amino-9H-purin-9-yl)-2-(phosphonomethoxy)propanoic acid (7a)
Method A: white solid (68%), dec >250 °C; method B: white crystals (46%), dec >250 °C; 1H NMR (D2O) δ: 8.17 (s, 1H, H-2), 8.16 (s, 1H, H-8), 4.55 (dd, Jgem=14.8 Hz, J 1′a–2′=4.0 Hz, 1H, H-1′a), 4.48 (dd, Jgem=14.8 Hz, J 1′b–2′=6.0 Hz, 1H, H-1′b), 4.20 (dd, J 2′–1′b=6.0 Hz, J 2′–1′a=4.0 Hz, 1H, H-2′), 3.71 (dd, Jgem=12.8 Hz, J H–C–P=8.6 Hz, 1H, CH2Pa), 3.45 (dd, Jgem=12.8 Hz, J H–C–P=9.8 Hz, 1H, CH2Pb); 13C NMR (D2O) δ: 177.20 (C-3′), 155.86 (C-6), 149.62 (C-4), 143.74 (C-8), 118.57 (C-5), 81.75 (d, J 2′–P=12.4 Hz, C-2′), 67.33 (d, J C–P=155.0 Hz, CH2P), 46.12 (C-1′); MS-ESI+ m/z (%) 318 (100, M+H+), 340 (83, M+Na+), 362 (65, M+2Na+); HRMS-ESI+: m/z calcd for C9H13O6N5P (M+H+) 318.0598, found 318.0598; FTIR (KBr, cm−1) ν: 3178, 3033, 2838, 1647, 1598, 1474, 1417, 1056. Anal. Calcd for C9H12N5O6P 0.75H2O: C, 32.69; H, 4.11; N, 21.18; P, 9.37. Found: C, 32.86; H, 3.99; N, 20.95; P, 9.61. [α]D 20 −6.1 (c 0.379, H2O).
4.8.4. (R)-3-(6-Amino-9H-purin-9-yl)-2-(phosphonomethoxy)propanoic acid (7b)
Method A: white crystals (50%), dec >250 °C; 1H NMR (D2O) δ: 8.17 (s, 1H, H-2), 8.16 (s, 1H, H-8), 4.55 (dd, Jgem=14.8 Hz, J 1′a–2′=4.0 Hz, 1H, H-1′a), 4.48 (dd, Jgem=14.8 Hz, J 1′b–2′=6.0 Hz, 1H, H-1′b), 4.20 (dd, J 2′–1′b=6.0 Hz, J 2′–1′a=4.0 Hz, 1H, H-2′), 3.71 (dd, Jgem=12.8 Hz, J H–C–P=8.6 Hz, 1H, CH2Pa), 3.45 (dd, Jgem=12.8 Hz, J H–C–P=9.8 Hz, 1H, CH2Pb); 13C NMR (D2O) δ: 177.20 (C-3′), 155.86 (C-6), 149.62 (C-4), 143.74 (C-8), 118.57 (C-5), 81.75 (d, J 2′-P=12.4 Hz, C-2′), 67.33 (d, J C–P=155.0 Hz, CH2P), 46.12 (C-1′); MS-ESI+ m/z (%) 318 (100, M+H+), 340 (83, M+Na+), 362 (65, M+2Na+); HRMS-ESI+: m/z calcd for C9H13O6N5P (M+H+) 318.0598, found 318.0598; FTIR (KBr, cm−1) ν: 3178, 3033, 2838, 1647, 1598, 1474, 1417, 1056. Anal. Calcd for C9H12N5O6P·0.75H2O: C, 32.69; H, 4.11; N, 21.18; P, 9.37. Found: C, 32.86; H, 3.99; N, 20.97; P, 9.63. [α]D 20 +6.2 (c 0.324, H2O).
