

Abstract
The SARS-coronavirus (SARS-CoV) is the etiological agent of severe acute respiratory syndrome (SARS). The SARS-CoV spike (S) glycoprotein mediates membrane fusion events during virus entry and virus-induced cell-to-cell fusion. To delineate functional domains of the SARS-CoV S glycoprotein, single point mutations, cluster-to-lysine and cluster-to-alanine mutations, as well as carboxyl-terminal truncations were investigated in transient expression experiments. Mutagenesis of either the coiled-coil domain of the S glycoprotein amino terminal heptad repeat, the predicted fusion peptide, or an adjacent but distinct region, severely compromised S-mediated cell-to-cell fusion, while intracellular transport and cell-surface expression were not adversely affected. Surprisingly, a carboxyl-terminal truncation of 17 amino acids substantially increased S glycoprotein-mediated cell-to-cell fusion suggesting that the terminal 17 amino acids regulated the S fusogenic properties. In contrast, truncation of 26 or 39 amino acids eliminating either one or both of the two endodomain cysteine-rich motifs, respectively, inhibited cell fusion in comparison to the wild-type S. The 17 and 26 amino-acid deletions did not adversely affect S cell-surface expression, while the 39 amino-acid truncation inhibited S cell-surface expression suggesting that the membrane proximal cysteine-rich motif plays an essential role in S cell-surface expression. Mutagenesis of the acidic amino-acid cluster in the carboxyl terminus of the S glycoprotein as well as modification of a predicted phosphorylation site within the acidic cluster revealed that this amino-acid motif may play a functional role in the retention of S at cell surfaces. This genetic analysis reveals that the SARS-CoV S glycoprotein contains extracellular domains that regulate cell fusion as well as distinct endodomains that function in intracellular transport, cell-surface expression, and cell fusion.
Keywords: SARS, Coronavirus, Spike, Heptad repeat, Fusion
Introduction
An outbreak of atypical pneumonia, termed severe acute respiratory syndrome (SARS), appeared in the Guangdong Province of southern China in November, 2002. The mortality rates of the disease reached as high as 15% in some age groups (Anand et al., 2003). The etiological agent of the disease was found to be a novel coronavirus (SARS-CoV), which was first isolated from infected individuals by propagation of the virus on Vero E6 cells (Drosten et al., 2003, Ksiazek et al., 2003, Peiris et al., 2003). Analysis of the viral genome has demonstrated that the SARS-CoV is phylogenetically divergent from the three known antigenic groups of coronaviruses (Drosten et al., 2003, Ksiazek et al., 2003). Analysis of the polymerase gene alone, however, has indicated that the SARS-CoV may be an early off-shoot from the group 2 coronaviruses (Snijder et al., 2003).
The coronaviruses are the largest of the enveloped RNA viruses with a positive-stranded RNA genome of 28 to 32 kb (Holmes, 2003). Coronaviruses possess a wide host range, capable of infecting mammalian and avian species. All identified coronaviruses have a common group of indispensable genes that encode nonstructural proteins including the RNA replicase gene open reading frame (ORF) 1ab and the structural proteins nucleocapsid (N), membrane protein (M), envelope protein (E), and spike glycoprotein (S), which are assembled into virus particles. A hemagglutinin-esterase (HE) protein is also encoded by some coronaviruses. Distributed among the major viral genes are a series of ORFs that are specific to the different coronavirus groups. Functions of the majority of these ORFs have not been determined.
The SARS spike glycoprotein, a 1255-amino-acid type I membrane glycoprotein (Rota et al., 2003), is the major protein present in the viral membrane forming the typical spike structure found on all coronavirions. The S glycoprotein is primarily responsible for entry of all coronaviruses into susceptible cells through binding to specific receptors on cells and mediating subsequent virus-cell fusion (Cavanagh, 1995). The S glycoprotein specified by mouse hepatitis virus (MHV) is cleaved into S1 and S2 subunits, although cleavage is not necessarily required for virus-cell fusion (Bos et al., 1997, Gombold et al., 1993, Stauber et al., 1993). Similarly, the SARS-CoV S glycoprotein seems to be cleaved into S1 and S2 subunits in Vero-E6-infected cells (Wu et al., 2004), while it is not known whether this cleavage affects S-mediated cell fusion. The SARS-CoV receptor has been recently identified as the angiotensin-converting enzyme 2 (ACE2) (Li et al., 2003). Although the exact mechanism by which the SARS-CoV enters the host cell has not been elucidated, it is most likely similar to other coronaviruses. Upon receptor binding at the cell membrane, the S glycoprotein is thought to undergo a dramatic conformational change causing exposure of a hydrophobic fusion peptide, which is subsequently inserted into cellular membranes. This conformational change of the S glycoprotein causes close apposition followed by fusion of the viral and cellular membranes resulting in entry of the virion nucleocapsids into cells (Eckert and Kim, 2001, Tsai et al., 2003, Zelus et al., 2003). This series of S-mediated virus entry events is similar to other class I virus fusion proteins (Baker et al., 1999, Melikyan et al., 2000, Russell et al., 2001).
Heptad repeat (HR) regions, a sequence motif characteristic of coiled-coils, appear to be a common motif in many viral and cellular fusion proteins (Skehel and Wiley, 1998). These coiled-coil regions allow the protein to fold back upon itself as a prerequisite step to initiating the membrane fusion event. There are usually two HR regions: an N terminal HR region adjacent to the fusion peptide and a C-terminal HR region close to the transmembrane region of the protein. Within the HR segments, the first amino acid (a) and fourth amino acid (d) are typically hydrophobic amino acids that play a vital role in maintaining coiled-coil interactions. Based on structural similarities, two classes of viral fusion proteins have been established. Class I viral fusion proteins contain two heptad repeat regions and an N-terminal or N-proximal fusion peptide. Class II viral fusion proteins lack heptad repeat regions and contain an internal fusion peptide (Lescar et al., 2001). The MHV S glycoprotein, which is similar to other coronavirus S glycoproteins, is a class I membrane protein that is transported to the plasma membrane after being synthesized in the endoplasmic reticulum (Bosch et al., 2003). Typically, the ectodomains of the S2 subunits of coronaviruses contain two regions with a 4, 3 hydrophobic (heptad) repeat the first being adjacent to the fusion peptide and the other being in close proximity to the transmembrane region (de Groot et al., 1987).
In the present study, we investigated the role of several predicted structural and functional domains of the SARS spike glycoprotein by introducing specific alterations within selected S glycoprotein regions. The results show that the SARS-CoV S glycoprotein conforms to the general structure and function relationships that have been elucidated for other coronaviruses, most notably the MHV (Chang et al., 2000, Ye et al., 2004). However, in contrast to the MHV endodomain, the carboxyl terminus of the SARS-CoV S glycoprotein contains multiple non-overlapping domains that function in intracellular transport, cell-surface expression, and endocytosis as well as in S glycoprotein-mediated cell-to-cell fusion.
Results
Genetic analysis of S glycoprotein functional domains
To delineate domains of the S glycoprotein that function in membrane fusion, intracellular transport, and cell-surface expression, two types of mutations were introduced within the S gene: (a) mutations were introduced within and adjacent to the predicted amino terminal heptad repeat (HR1) core and the predicted fusion peptide, which are known to play important roles in membrane fusion (Bosch et al., 2004, Bosch et al., 2003, Ingallinella et al., 2004, Tripet et al., 2004); (b) mutations and carboxyl-terminal truncations of the S glycoprotein were engineered to delineate S cytoplasmic domains that function in glycoprotein synthesis, intracellular transport, and membrane fusion (Fig. 1 ). Specifically, to investigate the amino-acid requirements of the HR1 of the SARS-CoV S glycoprotein, the a and d amino-acid positions L(898) and N(901) were both replaced by lysine residues in the cluster mutation CL2, effectively collapsing the predicted α-helical structure at the amino terminal terminus of the HR. This amino-acid sequence is thought to align with the L(1184) of HR2 in the formation of the HR1/HR2 core complex (Xu et al., 2004). In addition, cluster-to-lysine mutations CL3 and CL4 replaced the a and d positions within the HR1 region (Fig. 1B). The CL5 cluster mutation was placed adjacent to the HR1 region to investigate whether regions proximal to HR1 had any effect on S-mediated cell fusion. Similarly, the role of the a and d positions within the predicted fusion peptide, located immediately proximal to the N terminus of HR1, was investigated by constructing the CL1 cluster mutation (Fig. 1B).
