

Abstract
The small envelope (E) protein of porcine reproductive and respiratory syndrome virus (PRRSV) is a hydrophobic 73 amino acid protein encoded in the internal open reading frame (ORF) of the bicistronic mRNA2. As a first step towards understanding the biological role of E protein during PRRSV replication, E gene expression was blocked in a full-length infectious clone by mutating the ATG translational initiation to GTG, such that the full-length mutant genomic clone was unable to synthesize the E protein. DNA transfection of PRRSV-susceptible cells with the E gene knocked-out genomic clone showed the absence of virus infectivity. P129-ΔE-transfected cells however produced virion particles in the culture supernatant, and these particles contained viral genomic RNA, demonstrating that the E protein is essential for PRRSV infection but dispensable for virion assembly. Electron microscopy suggests that the P129-ΔE virions assembled in the absence of E had a similar appearance to the wild-type particles. Strand-specific RT-PCR demonstrated that the E protein-negative, non-infectious P129-ΔE virus particles were able to enter cells but further steps of replication were interrupted. The entry of PRRSV has been suggested to be via receptor-mediated endocytosis, and lysomotropic basic compounds and known ion-channel blocking agents both inhibited PRRSV replication effectively during the uncoating process. The expression of E protein in Escherichia coli-mediated cell growth arrests and increased the membrane permeability. Cross-linking experiments in cells infected with PRRSV or transfected with E gene showed that the E protein was able to form homo-oligomers. Taken together, our data suggest that the PRRSV E protein is likely an ion-channel protein embedded in the viral envelope and facilitates uncoating of virus and release of the genome in the cytoplasm.
Keywords: PRRSV, Reverse genetics, Infectious clone, Small envelope protein, Uncoating, Viroporin, Oligomerization, Ion channel activity
Introduction
Porcine reproductive and respiratory syndrome virus (PRRSV) has plagued the global swine industry leading to significant economic losses for pig production worldwide (Nuemann et al., 2005). PRRSV is a member of the family Arteriviridae, which includes equine arteritis virus (EAV), lactate dehydrogenase-elevating virus (LDV) of mice and simian hemorrhagic fever virus (SHFV) (Meulenberg et al., 1993, Snijder and Meulenberg, 1998). Along with the family Coronaviridae, arteriviruses are grouped in the new order of Nidovirales (Snijder et al., 2005). Despite the similar virion morphology and genome organization, PRRSV is divided into two genotypes, the European genotype (Lelystad virus; LV) and the North American genotype, based on their antigenic and genetic characteristics (Meng et al., 1995, Nelsen et al., 1999, Nelson et al., 1993, Wootton et al., 2000).
PRRSV is a small, enveloped virus possessing a single-stranded positive-sense RNA of ∼ 15 kb in size with a 5′ cap and a 3′ polyadenylated tail (Meulenberg et al., 1993, Sagripanti et al., 1986, Snijder and Meulenberg, 1998, Wootton et al., 2000). The PRRSV genome consists of the 5′ untranslated region (UTR), nine open reading frames (ORF1a, ORF1b, ORF2a, ORF2b and ORFs 3 through 7) and the 3′ UTR (Meulenberg et al., 1993, Snijder and Meulenberg, 1998, Wootton et al., 2000). Two large ORFs la and lb occupy the 5′ two-thirds of the genome and encode 13 non-structural proteins, which are suggested to be involved in the genome replication and transcription (Bautista et al., 2002, van Dinten et al., 1999). The remaining ORFs, 2a through 7 in the 3′ terminal 3 kb region, encode six membrane-associated proteins (GP2, E, GP3, GP4, GP5 and M) present in the envelope and nucleocapsid (N) protein (Meulenberg et al., 1995, Wootton et al., 2000). Mature virions are spherical, enveloped particles with a diameter of 50–65 nm and contain a 20- to 30-nm isometric core structure enclosing the genomic RNA (Benfield et al., 1992, Dea et al., 1995).
The small envelope (E) protein is a newly identified structural component in arteriviruses. The PRRSV E protein, also known as 2b protein, is translated from the internal ORF (ORF2b) starting from the + 6 nucleotide position in mRNA2 (Fig. 1A). The E protein is 73 and 70 amino acids for the North American and European type of PRRSV, respectively. The E protein is highly hydrophobic but contains a cluster of basic amino acids in the hydrophilic C-terminal region. The E protein is non-glycosylated and intracellular membrane-associated (Snijder et al., 1999, Wu et al., 2001). In PRRSV-infected pigs, the E protein induces specific antibody (Wu et al., 2001). Recent studies with a European PRRSV isolate showed that the E protein is incorporated into the virions in association with GP2–GP3–GP4 heterotrimers, suggesting a critical role for the heteromultimeric complex in the virus entry process (Wissink et al., 2005). Although the E protein of North American genotype PRRSV contains two cysteine residues at positions 49 and 54, a study has shown that E is unable to form disulfide-linked homodimers (Lee and Yoo, 2005). In that study, cysteine residues of the E protein were shown to be non-essential for virus multiplication. The function and significance of E in PRRSV replication remain to be determined.
Fig. 1.
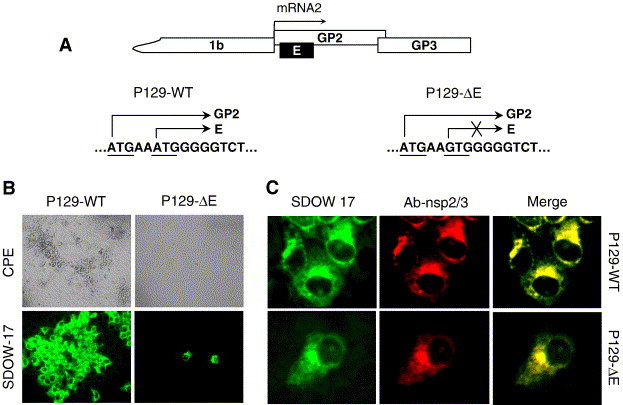
(A) The partial genome organization of PRRSV. Genomic locations of GP2 and E genes and the E gene-knockout are illustrated. (B) Absence of infectivity of the E gene-knockout full-length clone for PRRSV, P129-ΔE. Marc-145 cells were transfected with the full-length cDNA genomic clone of P129-WT or P129-ΔE and incubated for 5 days. PRRSV-specific CPEs were monitored daily and photographed 4 days post-transfection (upper panels). For immunofluorescence, cells were fixed with cold methanol at 2 days post-transfection and incubated with the N-specific MAb SDOW-17 (lower panels) (magnification 20×). (C) Double staining for N (green) and nsp2/3 (red) proteins for P129-WT (upper panels) or P129-ΔE (lower panels). Marc-145 cells transfected with P129-WT or P129-ΔE plasmid DNA were fixed at 2 days post-transfection and co-stained with nsp2/3-specific rabbit antiserum and N-specific MAb SDOW17. Yellow indicates merged images where both N and nsp2/3 are co-localized.
In the present study, we investigated the role of E protein during PRRSv replication. An infectious cDNA clone was used to generate an E gene-knockout mutant PRRSV, and we report here that the E protein is essential for virus infectivity but dispensable for virus particle formation. The E protein-negative, non-infectious virus particles were able to enter cells but unable to continue the further steps of replication. Furthermore, two ion channel blockers were shown to greatly affect PRRSV replication at early stages of infection, suggesting that ion channel activity was essential for virus uncoating. We also found that expression of the E protein enhanced membrane permeability of hygromycin B in bacterial cells. Cross-linking studies revealed that the E protein associated with itself into higher-order structures, including dimers, trimers and tetramers. Our study suggests that the PRRSV E protein may function as a viroporin in the virion envelope that facilitates uncoating of the virus in order to release the genomic RNA into the cytoplasm for subsequent replication.
Results
Generation of E gene-knockout PRRSV
To determine the biological significance of E protein during PRRSV infection, an E gene-knockout mutant virus was generated using an infectious cDNA clone. The start codon of ORF2b (E gene) was modified to abolish the E protein expression. With the shuttle plasmid by site-directed mutagenesis, ‘ATG’ for translation initiation of ORF2b was changed to ‘GTG’ at genomic positions 12,062 to 12,064. This mutation did not alter the amino acid sequence in ORF2a encoding GP2 protein. The A12062G modification was subsequently introduced into the full-length genomic cDNA clone by subcloning the Eco47III–BsrGI fragment obtained from the shuttle plasmid. Transformants were screened by restriction patterns using XmaI to determine the ligated clone, followed by nucleotides sequencing to verify the specific modification in the full-length genome. The resulting E gene-knockout genomic clone was designated P129-ΔE.
Absence of infectivity for P129-ΔE
The infectivity of the E gene-knockout clone P129-ΔE was examined by transfection of Marc-145 cells. Cells were transfected with P129-WT or P129-ΔE, and the appearance of CPE was monitored daily. P129-WT induced visible CPE at 3 days post-transfection and showed N-specific staining in clusters of cells, indicating the infection and spread of virus to neighboring cells (Fig. 1B, left panels). In contrast, transfection of P129-ΔE did not produce any visible CPE for up to at least 7 days post-transfection, suggesting the lack of infectivity. A few single cells exhibited N-specific fluorescence, and these cells represent individually transfected cells with P129-ΔE (Fig. 1B).
