

Abstract
Because phosphorylation of the infectious bronchitis virus (IBV) nucleocapsid protein (N) may regulate its multiple roles in viral replication, the dynamics of N phosphorylation were examined. 32P-orthophosphate labeling and Western blot analyses confirmed that N was the only viral protein that was phosphorylated. Pulse labeling with 32P-orthophosphate indicated that the IBV N protein was phosphorylated in the virion, as well as at all times during infection in either chicken embryo kidney cells or Vero cells. Pulse-chase analyses followed by immunoprecipitation of IBV N proteins using rabbit anti-IBV N polyclonal antibody demonstrated that the phosphate on the N protein was stable for at least 1 h. Simultaneous labeling with 32P-orthophosphate and 3H-leucine identified a 3.5-fold increase in the 32P:3H counts per minute (cpm) ratio of N in the virion as compared to the 32P:3H cpm ratio of N in the cell lysates from chicken embryo kidney cells, whereas in Vero cells the 32P:3H cpm ratio of N from the virion was 10.5-fold greater than the 32P:3H cpm ratio of N from the cell lysates. These studies are consistent with the phosphorylation of the IBV N playing a role in assembly or maturation of the viral particle.
Keywords: IBV, Pulse-chase, Phosphorylation, Nucleocapsid protein, Double labeling
Introduction
Infectious bronchitis virus (IBV) is a prototype of the Coronaviridae family (Collisson et al., 1992). Coronaviruses are positive-stranded RNA viruses that primarily cause respiratory and enteric infections. Infection with highly contagious IBV causes respiratory disease in chickens resulting in high mortality in young chicks. It also affects the enteric, reproductive, and urinary systems (Alexander and Gouch, 1978, Crinion and Hofstad, 1972).
IBV has four structural proteins, the membrane (M), spike (S), nucleocapsid (N), and small membrane associated (E) proteins. Similar to other coronavirus N proteins, the IBV N is a basic, phosphorylated protein. The 409 amino acid IBV N has a highly conserved region between residues 238 and 293 (Hogue and Brian, 1986, Wilbur et al., 1986, Williams et al., 1992). The N protein directly binds viral genomic RNA and forms a helical ribonucleoprotein complex (RNP) (Davies et al., 1981). It has been implicated in playing a role in genome replication, transcription of subgenomic RNA, translation, and formation of the nucleocapsid (Masters and Sturman, 1990, Tahara et al., 1998).
Murine hepatitis virus (MHV) N protein, the most abundant viral protein, has been reported to be synthesized almost 4–5 h post-infection (p.i.) in Sac (−) cells infected at a high multiplicity of infection (M.O.I.) (Siddell et al., 1980). In MHV, a 57-kDa (p57) non-phosphorylated form is the precursor to the 60-kDa (p60) phosphorylated form of the protein. The p57 is present only in the cytosol, whereas p60 is also found associated with membranes (Stohlman et al., 1983).
Analysis of the tryptic peptides of the 32P-labeled N proteins of two plaque morphology variants of the neurovirulent JHM strain of MHV and the non-neurovirulent prototype strain A59 indicated that the N protein of MHV strains differ in their sites of phosphorylation (Wilbur et al., 1986). No differences were detected in the tryptic peptide maps of the MHV N protein purified from the virion, the infected cytosol and the infected cellular membranes (Wilbur et al., 1986). Although two phosphorylated forms of the N protein, 50 kDa and 55 kDa, were detected in bovine coronavirus (BCoV)-infected cells, only the 50-kDa form was detected in the virion. The 55-kDa form was susceptible to calf intestinal alkaline phosphatase (CIAP) treatment, while the 50-kDa form was not susceptible to CIAP treatment (Hogue, 1995, King and Brian, 1982).
Whereas the IBV N protein has been shown to be phosphorylated in infected cells (Lomniczi and Morser, 1981), the current study demonstrates that the N protein is phosphorylated at all times in cell lysates, as well as in the virion. We also show that the phosphorylation of N is greater in the IBV viral particle.
Results
N from infected cell lysates and the virion differ in electrophoretic mobility
The N protein is the only coronavirus structural protein known to be phosphorylated, with the exception of the M protein in SARS CoV which has recently been shown by mass spectrometry to have a unique phosphorylation site. (Siddell, 1995, Zeng et al., 2004). Because phosphorylation is known to play critical roles in the function of proteins, the nature of the phosphorylation of N was compared in the cell at varying times p.i. and in the supernatant virion. At 8 h p.i., and 16 h p.i., 32P-orthophosphate (32Pi)-labeled IBV N protein was purified by immunoprecipitation of infected Vero cell lysates and from supernatant viral particles, respectively, using rabbit anti-N polyclonal antibody. The N protein from both the intracellular lysates and virions were found to be phosphorylated (Fig. 1A). Two phosphorylated forms of the cell lysate N were detected, as distinguished by electrophoretic mobilities (Fig. 1A). Using Western blot analysis, both bands reacted with chicken anti-IBV polyclonal antibody (Fig. 1B) or rabbit anti-N polyclonal antibody (not shown), suggesting that the protein with a faster electrophoretic mobility is either a degradation product or an isoform of N. In the case of the virion N, Western blot analyses indicated that the band corresponding to N protein consistently migrated somewhat slower than the cellular N (Fig. 1B).
Fig. 1.