4.8.5. (S)-3-(4-Amino-2-oxopyrimidin-1(2H)-yl)-2-(phosphonomethoxy)propanoic acid (7c)
Method A: yellowish crystals (62%), dec >250 °C; method B: yellowish crystals (39%), dec >250 °C; 1H NMR (D2O) δ: 7.64 (d, J 6–5=7.4 Hz, 1H, H-6), 5.98 (d, J 5–6=7.3 Hz, 1H, H-5), 4.21 (dd, Jgem=14.0 Hz, J 1′a–2′=3.9 Hz, 1H, H-1′a), 4.05 (dd, J 2′–1′b=7.7 Hz, J 2′–1′a=3.9 Hz, 1H, H-2′), 3.89 (dd, Jgem=14.0 Hz, J 1′b–2′=7.7 Hz, 1H, H-1′b), 3.71 (dd, Jgem=12.9 Hz, J H–C–P=8.7 Hz, 1H, CH2Pa), 3.42 (dd, Jgem=12.8 Hz, J H–C–P=10.2 Hz, 1H, CH2Pb); 13C NMR (D2O) δ: 177.57 (C-3′), 166.01 (C-4), 157.42 (C-2), 148.94 (C-6), 95.69 (C-5), 81.62 (d, J 2′-P=13.1 Hz, C-2′), 67.07 (d, J C–P=156.4 Hz, CH2P), 51.87 (C-1′); MS-ESI− m/z (%) 292 (100, M−H+), 314 (50, M+Na+), 336 (25, M+2Na+); HRMS-ESI−: m/z calcd for C8H11O7N3P (M−H+) 292.0340, found 292.0340; FTIR (KBr, cm−1) ν: 3385, 3171, 2795, 1650, 1613, 1494, 1400, 1051. [α]D 20 −7.0 (c 0.339, H2O).
4.8.6. (S)-3-(5-Methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-(phosphonomethoxy)propanoic acid (7d)
Method A: white crystals (64%), dec >250 °C; method B: white crystals (44%), dec >250 °C; 1H NMR (D2O) δ: 7.50 (q, J 6-CH3=1.2 Hz, 1H, H-6), 4.10 (dd, Jgem=13.9 Hz, J 1′a–2′=4.1 Hz, 1H, H-1′a), 4.05 (dd, J 2′–1′=6.6 and 4.1 Hz, 1H, H-2′), 3.96 (dd, Jgem=13.9 Hz, J 1′b–2′=6.6 Hz, 1H, H-1′b), 3.69 (dd, Jgem=12.8 Hz, J H–C–P=8.7 Hz, 1H, CH2Pa), 3.42 (dd, Jgem=12.8 Hz, J H–C–P=9.9 Hz, 1H, CH2Pb), 1.87 (d, J CH3-6=1.2 Hz, 3H, 5-CH3); 13C NMR (D2O) δ: 177.59 (COOH), 167.78 (C-4), 152.84 (C-2), 144.62 (C-6), 110.71 (C-5), 81.61 (d, J 2′-P=12.6 Hz, C-2′), 67.24 (d, J C–P=157.1 Hz, CH2P), 50.34 (C-1′), 11.93 (5-CH3); MS-ESI− m/z (%) 307 (100, M−H+), 329 (60, M+Na+), 351 (15, M+2Na+); HRMS-ESI−: m/z calcd for C9H12O8N2P (M−H+) 307.0326, found 307.0327; FTIR (KBr, cm−1) ν: 3191, 3036, 2833, 1691, 1601, 1475, 1432, 1359, 1110, 1062. [α]D 20 −2.7 (c 0.401, H2O).
4.8.7. (S)-3-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-(phosphonomethoxy)propanoic acid (7e)
Method B: white crystals (42%), dec >250 °C; 1H NMR (D2O+NaOD) δ: 7.59 (d, J 6–5=7.5 Hz, 1H, H-6), 5.74 (d, J 5–6=7.5 Hz, 1H, H-5), 3.97–4.05 (m, 3H, H-1′, 2′), 3.53 (dd, Jgem=11.8 Hz, J H–C–P=9.1 Hz, 1H, CH2P), 3.30 (dd, Jgem=11.8 Hz, J H–C–P=10.2 Hz, 1H, CH2P); 13C NMR (D2O+NaOD) δ: 178.47 (C-3′), 174.43 (C-4), 158.01 (C-2), 147.85 (C-6), 102.05 (C-5), 81.99 (d, J 2′-P=13.6 Hz, C-2′), 68.91 (d, J C–P=150.7 Hz, CH2P), 51.03 (C-1′); MS-ESI− m/z (%) 293 (100, M−H+); HRMS-ESI−: m/z calcd for C8H10O8N2P (M−H+): 293.0180, found: 293.0180; FTIR (KBr, cm−1) ν: 3428, 3168, 2976, 1692, 1617, 1402, 1356, 1070, 1050. [α]365 20 −50.0 (c 0.346, H2O).
