

Abstract
We investigated the mechanism by which equine herpesvirus-1 (EHV-1) enters primary cultured equine brain microvascular endothelial cells (EBMECs) and equine dermis (E. Derm) cells. EHV-1 colocalized with caveolin in EBMECs and the infection was greatly reduced by the expression of a dominant negative form of equine caveolin-1 (ecavY14F), suggesting that EHV-1 enters EBMECs via caveolar endocytosis. EHV-1 entry into E. Derm cells was significantly reduced by ATP depletion and treatments with lysosomotropic agents. Enveloped virions were detected from E. Derm cells by infectious virus recovery assay after viral internalization, suggesting that EHV-1 enters E. Derm cells via energy- and pH-dependent endocytosis. These results suggest that EHV-1 utilizes multiple endocytic pathways in different cell types to establish productive infection.
Keywords: Equine herpesvirus-1, Equine brain microvascular endothelial cells, Caveolar endocytosis
Introduction
Viruses deliver their genomes and accessory proteins into host cells in order to initiate their replication. Certain enveloped viruses, including retroviruses (Stein et al., 1987), enter cells through direct fusion of the virion envelope with the plasma membrane, a process that is followed by the release of the viral capsid or genome into the cytoplasm. Other enveloped viruses, such as influenza virus (Matlin et al., 1981) and Semliki Forest virus (Helenius et al., 1980), as well as most nonenveloped viruses rely on the cellular endocytic machinery for their entry into host cells.
Productive infection with alphaherpesviruses had been thought to be established only by direct fusion of the viral envelope with the plasma membrane, as demonstrated by electron microscopic analysis and the effects of treatment with neutralizing antibodies (Fuller and Spear, 1987, Fuller et al., 1989, Fuller and Lee, 1992). Agents that perturb endocytosis were found to have little or no effect on herpes simplex virus (HSV) infection in HEp-2 and Vero cells (Wittels and Spear, 1991). Furthermore, entry of HSV-1 via endocytic vesicles was shown to result in degradation of the virus particles (Campadelli-Fiume et al., 1988). However, it has recently become clear that HSV successfully infects HeLa, receptor-expressing CHO, and C10 murine melanoma cells as well as primary and transformed human epidermal keratinocytes via endocytosis (Nicola et al., 2003, Nicola et al., 2005, Nicola and Straus, 2004, Milne et al., 2005). The cellular and viral requirements for the endocytic entry of HSV into these cells have been characterized. In HeLa and receptor-expressing CHO cells, infectious entry of HSV requires trafficking of the virus to an acidic intracellular compartment, phosphatidylinositol 3-kinase activity, glycoprotein D (gD) receptors, as well as viral gB, gD, and gH–gL (Nicola et al., 2003, Nicola and Straus, 2004). The pathway into C10 murine melanoma cells is gD receptor dependent but independent of vesicles with a low pH (Milne et al., 2005). HSV enters primary and transformed human epidermal keratinocytes, an important target cell population in vivo, by a pH- and tyrosine phosphorylation-dependent mechanism (Nicola et al., 2005).
Equine herpesvirus-1 (EHV-1), an alphaherpesvirus of the family Herpesviridae, is distributed worldwide and causes rhinopneumonitis, abortion, and encephalomyelitis in horses (Storts and Montgomery, 2001). With the use of ultrastructural analysis, we have previously suggested that EHV-1 enters equine brain microvascular endothelial cells (EBMECs) via endocytosis (Hasebe et al., 2006). Similar ultrastructural observations were described for EHV-1 endocytosis in mouse fibroblast L-M cells (Abodeely et al., 1970). Frampton et al. (2007) demonstrated that EHV-1 strain L11ΔgIΔgE, which lacks gI and gE, enters CHO-K1 cells by endocytosis, while entry pathway into equine dermis (E. Derm) cells and rabbit kidney (RK13) cells is direct fusion of viral envelope with the plasma membrane. van de Walle et al. (2008) reported that integrin on the surface of the host cells is involved in the endocytosis of EHV-1. It has remained unclear, however, whether the other strains of EHV-1 utilize endocytosis to enter the susceptible cells.
Here, we have investigated the entry mechanism of EHV-1 into EBMECs and E. Derm cells. With the use of confocal immunofluorescence microscopy, we examined the localization both of EHV-1 and of the endocytic markers clathrin and caveolin during viral internalization. Moreover, we evaluated the involvement of caveolar endocytosis in EHV-1 entry with the cells expressing a dominant negative form of caveolin-1. We also assessed the role of tyrosine kinase activity and of low pH in the endosomal compartment in EHV-1 entry with the use of pharmacological approaches. We also performed energy depletion experiments and infectious virus recovery assay for direct indications of endocytosis. Our data identify caveolar endocytosis as an entry pathway for alphaherpesviruses. Moreover, our results demonstrate the existence of multiple endocytic pathways for EHV-1 entry.
Results
Susceptibility of EBMECs and E. Derm cells to EHV-1 infection
EBMECs and E. Derm cells were examined for their ability to support EHV-1 replication. Kinetics of viral growth in E. Derm cells was similar to those in EBMECs (Fig. 1 ), which were typical of a fully productive infection (Hasebe et al., 2006). These results demonstrated that both EBMECs and E. Derm cells were susceptible to EHV-1 infection and that there was no significant difference in viral replication between EBMECs and E. Derm cells.
Fig. 1.

Viral growth in EBMECs (solid circles) and E. Derm cells (open squares) infected at an m.o.i. of 5 p.f.u. per cell. Each point represents the amount of cell-associated virus as the mean of three determinations. The error bars indicate the SD. The results are representative of 2 independent experiments.
Ultrastructural analysis of the early stage of EHV-1 entry
We have previously suggested that the entry of EHV-1 in EBMECs occurs via endocytosis by using electron microscopy (Hasebe et al., 2006). Here, we further examined the mode of EHV-1 entry into EBMECs and E. Derm cells using electron microscopy. At 10 min post infection (p.i.), enveloped virions were detected in noncoated vesicles within the cytoplasm of EBMECs (Fig. 2A, Hasebe et al., 2006) and E. Derm cells (Figs. 2B, C). These observations imply that EHV-1 enters E. Derm cells as well as EBMECs via endocytosis. We were unable to quantify the numbers of enveloped viral particles in the endosomes, because we could not catch enough particles having the complete recognizable structure (i.e. core, capsid and envelope) for the quantification.
Fig. 2.

Ultrastructural analysis of EHV-1 entry into EBMECs (A) and E. Derm cells (B, C). Cells were incubated with EHV-1 at an m.o.i. of 150 p.f.u. per cell first for 2 h at 4 °C and then for 10 min at 37 °C. Scale bars represent 200 nm. PM, plasma membrane. N, nucleus.
Localization of EHV-1 and clathrin during viral internalization
Clathrin-dependent endocytosis plays a major role in the entry of many viruses, having been classically described for Semliki Forest virus, vesicular stomatitis virus, and influenza virus (Helenius et al., 1980, Matlin et al., 1981, Matlin et al., 1982). Although the vesicles containing the virions appeared not to possess clathrin coats by electron microscopy (Fig. 2; Hasebe et al., 2006), these observations did not exclude the possible involvement of clathrin-dependent endocytosis in EHV-1 entry. We therefore examined the EHV-1-infected cells by two-color immunofluorescence staining for EHV-1 and clathrin-heavy chain at 10 or 30 min p.i. Both in infected EBMECs (Figs. 3 A–F) and E. Derm cells (Figs. 3G–L), EHV-1 immunoreactivity did not colocalize with clathrin at any time points. Lack of cross reactivity of anti-EHV-1 antibodies with anti-clathrin antibody was confirmed by the double staining of uninfected EBMECs and E. Derm cells (data not shown). The specificity of the mouse monoclonal antibody to clathrin-heavy chain in EBMECs and E. Derm cells was examined by Western blotting, in which the antibody detected the specific band at 180 kDa (data not shown).