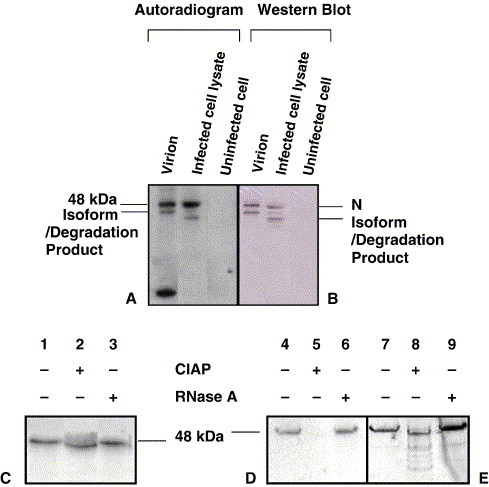
Calf intestinal alkaline phosphatase treatment (CIAP) of the IBV N protein. 32P-orthophosphate-labeled IBV N protein from lysates of infected Vero cells and from virions was immunoprecipitated with rabbit anti-IBV polyclonal antibody before separating by SDS–PAGE. Panel A represents the autoradiogram and panel B, the Western blot using chicken anti-IBV (Gray) polyclonal antibody. The labeled N protein was treated with 20 U CIAP and resolved by denaturing electrophoresis in a 15% polyacrylamide gel. Panel C represents an autoradiogram of the virion N. Panels D and E show the autoradiogram and Western blot, respectively, of cellular N obtained 8 h p.i.
IBV N protein mobility was affected by alkaline phosphatase treatment
32Pi-labeled N proteins from infected cells and virions were treated with calf intestinal alkaline phosphatase (CIAP), which removes phosphates from phosphoserine and phosphothreonine residues. As determined by autoradiography, the labeled cellular N protein was susceptible to CIAP treatment, resulting in the absence of detectable phosphorylation of N from infected cell lysates (Fig. 1D). The cellular N protein completely lost its phosphate (on treatment with alkaline phosphate) (Fig. 1D) and the dephosphorylated cellular N protein has an increased mobility as compared to the unphosphorylated cellular N (1E). The Western blot confirms that the IBV N protein was indeed present in the CIAP-treated immunoprecipitate from the cell lysate (Fig. 1E).
Susceptibility of the virion N protein to CIAP was indicated by a slight increase in mobility of the treated protein (Fig. 1C). Greater concentrations (up to 40 U) of CIAP did not remove the remaining labeled phosphate (data not shown). To confirm that the TRIZOL extraction had removed RNA and that the band observed in the autoradiograph was not due to the presence of labeled RNA bound to the N protein, the preparations from both sources were treated with 10 μg of RNase A. There was no detectable decrease in the intensity of the bands from the RNase A-treated samples as compared to the untreated controls (Figs. 1C and D). Therefore, RNA was not associated with the IBV N protein from the cellular and virion N protein, or alternatively any RNA present was protected from the enzyme activity.
IBV N protein was phosphorylated at all times during infection
The kinetics of the phosphorylation of the IBV N protein has not been previously reported, nor has it been shown as to whether N is phosphorylated or differentially phosphorylated during IBV infection and maturation. IBV Beaudette-infected Vero cells were treated with actinomycin-D (5 μg/ml), an inhibitor of RNA synthesis, immediately after infection and continued during the 1 h of labeling with 100 μCi 32Pi. One-eighth of the volume of the TRIZOL-treated cell lysate was used for denaturing gel electrophoresis. As shown by 32Pi labeling, IBV N was the only phosphorylated viral protein in infected cell lysates, as compared with the uninfected cell lysate control (Fig. 2A). N protein, as represented by a phosphorylated band at approximately a 48-kDa molecular mass, was phosphorylated throughout infection, with label appearing around 6 h p.i. until at least 18 h p.i., the last hour examined (Fig. 2A). A second, slower phosphorylated band, appearing to be slightly larger than the 36.4-kDa marker, was determined to be a cellular protein because it was also clearly present in the uninfected cell lysate control. Western blot analyses with rabbit anti-N polyclonal antibody, specific for IBV N protein, highlighted a band at approximately 48 kDa molecular mass (Fig. 2B).
Fig. 2.
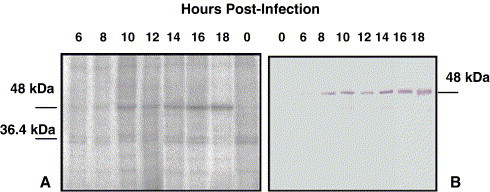
Time course of phosphorylation of the IBV N protein in the presence of actinomycin D. Phosphorylation of IBV N protein from IBV-infected Vero cells was evaluated by labeling with 100 μCi 32P-orthophosphate for 1 h in the presence of 5 μg/ml actinomycin-D. At varying times p.i. (as indicated above the figure), equal amounts of whole cell lysate were resolved by 15% SDS–PAGE and the labeled N visualized by autoradiography. Panel A represents the phosphor image, and panel B represents the Western blot using rabbit anti-N polyclonal antibody. IBV N protein was observed at approximately 48 kDa molecular mass.