4.8.8. (R)-3-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-(phosphonomethoxy)propanoic acid (7f)
Method B: white crystals (48%), dec >250 °C; 1H NMR (D2O+NaOD), 13C NMR (D2O+NaOD), MS and FTIR spectra are identical with compound 7e. [α]365 20 +46.0 (c 0.336, H2O).
4.8.9. (S)-3-(2-Amino-6-oxo-1H-purin-9(6H)-yl)-2-(phosphonomethoxy)propanoic acid (7g)
Method A: yellowish crystals (61%), dec >250 °C; method B: white solid (37%), dec >250 °C; 1H NMR (D2O) δ: 7.84 (s, 1H, H-8), 4.40 (dd, Jgem=14.7 Hz, J 1′a–2′=4.1 Hz, 1H, H-1′a), 4.32 (dd, Jgem=14.7 Hz, J 1′b–2′=6.1 Hz, 1H, H-1′b), 4.17 (dd, J 2′–1′=6.1 and 4.1 Hz, 1H, H-2′), 3.73 (dd, Jgem=13.0 Hz, J H–C–P=8.7 Hz, 1H, CH2Pa), 3.51 (dd, Jgem=13.0 Hz, J H–C–P=9.8 Hz, 1H, CH2Pb); 13C NMR (D2O) δ: 177.21 (C-3′), 159.57 (C-6), 154.24 (C-2), 152.37 (C-4), 141.38 (C-8), 116.08 (C-5), 81.73 (d, J 2′-P=12.5 Hz, C-2′), 66.85 (d, J C–P=156.1 Hz, CH2P), 45.82 (C-1′); MS-ESI− m/z (%) 332 (100, M−H+), 354 (50, M+Na+), 376 (15, M+2Na+); HRMS-ESI−: m/z calcd for C9H11O7N5P (M−H+): 332.0402, found: 332.0404; FTIR (KBr, cm−1) ν: 3405, 3128, 1693, 1607, 1540, 1481, 1409, 1108, 1069, 914. [α]D 20 −20.2 (c 0.316, H2O).
4.8.10. (S)-3-(2,6-Diamino-9H-purin-9-yl)-2-(phosphonomethoxy)propanoic acid (7h)
Method B (from 9j): white crystals (38%), dec >250 °C; 1H NMR (D2O+NaOD) δ: 7.94 (s, 1H, H-8), 4.33–4.39 (m, 2H, H-1′), 4.13 (t, J 2′–1′=5.1 Hz, 1H, H-2′), 3.58 (dd, Jgem=12.1 Hz, J H–C–P=8.9 Hz, 1H, CH2P), 3.32 (dd, Jgem=12.1 Hz, J H–C–P=9.8 Hz, 1H, CH2P); 13C NMR (D2O+NaOD) δ: 178.02 (C-3′), 160.60 (C-2), 456.66 (C-6), 151.92 (C-4), 141.68 (C-8), 113.20 (C-5), 81.90 (d, J 2′-P=12.7 Hz, C-2′), 68.82 (d, J C–P=150.5 Hz, CH2P), 45.75 (C-1′); MS-ESI+ m/z (%) 333 (100, M+H+), 355 (25, M+Na+), 377 (15, M+2Na+); HRMS-ESI+: m/z calcd for C9H14O6N6P (M+H+) 333.0707, found 333.0707; FTIR (KBr, cm−1) ν: 3194, 2470, 1605, 1560, 1551, 1482, 1407, 1119, 1087, 916. [α]D 20 −17.4 (c 0.259, H2O).
4.8.11. (S)-3-(2-Benzamido-6-oxo-1H-purin-9(6H)-yl)-2-(phosphonomethoxy)propanoic acid (7i)
Method A: white crystals (52%), dec >250 °C; 1H NMR (D2O) δ: 8.11 (s, 1H, H-8), 7.97 (m, 2H, H-2″), 7.62 (m, 1H, H-4″), 7.53 (m, 2H, H-3″), 4.45–4.52 (m, 2H, H-1′), 4.16 (m, 1H, H-2′), 3.59 (m, 1H, CH2P), 3.40 (m, 1H, CH2P); 13C NMR (D2O) δ: 177.97 (C-3′), 172.77 (CON), 162.48 (C-6), 153.12 (C-2), 151.42 (C-4), 142.76 (C-8), 135.04 (C-1″), 133.13 (C-4′), 129.28 (C-3′), 128.54 (C-2′), 119.73 (C-5), 81.83 (d, J 2′-P=12.9 Hz, C-2′), 68.76 (d, J C–P=150.8 Hz, CH2P), 45.76 (C-1′); MS-ESI+ m/z (%) 438 (100, M+H+), 460 (78, M+Na+); HRMS-ESI−: m/z calcd for C16H15O8N5P (M−H+) 436.0664, found 436.0664; FTIR (KBr, cm−1) ν: 3408, 3135, 3033, 1665, 1604, 1402, 1269, 1154, 1061. [α]D 20 −31.9 (c 0.244, H2O).