Fig. 3.

Distribution of EHV-1 immunoreactivity and clathrin during viral entry. EBMECs (A–F) and E. Derm cells (G–L) were infected with EHV-1 at an m.o.i. of 10 p.f.u. per cell for 10 (A–C, G–I) or 30 min (D–F, J–L) at 37 °C, fixed, and processed for double immunofluorescence staining of EHV-1 (green) and clathrin-heavy chain (red). Scale bars represent 5 μm.
Localization of EHV-1 and caveolae during viral internalization
Caveolar endocytosis has emerged as a route of entry for several viruses including simian virus 40 (SV40) (Anderson et al., 1996), mouse polyomavirus (Richterová et al., 2001, Gilbert et al., 2003, Gilbert and Benjamin, 2004), echovirus (Marjomäki et al., 2002), human papillomavirus type 31 (Bousarghin et al., 2003, Smith et al., 2007), human polyomavirus BK (BKV) (Eash et al., 2004), and species C human adenovirus (Colin et al., 2005). So we next examined the possible association of caveolae with EHV-1 during viral internalization. Two-color immunostaining revealed that EHV-1 gB and caveolin, the major component of caveolae, were colocalized in EBMECs at 10 (Figs. 4 A–C) and 30 min p.i. (Figs. 4D–F). Three fields were chosen at random and more than 50 signals for EHV-1 immunoreactivity were counted. Approximately 40% of the EHV-1 signal in EBMECs was colocalized with caveolin at 10 min p.i. The percentage of colocalization was reduced to 19% at 30 min p.i. Such colocalization was not detected in infected E. Derm cells at any time points (Figs. 4G–L). Lack of cross reactivity of anti-EHV-1 antibody with anti-caveolin antibodies was confirmed by the double staining of uninfected EBMECs and E. Derm cells (data not shown). The specificity of the rabbit polyclonal antibodies to caveolin in EBMECs and E. Derm cells was examined by Western blotting, in which the antibodies detected the specific bands at 24 kDa for alpha isoform and/or 21 kDa for beta isoform (data not shown).
Fig. 4.

Distribution of EHV-1 gB and caveolin during viral entry. EBMECs (A–F) and E. Derm cells (G–L) were infected with EHV-1 at an m.o.i. of 10 p.f.u. per cell for 10 (A–C, G–I) or 30 min (D–F, J–L) at 37 °C, fixed, and processed for double immunofluorescence staining of EHV-1 gB (green) and caveolin (red). Arrowheads show colocalization of EHV-1 gB and caveolin. Scale bars represent 5 μm.
Effects of a dominant negative form of caveolin-1 in EHV-1 entry
Previous studies have reported that the tyrosine phosphorylation of caveolin-1 at residue 14 mediates the release of caveolae from the plasma membrane and is an integral part of certain signaling pathways (Parton et al., 1994, Aoki et al., 1999, Orlichenko et al., 2006). To confirm the involvement of caveolae vesicles in EHV-1 entry into EBMECs, we constructed equine caveolin-1 mutant. This mutant encodes a protein in which tyrosine 14 is mutated to phenylalanine (ecavY14F). This mutant has been known to act as a dominant negative inhibitor of caveolin-1 (Orlichenko et al., 2006). The ecavY14F construct and wild type equine caveolin-1 (WT ecav) were expressed in EBMECs and E. Derm cells using a lentivirus vector. Cells expressing either WT ecav or ecavY14F were infected with EHV-1 strain Ab4-GFP (Ab4-GFP). The Ab4-GFP harbors a GFP expression cassette between gene 62 and gene 63 (Ibrahim et al., 2004). Therefore, the viral infected cells show GFP signal. Fewer EHV-1-infected EBMECs were observed in cells expressing ecavY14F than in cells expressing WT ecav (p < 0.01). However, there was no difference in the number of the EHV-1-infected E. Derm cells expressing ecavY14F or WT ecav (Figs. 5 A, B). Expression of WT ecav and ecavY14F was confirmed by Western blotting with rabbit anti-caveolin polyclonal antibodies (Fig. 5C).
Fig. 5.

Effects of the expression of a dominant negative form of equine caveolin-1 on EHV-1 infection. Cells expressing WT ecav or ecavY14F were infected with Ab4-GFP at an m.o.i. of 1 per cell for 16 h p.i. (A) The GFP signals in the infected cells were detected by fluorescent microscopy. Scale bars represent 500 μm. (B) The relative proportion of EHV-1 infected cells expressing either WT ecav or ecavY14F determined as described in Materials and methods. The graphs show the mean of three determinations. The error bars show SD. ⁎P < 0.01 versus the control value (Student's t test). The results are representative of 2 independent experiments. (C) Western blotting analysis of the expression of WT ecav (WT) and ecavY14F (Y14F) with rabbit polyclonal antibodies to caveolin. Mock indicates the endogenous expression of caveolin-1 in EBMECs and E. Derm cells.
Effects of a tyrosine kinase inhibitor on EHV-1 entry
The effects of endocytosis inhibitors on EHV-1 infection were assessed by quantification of ICP0 RNA, the product of an early gene of EHV-1. The production of ICP0 RNA is indicative of successful entry of the viral genome into the nucleus and is maximal at 3 h p.i. in RK13 cells (Kimura et al., 2004). Given that the onset of EHV-1 DNA synthesis in RK13 and L-M cells was detected at 4 h p.i. (Caughman et al., 1985, O'Callaghan et al., 1968), the abundance of ICP0 RNA at 3 h p.i. is thought to reflect the number of virions that have infected the cells. The ICP0 RNA could be detected in EHV-1-infected RK13 cells at a multiplicity of infection (m.o.i.) of 0.004 plaque forming unit (p.f.u.) per cell (data not shown).
Previous studies have suggested that cellular tyrosine kinase activity aids EHV-1 infection (Frampton et al., 2007). Cellular tyrosine kinase activity is important for receptor-mediated endocytosis (Greenberg et al., 1993, Lamaze et al., 1993, McPherson et al., 2001). Furthermore, tyrosine phosphorylation of caveolin-1 at residue 14 is important in signaling pathways mediating release of caveolae from the plasma membrane since caveolar fission is decreased by kinase inhibition (Parton et al., 1994, Aoki et al., 1999). We therefore examined the effects of genistein, a tyrosine kinase inhibitor (Akiyama et al., 1987), on the endocytosis of EHV-1. The amount of ICP0 RNA in EBMECs at 3 h p.i. was greatly reduced by treatment with genistein at a concentration of 50 μg/ml (Fig. 6A). In contrast, genistein had no effect on the abundance of ICP0 RNA in E. Derm cells. To eliminate the possibility that the results of E. Derm cells were due to the inefficient uptake of genistein, we assessed the effect of genistein on the tyrosine phosphorylation of caveolin-1 in E. Derm cells (Fig. 6B). Tyrosine phosphorylation of caveolin-1 at residue 14 was diminished by the treatment of genistein at 100 μg/ml, suggesting that the concentration of genistein used in this study was effective to down-regulate the tyrosine phosphorylation of caveolin-1. Neither the morphology of both cell types nor the level of expression of the cellular housekeeping gene for horse GAPDH was affected by genistein in both cell types at 50 μg/ml and 100 μg/ml (data not shown).
Fig. 6.

Role of tyrosine phosphorylation of caveolin in EHV-1 entry. (A) Effects of genistein on EHV-1 entry into EBMECs and E. Derm cells. Cells were incubated with the indicated concentrations of genistein for 1 h at 37 °C, infected with EHV-1 at an m.o.i. of 5 p.f.u. per cell for 1 h, and then incubated for an additional 2 h in the continued presence of genistein. The amount of EHV-1 ICP0 RNA was normalized by the amount of GAPDH mRNA and then expressed as a percentage of the value for infected cells not treated with genistein (control). ⁎P < 0.001 versus the corresponding control value (Student's t test). ND, not determined. The graphs show the mean of three determinations. The error bars show SD. The results are representative of 2 dependent experiments. (B) Effects of genistein on tyrosine phosphorylation in E. Derm cells. Cells were incubated with the indicated concentrations of genistein for 1 h at 37 °C, collected in lysis buffer. The cell lysates were immunoprecipitated with rabbit polyclonal antibodies to caveolin and the immunoprecipitates were subjected to Western blotting with a specific antibody to phosphorylated caveolin-1 at residue 14 (pcavY14).