To rule out a possibility that an additional mutation might have been introduced during construction of P129-ΔE, which may have resulted in the loss of infectivity, six individual mutant clones were independently generated. In these clones, no mutation was identified for the ORF2 gene, and upon transfection of cells, all six P129-ΔE mutant clones were non-infectious (data not shown). This confirmed the conclusion that P129-ΔE was non-viable and the absence of infectivity was due to the lack of E protein expression, demonstrating the essential role of E for PRRSV replication.
The transcription ability of P129-ΔE was examined by double staining of transfected cells using the Alexa green-labeled SDOW17 MAb specific for N and the Texas red-labeled rabbit antiserum specific for nsp2/3 non-structural proteins (Fig. 1C). In cells transfected with P129-WT DNA, clusters of cells were stained by both antibodies, indicating both N (green) and nsp2/3 (red) expressions (Fig. 1C, upper panels). Merging of the two images showed yellow regions where the two proteins co-localized. For P129-ΔE, dual-staining of N (green) and nsp2/3 (red) was also observed but limited to transfected cells only, and no evidence for the spread of infection was obtained (Fig. 1C, lower panels). The dual staining demonstrated the expression of nsp2/3 and N proteins in transfected cells, indicating the synthesis of both non-structural and structural proteins, which in turn suggests that the PRRSV genome replicated and mRNA transcription occurred upon transfection of P129-ΔE DNA. The results demonstrate that E protein expression is required for PRRSV infectivity, but genome replication and transcription may occur without E protein. It leads us to hypothesize that either virion assembly or virus entry is interrupted in the absence of E protein expression.
Complementation of P129-ΔE replication in cells expressing the E protein
We next examined whether the infectivity of P129-ΔE could be restored by provision of the E protein in trans. The E gene was expressed in BHK-T7 cells and the expression was confirmed by immunofluorescence staining with E-specific antiserum (Fig. 2A). BHK-T7 cells were also suitable for transfection of the full-length cDNA clone as N-specific staining was detected in many cells (Figs. 2B and E). To determine whether P129-ΔE in the E gene-transfected cells leads to the production of infectious virus particles, BHK-T7 cells were co-transfected with P129-ΔE and E gene. The culture supernatant was harvested at 48 h post-transfection from BHK-T7 cells and transferred to Marc-145 cells. At 24 h post-inoculation, Marc-145 cells were fixed, and virus infection was examined by immunofluorescent staining with N-specific MAb. Marc-145 cells inoculated with a culture fluid from cells co-transfected with P129-ΔE and E gene showed bright N-specific fluorescent signal, indicating that the P129-ΔE replication may be rescued by trans-complementation of the E protein (Fig. 2C). In contrast, in cells inoculated with the supernatant from co-transfection of P129-ΔE and N gene (Fig. 2F), or of P129-ΔE and an empty plasmid (data not shown), no staining was observed. The staining of Marc-145 cells inoculated with the supernatant from BHK cells co-transfected with P129-ΔE and N gene was however limited to single cells and no infectivity was produced for up to 7 days post-inoculation (Fig. 2C), indicating abortive infection of the P129-ΔE mutant virus in non-complementing Marc-145 cells. Attempts to pass the passage-1 culture supernatant to Marc-145 resulted in no virus replication, confirming that the P129-ΔE virus particles generated by the provision of E in trans was only capable of a single round of replication. These data demonstrated that the inability of P129-ΔE to produce infectious virus was due to the absence of E protein in the virions.
Fig. 2.
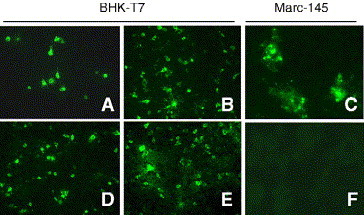
trans-complementation of P129-ΔE. BHK-T7 cells were co-transfected with P129-ΔE and pCITE-E and the culture supernatants were harvested at 48 h post-transfection followed by inoculation to Marc-145 cells. At 24 h post-inoculation, cells were stained with the Alexa green-labeled N-specific Mab, or E-specific rabbit antiserum and FITC-conjugated secondary antibody. Cells were visualized by fluorescent microscope (20×). (A) E gene (pCITE-E) transfection; (B) P129-ΔE and pCITE-E co-transfection; (C) inoculation of supernatant from (B) to Marc-145 cells; (D) N gene (pCITE-N) transfection; (E) P129-ΔE and pCITE-N co-transfection; (F) inoculation of supernatant from (E) to Marc-145 cells.
E protein is not required for virion assembly
Because P129-ΔE did not induce infectivity from transfection, it was of interest to examine whether PRRSV particles were not produced in the absence of E protein. Due to a low transfection efficiency in Marc-145 cells, BHK-21 cells were used to achieve higher transfection efficiency. When stained with N-specific MAb at 2 days post-transfection, numerous cells showed N-specific fluorescence (data not shown), indicating high levels of transfection in BHK-21 cells, which may be sufficient for the study of particle formation. BHK-21 cells were transfected with P129-WT or P129-ΔE, and at 30 h post-transfection, the transfected cells were radiolabeled for 1 day with [35S]methionine/cysteine. The cells were harvested and lysed with RIPA buffer. The culture supernatants were collected separately and centrifuged through a 20% (wt/vol) sucrose cushion, and the resulting pellets were dissolved in RIPA buffer. The lysates prepared from cells or supernatants were immunoprecipitated with a mixture of individual antibody specific for E, N, or M and resolved by SDS–PAGE. As shown in Fig. 3A, intracellular major viral-specific proteins were detectable in all lysates (lower panel), confirming the ability of P129-ΔE for transcription. Similarly, in the culture supernatants, the three major virion proteins GP5, M and N were clearly identified (upper panel), showing that, without E protein, virus particles can be produced and released. It was not possible to detect the E protein in the cell lysates or supernatants from P129-WT-transfected cells, and this is probably due to the low abundance of E protein in BHK-21 cells as E is a minor protein (Wu et al., 2005). This result suggests that the PRRSV E protein is not required for particle formation.
Fig. 3.
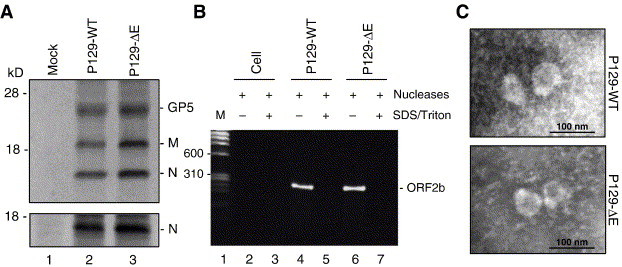
PRRSV particle assembly in the absence of E protein. (A) Radioimmunoprecipitation of virus particles lacking the E protein. BHK-21 cells were transfected with P129-WT or P129-ΔE genomic clone. At 30 h post-transfection, the cells were radiolabeled with [35S]methionine/cysteine for 24 h at 37 °C, and the supernatants and cells were separately collected. Cell lysates were prepared and subjected to immunoprecipitation with N-specific MAb (lower panel). The pellets were prepared from culture supernatants by ultracentrifugation, lysed in RIPA buffer and used for immunoprecipitation with a mixture of anti-M, anti-N and anti-E-specific antibodies, followed by SDS–15% PAGE under reducing conditions (upper panel). Lane 1, mock transfected; lane 2, P129-WT transfected; lane 3, P129-ΔE transfected. (B) Incorporation of genomic RNA in P129-ΔE virus particles. Culture supernatants were pelleted by ultracentrifugation and the pellets were treated with nucleases in the presence (+) or absence (−) of SDS and Triton X-100, followed by proteinase K treatment. RT-PCR was conducted for E gene amplification followed by electrophoresis in 1.5% agarose gel. Lane 1, molecular weight marker; lanes 2 and 3, culture supernatant spiked with the P129-ΔE full-length plasmid; lanes 4 and 5, supernatant from P129-WT-transfected cells; lanes 6 and 7, culture supernatant from P129-ΔE-transfected cells. (C) Electron microscopy of the culture supernatant from P129-ΔE-transfected cells. Particles in the culture supernatants released from BHK-21 cells transfected with P129-WT (upper panel) or P129-ΔE (lower panel) genomic clones were concentrated by ultracentrifugation through a 20% (wt/vol) sucrose cushion. Pellets were negatively stained by sodium phosphotungstate and visualized by electron microscopy. Scale bar, 100 nm.
Because the virus particles formed in the absence of E protein were non-infectious, these particles were examined for the presence of viral genome. The culture supernatants, prepared from BHK-21 cells transfected with P129-WT or P129-ΔE, were first incubated with both RNase A and DNase I to remove any possible contamination of viral RNA and the transfected DNA from cells. The digestion was carried out in the presence or absence of detergents, followed by proteinase K treatment to inactivate the nucleases. RNA was extracted from samples and treated again with DNase I, and the E gene region of PRRSV was RT-PCR-amplified and sequenced. In cell culture media mixed with the full-length cDNA clone as a negative control, no PCR fragment was amplified in the presence or absence of detergents (Fig. 3B, lanes 2 and 3), indicating the appropriateness of DNase I treatment. Although no amplification was identified for controls (Fig. 3B, lanes 5 and 7), a 260-bp product was specifically amplified for both samples of P129-WT and P129-ΔE treated with the nucleases but in the absence of detergents (Fig. 3B, lanes 4 and 6). Sequencing of the 260-bp product confirmed the stable incorporation of the ‘A12062G’ mutation at the start codon of the E gene in the genome from P129-ΔE particles. These studies demonstrate that particles formed in the absence of E protein contained the P129-ΔE genomic RNA, which in turn suggests that the lack of infectivity of P129-ΔE was not due to the improper packaging of the genome.