Acknowledgements
This study was performed as a part of research project RVO: 61388963 of the Institute of Organic Chemistry and Biochemistry, v.v.i. This study was supported by Ministry of the Interior of the Czech Republic (VG20102015046), and by Gilead Sciences and IOCB Research Centre.
Footnotes
Supplementary data associated with this article can be found in the online version, at doi:10.1016/j.tet.2012.03.066. These data include MOL files and InChiKeys of the most important compounds described in this article.
Supplementary data
The following ZIP file contains the MOL files of the most important compounds referred to in this article.
ZIP file containing the MOL files of the most important compounds in this article.
References and notes
- 1.(a) De Clercq E., Holý A., Rosenberg I., Sakuma T., Balzarini J., Maudgal P.C. Nature. 1986;323:464. doi: 10.1038/323464a0. [DOI] [PubMed] [Google Scholar]; (b) De Clercq E. J. Med. Microbiol. 1998;47:1. doi: 10.1099/00222615-47-1-1. [DOI] [PubMed] [Google Scholar]; (c) Holý A. Curr. Pharm. Des. 2003;9:2567. doi: 10.2174/1381612033453668. [DOI] [PubMed] [Google Scholar]; (d) De Clercq E., Holý A. Nat. Rev. Drug Discov. 2005;4:928. doi: 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]; (e) Holý A. Antiviral Res. 2006;71:248. doi: 10.1016/j.antiviral.2006.06.002. [DOI] [PubMed] [Google Scholar]; (f) De Clercq E. Antiviral Res. 2007;75:1. doi: 10.1016/j.antiviral.2006.10.006. [DOI] [PubMed] [Google Scholar]; (g) De Clercq E. Biochem. Pharmacol. 2007;73:911. doi: 10.1016/j.bcp.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 2.(a) Holý A., Votruba I., Tloušťová E., Masojídková M. Collect. Czech. Chem. Commun. 2001;66:1545. [Google Scholar]; (b) Zídek Z., Potměšil P., Holý A. Toxicol. Appl. Pharmacol. 2003;192:246. doi: 10.1016/s0041-008x(03)00215-1. [DOI] [PubMed] [Google Scholar]; (c) Reiser H., Wang J., Chong L., Watkins W.J., Ray A.S., Shibata R., Birkus G., Cihlar T., Wu S., Li B., Liu X., Henne I.N., Wolfgang G.H.I., Desai M., Rhodes G.R., Fridland A., Lee W.A., Plunkett W., Vail D., Thamm D.H., Jeraj R., Tumas D.B. Clin. Cancer. Res. 2008;14:2824. doi: 10.1158/1078-0432.CCR-07-2061. [DOI] [PubMed] [Google Scholar]
- 3.(a) Keough D.T., Hocková D., Holý A., Naesens L.M.J., Skinner-Adams T.S., de Jersey J., Guddat L.W. J. Med. Chem. 2009;52:4391. doi: 10.1021/jm900267n. [DOI] [PubMed] [Google Scholar]; (b) Hocková D., Holý A., Masojídková M., Keough D.T., de Jersey J., Guddat L.W. Bioorg. Med. Chem. 2009;17:6218. doi: 10.1016/j.bmc.2009.07.044. [DOI] [PubMed] [Google Scholar]
- 4.(a) Zídek Z., Franková D., Holý A. Int. J. Immunopharmacol. 2000;22:1121. doi: 10.1016/s0192-0561(00)00068-0. [DOI] [PubMed] [Google Scholar]; (b) Zídek Z., Potměšil P., Kmoníčková E., Holý A. Eur. J. Pharmacol. 2003;475:149. doi: 10.1016/j.ejphar.2005.11.037. [DOI] [PubMed] [Google Scholar]; (c) Potměšil P., Krečmerová M., Kmoníčková E., Holý A., Zídek Z. Eur. J. Pharmacol. 2006;540:191. doi: 10.1016/j.ejphar.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 5.(a) De Clercq E. Intervirology. 1997;40:295. doi: 10.1159/000150563. [DOI] [PubMed] [Google Scholar]; (b) De Clercq E. Clin. Microbiol. Rev. 2003;16:569. doi: 10.1128/CMR.16.4.569-596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) De Clercq E. Expert Rev. Anti-infect. Ther. 2003;1:21. doi: 10.1586/14787210.1.1.21. [DOI] [PubMed] [Google Scholar]