35S methionine pulse labeling followed by unlabeled media chase determined the stability of the IBV N protein
The stability of the IBV N protein was first determined. IBV Beaudette-infected Vero cells were pulse labeled for 0.5 h with 35S-methionine at 7.5 h p.i, before chasing with excess cold methionine for 0.5 h (that is, 8 h p.i.) to 3.5 h (that is, 11.5 h p.i.) when the infected cells were harvested. IBV N protein was isolated by immunoprecipitation at saturating concentrations of rabbit anti-N-polyclonal antibody, such that all N in the infected cell lysates was precipitated. N protein in cell lysates was consistently detected after 30 min of pulse labeling, whereas labeled N was not detected with a 15-min pulse-label with 35S-methionine. After labeling for 30 min, media containing an excess of unlabeled methionine were added for varying lengths of time in order to dilute or chase the label from N. Maximum incorporation of the label was observed after the 1.5 h of chase (Figs. 3A and B). As can be seen from the graphic representation of the densitometric analyses of the bands, using a phosphor imager (Fig. 3B), there was a slight decrease in the intensity of the band until the 2.5-h chase period when an increase in intensity was observed at the 3- and 3.5-h chase periods (Fig. 3A). The intensity of 35S-labeled band resulting from the 2- and 2.5-h chase corresponded, respectively, to 75% and 82% of the 1.5-h chase period band intensity. After the 3- and 3.5-h chase with unlabeled methionine, the band intensity corresponded, respectively, to 85% and 92% of the band intensity for the 1.5-h chase. The decrease in the band intensity was never greater than 50%. If we interpret the stability as beginning with 1.5 h, the time of maximum incorporation, it can be deduced that the N protein is stable for at least 2 h. N protein alone was immunoprecipitated as shown by Western blot analysis using chicken anti-IBV antibody (Fig. 1B). No bands were obtained when lysates from uninfected cells were used for immunoprecipitation, using rabbit-anti-N polyclonal antibody (Fig. 3A) or when using non-specific rabbit-anti-reticuloendotheliosis virus gag antibody as a control (data not shown). A faint band slightly larger than 48 kDa was obtained on exposure to an imaging plate when rabbit preimmune serum was used for immunoprecipitation of infected cell lysates. This band did not react with anti-IBV-Gray chicken serum as determined by the Western blot analysis (data not shown) and therefore was not considered a viral protein.
Fig. 3.
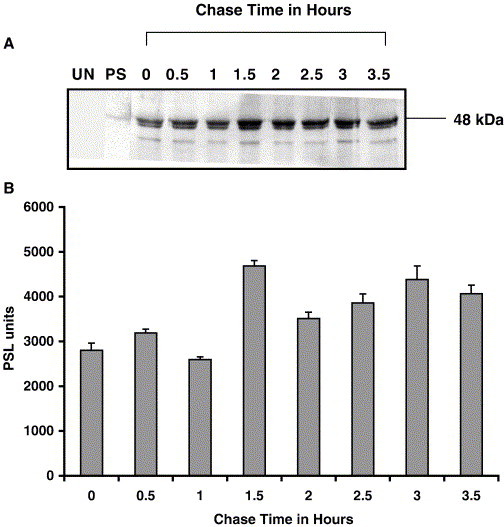
Stability of IBV N protein. Panel A represents a phosphor image, and panel B represents a graphical representation of the densitometric analysis of the bands obtained in the phosphor image. IBV-infected Vero cells were pulse labeled with 25 μCi 35S-methionine for 30 min and chased with a large excess of unlabeled methionine. The N was obtained from IBV-infected cell lysates by immunoprecipitation with rabbit anti-N polyclonal antibody. The 0-h chase time correlated with 8 h p.i. Lysates from uninfected cells precipitated with rabbit anti-N polyclonal antibody (0 h) and lysates from infected cell precipitated with preimmune serum (PS) are shown. Error bars represent standard deviations of two densitometric measurements from one experiment.
Stability of phosphate on IBV N protein or half-life of phosphate on IBV N protein in infected cell lysates
35S-methionine pulse-chase analysis indicated that the IBV N protein was stable for at least 2 h. The modulation of phosphate on a protein can be associated with function. In order to determine the stability of the phosphate on the N protein in terms of half-life, infected cells were labeled with 32Pi for 2 h and the chase analysis was started at 8 h p.i. The chase was continued until 11 h p.i., when N protein was immunoprecipitated from cell lysates at saturating amounts of the antibody and standardized so as to precipitate all the N protein. Figs. 4A and B show that phosphate incorporation continued throughout the 1.5-h chase period, reaching a maximum at the 2-h chase period and then dropped to 96% and 70% of this value observed in the 2.5- and 3-h chase period, respectively. Since phosphate was readily available in the cell, intracellular phosphate pools would be difficult to replace. Labeling on the N protein might increase during the chase period due to labeled cellular-derived phosphate. Similar results obtained for two experiments indicated that the amount of phosphorylation of N does not fall below 70% of the maximum label incorporation during a 1-h chase.
Fig. 4.