Fig. 1.
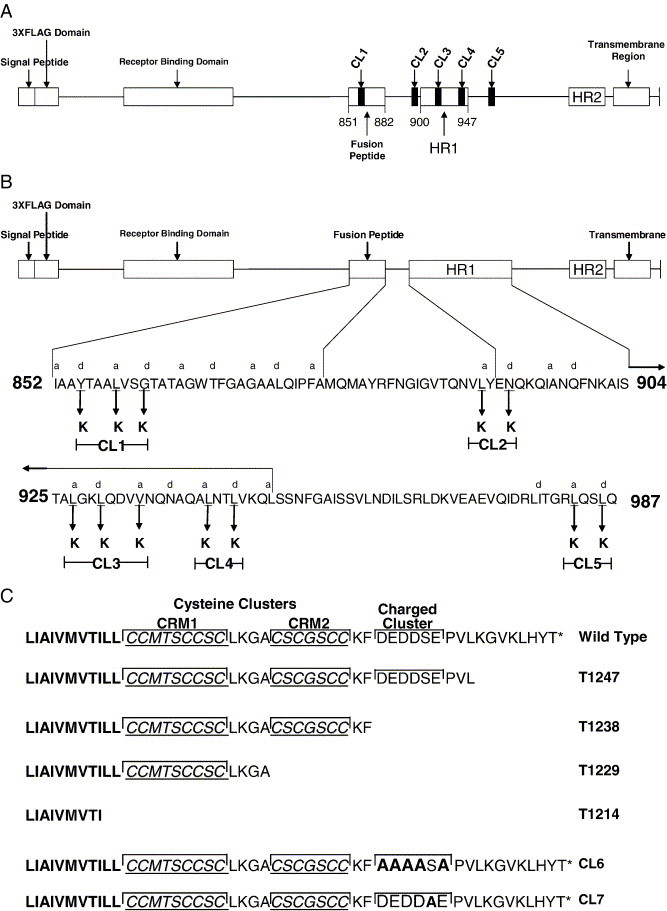
Schematic diagram of the SARS-CoV S glycoprotein. (A) Graphical representation of the S glycoprotein showing the approximate location of the cluster to lysine mutations CL1–CL5 relative to known and indicated functional domains. (B) Shown on the top of the diagram is a graphical representation of the SARS-CoV S glycoprotein. The predicted fusion peptide and the HR1 region are enlarged below to show the sets of amino acids replaced by lysines in the cluster mutations. The heptad repeat a and d positions are labeled above the corresponding amino acid. Amino acids changed to lysine are demarcated by arrows with the name of that particular mutation shown in brackets. (C) Amino-acid sequences of the carboxyl termini of the truncation and acidic cluster associated mutations. Cysteine clusters (CRM1 and CRM2) are denoted by underlined italicized text as well as a bracket encompassing their respective regions. The charged cluster is bracketed over the region. Amino acids mutated to alanines for the CL6 and CL7 cluster mutations are in bold.
It has been shown for other viral class I fusion proteins that the carboxyl terminus plays a regulatory role in membrane fusion (Bagai and Lamb, 1996, Sergel and Morrison, 1995, Seth et al., 2003, Tong et al., 2002, Yao and Compans, 1995). Specifically, for coronaviruses, the MHV S glycoprotein endodomain has been shown to contain charged-rich and cysteine-rich regions, which are critical for fusion of infected cells (Bos et al., 1995, Chang et al., 2000, Ye et al., 2004). The carboxyl-terminal portion of the S glycoprotein contains a consensus acidic amino-acid cluster with a motif that has been predicted by the NetPhos 2.0 software to be phosphorylated (Blom et al., 1999). To investigate the potential role of the acidic amino-acid cluster in synthesis, transport, and cell fusion, serial truncations of S were constructed. The acidic cluster was specifically targeted by mutagenizing the predicted phosphorylation site embedded within the acidic cluster as well as by replacing acidic residues of the acidic cluster with alanine residues. In addition, carboxyl-terminal truncations of 8, 17, 26, and 41 amino acids were engineered by insertion of stop codons within the S glycoprotein gene. The 8 aa truncation (T1247) was designed to bring the predicted charged cluster DEDDSE proximal to the carboxyl terminus of the mutated S glycoprotein (Fig. 1C). Similarly, the 17 aa truncation (T1238) was designed to delete the DEDDSE acidic cluster. The SARS-CoV S glycoprotein endodomain contains two cysteine residue clusters, a CCMTSCCSC (CRM1) cluster immediately adjacent to the membrane and a CSCGSCC (CRM2) downstream of the first cluster. To address the role of these domains in S glycoprotein-mediated cell-to-cell fusion, the 26 aa truncation (T1229) was designed to delete the CRM2 domain, while the 41 aa truncation (T1214) deleted both the CRM1 and CRM2 domains (Fig. 1C).
Effect of mutations on S synthesis
To investigate the effect of the different mutations on S synthesis, western immunoblot analysis was used to detect and visualize all of the constructed mutant glycoproteins as well as the wild-type S (Fig. 2 ). Cellular lysates prepared from transfected cells at 48 h post-transfection were electrophoretically separated by SDS-PAGE and the S glycoproteins were detected via chemiluminescence using a monoclonal antibody specific for the SARS-CoV S glycoprotein. Carbohydrate addition was shown to occur in at least four different locations of the SARS-CoV S glycoprotein (Krokhin et al., 2003, Ying et al., 2004). Furthermore, transiently expressed S glycoprotein in Vero E6 cells was proteolytically cleaved into S1 and S2 components (Wu et al., 2004). The anti-S monoclonal antibody SW-111 detected a protein species in cellular extracts from transfected cells, which migrated with an apparent molecular mass of approximately 180 kDa, as reported previously (Song et al., 2004). All mutated S glycoproteins produced similar S-related protein species to that of the wild-type S indicating that none of the engineered mutations adversely affect S synthesis and intracellular processing (Fig. 2A). The SARS S glycoprotein is known to form homotrimers in its native state (Song et al., 2004). To investigate the effect of the mutations on S oligomerization, cellular lysates from transfected cells were electrophoretically separated without prior boiling of the samples and in the absence of reducing agents (Song et al., 2004). The different S species were detected via chemiluminescence using the monoclonal SW-111 to the SARS S glycoprotein. An S protein species was detected that had an approximate apparent molecular mass of 500 kDa, which was consistent with previously published data (Song et al., 2004) (Fig. 2B). Although levels of oligomer expression seemed to vary slightly between mutant forms, all mutated S glycoproteins produced similar species to the wild type, indicating that none of the mutations blocked oligomerization from occurring.
Fig. 2.

Western blot analysis of the expressed mutant SARS-CoV S mutant glycoproteins. (A, B) Immunoblots of wild-type [3xFLAG So (WT)], cluster to lysine, cluster to alanine, and carboxyl truncation mutant S glycoproteins probed with monoclonal anti-SARS S antiserum. “Cells only” represents a negative control in which Vero cells with no protein transfected into them were probed with the monoclonal antibody to SARS S glycoprotein. (B) In order to detect trimer formation more efficiently, the protein extracts of the mutants were neither boiled nor subject to treatment with beta mercaptoethanol.
Ability of mutant S glycoproteins to be expressed on the cell surface
To determine if the mutant S glycoproteins were expressed on the surface of cells, immunohistochemical analysis was used to label cell-surface-expressed S under live cell conditions that restrict antibody binding to cell surfaces. In addition, immunohistochemistry was used to detect the total amount of S expressed in cells by fixing and permeabilizing the cells prior to reaction with the antibody. A recombinant S protein having the 3xFLAG added in-frame to the carboxyl terminus of S was used as a negative control, since it would not be stained by the live cell reaction conditions (see Materials and methods). Both wild-type versions of S having the 3xFLAG, either at the amino or carboxyl terminus of S, caused similar amounts of fusion (Fig. 3 ), which also was similar to that obtained with the untagged wild-type S (not shown). The relative amounts of cell-surface versus total cellular expression of S were obtained through the use of an ELISA. A ratio between the cell-surface localized S and total cellular S expression was then calculated and normalized to the corresponding ratio obtained with the wild-type S glycoprotein (see Materials and methods) (Fig. 4 ). The CL1 (95%), CL2 (78%), CL3 (86%), CL4 (80%), CL5 (92%), CL6 (86%) mutants as well as the T1229 (91%), T1238 (94%), and T1247 (79%) truncations were expressed on the cellular surface by the percentages indicated when compared to the cell-surface expression of the wild-type protein. In contrast, the 1214T mutant expressed 77% less S on cell surfaces in comparison to the wild-type S (Fig. 4).