To obtain further evidence for particle formation in the absence of E, electron microscopy (EM) was conducted and the microscopic appearance of P129-ΔE particles was compared to that of P129-WT virions. At 2 days post-transfection, the culture supernatant from BHK-21 cells was harvested and pelleted by ultracentrifugation through a 20% (wt/vol) sucrose cushion, followed by electron microscopy. PRRSV particles were identified in the supernatant of cells transfected with either P129-WT (Fig. 3C, upper panel) or P129-ΔE (Fig. 3C, lower panel). No significant morphologic differences between P129-WT and P129-ΔE particles were noted. Each virion for P129-WT or P129-ΔE was a roughly spherical enveloped particle of 50–60 nm in diameter, with a densely stained core. The EM study confirmed that PRRSV particles may be formed in the absence of E protein and also showed that the E protein-negative particles had similar appearance and size to wild-type PRRSV particles.
Viral RNA detection from P129-ΔE virus
To determine if the lack of infectivity of P129-ΔE particles was due to a low amount of virus produced from BHK-21 cells, the supernatants collected from BHK-21 cells were blindly passaged twice in Marc-145 cells to amplify the infectivity. Although extensive CPE was readily evident in Marc-145 cells inoculated with either passage-1 or passage-2 of P129-WT supernatant, no CPE was detectable with passage-1 or passage-2 of P129-ΔE supernatant, even after 5 days post-inoculation (data not shown). To further determine infectivity in Marc-145 cells inoculated with P129-ΔE virus, time course immunofluorescence was carried out with the N-specific antibody. As shown in Fig. 4A, distinct staining of N was first observed at 12 h post-infection in Marc-145 cells inoculated with P129-WT supernatant, and then in many clusters of cells by 48 h post-infection, showing the spread of infectivity to neighboring cells (upper panels). In contrast, cells inoculated with passage-1 of P129-ΔE showed no specific staining throughout the experiments, further indicating the lack of infectivity (lower panels).
Fig. 4.
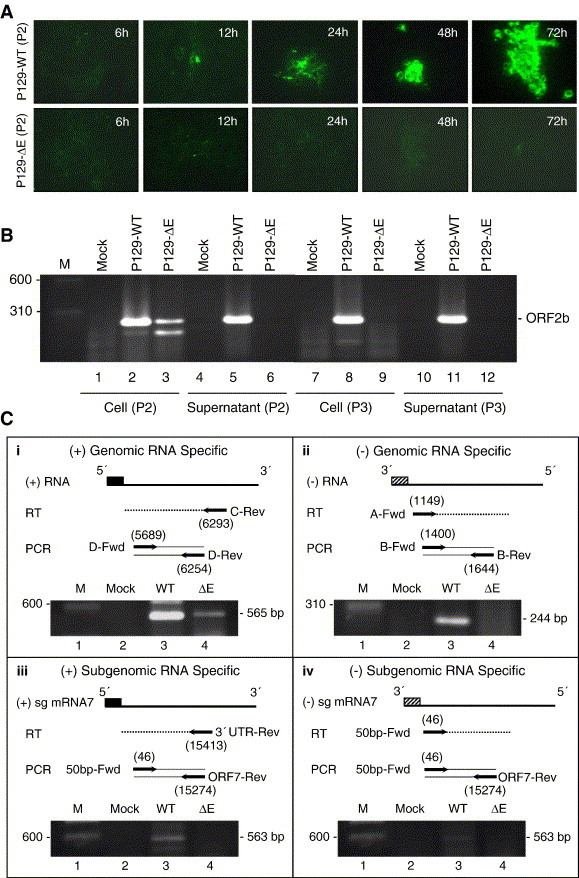
The E protein is essential for PRRSV replication. (A) Immunofluorescence of N protein in P129-ΔE inoculated cells. The ‘passage-1’ virus was prepared as culture supernatant harvested from BHK-21 cells transfected with P129-WT or P129-ΔE clone. Marc-145 cells were inoculated with ‘passage-1’ and fixed at the indicated times post-inoculation, followed by staining with N-specific MAb. (B) Detection of viral RNA in culture supernatants and in Marc-145 cells inoculated with ‘passage-1’ or ‘passage-2’ virus. Total RNA was extracted and treated with DNase I followed by RT-PCR for E gene. (C) Strand-specific detection of viral RNA in cells by RT-PCR. Marc-145 cells were inoculated with ‘passage-1’ P129-WT or ‘passage-1’ P129-ΔE, and total cellular RNA was extracted at 2 days post-inoculation. The RNA was treated by DNase I and RT-PCR was conducted to amplify the region as illustrated in the figure. Numbers in parenthesis indicate the 5′ most nucleotide position in each primer with respect to the PRRSV genome. Expected sizes of amplified products are indicated on the right of the gel.
Because neither CPE nor N-specific staining was detectable from serial passages of P129-ΔE virus, RT-PCR was conducted from cells and culture supernatants inoculated with passage-1 or passage-2 (Fig. 4B). To eliminate a possible carry-over contamination of the transfected DNA, the RNA preparations were treated with RNase-free DNase I prior to the E gene amplification. A 260-bp product was amplified from both culture media and cells that were inoculated with passage-1 or passage-2 P129-WT virus (Fig. 4B, lanes 2, 5, 8 and 11). The 260-bp product was also obtainable from cells inoculated with passage-1 P129-ΔE virus (Fig. 4B, lane 3), but no specific product was amplified from the supernatant (Fig. 4B, lane 6), nor from either supernatants or cells inoculated with passage-2 P129-ΔE virus (Fig. 4B, lanes 9 and 12). The amplified fragment was sequenced, and the sequencing results confirmed the ‘A12062G’ mutation at the start codon of ORF2b (data not shown). These results suggest that the E protein-negative, non-infectious P129-ΔE virus particles entered Marc-145 cells, but further steps beyond entry were interrupted, resulting in the absence of infectivity.
To investigate whether the P129-ΔE virus genome underwent replication following entry, strand-specific RT-PCR was performed from cells inoculated with passage-1 P129-ΔE virus. RT-PCR from P129-WT-inoculated cells yielded a specific product of the expected size for both positive- and negative-strand genomes and also for positive- and negative-strand mRNA7 (Fig. 4C, lanes 3 in all panels). In contrast, only a minimal amount was amplified for the positive-strand genome from cells inoculated with passage-1 P129-ΔE virus (Fig. 4C, panel i, lane 4). This product was likely derived from the incoming genomic RNA of P129-ΔE virus. These results indicate that P129-ΔE virus was unable to replicate, suggesting that the lack of RNA replication of P129-ΔE virus was likely due to an interruption during virus uncoating, a stage prior to genome replication.
Inhibition of PRRSV replication by ion channel blockers
The E-protein-negative PRRSV particles were shown to contain the viral genome and able to enter cells but unable to release the viral RNA for initiation of genome replication. This suggests that PRRS virions may contain ion channels and the E protein may function as an ion channel protein embedded in the viral envelope. It is postulated that PRRSV replication may be suppressed by ion channel blocking agents if ion channel activity is an essential requirement for PRRSV infection. This aspect was investigated in Marc-145 cells using amantadine and verapamil. Amantadine is known as a proton channel blocker and verapamil is the calcium channel blocking agent. Because it has been reported that an acidic environment is required for PRRSV infection (Kreutz and Ackermann, 1996), basic lysosomotropic agents including ammonium chloride and chloroquine were also included in the study to examine their inhibitory effects on PRRSV replication. Strand-specific RT-PCR experiments were carried out and demonstrated that PRRSV-infected Marc-145 cells produced reduced levels of positive-sense genomic RNA at 2 days post-infection in the presence of the drugs chloroquine, amantadine and verapamil (Fig. 5A, upper panel). A relatively moderate level of suppression was observed in ammonium chloride-treated cells (lane 4). Negative-sense genomic RNA was not detectable in cells treated with any of the four drugs until 2 days post-infection (middle and lower panels, lanes 4–7), showing that the initiation of viral RNA synthesis was inhibited by the ion channel blockers. Furthermore, the number of PRRSV-infected cells determined by the N protein staining was significantly reduced by individual drugs in comparison to untreated cells (Fig. 5B, upper panels). PRRSV-specific CPE was visible but significantly delayed by the ion channel blockers, indicating the negative effects on virus production (lower panels). The yields of virus production were determined by plaque assays from the supernatants in the presence of each drug. The virus titer in untreated cells reached 3 × 106 PFU/ml by 3 days post-infection (Fig. 5C). In contrast, there was no detectable virus production at 1 day post-infection in the presence of amantadine or verapamil, and the titers of virus production in the presence of these drugs reached only to a maximum of 5 to 8 × 101 PFU/ml by 3 days post-infection (Fig. 5C). These results showed that the treatment of infected cells with ion channel blockers greatly reduced the growth rate of PRRSV. However, when Marc-145 cells were treated with the drugs at 30 min post-infection, no significant inhibitory effect of virus production was observed (data not shown). Therefore, it seems that the ion channel blockers effectively interfered with virus uncoating, a step preceding genome replication and consequently affected PRRSV production. It is interesting to note that the inhibitory effect by amantadine are due to the blocking of pores that are necessary for post-internalization during uncoating of the virus (Wang et al., 1993).
Fig. 5.