- 6.(a) De Clercq E., Holý A. Antimicrob. Agents Chemother. 1991;35:701. doi: 10.1128/aac.35.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Neyts J., Snoeck R., Balzarini J., De Clercq E. Antiviral Res. 1991;16:41. doi: 10.1016/0166-3542(91)90057-x. [DOI] [PubMed] [Google Scholar]; (c) De Clercq E. Rev. Med. Virol. 1993;3:85. [Google Scholar]; (d) Hitchcock M.J.M., Jaffe H.S., Martin J.C., Stagg R.J. Antiviral Chem. Chemother. 1996;7:115. [Google Scholar]
- 7.(a) Holý A., Votruba I., Masojídková M., Andrei G., Snoeck R., Naesens L., De Clercq E., Balzarini J. J. Med. Chem. 2002;45:1918. doi: 10.1021/jm011095y. [DOI] [PubMed] [Google Scholar]; (b) Balzarini J., Pannecouque C., De Clercq E., Aquaro S., Perno C.-F., Egberink H., Holý A. Antimicrob. Agents Chemother. 2002;46:2185. doi: 10.1128/AAC.46.7.2185-2193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hocková D., Holý A., Masojídková M., Andrei G., Snoeck R., De Clercq E., Balzarini J. J. Med. Chem. 2003;46:5064. doi: 10.1021/jm030932o. [DOI] [PubMed] [Google Scholar]; (d) Hocková D., Holý A., Masojídková M., Andrei G., Snoeck R., De Clercq E., Balzarini J. Bioorg. Med. Chem. 2004;12:3197. doi: 10.1016/j.bmc.2004.04.002. [DOI] [PubMed] [Google Scholar]; (e) Ying C., Holý A., Hocková D., Havlas Z., De Clercq E., Neyts J. Antimicrob. Agents Chemother. 2005;49:1177. doi: 10.1128/AAC.49.3.1177-1180.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagner C.R., Iyer V.V., McIntee E.J. Med. Res. Rev. 2000;20:417. doi: 10.1002/1098-1128(200011)20:6<417::aid-med1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 9.Hecker S.J., Erion M.D. J. Med. Chem. 2008;51:2328. doi: 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]
- 10.(a) Holý A., Rosenberg I. Collect. Czech. Chem. Commun. 1987;52:2775. [Google Scholar]; (b) Rosenberg I., Holý A. Collect. Czech. Chem. Commun. 1987;52:2792. [Google Scholar]; (c) Krečmerová M., Masojídková M., Holý A. Collect. Czech. Chem. Commun. 2004;69:1889. [Google Scholar]; (d) Janeba Z., Masojídková M., Holý A. Collect. Czech. Chem. Commun. 2010;75:371. [Google Scholar]
- 11.(a) Jindřich J., Holý A., Dvořáková H. Collect. Czech. Chem. Commun. 1993;58:1645. [Google Scholar]; (b) Balzarini J., Holý A., Jindřich J., Dvořáková H., Hao Z., Snoeck R., Herdewijn P., John D.G., De Clercq E. Proc. Natl. Acad. Sci. U.S.A. 1991;88:4961. doi: 10.1073/pnas.88.11.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Balzarini J., Holý A., Jindřich J., Naesens L., Snoeck R., Schols D., De Clercq E. Antimicrob. Agents Chemother. 1993;37:332. doi: 10.1128/aac.37.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) De Clercq E., Descamps J., De Somer P., Holý A. Science. 1978;200:563. [PubMed] [Google Scholar]; (b) De Clercq E., Holý A. J. Med. Chem. 1979;22:510. doi: 10.1021/jm00191a010. [DOI] [PubMed] [Google Scholar]