Fig. 3.
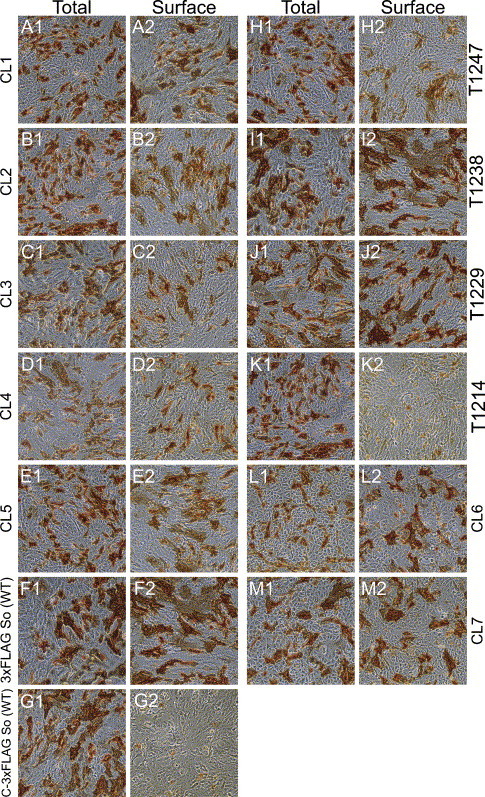
Immunohistochemical detection of cell-surface and total expression of the SARS-CoV S wild-type and mutant proteins. Vero cells were transfected with the wild-type SARS-CoV optimized S [3xFLAG So (WT)] (F1, F2), CL1 (A1, A2), CL2 (B1, B2), CL3 (C1, C2), CL4 (D1, D2), CL5 (E1, E2), CL6 (L1, L2), CL7 (M1, M2), T1214 (K1, K2), T1229 (J1, J2), T1238 (I1, I2), T1247(H1, H2) and a wild-type SARS-CoV optimized S labeled with a 3xFLAG carboxyl tag (G1, G2), which served as a negative control. At 48 h post-transfection, cells were immunohistochemically processed either under live conditions to show surface expression (A2, B2, C2, D2, E2, F2, G2, H2, I2, J2, K2, L2, and M2) or fixed and permeabilized conditions to show total expression (A1, B1, C1, D1, E1, F1, G1, H1, I1, J1, K1, L1, and M1).
Fig. 4.
Ratios of cell-surface to total cellular expression of mutant SARS-CoV S glycoproteins. Detection of cell-surface and total glycoprotein distribution was determined by immunohistochemistry and ELISA (see Materials and methods). Cell-surface expression of the S glycoprotein was measured by incubating the transfected cell monolayers with anti-FLAG antibody at room temperature before permeabilization. For total S glycoprotein detection, cells were fixed and permeabilized prior to incubation with the anti-FLAG antibody. A ratio between the surface localization and the total expression was calculated and normalized to the wild-type protein, then set to a percentage of the wild-type. The error bars represent the maximum and minimum surface to total ratios obtained from three independent experiments, and the bar height represents the average surface to total ratio.
Effect of mutations on S-mediated cell-to-cell fusion
Transiently expressed wild-type S causes extensive cell-to-cell fusion (syncytial formation), especially in the presence of the SARS-CoV ACE2 receptor (Li et al., 2003). To determine the ability of each mutant S glycoprotein to cause cell-to-cell fusion and the formation of syncytia, fused cells were labeled by immunohistochemistry using the anti-FLAG antibody (Fig. 5 ). The extent of cell-to-cell fusion caused by each mutant glycoprotein was calculated by obtaining the average size of approximately 300 syncytia. The average syncytium size for each mutant was then normalized to that found in wild-type S-transfected cells (see Materials and methods). The CL1 (73%), CL2 (75%), CL3 (68%), CL4 (71%), CL5 (76%), CL6 (51%) as well as the T1214 (86%) and T1247 (66%) mutants inhibited the formation of syncytia by the percentages indicated. The T1229 truncation and the cluster mutant CL7 produced syncytia, which were on the average 22% and 15% smaller, respectively, than that of the wild-type S. In contrast, the T1238 mutant produced on the average 43% larger syncytia than that of the wild-type S (Fig. 5).
Fig. 5.
Quantitation of the extent of S-mediated cell fusion. The average size of syncytia for each mutant was determined by digitally analyzing the area of approximately 300 syncytia stained by immunohistochemistry for S glycoprotein expression using the Image Pro Plus 5.0 software package (see Materials and methods). Error bars shown represent the standard deviations calculated through comparison of the data from each of three experiments.
Comparison of the membrane fusion and cell-surface expression results allowed the grouping of the different mutant S phenotypes into four distinct groups (Table 1 ): (1) S mutant forms in Group I (CL1–CL6 and T1247) resulted in high levels of cell-surface expression (78–95% of the wild-type S); however, the average size of syncytial formed by these mutated S glycoproteins was reduced substantially in comparison to the wild-type S (23–48% of the wild-type). CL1 affects the predicted fusion peptide, CL2–CL4 affect the HR1 domain, and CL5 affects a region downstream of the HR1 domain. The CL6 mutation is located within the S carboxyl-terminal acidic cluster. The T1247 mutation truncates the S carboxyl terminus by 8 amino acids; (2) S mutant forms in Group II produced high levels of S cell-surface expression and an average size of syncytia slightly smaller than that of the wild-type S. These mutations included CL7, which modified the acidic cluster and the T1229 truncations that deleted the cysteine-rich motif CRM2; (3) the single S mutant in Group III, T1214, produced significantly less cell-surface expression and concomitantly the average size of syncytia was substantially reduced in comparison to the wild-type S; (4) the T1238 truncation in Group IV produced high levels of cell-surface expression equivalent to that of the wild type (94% of the wild-type S), while the average syncytium size was 43% larger than that the syncytial produced by the wild-type S (Table 1).
Table 1.
Grouping of S mutants by their cell-surface expression and cell-fusion properties
| Group | Mutants | Surface % | Fusion % |
|---|---|---|---|
| Control | WT | 100 | 100 |
| I | T1247 | 79.15 | 34.01 |
| CL1 | 95.26 | 27.21 | |
| CL2 | 78.17 | 25.14 | |
| CL3 | 86.25 | 32.41 | |
| CL4 | 80.12 | 29.00 | |
| CL5 | 92.33 | 23.93 | |
| CL6 | 86.81 | 48.53 | |
| II | T1229 | 90.68 | 78.19 |
| CL7 | 88.26 | 85.17 | |
| III | T1214 | 23.40 | 15.52 |
| IV | T1238 | 93.77 | 143.09 |
Detection of the intracellular distribution of S mutant glycoproteins via confocal microscopy
To visualize the intracellular distribution of S mutant glycoproteins, cells were transfected with plasmids encoding the wild-type or S mutants and examined by confocal microscopy at 48 h post-transfection (Fig. 6 ). The wild-type and all the S mutants were detected throughout the cytoplasm of transfected cells and exhibited similar intracellular distribution patterns (Fig. 6, panels B, D, F, H, J, L). To determine and compare the endocytotic profiles of wild-type and S mutant forms, transfected cells were reacted with the anti-FLAG antibody under live conditions for 12 h at 37 °C and visualized by confocal microscopy. The majority of the wild-type S detected by the anti-FLAG antibody appeared to remain on cell surfaces (Fig. 6, panel A). In contrast, a significant fraction of cell-surface-expressed CL6 and CL7 as well as the T1229, T1238, and T1247 S mutants appeared to partially endocytose to cytoplasmic compartments (Fig. 6, panels C, E, G, I, K). The CL7 mutant, but not the other S mutants, appeared to colocalize with the early endosomal marker EEA-1 (Fig. 6, panel E). The S mutants CL1, CL2, CL3, CL4, and CL5 remained in plasma membranes exhibiting profiles similar to that of the S wild-type glycoprotein (data not shown).
Fig. 6.