Effects of lysosomotropic agents on EHV-1 entry
Low pH in endosome is important for many viruses to enter the host cells either via clathrin-dependent endocytosis or clathrin- and caveolae-independent pathway (Helenius et al., 1982, Yoshimura and Ohnishi, 1984, Blumenthal et al., 1987, Nicola et al., 2003). Intracellular low pH is involved in BKV entry by caveolae-dependent endocytosis (Eash et al., 2004) although SV40 infection by caveolar endocytosis is pH independent (Ashok and Atwood, 2003). To determine whether an acidic compartment is required for EHV-1 infectivity, we examined the effects of lysosomotropic agents on EHV-1 ICP0 RNA production. Cells were treated with bafilomycin A1 and ammonium chloride to neutralize the pH of acidic organelles (Tsiang and Superti, 1984, van Weert et al., 1995, Dröse and Altendorf, 1997) and infected with EHV-1 in the continued presence of the reagent. The abundance of ICP0 RNA in E. Derm cells was reduced in a concentration-dependent manner by treatment with bafilomycin A1 (Fig. 7A) and ammonium chloride (Fig. 7B). On the other hand, EHV-1 entry into EBMECs was not inhibited by bafilomycin A1 (Fig. 7A) and ammonium chloride at 10 mM (Fig. 7B). The effect of ammonium chloride at 20 mM in EBMECs was not determined, because the expression of horse GAPDH was significantly reduced (p < 0.01; data not shown) suggesting that 20 mM of ammonium chloride is toxic in EBMECs. A fluorescent pH indicator probe, LysoSensor™ Yellow/Blue DND-160, was used to confirm that the pH of the organelles was neutralized. This probe exhibits a pH-dependent increase in fluorescence intensity upon acidification when the cells are excited at 405 nm and the fluorescence is emitted at 490 nm. Untreated EBMECs and E. Derm cells exhibited punctuate staining. The staining of LysoSensor was diminished in EBMECs by the treatment with 0.2 μM of bafilomycin A1 and with 10 mM of ammonium chloride (Figs. 7C, D). In E. Derm cells, signals of LysoSensor were diminished by the treatment with 1 μM of bafilomycin A1 and 20 mM of ammonium chloride (Figs. 7C, D). The concentration of bafilomycin A1 and ammonium chloride used here seemed to be non-toxic, because neither the morphology of both cell types nor the level of expression of GAPDH was affected (data not shown).
Fig. 7.

Role of a low pH in intracellular organelles in EHV-1 entry. (A) Effects of bafilomycin A1 on EHV-1 entry into EBMECs and E. Derm cells. Cells were incubated with the indicated concentrations of bafilomycin A1 for 1 h at 37 °C, infected with EHV-1 at an m.o.i. of 5 p.f.u. per cell for 1 h, and then incubated for an additional 2 h in the continued presence of test agent. The amount of EHV-1 ICP0 RNA was normalized by the amount of GAPDH mRNA and then expressed as a percentage of the value for infected cells not treated with reagent (control). ⁎P < 0.01 versus the corresponding control value (Student's t test). The graphs show the mean of three determinations. The error bars show SD. The results are representative of 2 independent experiments. (B) Effects of ammonium chloride on EHV-1 entry into EBMECs and E. Derm cells. Viral entry was evaluated by the amount of EHV-1 ICP0 RNA with normalization by the amount of GAPDH mRNA. ⁎P < 0.01 versus the corresponding control value (Student's t test). ND, not determined. The graphs show the mean of three determinations. The error bars show SD. The results are representative of 2 independent experiments. (C) Evaluation of the effects of bafilomycin A1 on lysosomal pH in EBMECs and E. Derm cells. Cells were incubated with the indicated concentrations of bafilomycin A1 for 1 h at 37 °C and exposed to LysoSensor Yellow/Blue DND-160. The pH in acidic organelles was evaluated by the fluorescence intensity of LysoSensor Yellow/Blue DND-160. Scale bars represent 10 μm. (D) Evaluation of the effects of ammonium chloride on lysosomal pH in EBMECs and E. Derm cells with LysoSensor Yellow/Blue DND-160. Scale bars represent 10 μm.
Effects of ATP depletion on EHV-1 entry into E. Derm cells
EM studies and the effects of lysosomotropic agents suggest the possibility that EHV-1 enters E. Derm cells via endocytosis. However, these data are insufficient to demonstrate that endocytosis is involved in EHV-1 entry into E. Derm cells. To assess this possibility, we examined the effect of ATP depletion on EHV-1 infection to E. Derm cells. ATP depletion is known to inhibit endocytosis, but has no effect on herpesvirus entry by direct fusion of viral envelopes with plasma membranes (Nicola et al., 2003). E. Derm cells were pretreated with glucose-free media containing 2-deoxy-d-glucose for 1 h, and then infected with Ab4-GFP for 1 h in the continued presence of glucose-free media. After virus infection, the media were replaced with complete growth media. To confirm that ATP depletion affects the viral entry step, the cells were infected with EHV-1 with media including glucose for 1 h, and then cultured with glucose-free media for 2 h. The number of infected cells was later evaluated by counting GFP-positive cells at 12 h p.i. ATP depletion during viral entry greatly reduced EHV-1 infection (P < 0.001) whereas ATP depletion post viral entry did not significantly reduce EHV-1 infection (Fig. 8 ).
Fig. 8.
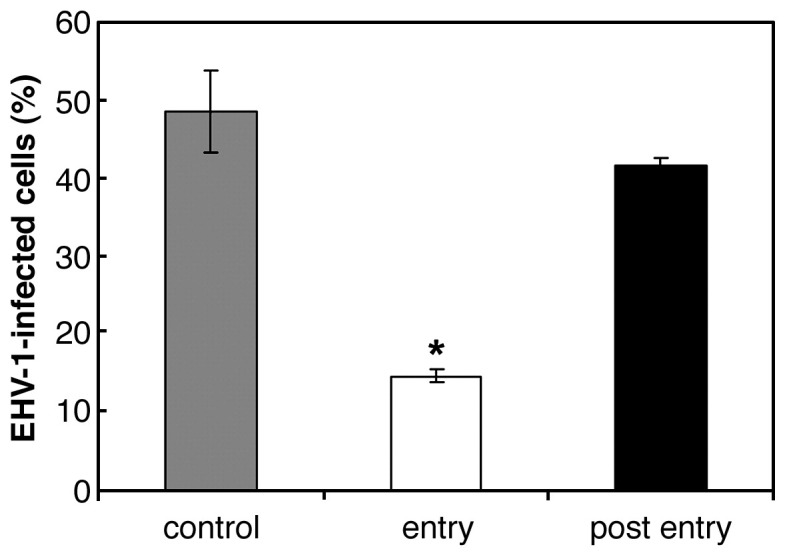
Effect of ATP depletion on EHV-1 entry into E. Derm cells. The cells were treated with ATP depletion media during (white bar) or post viral entry (black bar). For the untreated control (gray bar), the cells were incubated in FBS-free media. The cells were infected with Ab4-GFP at an m.o.i. of 5 per cell and EHV-1 infected cells were counted by FACS. ⁎P < 0.001 versus the corresponding control value (Student's t test). The graphs show the mean of three determinations. The error bars show SD. The results are representative of 2 independent experiments.