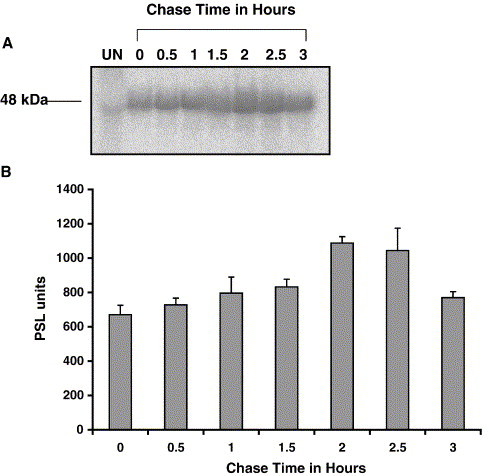
Pulse-chase analysis determined the stability of phosphorylation on the IBV N protein. IBV-infected Vero cells were labeled with 32P-orthophosphate for 2 h and chased with unlabeled phosphate starting at 8 h p.i. until 11 h p.i. Panel A represents a phosphor image of labeled N resolved by denaturing gel electrophoresis, and panel B represents the densitometric analysis of the bands using a Fuji2000 phosphor imager. Error bars represent standard deviations of two densitometric measurements from one experiment.
32P:3H ratios of IBV N protein differ in the virion compared to the cell lysates
To determine variations in the amount of phosphorylation of the N protein during replication, infected cells were labeled with 3H-leucine and 32Pi. The ratio of the counts per minute (cpm) of 32Pi to 3H-leucine provided a relative comparison of phosphate to the amount of protein. The ratios of 32Pi and 3H-leucine on the IBV N protein were determined in both IBV-infected Vero and CEK cells. Results for the one-step growth curve indicated that maximum plaque forming units (pfu) of virus was produced between 15 and 20 h p.i. (Fig. 5 ). Based on these results, dual labeling was determined with virions isolated at 16 h p.i. The 32P:3H cpm ratio for the collected virions was approximately 3.5-fold and 10-fold greater than the ratios obtained for the N protein precipitated 7 h p.i. from CEK cells or Vero cells, respectively (Table 1 ).
Fig. 5.
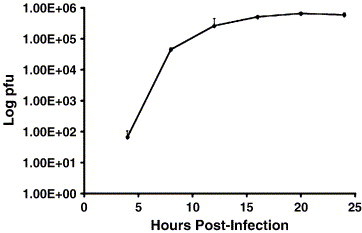
One-step growth curve of IBV in Vero cells. A one-step growth curve was determined by determining the plaque forming units from 200 μl of 10-fold dilutions of IBV collected at varying times p.i. of Vero cells. The experiment was done in triplicate and the averages of three experiments are reported. Bars represent standard deviations.
Table 1.
Dual labeling of IBV N protein in Vero and CEK cells indicated a significant linear increase in the 32P:3H ratio (P < 0.0001) of IBV N protein with the time post-infection
| Time of harvest: hours post-infection | Chicken embryo kidney cellsa | Vero cellsa |
|---|---|---|
| 7 | 0.35 ± 0.004 | 0.24 ± 0.004 |
| 9 | 0.83 ± 0.036 | 0.45 ± 0.003 |
| 11 | 0.80 ± 0.074 | 0.84 ± 0.001 |
| 13 | 0.73 ± 0.017 | 0.85 ± 0.056 |
| 16 (Virion N) | 1.22 ± 0.004 | 2.52 ± 0.006 |
Represents cpm ratios of 32P:3H ± standard deviation.
In order to establish the significance of the differences between the 32P:3H ratios of the N protein isolated at varying hours (7, 9, 11, 13 and 16) p.i. and to also determine the significance of the apparent increase in N phosphorylation with time, an orthogonal polynomial contrast test was calculated across the time points (Kuehl, 2000). The hours p.i. were calculated as the treatment variable and the ratio was used as the response. A significant linear trend (P < 0.0001) in the ratios was obtained (Fig. 6 ). Therefore, there was a direct correlation in the increase in phosphorylation of the N with time. The 32P:3H cpm ratios of the N protein (0.35 for CEK cells or 0.24 for Vero cells) at time point 1, that is 7 h p.i., were significantly different (P < 0.05) from the ratios (1.22 for CEK cells or 2.52 for Vero cells) at time point 5 (16 h p.i.) using the least significant difference (LSD) test (Table 1). The significance of these differences was also confirmed by the Student–Newman–Keuls test and Tukey's studentized range test multiple comparison procedures (Kuehl, 2000). The amount of phosphorylation was significantly greater in the virion N protein and the phosphorylation of the virion N was mostly resistant to CIAP treatment.
Fig. 6.
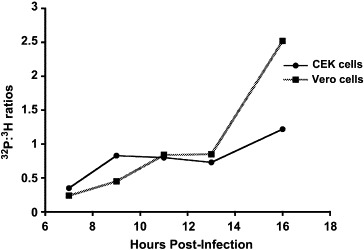
Graphic representation of the 32P:3H cpm ratios of N protein obtained at varying hours p.i. from Vero- and CEK-infected cells and obtained from the purified virion from Vero and CEK cells at 16 h pi. The N was obtained by immunoprecipitation with rabbit anti-N polyclonal antibody, followed by TRIZOL treatment to remove bound RNA and by TCA precipitation to remove unincorporated label before determining the 32P:3H cpm ratios. Standard deviations of two sets of experiments were less than 0.05. A significant linear trend (P < 0.0001) in the ratios was obtained.