Confocal microscopic visualization of endocytosed and intracellular distribution of SARS-CoV S glycoprotein mutants. Vero cells expressing wild-type SARS-CoV S glycoprotein [3xFLAG So (WT)] (A and B), CL6 (C and D), CL7 (E and F), T1229 (G and H), T1238 (I and J), and T1247 (K and L) were processed for confocal microscopy using two different methods in order to assess different properties of the mutants. Endocytosis patterns (A, C, E, G, I, and K) were visualized by adding anti-FLAG (green) antibody into the media 12 h prior to processing, enabling detection of the mutant protein after endocytosis from cellular surfaces. Early endosomes were also detected for these panels using a polyclonal anti-early endosomal antigen I antibody (red). For total glycoprotein detection (B, D, F, H, J, and L), cells were fixed and permeabilized prior to labeling with anti-FLAG (green).
Time-dependent endocytotic profiles of wild-type and mutant S proteins
A time-dependent endocytosis assay was utilized to better visualize the endocytotic patterns of the wild-type and mutant S glycoproteins as well as to exclude the possibility that the observed plasma membrane accumulation of the wild-type S and some of the S mutants was due to recirculation of endocytosed S to cell surfaces. In this assay, cell-surface-expressed S was reacted with anti-FLAG antibody at 4 °C and subsequently, cells were incubated at 37 °C for different time periods before processing for confocal microscopy (see Materials and methods). Generally, these time-dependent endocytosis studies were in agreement with the results shown in Fig. 6. Specifically, in cells that were not shifted to 37 °C, referred to as time zero cells, wild-type and mutant spike were detected exclusively at the surface of the cells (Fig. 7 , panels A1, B1, C1, D1, E1, F1). At 5 and 15 min after the shift to 37 °C, the wild-type S remained exclusively at the surface while the other S mutants were detected in numerous intracellular vesicles dispersed inside the cell (Fig. 7, panels A2 and A3 compared to panels B2 and B3, C2 and C3, D2 and D3, E2 and E3, F2 and F3). By 60 min after the shift to 37 °C, the wild-type S was still localized exclusively to the surface of the cell (Fig. 7, panel A4), while the T1229, T1238, CL6, and CL7 S mutants appeared to be present throughout the cytoplasm of the cell (Fig. 7, panels B4, C4, E4, F4). The T1247 S mutant seemed to undergo rapid and complete endocytosis during the 60 min observation and appeared to localize into punctuate structures in the cytoplasm of cells unlike the fairly even cellular distribution of all other S mutants (Fig. 7, panels D1–D4 compared to panels A4, B4, C4, D4, E4, and F4).
Fig. 7.
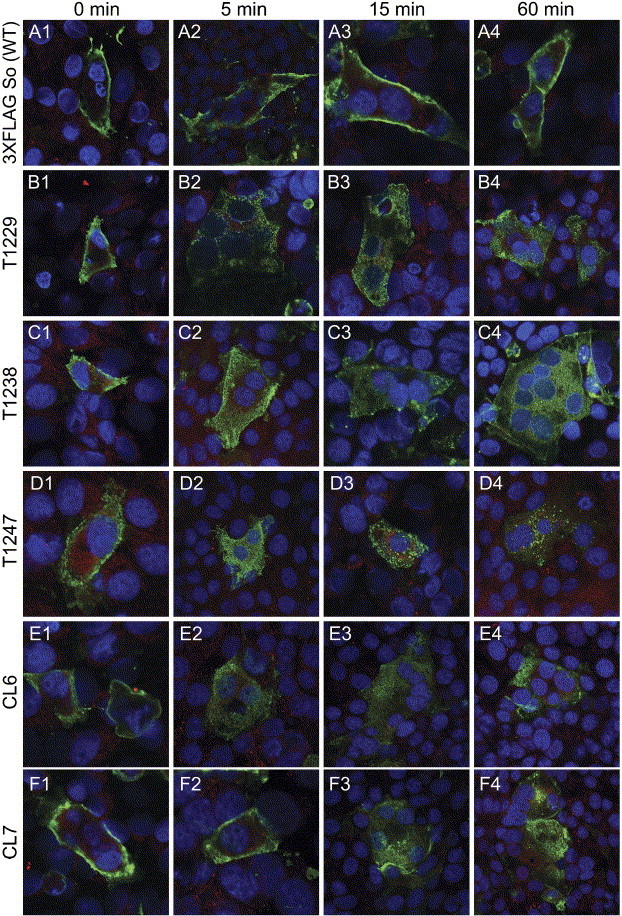
Analysis of the endocytotic kinetic profile of the truncation mutants and the acidic cluster mutants using confocal microscopy. After transfection, SARS-CoV S glycoprotein wild-type [3xFLAG So (WT)] (A1–A4), T1229 (B1–B4), T1238 (C1–C4), T1247 (D1–D4), CL6 (E1–E4), and CL7 (F1–F4) expressing cells were incubated with an anti-FLAG monoclonal antibody (green) for 1 h and then returned to 37 °C for different times. Early endosomes were also detected for these panels using a polyclonal anti-early endosomal antigen I antibody (red). Cell nuclei were labeled with To-Pro-3 Iodide (blue). Panels A1–A4, B1–B4, C1–C4, D1–D4, E1–E4, and F1–F4 correspond to 0-, 5-, 15-, and 60-min incubation times at 37 °C, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Discussion
The mechanism by which class I fusion proteins such as the coronavirus S glycoprotein, the hemagglutinin protein (HA) of influenza virus, the gp41 of human immunodeficiency virus (HIV), the Ebola virus surface glycoprotein (GP), and the fusion protein (F) of paramyxovirus facilitate membrane fusion during viral entry into cells has been extensively investigated (Eckert and Kim, 2001, Hernandez et al., 1996, Tsai et al., 2003, White, 1992, Zelus et al., 2003). Currently, specific membrane fusion models have been proposed all of which include the following general steps: (a) binding of a receptor through a receptor-specific domain located within the ectodomain of the viral glycoprotein; (b) induction of a conformational change via low pH or binding to the receptor that exposes a fusion peptide, typically a hydrophobic region in the membrane-anchored subunit, which inserts into the cellular lipid membrane; (c) formation of a trimer-of-hairpins-like structure by α-helical peptides, termed heptad repeat segments, via a transient pre-hairpin intermediate that facilitates the juxtaposition of the viral and cellular membranes which then leads to fusion of the viral envelope with cellular membranes (reviewed in Eckert and Kim, 2001, Hernandez et al., 1996). Although the most important domains of the class I fusion proteins are naturally located in their ectodomains, it has been reported that intracytoplasmic endodomains play an important role in intracellular transport and virus-induced cell fusion (Bagai and Lamb, 1996, Bos et al., 1995, Chang et al., 2000, Lontok et al., 2004, Schwegmann_Wessels et al., 2004, Sergel and Morrison, 1995, Seth et al., 2003, Tong et al., 2002, Waning et al., 2004, Yao and Compans, 1995).
In this paper, we show that mutations that alter the HR1 and predicted fusion peptide domains of the SARS-CoV S glycoprotein as well as mutations located within α-helical regions well separated from the HR1, HR2, and predicted fusion peptide domains drastically affected S-mediated cell fusion. Importantly, mutagenesis of the S cytoplasmic domains suggests that the carboxyl terminus of the S glycoprotein contains multiple but distinct regulatory domains that may function in virus-induced cell fusion through different mechanisms.
Functional domains of the S ectodomain
The CL1 cluster mutation is located within the predicted fusion peptide. The constructed cluster mutations replaced the amino terminal a and d positions of the predicted fusion peptide resulting in shortening the predicted α-helical portion of the fusion peptide. The S mutant glycoprotein carrying the CL1 mutation was apparently synthesized in comparable levels to the wild-type S glycoprotein. As expected, although this mutant S form was able to be expressed on cell surfaces (95% of wild-type S levels), its ability to cause cell fusion was inhibited by more than 70% (Table 1; Group I mutants). This result confirms that the predicted fusion peptide is absolutely essential for S-mediated cell fusion, although the engineered collapse of the predicted region does not significantly effect glycoprotein synthesis, processing, and cell-surface expression.
