

Graphical abstract
The synthesis of a new series of 2-S-pyridyl-6-thionucleosides, bearing natural and modified purines and pyrimidines, by developing highly efficient synthetic routes, is reported. Compound 7c, and in particular 7b, were endowed with significant and selective cytostatic activity and can be regarded as novel lead compounds for further modifications.
Keywords: 3-Fluoro-6-S-(2-S-pyridyl) nucleosides, Thiopyridinylation, Cytostatic agents
Abstract
The 3-deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-β-d-glucopyranosyl nucleoside analogs 7 were prepared via two facile synthetic routes. Their precursors, 3-fluoro-6-thio-glucopyranosyl nucleosides 5a-e, were obtained by the sequence of deacetylation of 3-deoxy-3-fluoro-β-d-glucopyranosyl nucleosides 2a-e, selective tosylation of the primary OH of 3 and finally treatment with potassium thioacetate. The desired thiolpyridine protected analogs 7a-c,f,g were obtained by the sequence of deacetylation of 5a-c followed by thiopyridinylation and/or condensation of the corresponding heterocyclic bases with the newly synthesized peracetylated 6-S-(2-S-pyridyl) sugar precursor 13, which was obtained via a novel synthetic route from glycosyl donor 12. None of the compounds 6 and 7 showed antiviral activity, but the 5-fluorouracil derivative 7c and particularly the uracil derivative 7b were endowed with an interesting and selective cytostatic action against a variety of murine and human tumor cell cultures.
1. Introduction
Nucleoside analogs markedly contribute to the chemotherapy of cancer and viral diseases [1], [2], [3], [4]. However, novel applications are still being explored and new nucleoside structures have reached the clinic as effective agents for the treatment of AIDS [5], herpes virus infections [6] and viral hepatitis [7].
Fluorine-containing nucleosides and their analogs are attractive compounds and have drawn special attention [8], [9], [10], [11], with the fluorine incorporated either on the base moiety or on the sugar part frequently leading to a drastic change in biological activity, stability and bioavailability [8], [12], [13]. Our recent data have revealed, that new classes of uncommon fluorinated pyranonucleosides have a promising potential in combating the rotaviral infections, in the treatment of colon cancer and are efficient antitumor growth inhibitors [10], [11], [14], [15]. Experimental data also suggested, that human Poly(A)-specific ribonuclease is among the molecular targets of these compounds and could act therapeutically by lowering the mRNA turnover rate [16].
In the field of nucleosides, sulfur chemistry appears particularly fruitful in the development of therapeutic agents [17], [18]. Considerable interest has been drawn in the synthesis of thionucleoside analogs [18], [19], [20], [21] and nucleosidyl disulfides [22], [23], [24], [25], [26], attractive candidates for use as both antiviral and antineoplastic agents [21], [24], [27], with the latter being assessed as prodrugs able to interfere with HIV reverse transcription [21], [22], [26]. Particularly, nucleosides in which the primary hydroxyl group is replaced by a thiol- or an alkylthio-group, are promising antiviral agents since they disrupt essential viral macromolecular methylation processes [28], [29].
In view of the above data and our recent findings on the biology of modified nucleosides, as templates for antiviral and anticancer activity [10], [11], [14], [15], [16], [30], [31], [32], we turned our attention to the possible improvement of the biological properties of such molecules. We thus synthesised a series of glucopyranosyl nucleoside analogs, where we combined both a pharmacologically compatible fluorine and a sulfur/thiopyridine moiety in the sugar part. In this paper, we report on two efficient methods for their preparation and supply data on their biological activity.
2. Experimental
2.1. General methods
Melting points were recorded in a Mel-Temp apparatus and are uncorrected. Thin layer chromatography (TLC) was performed on Merck precoated 60F254 plates. Reactions were monitored by TLC on silica gel, with detection by UV light (254 nm) or by charring with sulfuric acid. Flash column chromatography was performed using silica gel (240–400 mesh, Merck). 1H, 19F and 13C NMR spectra were obtained at room temperature with a Brucker 400 spectrometer at 400, 376 and 100 MHz, respectively, using CDCl3 and methanol-d4 (CD3OD) with internal tetramethylsilane (TMS) for 1H and 13C and internal trifluorotoluene (TFT) for 19F.
The chemical shifts are expressed in parts per million (δ) and following abbreviations were used: s = singlet, d = doublet, dd = doublet doublet, dtr = doublet triplet and m = multiplet. Mass spectra were obtained with a Micromass Platform LC (ESI-MS). Infrared spectra were obtained with a Perkin–Elmer Model 1600 FT-IR spectrophotometer. Optical rotations were measured using an Autopol I polarimeter. All reactions were carried out in dry solvents. Acetonitrile was distilled from calcium hydride and stored over 3E molecular sieves. Dimethylformamide (DMF) was also stored over 3E molecular sieves, and pyridine was stored over potassium hydroxide pellets.