Infectious virus recovery from E. Derm cells
Frampton et al. (2007) performed infectious virus recovery assays to demonstrate that EHV-1 L11ΔgIΔgE strain enters CHO-K1 cells via endocytosis. Since endocytosed virions possess envelopes in the early phase of entry, infectivity can be detected by titration on RK13 cells. On the other hand, when virus penetrates by direct fusion of envelope with plasma membrane, infectious virions cannot be detected due to the loss of the viral envelope. To confirm that EHV-1 enters E. Derm cells via endocytosis, we performed infectious virus recovery assay. After incubation at 4 °C for 5 min, the cells were infected with EHV-1 at an m.o.i. of 10 p.f.u. per cell for 2 h at 4 °C. The temperature was then shifted to 37 °C to allow the virus internalization. At 0, 7.5, 15, 30 and 45 min after the temperature shift, the viruses on the cell surface were inactivated by washing with acidic buffer. The internalized infectious viruses were titrated on RK13 cells. For a control to eliminate the possibility of detecting virions remaining on the cell surface, we used NIH3T3 cells, which seem to be resistant to EHV-1 entry because we could not detect viral RNA at 12 h p.i. by RT-PCR (data not shown). At 0 and 7.5 min, no infectious virus was detected from E. Derm and NIH3T3 cells. At 15 min, virus was recovered from E. Derm cells. The virus titer from E. Derm cells reached a peak at 30 min and slightly declined at 45 min. In contrast, no virus was recovered from NIH3T3 cells at any time point (Fig. 9 ).
Fig. 9.

Infectious virus recovery assay. E. Derm (open square) and NIH3T3 cells (solid circle) were infected with EHV-1 at an m.o.i. of 10 per cell at 4 °C for 2 h. Then the cells were shifted to 37 °C to allow viral internalization. At the indicated times, the remaining viruses on the cell surface were inactivated by acidic buffer. Internalized infectious viruses were detected by titration on RK13 cells. The graphs show the mean of three determinations. The error bars show SD.
Discussion
The colocalization of EHV-1 with caveolin at the early stage of infection and the significant effect of a dominant negative form of caveolin-1 on the EHV-1 infection suggests that the virus enters EBMECs via caveolar endocytosis. The results of double immunolabeling indicated that clathrin-dependent endocytosis plays a relatively minor role in EHV-1 entry into EBMECs. As far as we are aware, our study is the first to demonstrate alphaherpesvirus entry into cells via caveolar endocytosis.
The production of EHV-1 ICP0 RNA in EBMECs was blocked by the tyrosine kinase inhibitor genistein, indicating a requirement for tyrosine phosphorylation in the entry of EHV-1 into these cells although we cannot exclude the possibility that genistein might affect ICP0 gene expression after viral entry. Tyrosine phosphorylation initiates signal transduction events that lead to receptor-mediated endocytosis (McPherson et al., 2001). In caveolar endocytosis, tyrosine kinase activity is required for phosphorylation of caveolin at residue 14, which induces caveolar vesiculation and enclosure of ligands within caveolae (Aoki et al., 1999, Chen and Norkin, 1999). Our result indicated that EHV-1 may induce caveolin phosphorylation, which activates the subsequent signal transduction.
Caveolae have traditionally been described as smooth invaginations of the plasma membrane with a diameter of 50 to 80 nm (Palade, 1953, Yamada, 1955). Most viruses that have been shown to enter host cells via caveolae are nonenveloped and therefore smaller than typical caveolar invaginations. However, recent studies have shown that this traditional description of caveolar morphology is inadequate, as caveolae with flat or tubular forms have also been detected (Anderson, 1998). Caveolae-dependent endocytosis has also been found to contribute to the entry of enveloped viruses, such as filoviruses and human coronavirus (Empig and Goldsmith, 2002, Nomura et al., 2004). Our data now provide support for the notion that caveolar vesicles mediate the delivery of large enveloped viruses.
Infection of endothelial cells in the horse central nervous system (CNS) is required for establishment of EHV-1-induced encephalomyelitis, which is characterized by vasculitis, thrombosis, and secondary ischemia of neuronal tissue (Edington et al., 1986). We have previously proposed that EBMECs are an appropriate in vitro model for studies of the endotheliotropism of EHV-1 (Hasebe et al., 2006). Primary cultured brain microvascular endothelial cells (BMECs) retain several characteristics of CNS endothelial cells in vivo (Joó, 1996). Infection of human BMECs with Escherichia coli K1 results in the formation of abundant caveolae that mediate bacterial uptake (Sukumaran et al., 2002). Immunohistochemical studies of normal brain tissue have also shown the caveolar compartment to be pronounced in endothelial cells, suggesting an important physiological role for caveolar mediated endocytosis in vivo (Virgintino et al., 2002). It is therefore reasonable to propose that EHV-1 makes use of caveolar endocytosis to infect endothelial cells in the horse CNS.
Viral entry through caveolae has traditionally been considered to occur in a pH-neutral setting, bypassing the acidic endosome (Ashok and Atwood, 2003). Recently, caveolin-1-positive endosomes have been shown to deliver caveolae-internalized cargo to the Golgi complex (Nichols, 2003) and BKV enters Vero cells via this pathway (Eash et al., 2004). The effects of the lysosomotropic agent used in this study suggest that acidic intracellular organelles do not facilitate EHV-1 infection in EBMECs. Therefore, EHV-1 may be transported to pH-neutral organelles after internalization via caveolae.
Immunofluorescence microscopic analysis and the lack of an effect of the dominant negative form of caveolin-1 on EHV-1 infection in E. Derm cells suggested that EHV-1 entry occurred by a caveolae-independent route. Such a route might be operative in vivo for EHV-1 infection of certain cell types, such as lymphocytes, that do not appear to form caveolae. The principal pathway of EHV-1 entry into E. Derm cells also appears to be clathrin independent, given that EHV-1 immunoreactivity did not colocalize with clathrin-heavy chain in these cells.
Despite the lack of colocalization of EHV-1 with endocytic markers, our data from electron microscopy, lysosomotropic agent treatment, energy depletions and infectious virus recovery assays suggest that EHV-1 enters E. Derm cells via energy- and pH-dependent endocytosis. In contrast, Frampton et al. (2007) suggested that EHV-1 strain L11ΔgIΔgE entry into E. Derm cells occurs by direct fusion at the cell surface. The difference between Frampton's study and our own could be explained by the difference in virus strain. The strain L11ΔgIΔgE is a mutant which lacks gI and gE, resulting in attenuated virulence in mice and reduction of viral growth on RK13 cells compared to the parental virus strain RacL11 (Frampton et al., 2002). It has been thought that gE of varicella zoster virus (VZV), another Varicellovirus, is associated with viral entry. Notably, Li et al. (2006) reported that gE interacts with insulin-degrading enzyme, which acts as a cellular receptor mediating cell-free VZV infection and cell-to-cell spread. Therefore, the lack of gE might influence the EHV-1 entry mechanism. The dependence of entry mechanisms on virus strain has been reported for human papillomavirus by Bousarghin et al. (2003), who demonstrated that although they are very closely related viruses human papillomavirus types 16, 31 and 58 use different pathways to enter cells.
In conclusion, our results suggest that EHV-1 entry pathways are cell type dependent. Furthermore, they show that EHV-1 enters certain cell types via caveolar endocytosis, a pathway that has not previously been known to mediate the entry of alphaherpesviruses.