Recent studies have shown that interactions between HR1 and HR2 of SARS-CoV are critical in producing the necessary conformation changes that result in exposure of the fusion peptide and its insertion into apposed membranes (Ingallinella et al., 2004, Tripet et al., 2004, Xu et al., 2004). Biochemical and X-ray crystallography studies have shown that the HR1 and HR2 form a stable six-helix bundle, in which the HR1 helices form a central coiled-coil surrounded by three HR2 helices in an oblique, antiparallel manner termed the fusion core (Ingallinella et al., 2004, Tripet et al., 2004, Xu et al., 2004), which is consistent with other class I fusion proteins (Baker et al., 1999, Bullough et al., 1994, Caffrey et al., 1998, Chan et al., 1997, Lu et al., 1995, Tan et al., 1997, Weissenhorn et al., 1998a, Weissenhorn et al., 1998b, Weissenhorn et al., 1997). Amino-acid residues 902–947 in the SARS-CoV S HR1 domain fold into a predicted 12-turn α-helix (entire length of the fusion core) with hydrophobic amino acids predominantly occupying the a and d positions. The CL3 and CL4 mutations were designed to change the a and d hydrophobic residues to hydrophilic (lysine) residues. Both mutant glycoproteins were expressed on cell surfaces at reduced levels in comparison to the wild-type S glycoprotein (14% and 20% reduction, respectively). However, these mutations inhibited S-mediated fusion by 68% and 71%, respectively (Table 1; Group I mutants). Therefore, the inability of the CL3 and CL4 mutants to cause fusion is most likely due to ectodomain structural changes involving the HR1 domain. Inhibition of proper HR1 interaction with HR2 may be the primary cause of the observed inhibition of S-mediated cell fusion.
The CL2 and CL5 cluster mutations are located upstream and downstream of HR1 within a predicted α-helical portion of the S ectodomain extending from residues 875 to 1014. Both CL2 and CL5 S mutant forms exhibited reduced S-mediated cell fusion (75% and 76% reduction in comparison to the S wild-type, respectively), while they were synthesized and expressed on cell surfaces at levels similar to the S wild-type (Table 1; Group I mutants). These data suggest that the inability of these S mutants to cause extensive cell fusion was mostly due to structural alterations of the extracellular portion of the S glycoprotein. Furthermore, these results suggest that α-helical portions of the S ectodomain that are well separated from HR1 and HR2 or the predicted fusion peptide are important for S-mediated cell fusion. It is possible that these mutations affect HR1 interactions with HR2 by inhibiting ectodomain conformational changes required for their optimal interactions. Specifically, the CL2 mutation is only 16 amino acids downstream of the predicted fusion peptide. Therefore, this mutation may interfere with fusion peptide-associated functions.
Functional domains of the endodomain of S
The T1247 S mutation deletes 8 amino acids from the carboxyl terminus of S. This S mutant exhibited a 65% reduction in cell fusion in comparison to the wild-type S, while cell-surface expression was reduced by 20% in comparison to the wild-type S (Table 1; Group I mutants). Kinetic endocytosis experiments revealed that the T1247 S mutant also endocytosed much faster than the wild-type S glycoprotein. Acidic cluster motifs are known to serve as endocytotic signals (Brideau et al., 1999, Zhu et al., 1996). The rapid endocytosis of the T1247 S mutant form may be due to the relocation of the acidic cluster KFDEDDSE proximal to the carboxyl terminus of the S glycoprotein after removal of the terminal 8 amino acids resulting in more efficient endocytosis. Therefore, the inability of the S mutant form to cause extensive cell fusion may be due primarily to its rapid endocytosis from cell surfaces. It is worth noting that a similar deletion of 8 amino acids from the carboxyl terminus of the MHV S glycoprotein resulted in enhanced S-mediated cell fusion (Chang et al., 2000). A comparison of the carboxyl-terminal amino-acid sequences of these two glycoproteins reveals that the MHV S does not have a well-defined acidic cluster in the SARS-COV S homologous location; however, the last three amino acids of the MHV S are charged residues (HED). Therefore, the ability of the truncated MHV S to cause more cell fusion may be due to increased surface retention resulting from a reduction in endocytosis mediated by these charged residues. Alternatively, the MHV deletion may cause structural changes that enhance the MHV S fusogenicity by destabilizing the overall structure of the glycoprotein. Stabilization of the carboxyl terminus has been shown to decrease the fusion activity of the vesicular stomatitis virus G glycoprotein (Waning et al., 2004); therefore, conversely, it is possible that destabilization of the carboxyl terminus may cause an increase in fusion.
Of particular interest is the T1238 truncation which removed the S carboxyl-terminal acidic domain. This S mutant glycoprotein exhibited a more than 40% increase in cell fusion relative to the S wild type, while there was only a slight decrease in cell-surface expression (Table 1; Group IV mutants). The fact that the overall levels of S glycoprotein detected on cell surfaces as well as the endocytosis profile were not altered suggests that this deletion may enhance fusion via a structural destabilization of the glycoprotein. In contrast, changing acidic amino acids of the acidic motif to alanine residues inhibited cell fusion indicating that the acidic motif was important for S-mediated cell fusion. Specifically, the CL6 cluster mutation changed the acidic residues (DEDDSE) to alanine residues (AAAASA). This mutant protein, while being efficiently expressed at cellular surfaces (87% of the wild-type protein), exhibited a 51% reduction in fusion activity in comparison to the wild type (Table 1; Group I mutant). The observed reduction in S-mediated cell fusion suggests that the acidic cluster plays an important regulatory role in S-mediated cell fusion without appreciably affecting intracellular transport and cell-surface expression. This result is in sharp contrast to the T1238 truncation that deleted the acidic cluster and caused enhanced S-mediated cell fusion. A possible explanation for these seemingly disparate results is that modifications of the carboxyl terminus produce differential effects on the structure and function of the protein by rendering the S glycoprotein more or less prone to S-mediated fusion. Alternatively, it is possible that the acidic cluster plays important roles only in the context of the entire S glycoprotein by regulating binding to other viral or cellular proteins that may modify the S fusogenic properties.
The acidic cluster located in the cytoplasmic portion of the SARS CoV S contains a predicted phosphorylation site (DEDDSE). To address the role of this predicted phosphorylation site within the acidic cluster, the CL7 cluster mutation was constructed. The CL7 replaces the serine residue with an alanine residue within the acidic cluster. This mutant S protein fused cells extensively (85% of the wild type) and was expressed on cell surfaces at levels similar to that of the wild-type S (88% of the wild type) (Table 1; Group II mutants), suggesting that the potential phosphorylation site does not play an important role in S-mediated cell fusion and cell-surface expression. However, the CL7 mutant appeared to recycle to early endosomes in contrast to the wild-type S, which remained mostly on cell surfaces. Therefore, it is possible that the altered putative phosphorylation site within the acidic cluster may play a yet unknown role in S retention at cell surfaces. Conversely, lack of this signal may cause aberrant endocytosis to early endosomes. The overall charge of the carboxyl terminus may also play some role in the structure and function of S. In this regard, there are additional charged amino acids dispersed upstream and downstream of the mutated charged cluster that may play some role in S transport and S-mediated fusion through electrostatic interactions with other viral and cellular proteins. Additional alanine scanning mutations would be needed to resolve their potential contribution to S functions.
In contrast to the MHV S glycoprotein, in which the domains overlap, the charged region of the SARS-CoV S glycoprotein and the two cysteine-rich motifs CRM1 and CRM2 are separate distinct regions (Fig. 1, Fig. 8 ). The T1229 mutation deleted the CRM2 domain, while the T1214 truncation deleted both CRM1 and CRM2. Deletion of the CRM2 slightly inhibited surface expression (91% of the wild type) while reducing fusion activity by 22% (Table 1; Group II mutants). A similar truncation of the MHV S glycoprotein produced a comparable effect on cell fusion (Chang et al., 2000). The 1214 truncation severely inhibited cell-surface expression by 77%, when compared to the wild type, while cell-to-cell fusion activity was reduced by 84% (Table 1; Group III mutant). These results differ from previously published data on similar truncations of the MHV S glycoprotein. Specifically, it was found that the replacement of the entire cysteine-rich domain with amino-acid sequences derived from the cytoplasmic terminus of the herpes simplex virus 1 (HSV-1) glycoprotein D (gD) severely inhibited MHV S glycoprotein function without necessarily affecting cell-surface expression (Chang et al., 2000, Ye et al., 2004). In contrast, the SARS-CoV S T1214 mutant glycoprotein failed to be expressed on cell surfaces explaining the inability of this glycoprotein to cause cell fusion. These results suggest that the proximal cysteine residues of the SARS CoV S play crucial roles in intracellular transport and cell-surface expression. The discrepancy with the MHV S carboxyl-terminal replacements may be due to additional gD sequences that facilitated intracellular transport and cell-surface expression. It is worth noting that depending on the algorithm used to predict the membrane spanning domain of S, a few of the cysteine residues may be included in the membrane spanning region (Chang et al., 2000, Ye et al., 2004). Therefore, it is possible that deletion of these cysteine residues may lead to S misincorporation into membranes, resulting in the apparent transport defects.