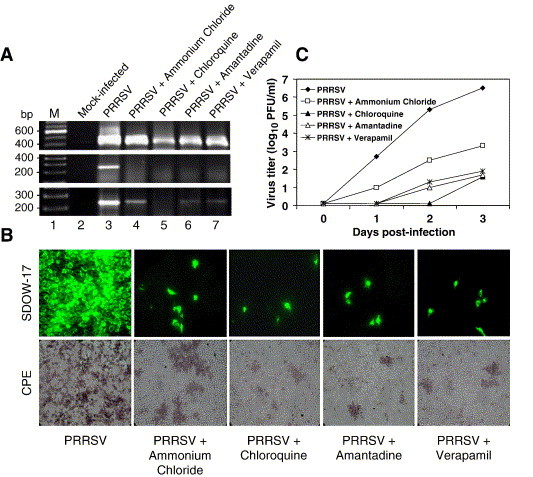
Inhibition of PRRSV replication by ion channel blockers. (A) Strand-specific RT-PCR for N gene. The drug concentrations were used according to the previously report (Kreutz and Ackermann, 1996). Verapamil concentration was determined by limiting dilution for cell cytotocixity in Marc-145 cells. Marc-145 cells were treated with ammonium chloride (5 mM), chloroquine (20 μM), amantadine (1 mM) or verapamil (50 μM) for 30 min prior to PRRSV infection and the virus-infected cells were incubated for 3 days in the presence of the drug. Total RNA was extracted from cells at 2 days (upper and middle panels) or 3 days (lower panel) post-infection, and strand-specific RT-PCR was conducted to detect positive-sense (upper panel) or negative-sense (middle and lower panels) genome. Lane 1, molecular weight marker; lane 2, mock-inoculated cells; lane 3, untreated; lanes 4 to 7, treated with the indicated drug. (B) PRRSV replication in the presence of the drug. Virus-infected, drug-treated Marc-145 cells were stained for immunofluorescence (upper panels) with N-specific MAb SDOW-17 at 2 days post-infection (magnification 20×). PRRSV-specific CPE was monitored and photographed at 3 days post-infection (lower panels). (C) Growth kinetics of PRRSV in the presence of drugs. Cells infected with PRRSV were incubated in the presence of the drug, and culture supernatants were harvested at the indicated times for plaque assays in duplicate.
Modification of membrane permeability by E protein and oligomerization
Because our data suggest that ion channel activity may be involved during uncoating and that the PRRSV E protein is likely responsible for this event, the E protein was assumed to function as a viroporin. We examined whether the E protein possessed general features commonly shared by other viroporins (ion channel proteins). Viroporins generally contain the properties of membrane permeability alteration and oligomerization (Liao et al., 2004, Maldarelli et al., 1993, Paterson et al., 2003, Sakaguchi et al., 1997, Torres et al., 2005), and so these two common properties were examined for the E protein. The effect of E expression on bacterial growth was first examined in an inducible protein expression system. When the inner bacterial membrane is intact, intracellular lysozymes cannot reach the cell wall. However, permeabilized membranes allow the lysozymes to gain access to the peptidoglycan, leading to cell lysis. This approach has been shown to be suitable as a permeabilization test of the inner bacterial membrane for viral proteins (Bodelón et al., 2002, Ciccaglione et al., 1998, Liao et al., 2004, Madan et al., 2005). The PRRSV E protein gene was first cloned in the GST-fusion vector and expressed in Escherichia coli. The growth rates of transformed cells were analyzed spectrophotometrically. A drastic arrest of cell growth was observed in the E gene-transformed cells upon IPTG induction, whereas bacteria carrying the empty plasmid or the PRRSV N gene had no effects on their growth during the 300-min period following induction (Fig. 6A). This finding suggests that the expression of E affected the cell growth negatively by altering the membrane permeability to intracellular lysozyme.
Fig. 6.
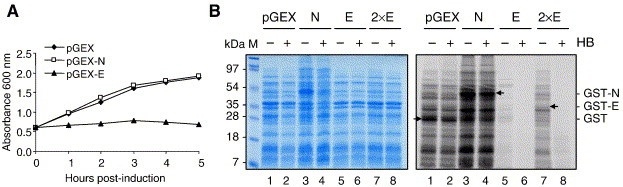
Alteration of membrane permeability by the E protein. (A) Growth kinetics of E. coli strain DH5α expressing GST, GST-N or GST-E. (B) Hygromycin B permeability assay in bacterial cells. Cultures of the gene-transformed E. coli were induced by IPTG to express GST (lanes 1, 2), GST-N (lanes 3, 4) or GST-E (lanes 5 to 8). At 1 h post-induction, cells were incubated in the presence (+) or absence (−) of 1 mM of hygromycin B for 15 min and radiolabeled with [35S]methionine/cysteine for 15 min. Cells were collected and lysed in sample buffer and analyzed by SDS–12% PAGE, followed by Coomassie blue staining (left panel) or autoradiography (right panel). Lanes 1 and 2, cells expressing GST alone; lanes 3 and 4, cells expressing PRRSV N fused with GST (GST-N); lanes 5 and 6, cells expressing PRRSV E fused with GST (GST-E); lanes 7 and 8, double amount of loading for cells expressing GST-E. Arrows indicate GST, GST-N or GST-E.
To further determine if the growth rate of cells expressing E was the consequence of permeability modification, a hygromycin B permeability assay was carried out. Hygromycin B is normally impermeable to the membrane barrier during a short period of time at low concentration, but it can readily penetrate the permeabilized membrane to cause strong inhibition of intracellular protein synthesis. The hygromycin B permeability assay therefore is widely used to study changes of membrane permeability as well as to identify proteins that can form pores in lipid membranes (Aldabe et al., 1996, Arroyo et al., 1995, Bodelón et al., 2002, Chang et al., 1999, Ciccaglione et al., 1998, de Jong et al., 2003, Doedens and Kirkegaard, 1995, Han and Harty, 2004, Liao et al., 2004, Madan et al., 2005). At 1 h induction for E protein expression, hygromycin B was added to the culture media. The cultures were further incubated for 15 min and metabolically labeled with [35S]methionine/cysteine for 15 min, followed by SDS–PAGE of cell lysates. The Coomassie blue staining showed that equal amounts of proteins were loaded on the gel (Fig. 6B, left panel). Autoradiography indicated that hygromycin B entered cells that expressed the E protein, and protein synthesis was completely blocked in those cells (Fig. 6B, right panel, compare lanes 5 and 6, lanes 7 and 8). For cells expressing the GST or PRRSV N protein, hygromycin B did not inhibit protein synthesis (compare lanes 1 and 2, lanes 3 and 4). These results indicate that the PRRSV E protein enhanced membrane permeability in bacterial cells.
A second common property of viroporins is oligomerization. The PRRSV E protein contains two well-conserved cysteine residues; however, the E protein was shown not to form a disulfide-linked homodimer (Lee and Yoo, 2005). The absence of covalently linked homodimers of E was confirmed using the recombinant E protein expressed in HeLa cells by vTF7-3 vaccinia virus (Fig. 7A). Although PRRSV N-N dimers of 30 kDa were readily detected in cells expressing the N protein (lane 1), no band corresponding to the predicted dimeric form of E was identified in non-reducing conditions (lane 3), concluding that the PRRSV E protein does not undergo cysteine-linked homodimerization.
Fig. 7.
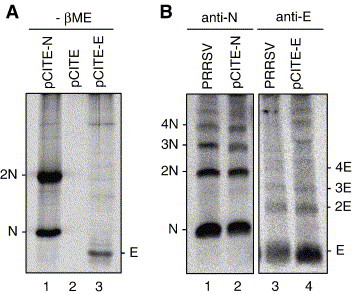
PRRSV E protein oligomerization by DSP cross-linking. (A) Absence of the disulfide-linked E protein oligomerization. HeLa cells were infected with vTF7-3 and transfected with pCITE-E for 16 h. Cells were radiolabeled for 5 h with [35S]methionine/cysteine, lysed with RIPA buffer and immunoprecipitated using anti-E-specific rabbit antiserum, followed by SDS–17% PAGE under non-reducing conditions in the absence of β-mercaptoethanol. Lane 1, N gene (pCITE-N) transfected cells; lane 2, pCITE empty vector transfected cells; lane 3, E gene (pCITE-E) transfected cells. 2N, dimeric form of PRRSV N protein. (B) Homo-oligomerization of the E protein by DSP cross-linking. PRRSV-infected Marc-145 cells (lanes 1, 3), or HeLa cells expressing recombinant N protein (lane 2) or E protein (lane 4) by vTF7-3 vaccinia virus were radiolabeled with [35S]methionine/cysteine and cross-linked in vivo with 2 mM DSP for 30 min. Cells were lysed and immunoprecipitated with anti-E-specific rabbit antiserum followed by SDS–17% PAGE under non-reducing conditions. The radiographic images were obtained using a PhosphorImager. Multimeric forms of the N or E protein are shown. Anti-N, N protein-specific MAb SDOW 17; anti-E, E protein-specific anti-rabbit antiserum.
Because the E protein may form a non-covalent association with itself as shown by GST-pull down assay (Lee and Yoo, 2005), the potential of E to form oligomers was further investigated by cross-linking experiments in the presence and absence of other viral constituents. Radiolabeled PRRSV-infected cells were treated with the membrane-permeable cross-linking reagent DSP, and the cell lysates were immunoprecipitated by N-specific MAb or E-specific antiserum followed by SDS–PAGE under non-reducing conditions (Fig. 7B). The PRRSV N protein formed a number of higher-order oligomers (lanes 1, 2), as reported previously (Wootton and Yoo, 2003). When the E protein in virus-infected cells was subjected to cross-linking, numerous multimeric forms of E protein were identified (lane 3). The E protein multimerization also was examined in the absence of other PRRSV proteins in HeLa cells expressing E protein by T7 vaccinia virus, and again higher-order multimeric forms of the E protein were observed (lane 4), indicating that oligomerization of E is independent of other viral proteins. Altogether, our data indicate that the PRRSV E protein exists as non-covalently linked oligomers in virus-infected cells and suggest that the multimerization may be the physical basis for viroporin formation of PRRSV E.