Materials and methods
Cells and virus
The EBMECs were isolated from the brain of a 6-month-old horse as described previously (Hasebe et al., 2006) and were cultured in Medium 199 Earl's (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Sigma, St. Louis, MO, USA) and both penicillin (100 U/ml) and streptomycin (100 μg/ml) (Invitrogen). The E. Derm cells were obtained from American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 0.1 mM nonessential amino acids (Invitrogen) and 10% FBS. The RK13 cells were cultured in minimum essential medium (MEM) supplemented with 10% FBS. The 293T cells were cultured in DMEM supplemented with 10% FBS. The EHV-1 strain HH1 was isolated from an aborted equine fetus in Japan (Kawakami et al., 1970). An EHV-1 mutant, Ab4-GFP, was generously provided by Dr. H. Fukushi. (Gifu University, Gifu, Japan). The Ab4-GFP was constructed by inserting a GFP expression cassette into the intergenic region between ORF62 and ORF63 of EHV-1 Ab4 strain (Ibrahim et al., 2004). Stock viruses were grown in confluent monolayers of RK13 cells. In preparation for RNA dot-blot analysis, viruses were treated with RNase (20 ng/ml) for 1 h at 37 °C to remove contaminating RNA in the stock virus. Viral titer was determined by a plaque formation assay using RK13 cells.
Chemicals and antibodies
Genistein was obtained from Sigma, and bafilomycin A1 and ammonium chloride were from Wako (Osaka, Japan). Bafilomycin A1 and genistein were dissolved in dimethyl sulfoxide at 1 mM and 100 mg/ml, respectively. Ammonium chloride was dissolved in distilled water at 5 M. The final concentration of dimethyl sulfoxide in culture medium was ≤ 0.1%, and the same concentration was also added to control incubations.
Rabbit polyclonal antibodies to EHV-1 were kindly provided by Dr. R. Kirisawa (Rakuno Gakuen University, Hokkaido, Japan), and a mouse monoclonal antibody to EHV-1 gB protein was kindly provided by Dr. T. Matsumura (Japan Racing Association, Tochigi, Japan). Rabbit polyclonal antibodies to caveolin, a mouse monoclonal antibody to caveolin (pY14) and a mouse monoclonal antibody to clathrin-heavy chain were obtained from BD Transduction Laboratories (San Jose, CA, USA). Alexa Fluor 488-conjugated goat antibodies to mouse immunoglobulin G, Alexa Fluor 594-conjugated goat antibodies to rabbit immunoglobulin G, 4′,6-diamidino-2-phenylindole (DAPI) and LysoSensor™ Yellow/Blue DND-160 were from Molecular Probes (Leiden, The Netherlands). Horse radish peroxidase (HRP)-conjugated goat antibodies to mouse immunoglobulin and HRP-conjugated goat antibodies to rabbit immunoglobulin were obtained from Biosource (Camarillo, CA, USA).
Viral growth
Confluent monolayers of EBMECs or E. Derm cells seeded into 24-well plates were infected with EHV-1 at an m.o.i. of 5 p.f.u. per cell. The infected cells were incubated at 37 °C for 1 h to allow attachment of the virus, then washed three times with phosphate-buffered saline (PBS), provided with fresh growth media, and incubated further at 37 °C. At 0 h (immediately after seeding the virus), 8 h, 16 h or 24 h p.i., the supernatants were removed and the cells were collected. The cells were suspended in 1 ml of MEM and lysed by three cycles of freezing and thawing. Viral titer was determined by the plaque formation on RK13 cells.
Electron microscopy
EBMECs or E. Derm cells cultured in six-well plates were exposed to EHV-1 at an m.o.i. of 150 p.f.u. per cell and incubated for 2 h at 4 °C to allow attachment of the virus to the cell surface. After subsequent incubation for 10 min at 37 °C, the cells were collected and fixed overnight at 4 °C with 2.5% glutaraldehyde. The cells were subsequently exposed to 2% osmic acid, dehydrated, and embedded in Epon 812 (Shell Chemical Company, New York, NY, USA). Sections were cut at a thickness of 70–80 nm, mounted on coated grids, stained with uranyl acetate and lead citrate, and examined with an electron microscope (JEM-1210; Japan Electron Optics Laboratory, Tokyo, Japan).
Indirect immunofluorescence staining
Confluent monolayers of EBMECs or E. Derm cells in eight-well chamber slides (BD Falcon, San Jose, CA, USA) were infected with EHV-1 at an m.o.i. of 10 p.f.u. per cell. After incubation at 37 °C for 10 or 30 min, the cells were fixed with 3.7% paraformaldehyde for 5 min and permeabilized with 0.1% Triton X-100 for 5 min. The cells were washed with PBS and incubated for 1 h at room temperature first with 2% bovine serum albumin (Sigma) and then with primary antibodies. For simultaneous detection of EHV-1 and clathrin, cells were stained with rabbit anti-EHV-1 polyclonal antibodies and mouse anti-clathrin-heavy chain monoclonal antibody. For simultaneous detection of EHV-1 and caveolae, cells were stained with mouse anti-EHV-1 gB monoclonal antibody and rabbit anti-caveolin polyclonal antibodies. Then the cells were incubated for 1 h at room temperature with secondary antibodies, mounted with the use of fluorescence mounting medium (Dako Cytomation, Carpinteria, CA, USA), and examined with a laser-scanning confocal microscope (Olympus, Tokyo, Japan). For all primary antibodies, control images were evaluated to ensure nonoverlapping binding of secondary antibodies and specific detection for each excitation channel. The images were processed with FV10-ASV 1.4 Viewer (Olympus). Coincidence of the immunoreactivity between rabbit anti-EHV-1 polyclonal antibodies and mouse anti-EHV-1 gB monoclonal antibody was confirmed (data not shown).
Preparation of lentiviral vector
The lentiviral vector system was provided by Dr. Hiroyuki Miyoshi, RIKEN BioResource Center, Ibaraki, Japan. Full-length equine caveolin-1 cDNA amplified by PCR from cDNA of EBMECs was cloned into pcDNA 3.1(−) (Invitrogen) and CSII-CMV-MCS-IRES2-Bsd (RIKEN BioResource Center). These constructs were designated as pcDNA 3.1(−)-ecav and CSII-CMV-ecav-IRES2-Bsd, respectively. A mutant form of equine caveolin-1 was generated from pcDNA 3.1(−)-ecav via PCR-based mutagenesis and subcloned into CSII-CMV-MCS-IRES2-Bsd, which was designated as CSII-CMV-ecavY14F-IRES2-Bsd. Lentiviral vectors pseudotyped with vesicular stomatitis virus G glycoprotein (VSV-G) were generated according to the instruction provided by the RIKEN BioResource Center. Briefly, 293T cells were transfected with the package construct (pCAG-HIVgp), the VSV-G-expressing construct (pCMV-VSV-G-RSV-Rev) and the self-inactivating vector construct (CSII-CMV-ecav-IRES2-Bsd or CSII-CMV-ecavY14F-IRES2-Bsd). After 48 h, the supernatant was concentrated by ultracentrifugation at 50,000 g for 2 h. The pellet was resuspended in Hanks' Balanced Salt Solutions (Invitrogen). Vector titer was determined using HIV p24 ELISA Kit (PerkinElmer Life Sciences, Inc., Wellesley, MA, USA).
Expression of wild type and mutant form of caveolin-1
The EBMECs and E. Derm cells were infected with lentiviral vectors at an m.o.i. of 0.01 infectious unit per cell and incubated for 24 h. Then the cells were infected with EHV-1 at an m.o.i. of 1 p.f.u. per cell. After 16 h, the cells were fixed with 3.7% paraformaldehyde, permeabilized with 0.1% Triton X-100 and counterstained with DAPI, a nuclear stain. The GFP and DAPI signal was evaluated with fluorescent microscopy (Olympus). The number of GFP expressing cells and the number of nuclei were counted by Image-J (NIH, Bethesda, MD, USA). The relative proportion of EHV-1-infected cells was calculated by dividing the number of the GFP-positive cells by the number of the nuclei. The number of the GFP-positive cells in wild type ecav expressing EBMECs or E. Derm cells was defined as 100%.