Discussion
Coronavirus N proteins are known to be phosphorylated though the mechanisms regulating the functions of the N gene are not known (Siddell, 1995). The sites of phosphorylation for the N protein expressed in Vero and SF9 have been identified though the sites of phosphorylation of N protein isolated from virus-infected cells or from the virion have not been determined (Chen et al., 2005). While the coronavirus N has been implicated in transcription, replication, translation, and packaging of the viral genome, it has been postulated that phosphorylation might functionally regulate the N protein (Masters et al., 1990). Following an M.O.I. of 5, synthesis of 35S-methionine-labeled MHV N was detected 4.5 h p.i. (Cheley and Anderson, 1981). With an M.O.I. between 2 and 3, we found that both synthesis of IBV N and phosphorylation could be detected by 6 h p.i. until at least 18 h p.i. Because differential phosphorylation might regulate the multiple functions of the N, it would be of interest to determine the sites of phosphorylation at varying times during infection.
Unlike the MHV N protein, that was found to be stable for only 1 h (Perlman et al., 1986) during pulse-chase labeling with 35S-methionine, the IBV N protein was stable for at least 2 h. The stability of N phosphorylation would decrease with removal of 32Pi from the protein. Our results indicated that the degree of phosphorylation of N was greatest only when chased for 2 h. The delay in removing the viral 32Pi may relate to the presence of the intracellular 32Pi obtained from other cellular processes. The intensity of labeled phosphate dropped down to 70% of this maximum value when chased for an additional hour. A consequence of the decrease to 70% of the phosphorylation, compared to the maximum level observed after 2 h labeling (7 h p.i.), could indicate a change or modification in protein function.
The IBV N protein was isolated during the pulse-chase analyses by immunoprecipitation using saturating amounts of rabbit polyclonal antibody to the N protein, such that both labeled and unlabeled N were precipitated from infected cell lysates. All analyses were based on the nearly identical results of duplicate sets of experiments. MHV M has been shown to co-precipitate with N using anti-MHV N monoclonal antibody (Narayanan et al., 2000). With BCoV, rabbit polyclonal antibody to N protein does not co-precipitate M in infected cells (Nguyen and Hogue, 1997). In our experiments, no proteins co-precipitated with IBV N using the rabbit-anti-N polyclonal antibody. Differences between such studies with MHV and IBV may be due to differences in the viruses or the conditions used for immunoprecipitation.
Dual labeling with 32Pi and 3H-leucine indicated that the virion N was more phosphorylated than the N from infected Vero or infected CEK cells. However, the 32P:3H cpm ratio of the virion N protein from Vero cells was greater than the ratio from CEK cells. The electrophoretic mobility of the virion N during SDS–PAGE was slightly slower than the cellular N, as might be expected with additional phosphorylation. In the case of measles virus, the nucleoprotein, phosphorylated on both serine and threonine, is preferentially assembled into the nucleocapsids (Gombart et al., 1995). Increased phosphorylation could relate to preferential binding of N protein to viral RNA for packaging or assembly into nucleocapsids. In the case of Chandipura virus, homodimerization of the phosphoprotein (P) occurs via the N-terminal 46 amino acids of the protein only after phosphorylation of this region (Raha et al., 2000). A global conformational change in the N-terminal domain of the P of the Chandipura virus is induced by phosphorylation (Raha et al., 1999). Similarly, phosphorylation of the IBV N protein could induce a conformational change that promotes both N–N interactions and RNA binding. N–N interactions may be important in nucleocapsid assembly and in compacting the nucleocapsid. The N associated with virions was mostly resistant to CIAP treatment which targets serine- and threonine-bound phosphates and to RNase treatment. It is likely that the hyperphosphorylated form of the IBV N protein binds genomic RNA thereby forming tight compact complexes that are resistant to enzyme treatment. Narayanan et al. (2003) showed that the MHV N protein in the virion nucleocapsid and in the intracellular genome-length RNP complexes that bound to the viral M protein was tightly self-associated, such that its association was retained even after extensive RNAse A treatment. However, this was not the case with RNP complexes formed with subgenomic RNA (Narayanan et al., 2003).
The mechanism accounting for hyperphosphorylation in virion N protein propagated in either Vero or CEK cells is not known, but it is of interest that a protein kinase activity was reported to be associated with purified virus preparations of MHV (Siddell et al., 1981a). However, the degree of phosphorylation may depend on the cellular functions since the N packaged in Vero cells apparently was associated with 7-fold more labeled phosphate than the N in virions assembled from CEK cells. In addition to the differences in avian versus primate origin, respectively, the CEK cells are not immortalized and make interferon, as compared with Vero cells that are deficient in interferon genes. Because the hyperphosphorylated state of N was consistently observed in viral particles from both cell types, it may be essential in packaging and assembly of genomic RNA into infectious virions.
Materials and methods
Virus and cells
The IBV Beaudette strain, used for all experiments in this study, was propagated in Vero and chicken embryo kidney (CEK) cells in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) or 10% chicken serum, respectively. Virus infections were carried out with DMEM containing 2% serum. Growth kinetics of IBV replication in Vero cells were performed by infecting Vero cells with virus at a multiplicity of infection (M.O.I) between 2 and 3 for 1 h. Virus was harvested at 4, 8, 12, 16, 20, and 24 h p.i. and quantified in terms of plaque forming units (pfu) per 200 μl of media (Darbyshire et al., 1978). Two hundred microliters of 10-fold serial dilutions of the supernatant containing the virus were used for the plaque assay. The experiments were done in triplicates.