Fig. 8.
Cluster alignment of the carboxyl terminus of the SARS-CoV S and the MHV S glycoproteins. The shaded residues indicate the position of the cysteine-rich motif in their respective protein. The cysteine residues are bolded. The charged clusters are indicated by a bracket over the corresponding regions.
Overall, these results suggest that the S-mediated cell fusion is regulated by both the ecto- and endodomains, which play important roles in cell-surface expression. Furthermore, the data suggest that the 17 carboxyl-terminal amino-acid residues of S exert a negative regulatory (repressor) effect on S-mediated cell fusion, while both the carboxyl-terminal acidic cluster and CRM2 domains exert secondary regulatory roles in S-mediated cell fusion. Additional studies are required to elucidate the specific amino-acid requirements of the S endodomain that can affect S-mediated cell fusion potentially via a transmembrane signal transduction process that leads to destabilization of the S ectodomain.
Materials and methods
Cells
African green monkey kidney (Vero) cells were obtained from the American Type Culture Collection (Rockville, MD). Cells were propagated and maintained in Dulbecco modified Eagle medium (Sigma Chemical Co., St. Louis, MO) containing sodium bicarbonate and 15 mM HEPES supplemented with 10% heat-inactivated fetal bovine serum.
Plasmids
The parental plasmid used in the present study, SARS-S-Optimized, has been previously described (Li et al., 2003). The Spike-3XFLAG-N gene construct was generated by cloning the codon-optimized S gene, without the DNA sequence coding for the signal peptide, into the p3XFLAG-CMV-9 plasmid vector (Sigma). PCR overlap extension (Aiyar et al., 1996) was used to construct the alanine mutants, cluster mutants, and the single point mutants. In order to construct the truncation mutants, primers were designed that incorporated a stop codon and a BamHI restriction site at the appropriate gene site. Restriction endonuclease sites HindIII and BamHI were then used to clone the gene construct into the Spike-3XFLAG-N plasmid.
The constructed cluster mutants targeting the S ectodomain changed the following sets of amino acids to lysine residues: CL:1 Y(855), L(859), G(862); CL2: L(898), N (901); CL3: L(927), L(930), V(934); CL4: L(941), L(944); CL5: L(983), L(986). The cluster mutants targeting the S endodomain changed the following amino acids to alanine residues: CL6: D(1239), E(1240), D(1241), D(1242), E(1244); CL7: S(1243) (Figs. 1B and C).
Production of SARS-CoV S monoclonal antibodies
The monoclonal antibodies SW-111 was raised against the Spike envelope glycoprotein of the SARS virus. A synthetic, codon-optimized gene corresponding to SAR-CoV S glycoprotein coding sequences (Li et al., 2003) was engineered to produce truncated, secreted proteins containing C-terminal His-tags which encode either the entire ectodomain or just the receptor binding domain of S1. These genes were cloned into baculoviral vectors, and the resulting virus was used to infect insect cell lines (High Five) (Invitrogen, Carlsbad, CA). The supernatants were then tested by Western blotting using anti-6-His antibodies. Protein was partially purified from supernatants by passage through a nickel column and then concentrated by ultrafiltration. Additional protein derived in an analogous fashion was provided by the laboratory of Stephen Harrison. Standard protocols for mouse immunization were used. The animals were maintained in the animal facility of Dana Farber Cancer Institute, and hybridomas and monoclonal antibodies were produced in the Dana Farber/Harvard Cancer Center Monoclonal Antibody Core. Spleenocytes from immunogenized mice were fused with NS-1 myeloma cells (ATCC) using standard protocols. Antibody-producing clones were identified by Western blot analysis, using purified SARS-CoV S. The positive cells were then sub-cloned and re-tested against the purified SARS-CoV S glycoprotein. Isotype analysis revealed that the antibody belonged to the IgG-1 class.
Western blot and S oligomerization analysis
Vero cell monolayers in six-well plates were transfected with the indicated plasmids utilizing the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's directions. At 48 h post-transfection, cells were collected by low-speed centrifugation, washed with Tris-buffered saline (TBS), and lysed at on ice for 15 min in mammalian protein extraction reagent supplemented with a cocktail of protease inhibitors (Invitrogen/Life Technologies). Insoluble cell debris was pelleted, samples were electrophoretically separated by SDS-PAGE, transferred to nitrocellulose membranes, and probed with anti-SARS CoV monoclonal antibody at a 1:10 dilution. Samples being analyzed for transport and processing were boiled for 5 min and treated with beta mercaptoethanol, while samples being analyzed for trimer formation were not. Subsequently, blots were incubated for 1 h with a peroxidase-conjugated secondary antibody at a 1:50,000 dilution and then visualized on X-ray film by chemiluminescence (Pierce Chemicals, Rockford, IL). All antibody dilutions and buffer washes were performed in TBS supplemented with 0.135 M CaCl2 and 0.11 M MgCl2 (TBS–Ca/Mg).
Cell-surface immunohistochemistry
Vero cell monolayers in six-well plates were transfected with the indicated plasmids utilizing the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's directions. At 48 h post-transfection, the cells were washed with TBS–Ca/Mg and either fixed with ice-cold methanol or left unfixed (live). Immunohistochemistry was performed by utilizing the Vector Laboratories Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA) essentially as described in the manufacturer's directions. Briefly, cells were washed with TBS–Ca/Mg and incubated in TBS supplemented with 5% normal horse serum and 5% bovine serum albumin at room temperature for 1 h. After blocking, cells were reacted with anti-FLAG antibody (1:500) in TBS blocking buffer for 3 h, washed four times with TBS–Ca/Mg, and incubated with biotinylated horse anti-mouse antibody. Excess antibody was removed by four washes with TBS–Ca/Mg and subsequently incubated with Vectastain Elite ABC reagent for 30 min. Finally, cells were washed three times with TBS–Ca/Mg, and reactions were developed with NovaRed substrate (Vector Laboratories) according to the manufacturer's directions.
Determination of cell-surface to total cell S glycoprotein expression
Vero cell monolayers in six-well plates were transfected with the indicated plasmids and processed for immunohistochemistry as described above with the exception that the ABTS Substrate Kit, 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (Vector Laboratories) was used instead of the NovaRed substrate. After the substrate was allowed to develop for 30 min, 100 μl of the developed substrate was transferred, in triplicate, to a 96-well plate. The samples were then analyzed for color change at a wavelength of 405 nm. The absorbance reading from cell-surface labeling experiments obtained from live cells was divided by the total labeled absorbance readings obtained from fixed cells which were then normalized to the wild-type protein values. The measurements were then converted to percentages reflecting the ratio of S present on cell surfaces versus the total S expressed in the transfected cells.
Confocal microscopy
Vero cell monolayers grown on coverslips in six-well plates were transfected with the indicated plasmids utilizing the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's directions. For the endocytosis analysis of the mutants, anti-FLAG diluted 1:500 in TBS supplemented with 5% normal goat serum and 5% bovine serum albumin (TBS blocking buffer) was added to the cell culture media for 12 h before processing. At 48 h post-transfection, cells were washed with TBS and fixed with electron microscopy-grade 3% paraformaldehyde (Electron Microscopy Sciences, Fort Washington, PA) for 15 min, washed twice with TBS–Ca/Mg, and permeabilized with 0.1% Triton X-100. Monolayers were blocked for 1 h with TBS blocking buffer before incubation for 3 h with anti-FLAG antibody (Sigma) diluted 1:500 in TBS blocking buffer. Cells were then washed extensively and subsequently incubated for 1 h with Alexafluor 488-conjugated anti-immunoglobulin G (IgG) (Molecular Probes) diluted 1:750 in TBS blocking buffer. To visualize the early endosomes, cells were stained with a 1:500 dilution of anti-early endosomal antigen I antibody (Affinity Bioreagents Inc.). Cells were examined by using a Leica TCS SP2 laser-scanning microscope (Leica Microsystems, Exton, PA) fitted with a 63× Leica objective lens (Planachromatic; 1.4 numerical aperture). Individual optical sections in the z axis, averaged eight times, were collected simultaneously in the different channels at a 512 × 512 pixel resolution as described previously (Foster et al., 2004, Lee et al., 2000). Images were compiled and rendered in Adobe Photoshop.