2.2. Synthesis of 1-[3-deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-β-d-glucopyranosyl] nucleosides (7a-c) from 5a-c and/or 6a-c
2.2.1. 1-(3-Deoxy-3-fluoro-β-d-glucopyranosyl)cytosine (3f)
A mixture of methanolic ammonia (140 mL) and N 4-benzoyl-cytosine derivative 2d [11] (1.70 g, 3.36 mmol) stirred for 4 h at room temperature. The reaction mixture was concentrated and crude 3f was obtained (0.83 g, 90%, R f = 0.15 in AcOEt) as colorless oil. Product 3f was used without further purification. + 5.00 (c 0.1, MeOH); λ max 268 nm (ε 7162); ESI-MS (m/z): 276.26 (M+H+).
2.2.2. 9-(3-Deoxy-3-fluoro-β-d-glucopyranosyl)adenine (3g)
Adenine derivative 3g was synthesized from N 6-benzoyl adenine derivative 2e [11] by the similar procedure as described for 3f. Compound 3g was obtained (0.85 g, 88%, R f = 0.12 in AcOEt) as colorless oil and it was used without further purification. + 4.00 (c 0.1, MeOH); λ max 259 nm (ε 4930); ESI-MS (m/z): 300.27 (M+H+).
2.2.3. 1-(2,4-Di-O-acetyl-3-deoxy-3-fluoro-6-O-p-toluenesulfonyl-β-d-glucopyranosyl)thymine (4a)
A solution of compound 3a [15] (1.10 g, 3.70 mmol) in 4.50 mL of dry pyridine was cooled to 0 °C, and p-toluenesulfonyl chloride (1.10 g, 5.58 mmol) in 4.50 mL of dry pyridine was added dropwise with stirring. The reaction mixture was stored at room temperature overnight, and then was concentrated. The residual gum was dissolved in pyridine (49 mL), acetic anhydride was added (25 mL) and the resulted mixture stirred for 3 h at room temperature. MeOH (0.40 mL) was added to quench the reaction and the mixture was concentrated under high vacuum to remove the solvents. The mixture was extracted with two 200 mL portions of ethyl acetate and neutralized with aqueous NaHCO3. The organic layer was washed with NaHSO4 (20 mL), dried over anhydrous sodium sulfate and evaporated to dryness. Purification by flash chromatography (hexane/AcOEt, 4:6), gave 4a (1.20 g, 62%, R f = 0.35 in hexane/AcOEt, 4:6) as white solid. m.p. 145–147 °C; + 10.52 (c 0.1, CHCl3); λ max 260 nm (ε 8836); 1H NMR (CDCl3): δ 8.60 (br s, NH), 7.37–7.20 (m, 4H, tosyl group), 7.19 (s, 1H, H-6), 5.75 (d, 1H, J 1′,2′ = 9.5 Hz, H-1′), 5.29–5.20 (m, 2H, H-2′ and H-4′), 4.70 (dtr, 1H, J F,3′ = 51.7 Hz, J 2′,3′ = 9.1, J 3′,4′ = 9.0 Hz, H-3′), 4.21–4.07 (m, 2H, H-6a′,6b′), 3.84 (m, 1H, Η-5′), 2.45 (s, 3H, ArCH3), 2.10 and 2.05 (2s, 6H, 2OAc), 1.96 (s, 3H, 5-CH3); Anal. Calcd for C22H25FN2O10S: C, 50.00; H, 4.77; N, 5.30. Found: C, 50.26; H, 4.92; N, 5.47. ESI-MS (m/z): 529.51 (M+H+).
2.2.4. 1-(2,4-Di-O-acetyl-3-deoxy-3-fluoro-6-O-p-toluenesulfonyl-β-d-glucopyranosyl)uracil (4b)
Uracil derivative 4b was synthesized from 3b [15] by the similar procedure as described for 4a. Purified by flash chromatography (hexane/AcOEt, 3:7) compound 4b was obtained (1.10 g, 58%, R f = 0.4 in hexane/AcOEt, 3:7) as white solid. m.p. 175–177 °C; + 5.26 (c 0.1, CHCl3); λ max 260 nm (ε 6760); 1H NMR (CDCl3): δ 8.20 (br s, NH), 7.76 (d, 1H, J 5,6 = 8.2 Hz, H-6), 7.38–7.22 (m, 4H, tosyl group), 5.79 (d, 1H, H-5), 5.73 (d, 1H, J 1′,2′ = 9.6 Hz, H-1′), 5.26–5.15 (m, 2H, H-2′ and H-4′), 4.70 (dtr, 1H, J F,3′ = 51.7 Hz, J 2′,3′ = J 3′,4′ = 9.0 Hz, H-3′), 4.22–4.06 (m, 2H, H-6a′,6b′), 3.85 (m, 1H, Η-5′), 2.45 (s, 3H, ArCH3), 2.10 and 2.06 (2s, 6H, 2OAc); Anal. Calcd for C21H23FN2O10S: C, 49.03; H, 4.51; N, 5.45. Found: C, 49.19; H, 4.65; N, 5.62. ESI-MS (m/z): 515.49 (M+H+).