Expression of the transgene products was confirmed by Western blotting. The cells infected with lentiviral vector at 24 h p.i. were lysed in RIPA buffer [10 mM Tris–HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA, 50 mM NaF, 10% glycerol, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 5 mM phenylmethylsulfonyl fluoride], and mixed with Complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). The cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-page) and transferred to PVDF membrane (Millipore, Billerica, MA, USA). The membrane was blocked with 2% low-fat milk in Tris-buffered saline containing 0.05% Tween 20 (TBS-T) at 4 °C overnight, incubated with a rabbit polyclonal antibodies to caveolin (1:1,000 dilution in 2% low-fat milk in TBS-T) for primary antibody, and then with HRP-conjugated goat antibodies to rabbit immunoglobulin (1:5000 dilution in 2% low-fat milk in TBS-T) for secondary antibody. The immunocomplex was visualized with Immobilon™ western chemiluminescent HRP substrate (Millipore) and LAS-1000mini (FUJIFILM, Tokyo, Japan).
Preparation of RNA probes
Complementary DNA fragments corresponding to EHV-1 ICP0 RNA and horse glyceraldehydes-3-phosphate dehydrogenase (GAPDH) were cloned by reverse transcription and PCR from total RNA extracted either from EHV-1-infected RK13 cells or from uninfected equine placenta, respectively, using PCR primers 5′-TTTTTGGCCGTGGATTCTGG-3′ and 5′-AGTTCTGCTTGGACGATGAG-3′ for ICP0 RNA and 5′-AGTTCCATGGCACAGTCAAG-3′ and 5′-ACAAACATTGGGGCATCAGC-3′ for GAPDH. The PCR products were cloned into the pGEM-T vector (Promega, Madison, WI, USA), and the resultant plasmids were sequenced to verify their identity.
Antisense RNA probes were prepared with a digoxigenin-based RNA labeling kit (SP6/T7; Roche Diagnostics). Plasmids containing cloned cDNA were linearized with Not I for synthesis of RNA in the presence of digoxigenin-11-UTP. The labeled probes generated from 1 μg of plasmid DNA were precipitated with ethanol, dissolved in 50 μl of RNase-free water, and stored at − 80 °C.
Dot-blot analysis
Confluent monolayers of EBMECs or E. Derm cells in six-well plates were treated with inhibitor at 37 °C for 1 h and then infected with RNase-treated EHV-1 at an m.o.i. of 5 p.f.u. per cell for 1 h at 37 °C in the continued presence of an inhibitor. After additional 2 h incubation in the presence of inhibitor, total RNA was extracted with using Trizol reagent (Invitrogen), treated with DNase with the use of a kit (Ambion, Austin, TX, USA), and diluted to a concentration of 2 mg/ml. Hybridization was performed as previously described (Kimura et al., 2004). In brief, 1 μl of each sample was spotted onto a dry positively charged nylon membrane (Roche) and allowed to dry in air. The RNA was fixed to the membrane with a UV cross-linker (XL-1000; Spectronics, Lincoln, NE) and baked for 30 min at 80 °C. The membrane was then incubated for 3 h at 68 °C in a solution containing 0.25 M sodium phosphate buffer (pH 7.2), 10% SDS, 1 mM EDTA, and 2% blocking reagent. Hybridization was performed for 12 h at 68 °C in the same solution containing the digoxigenin-labeled cRNA probe (20 ng/ml), after which the membrane was washed three times (each for 20 min) with 25 mM sodium phosphate buffer (pH 7.2) containing 10% SDS and 1 mM EDTA. Hybridization complexes were detected with alkaline phosphatase-conjugated antibodies to digoxigenin and disodium 3-(4-methoxyspiro{1,2-dioxetane-3,2′-(5′-chloro)tricyclo[3.3.1.1.3,7]decan}-4-yl) phenyl phosphate (CSPD) as the chemiluminescent substrate (Roche). Quantitative analysis of autoradiograms was performed with Scion Image software. The relative amount of EHV-1 ICP0 RNA was calculated by dividing the intensity of the signal for ICP0 RNA by that of the signal for GAPDH mRNA. The adjusted signal intensity for infected but mock-treated cells was defined as 100%.
Immunoprecipitation and Western blot analysis
Confluent monolayers of E. Derm cells in 60 mm dishes were treated with genistein at 37 °C for 1 h, washed once with PBS and lysed in RIPA buffer with Complete protease inhibitor cocktail. The cell lysates were mixed at 4 °C with rabbit polyclonal antibodies to caveolin for 1 h and collected on protein A-Sepharose beads (GE Healthcare Bio-Science Corp, NJ, USA). The immunoprecipitates were subjected to SDS-page and Western blotting with a mouse monoclonal antibody to caveolin (pY14) (1:1000 dilution in 2% low-fat milk in TBS-T) or rabbit polyclonal antibodies to caveolin for primary antibodies and HRP-conjugated goat antibodies to mouse immunoglobulin (1:5000 dilution in 2% low-fat milk in TBS-T) or HRP-conjugated goat antibodies to rabbit immunoglobulin for secondary antibodies.
Vital staining with pH indicator
Cells seeded on 35 mm glass bottom dishes were incubated with or without bafilomycin A1 or ammonium chloride at 37 °C for 1 h, and then exposed to 2 μM of a pH sensitive fluorescence dye, LysoSensor Yellow/Blue DND-160, in pre-warmed growth medium. After incubation at 37 °C for 5 min, the medium was replaced with fresh medium. The fluorescence was observed using a confocal laser-scanning microscope with excitation at 405 nm and emission was measured at 480–510 nm.
ATP depletion
Cells were incubated in ATP depletion media composed of glucose-free, FBS-free DMEM (Invitrogen) with 10 mM 2-deoxyglucose (Sigma) for 1 h and infected with Ab4-GFP at an m.o.i. of 5 per cell for 1 h in ATP depletion media. After EHV-1 infection, the cells were treated with citrate buffer pH 3.0 to inactivate remaining viruses on cell surface. The media were replaced with regular culture media and the cells were cultured at 37 °C. For untreated controls, the cells were incubated with FBS-free DMEM for 1 h, infected with Ab4-GFP in FBS-free DMEM for 1 h and the media were replaced with regular culture media. For samples depleted of ATP after post entry, the cells were infected with Ab4-GFP in FBS-free DMEM. After viral infection for 1 h, the cells were incubated in ATP depletion media for 2 h and the media were replaced with regular culture media. At 12 h p.i., the cells were harvested, fixed with 4% paraformaldehyde and GFP-positive cells were counted by FACS Canto (BD Biosciences, Sun Jose, CA, USA).
Infectious virus recovery assay
Infectious virus recovery assay was performed as previously described in Frampton et al. (2007) with some modifications. E. Derm and NIH3T3 cells seeded in 24 well plates were washed with ice-cold DMEM supplemented with 25 mM HEPES and 1% FBS and incubated on ice for 5 min. The cells were infected with EHV-1 HH1 strain at an m.o.i. of 10 p.f.u. per cell for 2 h at 4 °C. The media were replaced with pre-warmed fresh DMEM containing 25 mM HEPES and 1% FBS at 37 °C. At 0, 7.5, 15, 30 and 45 min after incubation at 37 °C, the cells were washed with glycine pH 3.0 for 1 min at room temperature, washed with DMEM containing 25 mM HEPES and harvested. The cells were freeze–thawed once and sonicated three times for 15 s each. The infectious virus was detected by titration on RK13 cells. Triplicate samples were measured for each time point.
Statistical analysis
Quantitative data are expressed as means ± SD and were compared with Student's t test. A P value of < 0.05 was considered statistically significant.
Acknowledgments
The authors gratefully acknowledge the invaluable suggestions by Dr. B. Caughey, senior investigator, chief of TSE/Prion Biochemistry Section, Rocky Mountain Laboratories, NIAID, NIH. The authors thank Ms. S. Yamanouchi and Ms. M. Sasada for technical assistance. This work was supported in part by Grant-in-Aids for young scientist (T. K.) and for JSPS Fellows (R. H.) and for the Program of Founding Research Centers for Emerging and Reemerging Infectious Diseases (R. H., T. K. and H. S.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, by a grant from the Akiyama foundation (T. K.) and by a grant from the Japan Racing Association (T. U.).