S glycoprotein endocytosis assay
Vero cell monolayers grown on coverslips in six-well plates were transfected with the indicated plasmids utilizing the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's directions. At 48 h post-transfection, the cells were washed once with room temperature TBS–Ca/Mg. The plates were then moved to a 4 °C cold room and washed with 4 °C TBS–Ca/Mg. The cells were labeled for 1 h at 4 °C with anti-FLAG antibody diluted 1:500 in TBS blocking buffer. The cells were washed with 4 °C TBS three times and then brought back to 37 °C and allowed to incubate for their respective time points. Then, the cells were immediately fixed and permeabilized with ice-cold methanol. Monolayers were blocked for 2 h in TBS blocking buffer before incubation for 1 h with Alexafluor 488-conjugated anti-immunoglobulin G (IgG) (Molecular Probes) diluted 1:750 in TBS–Ca/Mg blocking buffer. After incubation, excess antibody was removed by washing five times with TBS–Ca/Mg. The nuclei were counterstained for 15 min with TO-PRO-3 iodide (1:5000 dilution) and visualized at 647 nm. Cells were examined by using a Leica TCS SP2 laser-scanning microscope (Leica Microsystems, Exton, Pa.) fitted with a 63× Leica objective lens (Planachromatic; 1.4 numerical aperature). Individual optical sections in the z axis, averaged eight times, were collected simultaneously in the different channels at 512 × 512 pixel resolution as described previously (Foster et al., 2004, Lee et al., 2000). Images were compiled and rendered in Adobe Photoshop.
Quantitation of the extent of S-mediated cell fusion
Vero cell monolayers in six-well plates were transfected in triplicate with the indicated plasmids utilizing the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's directions. Concurrently, Vero cell monolayers in six-well plates were transfected with the plasmid encoding the ACE2 receptor protein utilizing the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's directions. At 24 h post-transfection, cells containing the mutant plasmids, the ACE2 receptor, and normal untransfected cells were washed with TBS–Ca/Mg, trypsinized, and overlayed in a single well of a six-well plate at a ratio of 2 ml (cells transfected with the ACE2 receptor):0.5 ml (cells transfected with the mutant):1.5 ml (untransfected cells). All of the cells transfected with ACE2 were pooled to ensure that every well had an equal amount of cells with receptor expressed on their surface. After incubation for 24 h, the cells were washed with TBS–Ca/Mg and fixed with ice-cold methanol. Immunohistochemistry was performed by utilizing the Vector Laboratories Vectastain Elite ABC kit essentially as described in the manufacturer's directions. Briefly, cells were washed with TBS–Ca/Mg and incubated in TBS blocking buffer supplemented with normal horse serum at room temperature for 1 h. After blocking, cells were reacted with anti-FLAG antibody (1:500) in TBS blocking buffer for 3 h, washed four times with TBS–Ca/Mg, and incubated with biotinylated horse anti-mouse antibody. Excess antibody was removed by four washes with TBS–Ca/Mg and subsequently incubated with Vectastain Elite ABC reagent for 30 min. Finally, cells were washed three times with TBS–Ca/Mg, and reactions were developed with NovaRed substrate (Vector Laboratories) according to the manufacturer's directions. The average size of syncytia for each mutant was determined by analyzing the area of approximately 300 syncytia, from digital images, using the Image Pro Plus 5.0 software package. The averages were then converted to percentages of the average syncytia size of the wild-type SARS-CoV S. Error bars shown represent the standard deviations calculated through comparison of the data from each of three experiments.
Acknowledgments
C.M.P. was supported by a Louisiana Economic Development graduate assistantship from the Louisiana Board of Regents. This research was supported by a USDA NRI grant to K.G.K. and by a grant from the New England Regional Center for Excellence in Biodefense and Emerging Infectious Diseases (D.M.K.). We thank Anton Chestukhin and the Dana Farber/Harvard Cancer Center Monoclonal Antibody Core for production of the SW-111 monoclonal antibody.
References
- Aiyar A., Xiang Y., Leis J. Site-directed mutagenesis using overlap extension PCR. Methods Mol. Biol. 1996;57:177–191. doi: 10.1385/0-89603-332-5:177. [DOI] [PubMed] [Google Scholar]
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Bagai S., Lamb R.A. Truncation of the COOH-terminal region of the paramyxovirus SV5 fusion protein leads to hemifusion but not complete fusion. J. Cell Biol. 1996;135(1):73–84. doi: 10.1083/jcb.135.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K.A., Dutch R.E., Lamb R.A., Jardetzky T.S. Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell. 1999;3(3):309–319. doi: 10.1016/s1097-2765(00)80458-x. [DOI] [PubMed] [Google Scholar]
- Blom N., Gammeltoft S., Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999;294(5):1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- Bos E.C., Heijnen L., Luytjes W., Spaan W.J. Mutational analysis of the murine coronavirus spike protein: effect on cell-to-cell fusion. Virology. 1995;214(2):453–463. doi: 10.1006/viro.1995.0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos E.C., Luytjes W., Spaan W.J. The function of the spike protein of mouse hepatitis virus strain A59 can be studied on virus-like particles: cleavage is not required for infectivity. J. Virol. 1997;71(12):9427–9433. doi: 10.1128/jvi.71.12.9427-9433.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch B.J., van der Zee R., de Haan C.A., Rottier P.J. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 2003;77(16):8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch B.J., Martina B.E., Van Der Zee R., Lepault J., Haijema B.J., Versluis C., Heck A.J., De Groot R., Osterhaus A.D., Rottier P.J. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. U.S.A. 2004;101(22):8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brideau A.D., del Rio T., Wolffe E.J., Enquist L.W. Intracellular trafficking and localization of the pseudorabies virus Us9 type II envelope protein to host and viral membranes. J. Virol. 1999;73(5):4372–4384. doi: 10.1128/jvi.73.5.4372-4384.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullough P.A., Hughson F.M., Skehel J.J., Wiley D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371(6492):37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- Caffrey M., Cai M., Kaufman J., Stahl S.J., Wingfield P.T., Covell D.G., Gronenborn A.M., Clore G.M. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 1998;17(16):4572–4584. doi: 10.1093/emboj/17.16.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. Plenum Press, Inc.; New York, NY: 1995. The Coronavirus Surface Glycoprotein. [Google Scholar]
- Chan D.C., Fass D., Berger J.M., Kim P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89(2):263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- Chang K.W., Sheng Y., Gombold J.L. Coronavirus-induced membrane fusion requires the cysteine-rich domain in the spike protein. Virology. 2000;269(1):212–224. doi: 10.1006/viro.2000.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot R.J., Luytjes W., Horzinek M.C., van der Zeijst B.A., Spaan W.J., Lenstra J.A. Evidence for a coiled-coil structure in the spike proteins of coronaviruses. J. Mol. Biol. 1987;196(4):963–966. doi: 10.1016/0022-2836(87)90422-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348(20):1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Eckert D.M., Kim P.S. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- Foster T.P., Melancon J.M., Baines J.D., Kousoulas K.G. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J. Virol. 2004;78(10):5347–5357. doi: 10.1128/JVI.78.10.5347-5357.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gombold J.L., Hingley S.T., Weiss S.R. Fusion-defective mutants of mouse hepatitis virus A59 contain a mutation in the spike protein cleavage signal. J. Virol. 1993;67(8):4504–4512. doi: 10.1128/jvi.67.8.4504-4512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez L.D., Hoffman L.R., Wolfsberg T.G., White J.M. Virus–cell and cell–cell fusion. Annu. Rev. Cell Dev. Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- Holmes K.V. SARS-associated coronavirus. N. Engl. J. Med. 2003;348(20):1948–1951. doi: 10.1056/NEJMp030078. [DOI] [PubMed] [Google Scholar]