Phosphate labeling
Vero or CEK cells were infected with IBV Beaudette at an M.O.I. between 2 and 3 for 1 h. Fresh DMEM containing 2% serum was used for virus-infected cells. For 1 h prior to labeling with 100 μCi 32P-orthophosphate (32Pi), cells were starved in phosphate-free DMEM containing 2% phosphate-free serum. The cells were labeled for 1 h (Lomniczi and Morser, 1981). The cells were washed with Tris-buffered saline (TBS) (Sambrook et al., 1989) and either 0.5 ml of lysis buffer [1 mM iodoacetamide, 1 mM phenyl methyl sulfonyl fluoride (PMSF), 100 μM sodium orthovanadate, 4 μg/ml aprotinin, 1 mM EDTA, 1% NP40, 10 mM Tris (pH 8), 0.1% SDS, 150 mM NaCl, and 0.025% sodium azide] was added to the 60-mm tissue culture dishes or 1 ml lysis buffer was added to 100 mm tissue culture dishes. Cells were scraped off the dish surface and lysis was carried out at 4 °C for 30 min, before immunoprecipitation.
Pulse-chase analysis
Vero and CEK cells were infected with IBV Beaudette strain at an M.O.I. between 2 and 3 for 1 h, after which fresh DMEM containing 2% serum was added. To determine the stability of IBV N protein, at 7 h p.i., 2 ml of hypertonic medium containing 2% serum (DMEM containing 0.335 M NaCl) was added to the cells to dissociate the ribosomes and thereby synchronize the cells for protein synthesis. After 1 h, the cells were washed with phosphate-buffered saline (PBS) (Sambrook et al., 1989) and 1 ml serum-free, methionine-free DMEM (Gibco, Carlsbad, CA) containing 100 μCi 35S-methionine (Perkin Elmer, Wellesley, MA) was added to the virus-infected cells. Labeling was carried out for 30 min. To determine the rate of phosphate removal on IBV N, the cells were washed with TBS and starved in phosphate-free DMEM containing 2% serum (phosphate free) for 1 h before labeling the virus-infected cells with 100 μCi 32P-orthophosphate for 2 h (Siddell et al., 1981b).
Chase analysis was carried out by removing the labeling medium, washing the cells twice with PBS, and adding 3 ml of chase medium, either DMEM containing 2% serum and 1 mM l-methionine, or DMEM containing 2% serum and 10 mM phosphate buffer. The 32Pi label in cells was chased for 2.5–3 h depending on the experiment. At the end of the chase period, the cells were washed with TBS, and after scraping the cells with a rubber policeman, the cells were incubated for 30 min at 4 °C in 0.5 ml/plate of lysing buffer and processed for immunoprecipitation (Siddell et al., 1981b).
Immunoprecipitation
The lysates were centrifuged at 1000×g to pellet the nuclei and cell debris. Rabbit anti-N polyclonal antibody was added to the lysate at saturating concentrations (as previously determined) to precipitate all the N protein. The mixture was incubated on a shaker at 4 °C for 1 h. Fifty microliters of a 50% slurry of protein-A-agarose (Pierce, Rockford, IL) in dilution buffer – 10 mM Tris, pH 8; 140 mM NaCl; 0.025% sodium azide (Tris saline azide, TSA); and 1% NP40 – was added to capture the antigen–antibody complex. This mix was incubated at 4 °C for 1 h. The protein-A-agarose antigen–antibody complex was washed three times with TSA and eluted by adding 20 μl of 2× SDS–PAGE loading buffer and heating at 100 °C for 10 min (Ausubel et al., 1987).
Western blot analyses
After resolving by denaturing gel electrophoresis, the proteins were transferred from the gel to a nitrocellulose membrane (Sambrook et al., 1989). The membrane was blocked with blocking solution (5% skim milk, 0.02% Tween 20 in TBS) for 2 h at room temperature (R.T.). Chicken anti-IBV (Gray strain) antibody (1:1000 dilution in blocking solution) was added to the blot and allowed to react with the proteins for 1 h at R.T. After two washes with washing buffer, TBS-0.02% Tween 20 (TBS-T), alkaline phosphatase-conjugated goat anti-chicken antibody (KPL) (1:10,000 dilution in blocking solution) was added to the blot and allowed to react for 1 h at R.T. After two washes with washing buffer, alkaline phosphatase substrate, 5-bromo-4-choloro-3-indolyly phosphate (BCIP), and nitroblue tetrazolium (NBT), alkaline phosphatase conjugate substrate (Bio-Rad, Richmond, CA), were added to the blot for color development before removing the substrate and washing with wash buffer (Sambrook et al., 1989).
Dual labeling of IBV N
One hour prior to labeling, virus-infected Vero or CEK cells were starved of leucine and phosphate in serum-free, leucine-, and phosphate-free DMEM (Specialty Media, Phillipsburg, NJ). Cells were labeled with 100 μCi 32Pi and 10 μCi 3H-leucine (Perkin Elmer, Wellesley, MA) for 1 h (Stohlman and Lai, 1979). At the end of the labeling period, the cells were harvested as described above and IBV N was immunoprecipitated using rabbit anti-N polyclonal antibody. RNA was extracted from the protein using TRIZOL (Invitrogen, Carlsbad, CA). IBV N was isolated from the organic phase by precipitating with isopropyl alcohol. The precipitate was dissolved in 2× SDS–PAGE gel loading buffer and used for TCA precipitation to determine the 3H and 32P counts per minute (cpm) using a scintillation counter and the 32P:3H ratios were determined (Ausubel et al., 1987). The experiments were done in duplicate. Statistical analysis was performed using the orthogonal polynomial contrast test, least significant difference, Student–Newman–Keuls test, and Tukey's studentized range test (Table 1) (Kuehl, 2000).