Discussion
The present study was conducted to investigate the role of PRRSV E protein during virus infection. A reverse genetics approach was used to modify the translation initiation of ORF2b, so that the modified genome was unable to express the E protein. Our experiments show that the absence of E protein expression does not affect genome replication transcription but impairs the production of infectious virus. These data indicate that the PRRSV E protein is an essential structural component for infectivity. The E protein however appears not to be an essential component for particle assembly nor genomic encapsidation. The virion particles devoid of E still contained the viral genome and had a similar appearance to that of wild-type virions. For infection, virus particles must proceed through a multiple-step cycle of entry and uncoating, replication and transcription and assembly and release. According to our findings, the E protein does not exert an important function at later events in the infection process such as genome replication, particle assembly and release of virus. We have observed that the E protein-negative, non-infectious virion particles are able to enter cells but subsequent steps of replication are inhibited. This suggests that the particles lacking the E protein most likely remain in the endosome and the viral genome is not released, which is the step between virus entry and genome replication. The uncoating is a critical step for virus infection, during which the lipid envelope is shed and the viral capsid is disassembled to release the genome to initiate a replication cycle in the cytoplasm. For viruses entering cells by receptor-mediated endocytosis, the uncoating process occurs in the endosome where an acidic environment triggers fusion between the viral membrane and the endosomal membrane (reviewed by Smith and Helenius, 2004). PRRSV enters cells through receptor-mediated microfilament-dependent endocytosis in which a low pH is required to trigger fusion for proper uncoating by an unknown mechanism (Kreutz and Ackermann, 1996, Nauwynck et al., 1999). It is possible that an acidic pH in the endosome may lead to conformational changes of viral protein(s) to expose hidden fusogenic domains that facilitate the fusion between viral and endosomal membranes. However, no direct fusion of PRRSV with the cell membrane, and no arterivirus membrane proteins possessing the fusogenic property have been reported. It is therefore tempting to speculate that PRRSV may contain a viral ion channel protein to promote the uncoating process in the endosome during the early stage of infection. In the present study, we have shown the inhibitory effect of ion channel blockers on PRRSV replication, indicating that PRRSV may indeed possess virus-coded ion channels whose activity is essential for proper uncoating for infection.
Virus-coded ion channels, or viroporins, consist of small, highly hydrophobic proteins generally composed of 60–120 amino acids. The insertion of these proteins into membranes creates typical hydrophilic pores or selective ion channels with (a) hydrophobic transmembrane domain(s) facing the lipid bilayer, leading to an alteration of membrane permeability to ions or other small molecules (for a review, see Fischer and Sansom, 2002, Gonzalez and Carrasco, 2003). Expression of these membrane-active proteins may cause cellular membrane leakiness further resulting in the development of cytopathic changes to facilitate particle release late in infection, or may be required to promote virus uncoating at an early stage of infection (reviewed by Carrasco, 1995).
The PRRSV E protein structurally resembles a number of viroporins found in other RNA viruses, in that it consists of 70–73 amino acid residues and contains central hydrophobic sequences with a cluster of basic amino acids at the C-terminus. The E protein localizes mainly in perinuclear regions including the ER and the Golgi complex and associates with intracellular membranes (Wu et al., 2001). Interestingly, coronaviruses, another family member of the order Nidovirales, also code for a small hydrophobic membrane, designated E protein, which may play a crucial role during virus morphogenesis (Fischer et al., 1998, Liu and Inglis, 1991). In recent studies, the coronavirus E protein has been shown to modify membrane permeability (Liao et al., 2004, Madan et al., 2005) as well as to form cation-selective ion channels in an artificial membrane (Wilson et al., 2004). Our data in the current study also demonstrate the alteration of membrane permeability by the PRRSV E protein and the inhibition of bacterial growth by the increase of hygromycin B penetration into bacterial cells. However, we were not able to show similar results in mammalian cells by infection or E gene transfection, which may be due to the different intracellular localization of E in mammalian cells. In PRRSV-infected cells, the E protein appears to remain in the ER and Golgi complex, where it likely participates in assembly of infectious progeny virus, rather than traveling to the plasma membrane. Bacterial cells do not possess such intracellular organelles and the expressed E protein may accumulate at the inner bacterial membrane, leading to membrane perturbation, and thus enhancing membrane permeabilization. Failure to observe the direct alteration of membrane permeability by E in mammalian cells implies that the role of E is not linked to membrane disorganization and cell lysis in facilitating virus release. The cross-linking studies show that the E protein can form homo-oligomers, including dimers, trimers and tetramers by non-covalent interactions. All information obtained in the present study support the hypothesis that the PRRSV E protein contains a potential for a pore-forming activity and thus may function as an ion channel for virus uncoating. The functional and structural features of the PRRSV E protein resemble the influenza A virus M2 protein that is best characterized as an ion-channel protein. The M2 protein forms a homo-tetramer in the viral membrane and functions as an ion channel. The M2 channel allows the translocation of protons from the acidic environment of the endosome to the inner space of virions and alters the pH in the virion. As a consequence, the M1 protein in the virion is dissociated from the viral ribonucleoprotein complex, which promotes the ribonucleoprotein complex traveling to the nucleus where the influenza replication takes place (Pinto et al., 1992, Sakaguchi et al., 1997). It is noteworthy that PRRSV replication could be inhibited by amantadine, an antiviral drug for influenza virus (Kreutz and Ackermann, 1996; Fig. 5). In summary, we propose a model for PRRSV uncoating based on our findings. In this model, E proteins form pores (ion channels) in the viral envelope. Upon internalization by receptor-mediated endocytosis through small clathrin-like coated vesicles, the virion particles are transported to the endosome. There, the E-protein ion channels in the viral membrane undergo conformational changes upon exposure to low pH in the endosome, and allow ions to enter the virion, which triggers the disassembly of inner capsid and the release of viral genome in the cytoplasm, such that further steps of genome replication and infection cycle can proceed.
Materials and methods
Cells, viruses, antibodies and plasmids
Marc-145 (Kim et al., 1993), HeLa and BHK-21 cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 8% fetal bovine serum (FBS; Invitrogen), penicillin (100 U/ml) and streptomycin (50 μg/ml). BHK-T7 cells stably expressing the bacteriophage T7 RNA polymerase (generously provided by C.Y. Kang, University of Western Ontario, London, Ontario, Canada) were grown in DMEM supplemented with 5% FBS, 100 U/ml of penicillin, 50 μg/ml of streptomycin and 500 μg/ml of G418 (Geneticin; Invitrogen). The cells were maintained at 37 °C with 5% CO2. Stocks of PRRSV (strain PA8) and recombinant vaccinia virus expressing T7 RNA polymerase (vTF7-3; Fuerst et al., 1986) were prepared in Marc-145 or HeLa cells, respectively, as described previously (Wootton et al., 2001). The N protein-specific monoclonal antibody (MAb) SDOW17, the E protein-specific anti-peptide rabbit antiserum and non-structural proteins 2 and 3-specific polyclonal rabbit antiserum are described elsewhere (Lee et al., 2005, Lee and Yoo, 2005, Nelson et al., 1993, Wootton et al., 2001). E. coli strains XL1-Blue (Stratagene) and DH5α were used as hosts for site-directed mutagenesis and general cloning, respectively. cDNA cloning of the PRRSV N or E gene to produce pCITE-N; pGEX-N or pCITE-E; pGEX-E, respectively, is described elsewhere (Wootton and Yoo, 2003, Lee and Yoo, 2005).
Generation of the E gene-knockout full-length PRRSV cDNA clone
To modify the translational initiation codon of PRRSV ORF2b (E), PCR-based site-directed mutagenesis was first conducted to mutate the ATG start codon of the E gene to GTG at genomic nucleotide positions 12,062 to 12,064 using the shuttle vector pTB-shuttle-PRRSV-3997 (Lee et al., 2005) with the following primer pairs; for A12062G mutation, E-KO-Fwd (5′-GAATTGAAATGAAgTGGGGTCTATAC-3′: nucleotide positions 12,049 to 12,074) and E-KO-Rev (5′-GTATAGACCCCAcTTCATTTCAATTC-3′: nucleotide positions 12,049 to 12,074), where lowercase letters indicate mutated nucleotides. The A12062G mutation was translationally silent with respect to ORF2a encoding the GP2 protein. PCR-based mutagenesis and screening of mutants were performed as described previously (Wootton et al., 2001). The shuttle plasmid carrying the A12062G mutation was digested with Eco47III and BsrGI, and a 908-bp fragment was purified. The wild-type full-length genomic cDNA clone was digested with Eco47III and BsrGI, and the 908-bp Eco47III–BsrGI fragment was replaced with the corresponding fragment obtained from the shuttle plasmid. The ligated full-length plasmid DNA was screened by XmaI digestion, and based on the XmaI digestion pattern, positive clones were selected. DNA manipulation and cloning were performed according to standard procedures (Sambrook and Russell, 2001). The selected clones were sequenced to confirm the presence of the A12062G mutation in the full-length genomic cDNA clone. The resulting plasmid was designated pCMV-S-P129-ΔE.