- 13.Holý A., Votruba I., De Clercq E. Collect. Czech. Chem. Commun. 1982;47:1392. [Google Scholar]
- 14.(a) Holý A., Votruba I., De Clercq E. Collect. Czech. Chem. Commun. 1985;50:262. [Google Scholar]; (b) De Clercq E., Holý A. J. Med. Chem. 1985;28:282. doi: 10.1021/jm00381a004. [DOI] [PubMed] [Google Scholar]
- 15.De Clercq E. Nucleosides Nucleotides Nucleic Acids. 2005;24:1395. doi: 10.1080/15257770500265638. [DOI] [PubMed] [Google Scholar]
- 16.Votruba I., Hasobe M., Holý A., Borchardt R.T. Biochem. Pharmacol. 1990;39:1573. doi: 10.1016/0006-2952(90)90523-n. [DOI] [PubMed] [Google Scholar]
- 17.Tuske S., Sarafianos S.G., Clark A.D., Jr., Ding J., Naeger L.K., White K.L., Miller M.D., Gibbs C.S., Boyer P.L., Clark P., Wang G., Gaffney B.L., Jones R.A., Jerina D.M., Hughes S.H., Arnold E. Nat. Struct. Mol. Biol. 2004;11:469. doi: 10.1038/nsmb760. [DOI] [PubMed] [Google Scholar]
- 18.(a) Webb R.R., Martin J.C. Tetrahedron Lett. 1987;28:4963. [Google Scholar]; (b) Holý A. Collect. Czech. Chem. Commun. 1993;58:649. [Google Scholar]
- 19.Bäckvall J.-E., editor. Modern Oxidation Methods. WILEY-VCH; Weinheim: 2004. [Google Scholar]
- 20.(a) Hutchison A.J., Williams M., De Jesus R., Yokoyama R., Oei H.H., Ghai G.R., Webb R.L., Zoganas H.C., Stone G.A., Jarvis M.F. J. Med. Chem. 1990;33:1919. doi: 10.1021/jm00169a015. [DOI] [PubMed] [Google Scholar]; (b) Ha S.B., Nair V. Tetrahedron Lett. 1996;37:1567. [Google Scholar]
- 21.Mori N., Togo H. Tetrahedron. 2005;61:5915. [Google Scholar]
- 22.(a) Singh A.K., Varma R.S. Tetrahedron Lett. 1992;33:2307. [Google Scholar]; (b) Varma R.S., Hogan M.E. Tetrahedron Lett. 1992;33:7719. [Google Scholar]
- 23.Homma H., Watanabe Y., Abiru T., Murayama T., Nomura Y., Matsuda A. J. Med. Chem. 1992;35:2881. doi: 10.1021/jm00093a022. [DOI] [PubMed] [Google Scholar]
- 24.(a) Vogler T., Studer A. Synthesis. 2008:1979. [Google Scholar]; (b) De Souza M.V.N. Mini-Rev. Org. Chem. 2006;3:155. [Google Scholar]; (c) Nooy A.E.J., Besemer A.C., Van Bekkum H. Synthesis. 1996:1153. [Google Scholar]
- 25.De Mico A., Margarita R., Parlanti L., Vescovi A., Piancatelli G. J. Org. Chem. 1997;62:6974. [Google Scholar]
- 26.Epp J.B., Widlanski T.S. J. Org. Chem. 1999;64:293. doi: 10.1021/jo981316g. [DOI] [PubMed] [Google Scholar]
- 27.Zhao M., Li J., Mano E., Song Z., Tschaen D.M., Grabowski E.J.J., Reider P.J. J. Org. Chem. 1999;64:2564. [Google Scholar]
- 28.Holý A., Rosenberg I. Collect. Czech. Chem. Commun. 1982;47:3447. [Google Scholar]
- 29.(a) Jansa P., Holý A., Dračínský M., Baszczyňski O., Česnek M., Janeba Z. Collect. Symp. Ser. 2011;12:347. [Google Scholar]; (b) Jansa, P.; et al., Green. Chem., in press.
- 30.ArgusLab 4.0, M. A. Thompson, Planaria Software LLC, Seattle, http://www.ArgusLab.com.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ZIP file containing the MOL files of the most important compounds in this article.