Purification of IBV virion
Virus from supernatants of five 100 mm tissue culture plates, infected at an M.O.I. between 2 and 3, were concentrated on a 20% sucrose cushion, by centrifuging at 35,000 rpm in an SW 55 Ti Beckmann rotor at 4 °C for 2.5 h. The virus pellets were suspended in 100 μl TEN buffer (10 mM Tris pH 7.4, 1 mM EDTA, 150 mM NaCl) and centrifuged through a linear 30–50% glycerol–tartrate gradient at 35,000 rpm, in an SW 55 Ti Beckmann rotor at 4 °C for 18 h. A distinct band corresponding to virions was obtained at approximately 1.18 g/ml density. The virus from this band was used for various analyses (Bingham, 1975).
Alkaline phosphatase treatment
IBV N isolated from cells or from virions after immunoprecipitation with rabbit anti-N polyclonal antibody was treated with TRIZOL to remove bound RNA. The purified protein was then dialyzed against PBS for 4 h at 4 °C. The eluted and dialyzed protein was aliquotted into three parts; as an untreated control, for treatment with CIAP, and for treatment with RNase A as a second control. The protein was treated with 20 U of calf intestinal alkaline phosphatase (CIAP) (Promega, Madison, WI) in 20 μl reaction volume (Hogue, 1995). The reaction was incubated at 37 °C for 30 min and stopped by heating at 70 °C for 15 min. The protein was resolved by 15% SDS–PAGE followed by a Western blot analyses with chicken anti-IBV (Gray strain) antibody (Sambrook et al., 1989).
Acknowledgments
This work was supported by grants from the U.S. Poultry and Egg Association (297), USDA Formula Animal Health Funds (1433), and NIH grant AI 51493 and the Institute of Food Safety and Engineering at Texas A&M University. We also thank Mr. Veerabhadran Baladandayuthapani for his help with the statistical analysis.
References
- Alexander D.J., Gouch R.E. A long-term study of the pathogenesis of infections of fowls with three strains of avian infectious bronchitis virus. Res. Vet. Sci. 1978;24:228–233. [PubMed] [Google Scholar]
- Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Smith J.A., Kevin S. Current Protocols in Molecular Biology. John Wiley and Sons, Inc.; New York: 1987. pp. 10.18.7–10.18.8. [Google Scholar]
- Bingham R.W. The polypeptide compositions of avian infectious bronchitis virus. Arch. Virol. 1975;49:207–216. doi: 10.1007/BF01317539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheley S., Anderson R. Cellular synthesis and modification of murine hepatitis virus polypeptides. J. Gen. Virol. 1981;54:301–311. doi: 10.1099/0022-1317-54-2-301. [DOI] [PubMed] [Google Scholar]
- Chen H., Gill A., Dove B.K., Emmett S.R., Kemp C.F., Ritchie M.A., Dee M., Hiscox J.A. Mass spectroscopic characterization of the coronavirus infectious bronchitis virus nucleoprotein and elucidation of the role of phosphorylation in RNA binding by using surface plasmon resonance. J. Virol. 2005;79:1164–1179. doi: 10.1128/JVI.79.2.1164-1179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collisson E.W., Parr R.L., Wang L., Williams A.K. An overview of the molecular characteristics of avian infectious bronchitis virus. Poult. Sci. Rev. 1992;4:41–55. [Google Scholar]
- Crinion R.A., Hofstad M.S. Pathogenicity of two embryo-passage levels of avian infectious bronchitis virus for the oviduct of young chickens of various ages. Avian Dis. 1972;16:967–973. [PubMed] [Google Scholar]
- Darbyshire J.H., Cook J.K., Peters R.W. Growth comparisons of avian infectious bronchitis virus strains in organ cultures of chicken tissues. Arch. Virol. 1978;56:317–325. doi: 10.1007/BF01315282. [DOI] [PubMed] [Google Scholar]
- Davies H.A., Dourmashkin R.R., Macnaughton M.R. Ribonucleoprotein of avian infectious bronchitis virus. J. Gen. Virol. 1981;53:67–74. doi: 10.1099/0022-1317-53-1-67. [DOI] [PubMed] [Google Scholar]
- Gombart A.F., Hirano A., Wong T.C. Nucleoprotein phosphorylated on both serine and threonine is preferentially assembled into the nucleocapsids of measles virus. Virology. 1995;37:63–73. doi: 10.1016/0168-1702(95)00020-q. [DOI] [PubMed] [Google Scholar]
- Hogue B.G. Bovine coronavirus nucleocapsid protein processing and assembly. Adv. Exp. Med. Biol. 1995;380:259–263. doi: 10.1007/978-1-4615-1899-0_41. [DOI] [PubMed] [Google Scholar]
- Hogue B.G., Brian D.A. Structural proteins of human respiratory coronavirus OC43. Virus Res. 1986;5:131–144. doi: 10.1016/0168-1702(86)90013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B., Brian D.A. Bovine coronavirus structural proteins. J. Virol. 1982;42:700–707. doi: 10.1128/jvi.42.2.700-707.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehl, R.O., 2000. Design of Experiments: Statistical Principles of Research Design and Analysis, 2nd edition. Duxbur Press, CA, pp. 85–91, 110–115.