Contributor Information
Rie Hasebe, Email: r-hasebe@vetmed.hokudai.ac.jp.
Michihito Sasaki, Email: m-sasaki@czc.hokudai.ac.jp.
Hirofumi Sawa, Email: h-sawa@czc.hokudai.ac.jp.
Ryuichi Wada, Email: Ryuichi_Wada@jra.go.jp.
Takashi Umemura, Email: umemura@vetmed.hokudai.ac.jp.
Takashi Kimura, Email: kimura@czc.hokudai.ac.jp.
References
- Abodeely R.A., Lawson L.A., Randall C.C. Morphology and entry of enveloped and deenveloped equine abortion (herpes) virus. J. Virol. 1970;5:513–523. doi: 10.1128/jvi.5.4.513-523.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama T., Ishida J., Nakagawa S., Ogawara H., Watanabe S., Itoh N., Shibuya M., Fukami Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J. Biol. Chem. 1987;262:5592–5595. [PubMed] [Google Scholar]
- Anderson R.G. The caveolae membrane system. Annu. Rev. Biochem. 1998;67:199–225. doi: 10.1146/annurev.biochem.67.1.199. [DOI] [PubMed] [Google Scholar]
- Anderson H.A., Chen Y., Norkin L.C. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell. 1996;7:1825–1834. doi: 10.1091/mbc.7.11.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki T., Nomura R., Fujimoto T. Tyrosine phosphorylation of caveolin-1 in the endothelium. Exp. Cell Res. 1999;253:629–636. doi: 10.1006/excr.1999.4652. [DOI] [PubMed] [Google Scholar]
- Ashok A., Atwood W.J. Contrasting roles of endosomal pH and the cytoskeleton in infection of human glial cells by JC virus and simian virus 40. J. Virol. 2003;77:1347–1356. doi: 10.1128/JVI.77.2.1347-1356.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal R., Bali-Puri A., Walter A., Covell D., Eidelman O. pH-dependent fusion of vesicular stomatitis virus with Vero cells. Measurement by dequenching of octadecyl rhodamine fluorescence. J. Biol. Chem. 1987;262:13614–13619. [PubMed] [Google Scholar]
- Bousarghin L., Touze A., Sizaret P.Y., Coursaget P. Human papillomavirus types 16, 31, and 58 use different endocytosis pathways to enter cells. J. Virol. 2003;77:3846–3850. doi: 10.1128/JVI.77.6.3846-3850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campadelli-Fiume G., Arsenakis M., Farabegoli F., Roizman B. Entry of herpes simplex virus 1 in BJ cells that constitutively express viral glycoprotein D is by endocytosis and results in degradation of the virus. J. Virol. 1988;62:159–167. doi: 10.1128/jvi.62.1.159-167.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughman G.B., Staczek J., O'Callaghan D.J. Equine herpesvirus type 1 infected cell polypeptides: evidence for immediate early/early/late regulation of viral gene expression. Virology. 1985;145:49–61. doi: 10.1016/0042-6822(85)90200-4. [DOI] [PubMed] [Google Scholar]
- Chen Y., Norkin L.C. Extracellular simian virus 40 transmits a signal that promotes virus enclosure within caveolae. Exp. Cell Res. 1999;246:83–90. doi: 10.1006/excr.1998.4301. [DOI] [PubMed] [Google Scholar]
- Colin M., Mailly L., Rogee S., D'Halluin J.C. Efficient species C HAdV infectivity in plasmocytic cell lines using a clathrin-independent lipid raft/caveola endocytic route. Molec. Ther. 2005;11:224–236. doi: 10.1016/j.ymthe.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Dröse S., Altendorf K. Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J. Exp. Biol. 1997;200:1–8. doi: 10.1242/jeb.200.1.1. [DOI] [PubMed] [Google Scholar]
- Eash S., Querbes W., Atwood W.J. Infection of vero cells by BK virus is dependent on caveolae. J. Virol. 2004;78:11583–11590. doi: 10.1128/JVI.78.21.11583-11590.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edington N., Bridges C.G., Patel J.R. Endothelial cell infection and thrombosis in paralysis caused by equid herpesvirus-1: equine stroke. Arch. Virol. 1986;90:111–124. doi: 10.1007/BF01314149. [DOI] [PubMed] [Google Scholar]
- Empig C.J., Goldsmith M.A. Association of the caveola vesicular system with cellular entry by filoviruses. J. Virol. 2002;76:5266–5270. doi: 10.1128/JVI.76.10.5266-5270.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton A.R., Jr., Smith P.M., Zhang Y., Matsumura T., Osterrieder N., O'Callaghan D.J. Contribution of gene products encoded within the unique short segment of equine herpesvirus 1 to virulence in a murine model. Virus Res. 2002;90:287–301. doi: 10.1016/s0168-1702(02)00245-9. [DOI] [PubMed] [Google Scholar]
- Frampton A.R., Jr., Stolz D.B., Uchida H., Goins W.F., Cohen J.B., Florioso J.C. Equine herpesvirus 1 enters cells by two different pathways, and infection requires the activation of the cellular kinase ROCK1. J. Virol. 2007;81:10879–10889. doi: 10.1128/JVI.00504-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller A.O., Spear P.G. Anti-glycoprotein D antibodies that permit adsorption but block infection by herpes simplex virus 1 prevent virion–cell fusion at the cell surface. Proc. Natl. Acad. Sci. U.S.A. 1987;84:5454–5458. doi: 10.1073/pnas.84.15.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller A.O., Lee W.C. Herpes simplex virus type 1 entry through a cascade of virus–cell interactions requires different roles of gD and gH in penetration. J. Virol. 1992;66:5002–5012. doi: 10.1128/jvi.66.8.5002-5012.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller A.O., Santos R.E., Spear P.G. Neutralizing antibodies specific for glycoprotein H of herpes simplex virus permit viral attachment to cells but prevent penetration. J. Virol. 1989;63:3435–3443. doi: 10.1128/jvi.63.8.3435-3443.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J., Benjamin T. Uptake pathway of polyomavirus via ganglioside GD1a. J. Virol. 2004;78:12259–12267. doi: 10.1128/JVI.78.22.12259-12267.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J.M., Goldberg I.G., Benjamin T.L. Cell penetration and trafficking of polyomavirus. J. Virol. 2003;77:2615–2622. doi: 10.1128/JVI.77.4.2615-2622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg S., Chang P., Silverstein S.C. Tyrosine phosphorylation is required for Fc receptor-mediated phagocytosis in mouse macrophages. J. Exp. Med. 1993;177:529–534. doi: 10.1084/jem.177.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasebe R., Kimura T., Nakamura K., Ochiai K., Okazaki K., Wada R., Umemura T. Differential susceptibility of equine and mouse brain microvascular endothelial cells to equine herpesvirus 1 infection. Arch. Virol. 2006;151:775–786. doi: 10.1007/s00705-005-0653-3. [DOI] [PubMed] [Google Scholar]
- Helenius A., Kartenbeck J., Simons K., Fries E. On the entry of Semliki forest virus into BHK-21 cells. J. Cell Biol. 1980;84:404–420. doi: 10.1083/jcb.84.2.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A., Marsh M., White J. Inhibition of Semliki forest virus penetration by lysosomotropic weak bases. J. Gen. Virol. 1982;58:47–61. doi: 10.1099/0022-1317-58-1-47. [DOI] [PubMed] [Google Scholar]
- Ibrahim E.S.M., Pagmajav O., Yamaguchi T., Matsumura T., Fukushi H. Growth and virulence alterations of equine herpesvirus 1 by insertion of a green fluorescent protein gene in the intergenic region between ORFs 62 and 63. Microbiol. Immunol. 2004;48:831–842. doi: 10.1111/j.1348-0421.2004.tb03615.x. [DOI] [PubMed] [Google Scholar]