Immunofluorescence
Marc-145 cells or BHK-21 cells were seeded on microscope coverslips placed in 35-mm-diameter dishes and grown overnight to 70% confluence. The cells were transfected with 2 μg of plasmid DNA using Lipofectin (Invitrogen) according to the manufacturer's instructions. At 48 h post-transfection, cell monolayers were washed twice in PBS and fixed immediately with cold methanol for 10 min at − 20 °C. For time course experiments, Marc-145 cells inoculated with purified passage-1 viruses prepared in the transfected BHK-21 cells were fixed at various time points after infection. Cells were blocked using 1% bovine serum albumin (BSA) in PBS for 30 min at room temperature (RT) and then incubated with N-specific MAb SDOW17 for 2 h. After washing five times in PBS, the cells were incubated for 1 h at room temperature with goat anti-mouse secondary antibody conjugated with Alexa green dye (Molecular Probes). For dual immunofluorescence, cells were co-stained with nsp2/3-specific rabbit antiserum and N-specific MAb SDOW17, followed by incubation with goat anti-rabbit antibody conjugated with Texas red (Molecular Probes) and goat anti-mouse antibody conjugated with Alexa green. The coverslips were washed five times in PBS and mounted on microscope glass slides in mounting buffer (60% glycerol and 0.1% sodium azide in PBS). Cell staining was visualized using a fluorescent microscope (model AX70; Olympus).
Preparation of radiolabeled viral particles and immunoprecipitation
BHK cells were seeded in 100 mm-diameter dishes and grown to 70% confluence. Cells were transfected for 20 h with 10 μg of the full-length cDNA plasmid using Lipofectin. The transfected cells were continued for incubation at 37 °C in DMEM supplemented with 8% FBS for 10 h. At 30 h post-transfection, the cells were starved for 30 min in methionine-deficient MEM (Invitrogen) and metabolically labeled for 24 h with 100 μCi/ml of EasyTag EXPRESS protein labeling mix ([35S]methionine and [35S]cysteine, specific activity, 407 MBq/ml) (Perkin-Elmer). After a 1-day labeling period, the cells were washed twice with cold PBS and lysed with RIPA buffer (1% Triton X-100, 1% sodium deoxycholate, 150 mM NaCl, 50 mM Tris–HCl [pH 7.4], 10 mM EDTA, 0.1% SDS) containing 1 mM phenylmethylsulfonyl fluoride (PMSF). To prepare radiolabeled viral particle samples, the culture supernatant was harvested, and the cell debris was removed by a low-speed centrifugation at 3500 rpm (model 5415; Eppendorf) for 10 min at RT. The virus particles were purified through a 20% sucrose cushion (wt/vol) prepared in TE buffer (10 mM Tris HCl [pH 8], 1 mM EDTA) at 40,000 rpm for 2 h at 4 °C in an SW41 rotor (model XL-90; Beckman). The resulting pellets were resuspended in 50 μl of RIPA buffer containing 1 mM PMSF.
For immunoprecipitation, the dissolved pellets or cell lysates equivalent to 1 in 15 of a 100-mm-diameter dish were adjusted with RIPA buffer to a final volume of 100 μl and incubated for 2 h at RT with a mixture of E-specific rabbit antiserum, N-specific MAb SDOW17, and M-specific rabbit antiserum. The immune complexes were adsorbed to 7 mg of protein-A Sepharose CL-4B beads (Amersham Biosciences) for 16 h at 4 °C. The beads were collected by centrifugation at 6000 rpm for 5 min, washed twice with RIPA buffer and once with wash buffer (50 mM Tris–HCl [pH 7.4], 150 mM NaCl). The beads were resuspended in 20 μl of SDS–PAGE sample buffer (10 mM Tris–HCl [pH 6.8], 25% glycerol, 10% SDS, 0.12% [wt/vol] bromophenol blue) with 10% β-mercaptoethanol (βME), boiled for 5 min and analyzed by 15% SDS–polyacrylamide gel electrophoresis (PAGE). Gels were dried on filter paper and radiographic images were obtained using a phosphorimager (model PhosphorImager SI; Molecular Dynamics).
Virus preparation from full-length cDNA clones
BHK-21 cells were transfected with the full-length cDNA plasmid as described above. After washing of cells with DMEM at 20 h post-transfection, the transfected cells were further maintained in DMEM supplemented with 8% FBS for 2 days and the culture supernatants were harvested at 2 days post-transfection. The viral particles were purified as described above and the pellets were suspended in DMEM. The resulting virus suspension was designated ‘passage-1’. The passage-1 virus was used to inoculate fresh Marc-145 cells and the 5-day harvest was designated ‘passage-2’. The ‘passage-3’ virus was prepared in the same way as for passage-2. Each passage virus was aliquoted and stored at − 80 °C until use.
Electron microscopy of viral particles
Culture supernatants of BHK-21 cells transfected with the full-length cDNA plasmid were harvested at 2 days post-transfection as described above. The culture fluids were centrifuged at 3500 rpm (model 5415; Eppendorf) for 10 min to remove cellular debris. The cleared supernatants were purified as described above, and the pellets were suspended in 50 μl of PBS and subsequently stored at 4 °C until use. Twenty microliters of the virion suspension was mounted on a Formar-coated copper grid. The grids were placed at RT for 5 min, and excess liquid was removed by wicking with filter paper. For negative staining, 20 μl of 2% sodium phosphotungstate (pH 6.8) was dropped onto the grids and incubated at RT for 30 s. The samples were viewed with a transmission electron microscope (model H-7600 120 keV PC-TEM; Hitachi) operating at 75 keV.
RT-PCR and sequencing
Viral RNA was extracted from either supernatants or lysates of infected cells using the QiaAmp viral RNA mini-kit (Qiagen). To remove any contaminated DNA in the RNA preparations, samples were treated with 1 U of RQ DNase I (Promega) at 37 °C for 30 min in 50 mM Tris–HCl [pH 7.5] and 1 mM MgCl2. For detection of viral RNA in the virions, samples were prepared as previously described (Wieringa et al., 2004) with some modifications. The viral particles in the culture supernatant from BHK-21 cells transfected with the full-length cDNA plasmid were pelleted by as described above. The pellet was resuspended in TNM buffer (20 mM Tris–HCl [pH 7.5], 50 mM NaCl, 5 mM MgCl2). Seventy microliters of each sample suspension were added with 2 μl of RQ DNase I (1 U/μl; Promega) and 3.6 μl of RNase A (10 mg/ml; Sigma), and the mixture was incubated for 1 h at 37 °C in the presence or absence of detergents (1 μl of Triton X-100, and 3.5 μl of 10% SDS). After incubation for 1 h at 37 °C, the nucleases were inactivated by the addition of 5 μl of proteinase K (20 mg/ml; Qiagen) and incubation for 30 min at 50 °C. RNA was isolated from each sample by using a QIAamp viral RNA mini-kit and subsequently, treated one more time with 1 unit of RQ DNase I. After DNase I treatment, RNA was re-extracted with an equal volume of phenol:chloroform:isoamyl alcohol (25:24:1) mixture and precipitated at − 80 °C for 1 h by adding 0.1 volume of 2.5 M sodium acetate (pH 5.2) and 2 volumes of ethanol. The pellets were washed once with 70% ethanol and dissolved in UltraPure DNase/RNase-Free Distilled Water (Invitrogen). The resulting RNA samples were used for first-strand cDNA synthesis by Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase (Invitrogen) using the reverse primer ORF2b-Rev (5′-TCATAAGATCTTCTGTAATTGCTC-3′). The E gene was amplified by Taq DNA polymerase (Invitrogen) using mRNA2-Fwd (5′-CCGTCATTGAACCAACTTTA-3′) and ORF2b-Rev. For strand-specific RT-PCR, the specific primer pairs (Table 1 ) were used to amplify DNA fragments representing positive-sense genomic RNA, negative-sense genomic RNA, positive-sense subgenomic mRNA7 or negative-sense subgenomic mRNA7. PCR was conducted under the following conditions; initial denaturation at 95 °C for 5 min, 35 cycles of denaturation at 95 °C for 30 s, annealing at 56 °C for 30 s and extension at 72 °C for 1 min, followed by a final extension at 72 °C for 10 min. PCR products were analyzed by 0.8% or 1.5% agarose gel electrophoresis depending on size of the fragment. Amplified products were purified using the PCR purification kit (Qiagen) and sequenced.
Table 1.