- Lomniczi B., Morser J. Polypeptides of infectious bronchitis virus. I. Polypeptides of the virion. J. Gen. Virol. 1981;55:155–164. doi: 10.1099/0022-1317-55-1-155. [DOI] [PubMed] [Google Scholar]
- Masters P.S., Sturman L.S. Background paper. Functions of the coronavirus nucleocapsid protein. Adv. Exp. Med. Biol. 1990;276:235–238. doi: 10.1007/978-1-4684-5823-7_32. [DOI] [PubMed] [Google Scholar]
- Masters P.S., Parker M.M., Ricard C.S., Duchala C., Frana M.F., Holmes K.V., Sturman L.S. Structure and function studies of the nucleocapsid protein of mouse hepatitis virus. Adv. Exp. Med. Biol. 1990;276:239–246. doi: 10.1007/978-1-4684-5823-7_33. [DOI] [PubMed] [Google Scholar]
- Narayanan K., Maeda A., Maeda J., Makino S. Characterization of the coronavirus M protein and nucleocapsid interaction in infected cells. J. Virol. 2000;74:8127–8134. doi: 10.1128/jvi.74.17.8127-8134.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Kim K.H., Makino S. Characterization of N protein self-association in coronavirus ribonucleoprotein complexes. Virus Res. 2003;98:131–140. doi: 10.1016/j.virusres.2003.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen V.P., Hogue B.G. Protein interactions during coronavirus assembly. J. Virol. 1997;71:9278–9284. doi: 10.1128/jvi.71.12.9278-9284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman S., Ries D., Bogler E., Chang L.J., Stolfus C.M. MHV nucleocapsid synthesis in the presence of cycloheximide and the accumulation of negative stranded RNA. Virus Res. 1986;6:261–272. doi: 10.1016/0168-1702(86)90074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha T., Chattopadhyay D., Roy S. A phosphorylation-induced major structural change in the N-terminal domain of the P protein of Chandipura virus. Biochemistry. 1999;38:2110–2116. doi: 10.1021/bi980734c. [DOI] [PubMed] [Google Scholar]
- Raha T., Samal E., Majumdar A., Basak S., Chattopadhyay D., Chattopadhyay D.J. N-terminal region of P protein of Chandipura virus is responsible for phosphorylation-mediated homodimerization. Protein Eng. 2000;13:437–444. doi: 10.1093/protein/13.6.437. [DOI] [PubMed] [Google Scholar]
- Sambrook S., Fritsch E.F., Maniatis T. 2nd ed. Cold Spring Laboratory Press; New York: 1989. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- Siddell S.G. Plenum Press; New York: 1995. The Coronaviridae. [Google Scholar]
- Siddell S.G., Wege H., Barthel A., ter Meulen V. Coronavirus JHM: cell-free synthesis of structural protein p60. J. Virol. 1980;33:10–17. doi: 10.1128/jvi.33.1.10-17.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddell S.G., Barthel A., ter Meulen V. Coronavirus JHM: a virion-associated protein kinase. J. Gen. Virol. 1981;52:235–243. doi: 10.1099/0022-1317-52-2-235. [DOI] [PubMed] [Google Scholar]
- Siddell S.G., Wege H., Barthel A., ter Meulen V. Coronavirus JHM: intracellular protein synthesis. J. Gen. Virol. 1981;53:145–155. doi: 10.1099/0022-1317-53-1-145. [DOI] [PubMed] [Google Scholar]
- Stohlman S.A., Lai M.M.C. Phosphoproteins of murine hepatitis virus. J. Virol. 1979;32:672–675. doi: 10.1128/jvi.32.2.672-675.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohlman S.A., Fleming J.O., Patton C.D., Lai M.M. Synthesis and subcellular localization of the murine coronavirus nucleocapsid protein. Virology. 1983;130:527–532. doi: 10.1016/0042-6822(83)90106-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara S.M., Dietlin T.A., Nelson G.W., Stohlman S.A., Manno D.J. Mouse hepatitis virus nucleocapsid protein as a translational effector of viral mRNAs. Adv. Exp. Med. Biol. 1998;440:313–318. doi: 10.1007/978-1-4615-5331-1_41. [DOI] [PubMed] [Google Scholar]
- Wilbur S.M., Nelson G.W., Lai M.M., McMillan M., Stohlman S.A. Phosphorylation of the mouse hepatitis virus nucleocapsid protein. Biochem. Biophys. Res. Commun. 1986;141:7–12. doi: 10.1016/S0006-291X(86)80326-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A.K., Wang L., Sneed L.W., Collisson E.W. Comparative analyses of the nucleocapsid genes of several strains of infectious bronchitis virus and other coronaviruses. Virus Res. 1992;25:213–222. doi: 10.1016/0168-1702(92)90135-V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng R., Ruan H.Q., Jiang X.S., Zhou H., Shi L., Zhang L., Sheng Q.H., Tu Q., Xia Q.C., Wu J.R. Proteomic analysis of SARS associated coronavirus using two-dimensional liquid chromatography mass spectrometry and one-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis followed by mass spectrometric analysis. Proteome Res. 2004;3:549–555. doi: 10.1021/pr034111j. [DOI] [PubMed] [Google Scholar]