- Joó F. Endothelial cells of the brain and other organ systems: some similarities and differences. Prog. Neurobiol. 1996;48:255–273. doi: 10.1016/0301-0082(95)00046-1. [DOI] [PubMed] [Google Scholar]
- Kawakami Y., Tokui T., Nakano K., Kume T., Hiramune T., Murase N. An outbreak of abortion due to equine rhinopneumonitis virus among mares in the Hidaka district, Hokkaido. I. Epizootiological survey and virus isolation. Bull. Natl. Inst. Anim. Hlth. 1970;61:9–16. (in Japanese) [Google Scholar]
- Kimura T., Hasebe R., Mukaiya R., Ochiai K., Wada R., Umemura T. Decreased expression of equine herpesvirus-1 early and late genes in the placenta of naturally aborted equine fetuses. J. Comp. Pathol. 2004;130:41–47. doi: 10.1016/s0021-9975(03)00068-9. [DOI] [PubMed] [Google Scholar]
- Lamaze C., Baba T., Redelmeier T.E., Schmid S.L. Recruitment of epidermal growth factor and transferring receptors into coated pits in vitro: differing biochemical requirements. Mol. Biol. Cell. 1993;4:715–727. doi: 10.1091/mbc.4.7.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Ali M.A., Cohen J.I. Insulin degrading enzyme is a cellular receptor mediating varicella-zoster virus infection and cell-to-cell spread. Cell. 2006;127:305–316. doi: 10.1016/j.cell.2006.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjomäki V., Pietiainen V., Matilainen H., Upla P., Ivaska J., Nissinen L., Reunanen H., Huttunen P., Hyypia T., Heino J. Internalization of echovirus 1 in caveolae. J. Virol. 2002;76:1856–1865. doi: 10.1128/JVI.76.4.1856-1865.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin K.S., Reggio H., Helenius A., Simons K. Infectious entry pathway of influenza virus in a canine kidney cell line. J. Cell Biol. 1981;91:601–613. doi: 10.1083/jcb.91.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin K.S., Reggio H., Helenius A., Simons K. Pathway of vesicular stomatitis virus entry leading to infection. J. Mol. Biol. 1982;156:609–631. doi: 10.1016/0022-2836(82)90269-8. [DOI] [PubMed] [Google Scholar]
- McPherson P.S., Kay B.K., Hussain N.K. Signaling on the endocytic pathway. Traffic. 2001;2:375–384. doi: 10.1034/j.1600-0854.2001.002006375.x. [DOI] [PubMed] [Google Scholar]
- Milne R.S., Nicola A.V., Whitbeck J.C., Eisenberg R.J., Cohen G.H. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 2005;79:6655–6663. doi: 10.1128/JVI.79.11.6655-6663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols B. Caveosomes and endocytosis of lipid rafts. J. Cell Sci. 2003;116:4707–4714. doi: 10.1242/jcs.00840. [DOI] [PubMed] [Google Scholar]
- Nicola A.V., Straus S.E. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J. Virol. 2004;78:7508–7517. doi: 10.1128/JVI.78.14.7508-7517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola A.V., McEvoy A.M., Straus S.E. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 2003;77:5324–5332. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola A.V., Hou J., Major E.O., Straus S.E. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 2005;79:7609–7616. doi: 10.1128/JVI.79.12.7609-7616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura R., Kiyota A., Suzaki E., Kataoka K., Ohe Y., Miyamoto K., Senda T., Fujimoto T. Human coronavirus 229E binds to CD13 in rafts and enters the cell through caveolae. J. Virol. 2004;78:8701–8708. doi: 10.1128/JVI.78.16.8701-8708.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Callaghan D.J., Cheevers W.P., Gentry G.A., Randall C.C. Kinetics of cellular and viral DNA synthesis in equine abortion (herpes) virus infection of L-M cells. Virology. 1968;36:104–114. doi: 10.1016/0042-6822(68)90120-7. [DOI] [PubMed] [Google Scholar]
- Orlichenko L., Huang B., Krueger E., McNiven M.A. Epithelial growth factor-induced phosphorylation of caveolin 1 at tyrosine 14 stimulates caveolae formation in epithelial cells. J. Biol. Chem. 2006;281:4570–4579. doi: 10.1074/jbc.M512088200. [DOI] [PubMed] [Google Scholar]
- Palade G.E. Fine structure of blood capillaries. J. Appl. Phys. 1953;24:1424. [Google Scholar]
- Parton R.G., Joggerst B., Simons K. Regulated internalization of caveolae. J. Cell Biol. 1994;127:1199–1215. doi: 10.1083/jcb.127.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richterová Z., Liebl D., Horak M., Palkova Z., Stokrova J., Hozak P., Korb J., Forstova J. Caveolae are involved in the trafficking of mouse polyomavirus virions and artificial VP1 pseudocapsids toward cell nuclei. J. Virol. 2001;75:10880–10891. doi: 10.1128/JVI.75.22.10880-10891.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.L., Campos S.K., Ozbun M.A. Human papillomavirus type 31 uses a caveolin-1 and dynamin 2-mediated entry pathway for infection of human keratinocytes. J. Virol. 2007;81:9922–9931. doi: 10.1128/JVI.00988-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein B.S., Gowda S.D., Lifson J.D., Penhallow R.C., Bensch K.G., Engleman E.G. pH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell. 1987;49:659–668. doi: 10.1016/0092-8674(87)90542-3. [DOI] [PubMed] [Google Scholar]
- Storts R.W., Montgomery D.L. The nervous system. In: MacGavin M.D., Carlton W.W., Zachary J.F., editors. Tomson's Special Veterinary Pathology. 3rd ed. Mosby-Year Book.; St Louis: 2001. pp. 430–431. [Google Scholar]
- Sukumaran S.K., Quon M.J., Prasadarao N.V. Escherichia coli K1 internalization via caveolae requires caveolin-1 and protein kinase Calpha interaction in human brain microvascular endothelial cells. J. Biol. Chem. 2002;277:50716–50724. doi: 10.1074/jbc.M208830200. [DOI] [PubMed] [Google Scholar]
- Tsiang H., Superti F. Ammonium chloride and chloroquine inhibit rabies virus infection in neuroblastoma cells. Brief report. Arch. Virol. 1984;81:377–382. doi: 10.1007/BF01310010. [DOI] [PubMed] [Google Scholar]
- van de Walle G.R., Peters S.T., VanderVen B.C., O'Callaghan D.J., Osterrieder N. Equine herpesvirus 1 entry via endocytosis is facilitated by αV integrins and an RSD motif in glycoprotein D. J. Virol. 2008;82:11859–11868. doi: 10.1128/JVI.00868-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Weert A.W.M., Dunn K.W., Geuze H.J., Maxfield F.R., Stoorvogel W. Transport from late endosomes to lysosomes, but not sorting of integral membrane proteins in endosomes, depends on the vacuolar proton pump. J. Cell Biol. 1995;130:821–834. doi: 10.1083/jcb.130.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgintino D., Robertson D., Errede M., Benagiano V., Tauer U., Roncali L., Bertossi M. Expression of caveolin-1 in human brain microvessels. Neuroscience. 2002;115:145–152. doi: 10.1016/s0306-4522(02)00374-3. [DOI] [PubMed] [Google Scholar]
- Wittels M., Spear P.G. Penetration of cells by herpes simplex virus does not require a low pH-dependent endocytic pathway. Virus Res. 1991;18:271–290. doi: 10.1016/0168-1702(91)90024-p. [DOI] [PubMed] [Google Scholar]
- Yamada E. The fine structure of the gall bladder epithelium of the mouse. J. Biophys. Biochem. Cytol. 1955;1:445–458. doi: 10.1083/jcb.1.5.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A., Ohnishi S. Uncoating of influenza virus in endosomes. J. Virol. 1984;51:497–504. doi: 10.1128/jvi.51.2.497-504.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]