List of primers used for strand-specific RT-PCR
| Primer name | Nucleotide sequence | Purpose | Location (nt) |
|---|---|---|---|
| A-Fwd | 5′-CCTAAACGGACCTATCGTCG-3′ | Strand-specific RT | 1149–1168 |
| B-Fwd | 5′-TGTCCGTTCGTGAAACCC-3′ | Strand-specific PCR | 1400–1417 |
| B-Rev | 5′-TGCAGGGAGTCTGAGGATTTGG-3′ | Strand-specific PCR | 1623–1644 |
| C-Rev | 5′-GCATGTCCCATCATTCTCCACAGG-3′ | Strand-specific RT | 6270–6293 |
| D-Fwd | 5′-GTGTTTACCATCGACGGG-3′ | Strand-specific PCR | 5689–5706 |
| D-Rev | 5′-AGAAGTTGGACGGTGGAGAGGC-3′ | Strand-specific PCR | 6233–6254 |
| 50 bp-Fwd | 5′-GGAGCTGTGACCATTGGCA-3′ | Strand-sp. RT and PCR | 46–64 |
| ORF7-Rev | 5′-AGAATGCCAGCCCATCA-3′ | Strand-specific PCR | 15258–15274 |
| 3′UTR-Rev | 5′-TTAATTTCGGCCGCATGGTTCT-3′ | Strand-specific RT | 15392–15413 |
The effect of ion channel inhibitors on PRRSV infection
Stock solutions of ammonium chloride, chloroquine, amantadine and verapamil (Sigma) were prepared in water at concentrations of 50 mM, 2 mM, 10 mM and 1 mM, respectively. Marc-145 cells grown in 6-well plates were pre-incubated with different concentrations of the reagents for 30 min and subsequently infected with PRRSV at a multiplicity of infection (MOI) of 1 for 1 h at 37 °C in the presence or absence of the drugs. The virus inoculum was removed and the cells washed three times with MEM. The inoculated cells were then incubated in fresh medium in the presence or absence of the reagents and monitored daily for the appearance of CPE. Culture supernatants were harvested daily from cells for 3 days and virus titer in the supernatant was determined by plaque assay. At 1 day or 2 days post-infection, Marc-145 cells infected with PRRSV in presence or absence of the drugs were fixed with cold methanol and subjected to an immunofluorescence assay as described above. The effect of ion channel blockers on PRRSV replication was also determined by strand-specific RT-PCR. Total cellular RNA was extracted from mock-infected or PRRSV-infected cells in presence or absence of the drugs using TRIzol (Invitrogen) and DNA fragments representing positive-sense or negative-sense viral genomic RNA were RT-PCR amplified using ORF7-specific primers (Lee et al., 2006) or negative-sense-specific primers (Table 1), respectively, as described above.
Protein expression in E. coli and hygromycin B permeability assay
An inducible E. coli expression system was used to express the PRRSV E protein fused with glutathione S-transfererase (GST). Plasmids were transformed into E. coli strain DH5α to express GST fusion proteins. A single colony was grown in Luria Bertani (LB) media containing 100 μg/ml ampicillin overnight. One hundred ml of LB media containing 100 μg/ml ampicillin was inoculated with 1/100 of overnight culture. When the absorbance of cultures reached at an OD600 of 0.6, 1 mM isopropylthio-β-d-galactoside (IPTG) was added to the media to induce protein synthesis. At indicated times after induction, the densities of bacterial cultures were determined by measuring the light scattering at 600 nm.
Permeability of the plasma membrane of bacterial cells expressing the PRRSV E protein to hygromycin B was determined as described previously (Liao et al., 2004). Briefly, bacterial cultures were incubated in the presence or absence of 1 mM of hygromycin B for 15 min at 1 h after IPTG induction, and aliquots of l ml were labeled with for 15 min with 4 μCi/ml of [35S]methionine/cysteine. The labeled bacterial cells were then harvested and lysed in equal volumes of SDS–PAGE sample buffer. Proteins were resolved on a 12% SDS–PAGE gel and visualized by Coomassie blue staining or autoradiography.
Chemical cross-linking
The PRRSV N or E protein was independently expressed in HeLa cells using the T7-based vaccinia virus vTF7-3. HeLa cells seeded on 60-mm-diameter dishes were grown to 90% confluence and infected for 1 h at 37 °C with vTF7-3 at an MOI of 10. Following infection, fresh medium was added and incubation continued for an additional 1 h. The cells were washed in OPTI-MEM and transfected with 1.5 μg of a plasmid for 16 h using the reagent Lipofectin. The transfected cells were then starved for 30 min in methionine-deficient MEM and incubated for 5 h with 100 μCi/ml of [35S]methionine/cysteine. To prepare radiolabeled PRRSV-infected cells, Marc-145 cells seeded on a 100-mm-diameter dish were infected with PRRSV PA8 strain. At 40 h post-infection, PRRSV-infected cells were starved for 30 min in methionine-deficient MEM and metabolically labeled for 10 h with 100 μCi/ml of [35S]methionine/cysteine. For cross-linking studies, cells were washed twice with cold PBS at the end of the labeling period, and samples were incubated with 2 mM of the membrane-permeable and thiol-cleavable cross-linker, 3,3′-dithiobis (succinimidylpropionate) (DSP, Pierce) prepared in 10% DMSO (vol/vol in PBS) at room temperature for 30 min. The reaction was quenched with 50 mM Tris–HCl [pH 7.5] and incubated for an additional 15 min. The cells were lysed with lysis buffer (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, 1% NP-40) containing 1 mM PMSF. The resultant cell lysates were then used in immunoprecipitation with 1 μl of the N-specific MAb SDOW 17 or 3 μl of the E-specific rabbit antiserum followed by SDS–17% PAGE analysis and autoradiography as described above.
Acknowledgments
This study was supported by funding to DY by NSERC Canada, Ontario Pork, OMAF and the USDA NRI PRRS CAP program of the USA. The authors are grateful to Pfizer Animal Health USA for providing the infectious cDNA clone for this study.
References
- Aldabe R., Barco A., Carrasco L. Membrane permeabilization by poliovirus proteins 2B and 2BC. J. Biol. Chem. 1996;271:23134–23137. doi: 10.1074/jbc.271.38.23134. [DOI] [PubMed] [Google Scholar]
- Arroyo J., Boceta M., Gonzalez M.E., Michel M., Carrasco L. Membrane permeabilization by different regions of the human immunodeficiency virus type 1 transmembrane glycoprotein gp41. J. Virol. 1995;69:4095–4102. doi: 10.1128/jvi.69.7.4095-4102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista E.M., Faaberg K.S., Mickelson D., McGruder E.D. Functional properties of the predicted helicase of porcine reproductive and respiratory syndrome virus. Virology. 2002;298:258–270. doi: 10.1006/viro.2002.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfield D.A., Nelson E., Collins J.E., Harris L., Goyal S.M., Robison D., Christianson W.T., Morrison R.B., Gorcyca D., Chladek D. Characterization of swine infertility and respiratory syndrome (SIRS) virus (isolate ATCC VR-2332) J. Vet. Diagn. Invest. 1992;4:127–133. doi: 10.1177/104063879200400202. [DOI] [PubMed] [Google Scholar]
- Bodelón G., Labrada L., Martínez-Costas J., Benavente J. Modification of late membrane permeability in avian Reovirus-infected cells. J. Biol. Chem. 2002;277:17789–17796. doi: 10.1074/jbc.M202018200. [DOI] [PubMed] [Google Scholar]
- Carrasco L. Modification of membrane permeability by animal viruses. Adv. Virus Res. 1995;45:61–112. doi: 10.1016/S0065-3527(08)60058-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.-S., Liao C.-L., Tsao C.-H., Chen M.-C., Liu C.-I., Chen L.-K., Lin Y.-L. Membrane permeabilization by small hydrophobic nonstructural proteins of Japanese encephalitis virus. J. Virol. 1999;73:6257–6264. doi: 10.1128/jvi.73.8.6257-6264.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccaglione A.R., Marcantonio C., Costantino A., Equestre M., Geraci A., Rapicetta M. Hepatitis C virus E1 protein induces modification of membrane permeability in E. coli cells. Virology. 1998;250:1–8. doi: 10.1006/viro.1998.9380. [DOI] [PubMed] [Google Scholar]
- Dea S., Sawyer N., Alain R., Athanassious R. Ultrastructural characteristics and morphogenesis of porcine reproductive and respiratory syndrome virus propagated in the highly permissive MARC-145 cell clone. Adv. Exp. Med. Biol. 1995;380:95–98. doi: 10.1007/978-1-4615-1899-0_13. [DOI] [PubMed] [Google Scholar]
- de Jong A.S., Wessels E., Dijkman H.B., Galama J.M., Melchers W.J., Willems P.H., van Kuppeveld F.J. Determinants for membrane association and permeabilization of the coxsackievirus 2B protein and the identification of the Golgi complex as the target organelle. J. Biol. Chem. 2003;278:1012–1021. doi: 10.1074/jbc.M207745200. [DOI] [PubMed] [Google Scholar]
- Doedens J.R., Kirkegaard K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 1995;14:894–907. doi: 10.1002/j.1460-2075.1995.tb07071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F., Stegen C.F., Masters P.S., Samsonoff W.A. Analysis of constructed E gene mutants of mouse hepatitis virus confirms a pivotal role for E protein in coronavirus assembly. J. Virol. 1998;72:7885–7894. doi: 10.1128/jvi.72.10.7885-7894.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W.B., Sansom M.S.P. Viral ion channels: structure and function. Biochim. Biophys. Acta. 2002;156:27–45. doi: 10.1016/s0304-4157(01)00009-0. [DOI] [PubMed] [Google Scholar]
- Fuerst T.R., Niles E.G., Studier F.W., Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M.E., Carrasco L. Viroporins. FEBS Lett. 2003;552:28–34. doi: 10.1016/s0014-5793(03)00780-4. [DOI] [PubMed] [Google Scholar]
- Han Z., Harty R.N. The NS3 protein of bluetongue virus exhibits viroporin-like properties. J. Biol. Chem. 2004;279:43092–43097. doi: 10.1074/jbc.M403663200. [DOI] [PubMed] [Google Scholar]