- Ingallinella P., Bianchi E., Finotto M., Cantoni G., Eckert D.M., Supekar V.M., Bruckmann C., Carfi A., Pessi A. Structural characterization of the fusion-active complex of severe acute respiratory syndrome (SARS) coronavirus. Proc. Natl. Acad. Sci. U.S.A. 2004;101(23):8709–8714. doi: 10.1073/pnas.0402753101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokhin O., Li Y., Andonov A., Feldmann H., Flick R., Jones S., Stroeher U., Bastien N., Dasuri K.V., Cheng K., Simonsen J.N., Perreault H., Wilkins J., Ens W., Plummer F., Standing K.G. Mass spectrometric characterization of proteins from the SARS virus: a preliminary report. Mol. Cell Proteomics. 2003;2(5):346–356. doi: 10.1074/mcp.M300048-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348(20):1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Lee B.S., Alvarez X., Ishido S., Lackner A.A., Jung J.U. Inhibition of intracellular transport of B cell antigen receptor complexes by Kaposi's sarcoma-associated herpesvirus K1. J. Exp. Med. 2000;192(1):11–21. doi: 10.1084/jem.192.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescar J., Roussel A., Wien M.W., Navaza J., Fuller S.D., Wengler G., Rey F.A. The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell. 2001;105(1):137–148. doi: 10.1016/s0092-8674(01)00303-8. [DOI] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lontok E., Corse E., Machamer C.E. Intracellular targeting signals contribute to localization of coronavirus spike proteins near the virus assembly site. J. Virol. 2004;78(11):5913–5922. doi: 10.1128/JVI.78.11.5913-5922.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M., Blacklow S.C., Kim P.S. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol. 1995;2(12):1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- Melikyan G.B., Markosyan R.M., Hemmati H., Delmedico M.K., Lambert D.M., Cohen F.S. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 2000;151(2):413–423. doi: 10.1083/jcb.151.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361(9366):1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300(5624):1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Russell C.J., Jardetzky T.S., Lamb R.A. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 2001;20(15):4024–4034. doi: 10.1093/emboj/20.15.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwegmann-Wessels C., Al-Falah M., Escors D., Wang Z., Zimmer G., Deng H., Enjuanes L., Naim H.Y., Herrler G. A novel sorting signal for intracellular localization is present in the S protein of a porcine coronavirus but absent from severe acute respiratory syndrome-associated coronavirus. J. Biol. Chem. 2004;279(42):43661–43666. doi: 10.1074/jbc.M407233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergel T., Morrison T.G. Mutations in the cytoplasmic domain of the fusion glycoprotein of Newcastle disease virus depress syncytia formation. Virology. 1995;210(2):264–272. doi: 10.1006/viro.1995.1343. [DOI] [PubMed] [Google Scholar]
- Seth S., Vincent A., Compans R.W. Mutations in the cytoplasmic domain of a paramyxovirus fusion glycoprotein rescue syncytium formation and eliminate the hemagglutinin-neuraminidase protein requirement for membrane fusion. J. Virol. 2003;77(1):167–178. doi: 10.1128/JVI.77.1.167-178.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel J.J., Wiley D.C. Coiled coils in both intracellular vesicle and viral membrane fusion. Cell. 1998;95(7):871–874. doi: 10.1016/s0092-8674(00)81710-9. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331(5):991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.C., Seo M.Y., Stadler K., Yoo B.J., Choo Q.L., Coates S.R., Uematsu Y., Harada T., Greer C.E., Polo J.M., Pileri P., Eickmann M., Rappuoli R., Abrignani S., Houghton M., Han J.H. Synthesis and characterization of a native, oligomeric form of recombinant severe acute respiratory syndrome coronavirus spike glycoprotein. J. Virol. 2004;78(19):10328–10335. doi: 10.1128/JVI.78.19.10328-10335.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauber R., Pfleiderera M., Siddell S. Proteolytic cleavage of the murine coronavirus surface glycoprotein is not required for fusion activity. J. Gen. Virol. 1993;74(Pt. 2):183–191. doi: 10.1099/0022-1317-74-2-183. [DOI] [PubMed] [Google Scholar]
- Tan K., Liu J., Wang J., Shen S., Lu M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc. Natl. Acad. Sci. U.S.A. 1997;94(23):12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong S., Li M., Vincent A., Compans R.W., Fritsch E., Beier R., Klenk C., Ohuchi M., Klenk H.D. Regulation of fusion activity by the cytoplasmic domain of a paramyxovirus F protein. Virology. 2002;301(2):322–333. doi: 10.1006/viro.2002.1594. [DOI] [PubMed] [Google Scholar]
- Tripet B., Howard M.W., Jobling M., Holmes R.K., Holmes K.V., Hodges R.S. Structural characterization of the SARS-coronavirus spike S fusion protein core. J. Biol. Chem. 2004;279(20):20836–20849. doi: 10.1074/jbc.M400759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J.C., Zelus B.D., Holmes K.V., Weiss S.R. The N-terminal domain of the murine coronavirus spike glycoprotein determines the CEACAM1 receptor specificity of the virus strain. J. Virol. 2003;77(2):841–850. doi: 10.1128/JVI.77.2.841-850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waning D.L., Russell C.J., Jardetzky T.S., Lamb R.A. Activation of a paramyxovirus fusion protein is modulated by inside–out signaling from the cytoplasmic tail. Proc. Natl. Acad. Sci. U.S.A. 2004;101(25):9217–9222. doi: 10.1073/pnas.0403339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenhorn W., Dessen A., Harrison S.C., Skehel J.J., Wiley D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387(6631):426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- Weissenhorn W., Calder L.J., Wharton S.A., Skehel J.J., Wiley D.C. The central structural feature of the membrane fusion protein subunit from the Ebola virus glycoprotein is a long triple-stranded coiled coil. Proc. Natl. Acad. Sci. U.S.A. 1998;95(11):6032–6036. doi: 10.1073/pnas.95.11.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenhorn W., Carfi A., Lee K.H., Skehel J.J., Wiley D.C. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol. Cell. 1998;2(5):605–616. doi: 10.1016/s1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- White J.M. Membrane fusion. Science. 1992;258(5084):917–924. doi: 10.1126/science.1439803. [DOI] [PubMed] [Google Scholar]
- Wu X.D., Shang B., Yang R.F., Yu H., Ma Z.H., Shen X., Ji Y.Y., Lin Y., Wu Y.D., Lin G.M., Tian L., Gan X.Q., Yang S., Jiang W.H., Dai E.H., Wang X.Y., Jiang H.L., Xie Y.H., Zhu X.L., Pei G., Li L., Wu J.R., Sun B. The spike protein of severe acute respiratory syndrome (SARS) is cleaved in virus infected Vero-E6 cells. Cell Res. 2004;14(5):400–406. doi: 10.1038/sj.cr.7290240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Lou Z., Liu Y., Pang H., Tien P., Gao G.F., Rao Z. Crystal structure of severe acute respiratory syndrome coronavirus spike protein fusion core. J. Biol. Chem. 2004;279(47):49414–49419. doi: 10.1074/jbc.M408782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Q., Compans R.W. Differences in the role of the cytoplasmic domain of human parainfluenza virus fusion proteins. J. Virol. 1995;69(11):7045–7053. doi: 10.1128/jvi.69.11.7045-7053.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye R., Montalto-Morrison C., Masters P.S. Genetic analysis of determinants for spike glycoprotein assembly into murine coronavirus virions: distinct roles for charge-rich and cysteine-rich regions of the endodomain. J. Virol. 2004;78(18):9904–9917. doi: 10.1128/JVI.78.18.9904-9917.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W., Hao Y., Zhang Y., Peng W., Qin E., Cai Y., Wei K., Wang J., Chang G., Sun W., Dai S., Li X., Zhu Y., Li J., Wu S., Guo L., Dai J., Wan P., Chen T., Du C., Li D., Wan J., Kuai X., Li W., Shi R., Wei H., Cao C., Yu M., Liu H., Dong F., Wang D., Zhang X., Qian X., Zhu Q., He F. Proteomic analysis on structural proteins of Severe Acute Respiratory Syndrome coronavirus. Proteomics. 2004;4(2):492–504. doi: 10.1002/pmic.200300676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelus B.D., Schickli J.H., Blau D.M., Weiss S.R., Holmes K.V. Conformational changes in the spike glycoprotein of murine coronavirus are induced at 37 degrees C either by soluble murine CEACAM1 receptors or by pH 8. J. Virol. 2003;77(2):830–840. doi: 10.1128/JVI.77.2.830-840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Hao Y., Gershon M.D., Ambron R.T., Gershon A.A. Targeting of glycoprotein I (gE) of varicella-zoster virus to the trans-Golgi network by an AYRV sequence and an acidic amino acid-rich patch in the cytosolic domain of the molecule. J. Virol. 1996;70(10):6563–6575. doi: 10.1128/jvi.70.10.6563-6575.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]