- Kim H.S., Kwang J., Yoon I.J., Joo H.S., Frey M.L. Enhanced replication of porcine reproductive and respiratory syndrome (PRRS) virus in a homogeneous subpopulation of MA-104 cell line. Arch. Virol. 1993;133:477–483. doi: 10.1007/BF01313785. [DOI] [PubMed] [Google Scholar]
- Kreutz L.C., Ackermann M.R. Porcine reproductive and respiratory syndrome virus enters cells through a low pH dependent endocytic pathway. Virus Res. 1996;42:137–147. doi: 10.1016/0168-1702(96)01313-5. [DOI] [PubMed] [Google Scholar]
- Lee C., Yoo D. Cysteine residues of the porcine reproductive and respiratory syndrome virus small envelope protein are non-essential for virus infectivity. J. Gen. Virol. 2005;86:3091–3096. doi: 10.1099/vir.0.81160-0. [DOI] [PubMed] [Google Scholar]
- Lee C., Calvert J.G., Welch S.W., Yoo D. A DNA-launched reverse genetics system for porcine reproductive and respiratory syndrome virus reveals that homodimerization of the nucleocapsid protein is essential for virus infectivity. Virology. 2005;331:47–62. doi: 10.1016/j.virol.2004.10.026. [DOI] [PubMed] [Google Scholar]
- Lee C., Hodgins D., Calvert J.G., Welch S.W., Jolie R., Yoo D. Mutations within the nuclear localization signal of the porcine reproductive and respiratory syndrome virus nucleocapsid protein attenuate virus replication. Virology. 2006;346:238–250. doi: 10.1016/j.virol.2005.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Lescar J., Tam J.P., Liu D.X. Expression of SARS-coronavirus envelope protein in Escherichia coli alters membrane permeability. Biochem. Biophys. Res. Commun. 2004;325:374–380. doi: 10.1016/j.bbrc.2004.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.X, Inglis S.C. Association of the infectious bronchitis virus 3c protein with the virion envelope. Virology. 1991;185:911–917. doi: 10.1016/0042-6822(91)90572-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan V., de García M.J., Sanz M.S., Carrasco L. Viroporin activity of murine hepatitis virus E protein. FEBS Lett. 2005;579:3607–3612. doi: 10.1016/j.febslet.2005.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldarelli F., Chen M.Y., Willey R.L., Strebel K. Human immunodeficiency virus type 1 Vpu protein is an oligomeric type I integral membrane protein. J. Virol. 1993;67:5056–5061. doi: 10.1128/jvi.67.8.5056-5061.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X.J., Paul P.S., Halbur P.G., Lum M.A. Phylogenetic analysis of the putative M (ORF 6) and N (ORF 7) genes of porcine reproductive and respiratory syndrome virus (PRRSV): implication for the existence of two genotypes of PRRSV in the U.S.A. and Europe. Arch. Virol. 1995;140:745–755. doi: 10.1007/BF01309962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulenberg J.J.M., Hulst M.M., de Meijer E.J., Moonen P.J.M., den Besten A., De Kluyver E.P., Wensvoort G., Moormann R.J.M. Lelystad virus, the causative agent of porcine epidemic abortion and respiratory syndrome (PEARS), is related to LDV and EAV. Virology. 1993;192:62–72. doi: 10.1006/viro.1993.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulenberg J.J., Petersen-denBesten A., De Kluyver E.P., Moormann R.J., Schaaper W.M., Wensvoort G. Characterization of proteins encoded by ORFs 2 to 7 of Lelystad virus. Virology. 1995;206:155–163. doi: 10.1016/S0042-6822(95)80030-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauwynck H.J., Duan X., Favoreel H.W., Van Oostveldt P., Pensaert M.B. Entry of porcine reproductive and respiratory syndrome virus into porcine alveolar macrophages via receptor-mediated endocytosis. J. Gen. Virol. 1999;80:297–305. doi: 10.1099/0022-1317-80-2-297. [DOI] [PubMed] [Google Scholar]
- Nelsen C.J., Murtaugh M.P., Faaberg K.S. Porcine reproductive and respiratory syndrome virus comparison: divergent evolution on two continents. J. Virol. 1999;73:270–280. doi: 10.1128/jvi.73.1.270-280.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson E.A., Christopher-Hennings J., Drew T., Wensvoort G., Collins J.E., Benfield D.A. Differentiation of US and European isolates of porcine reproductive and respiratory syndrome virus by monoclonal antibodies. J. Clin. Microbiol. 1993;31:3184–3189. doi: 10.1128/jcm.31.12.3184-3189.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuemann E.J., Kliebenstein J.B., Johnson C.D., Mabry J.W., Bush E.J., Seitzinger A.h., Green A.L., Zimmerman J.J. Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. J. Am. Vet. Med. Assoc. 2005;227:385–392. doi: 10.2460/javma.2005.227.385. [DOI] [PubMed] [Google Scholar]
- Paterson R.G., Takeda M., Ohigashi Y., Pinto L.H., Lamb R.A. Influenza B virus BM2 protein is an oligomeric integral membrane protein expressed at the cell surface. Virology. 2003;306:7–17. doi: 10.1016/s0042-6822(02)00083-1. [DOI] [PubMed] [Google Scholar]
- Pinto L.H., Holsinger L.J., Lamb R.A. Influenza virus M2 protein has ion channel activity. Cell. 1992;69:517–528. doi: 10.1016/0092-8674(92)90452-i. [DOI] [PubMed] [Google Scholar]
- Sagripanti J.L., Zandomeni R.O., Weinmann R. The cap structure of simian hemorrhagic fever virion RNA. Virology. 1986;151:146–150. doi: 10.1016/0042-6822(86)90113-3. [DOI] [PubMed] [Google Scholar]
- Sakaguchi T., Tu Q., Pinto L.H., Lamb R.A. The active oligomeric state of the minimalistic influenza virus M2 ion channel is a tetramer. Proc. Natl. Acad. Sci. U.S.A. 1997;94:5000–5005. doi: 10.1073/pnas.94.10.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Russell D.W. 3rd ed. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- Smith A.E., Helenius A. How viruses enter animal cells. Science. 2004;304:237–242. doi: 10.1126/science.1094823. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., Meulenberg J.J. The molecular biology of arteriviruses. J. Gen. Virol. 1998;79:961–979. doi: 10.1099/0022-1317-79-5-961. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., van Tol H., Pedersen K.W., Raamsman M.J., de Vries A.A. Identification of a novel structural protein of arteriviruses. J. Virol. 1999;73:6335–6345. doi: 10.1128/jvi.73.8.6335-6345.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Siddell S.G., Gorbalenya A.E. The order Nidovirales. In: Mahy B.W., terMeulen V., editors. Topley and Wilson's Microbiology and Microbial Infections: Virology Volume. Hodder Arnold; London, UK: 2005. pp. 390–404. [Google Scholar]
- Torres J., Wang J., Parthasarathy K., Liu D.X. The transmembrane oligomers of coronavirus protein E. Biophys. J. 2005;88:1283–1290. doi: 10.1529/biophysj.104.051730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dinten L.C., Rensen S., Gorbalenya A.E., Snijder E.J. Proteolytic processing of the open reading frame 1b-encoded part of arterivirus replicase is mediated by nsp4 serine protease and is essential for virus replication. J. Virol. 1999;73:2027–2037. doi: 10.1128/jvi.73.3.2027-2037.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Takeuchi K., Pinto L.H., Lamb R.A. Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J. Virol. 1993;67:5585–5594. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieringa R., de Vries A.A., van der Meulen J., Godeke G.J., Onderwater J.J.M., van Tol H., Koerten H.K., Mommaas A.M., Snijder E.J., Rottier P.J. Structural protein requirements in equine arteritis virus assembly. J. Virol. 2004;78:13019–13027. doi: 10.1128/JVI.78.23.13019-13027.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson L., Mckinlay C., Gage P., Ewart G. SARS coronavirus E protein forms cation-selective ion channels. Virology. 2004;330:322–331. doi: 10.1016/j.virol.2004.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissink E.H., Kroese M.V., van Wijk H.A., Rijsewijk F.A.M., Meulenberg J.J., Rottier P.J. Envelope protein requirements for the assembly of infectious virions of porcine reproductive and respiratory syndrome virus. J. Virol. 2005;79:12495–12506. doi: 10.1128/JVI.79.19.12495-12506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton S.K., Yoo D. Homo-oligomerization of the porcine reproductive and respiratory syndrome virus nucleocapsid protein and the role of disulfide linkages. J. Virol. 2003;77:4546–4557. doi: 10.1128/JVI.77.8.4546-4557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton S.K., Yoo D., Rogan D. Full-length sequence of a Canadian porcine reproductive and respiratory syndrome virus (PRRSV) isolate. Arch. Virol. 2000;145:2297–2323. doi: 10.1007/s007050070022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton S.K., Koljesar G., Yang L., Yoon K.J., Yoo D. Antigenic importance of the carboxy-terminal beta-strand of the porcine reproductive and respiratory syndrome virus nucleocapsid protein. Clin. Diagn. Lab. Immunol. 2001;8:598–603. doi: 10.1128/CDLI.8.3.598-603.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.H., Fang Y., Farwell R., SteffenBien M., Rowland R.R., ChristopherHennings J., Nelson E.A. A 10-kDa structural protein of porcine reproductive and respiratory syndrome virus encoded by ORF2b. Virology. 2001;287:183–191. doi: 10.1006/viro.2001.1034. [DOI] [PubMed] [Google Scholar]
- Wu W.H., Fang Y., Rowland R.R., Lawson S.R., Christopher-Hennings J., Yoon K.J., Nelson E.A. The 2b protein as a minor structural component of PRRSV. Virus Res. 2005;114:177–181. doi: 10.1016/j.virusres.2005.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]