2.2.5. 1-(2,4-Di-O-acetyl-3-deoxy-3-fluoro-6-O-p-toluenesulfonyl-β-d-glucopyranosyl)5-fluorouracil (4c)
5-Fluorouracil derivative 4c was synthesized from 3c [15] by the similar procedure as described for 4a. Purified by flash chromatography (hexane/AcOEt, 4:6) derivative 4c was obtained (1.18 g, 60%, R f = 0.4 in hexane/AcOEt, 4:6) as white solid. m.p. 198–200 °C; + 5.32 (c 0.1, CHCl3); λ max 260 nm (ε 7486); 1H NMR (CDCl3): δ 8.28 (br s, NH), 7.50 (d, 1H, J 6,F5 = 5.85 Hz, H-6), 7.37–7.20 (m, 4H, tosyl group), 5.74 (d, 1H, J 1′,2′ = 9.4 Hz, H-1′), 5.27–5.15 (m, 2H, H-2′ and H-4′), 4.75 (dtr, 1H, J F,3′ = 51.5 Hz, J 2′,3′ = 9.2, J 3′,4′ = 9.0 Hz, H-3′), 4.21–4.06 (m, 2H, H-6a′,6b′), 3.90 (m, 1H, Η-5′), 2.45 (s, 3H, ArCH3), 2.11 and 2.07 (2s, 6H, 2OAc); Anal. Calcd for C21H22F2N2O10S: C, 47.37; H, 4.16; N, 5.26. Found: C, 47.54; H, 4.29; N, 5.42. ESI-MS (m/z): 533.48 (M+H+).
2.2.6. 1-(2,4-Di-O-acetyl-3-deoxy-3-fluoro-6-O-p-toluenesulfonyl-β-d-glucopyranosyl)-N4-benzoyl cytosine (4d)
N 4-benzoyl cytosine derivative 4d was synthesized from 3d [11] by the similar procedure as described for 4a. Purified by flash chromatography (hexane/AcOEt, 4:6) derivative 4d was obtained (1.41 g, 62%, R f = 0.4 in hexane/AcOEt, 4:6) as white foam. + 12.00 (c 0.5, CHCl3); λ max 263 nm (ε 21,906); 1H NMR (CDCl3): δ 8.66 (br s, NH), 7.88–7.31 (m, 11H, Bz, H-6, H-5 and tosyl group), 6.01 (d, 1H, J 1′,2′ = 9.4 Hz, H-1′), 5.30–5.14 (m, 2H, H-2′ and H-4′), 4.74 (dtr, 1H, J F,3′ = 51.6 Hz, J 2′,3′ = 9.1, J 3′,4′ = 9.0 Hz, H-3′), 4.24–4.08 (m, 2H, H-6a′,6b′), 3.86 (m, 1H, Η-5′), 2.45 (s, 3H, ArCH3), 2.12 and 2.04 (2s, 6H, 2OAc); Anal. Calcd for C28H28FN3O10S: C, 54.45; H, 4.57; N, 6.80. Found: C, 54.84; H, 4.29; N, 6.62. ESI-MS (m/z): 618.62 (M+H+).
2.2.7. 9-(2,4-Di-O-acetyl-3-deoxy-3-fluoro-6-O-p-toluenesulfonyl-β-d-glucopyranosyl)-N6-benzoyl adenine (4e)
N 6-benzoyl adenine derivative 4e was synthesized from 3e [11] by the similar procedure as described for 4a. Purified by flash chromatography (hexane/AcOEt, 3:7) compound 4e was obtained (1.52 g, 64%, R f = 0.3 in hexane/AcOEt, 3:7) as white foam. − 2.00 (c 0.2, CHCl3); λ max 278 nm (ε 17,393); 1H NMR (CDCl3): δ 9.05 (br s, NH), 8.84 and 8.15 (2s, 2H, H-2,8), 8.14–7.51 (m, 9H, Bz and tosyl group), 5.84 (d, 1H, J 1′,2′ = 9.5 Hz, H-1′), 5.64 (m, 1H, H-2′), 5.35 (m, 1H, H-4′), 4.81 (dtr, 1H, J F,3′ = 51.5 Hz, J 2′,3′ = J 3′,4′ = 9.1 Hz, H-3′), 4.23–4.11 (m, 2H, H-6a′,6b′), 4.01 (m, 1H, Η-5′), 2.42 (s, 3H, ArCH3), 2.16 and 1.84 (2s, 6H, 2OAc); Anal. Calcd for C29H28FN5O9S: C, 54.29; H, 4.40; N, 10.92. Found: C, 54.34; H, 4.59; N, 10.78. ESI-MS (m/z): 642.64 (M+H+).
2.2.8. 1-(2,4-Di-O-acetyl-6-S-acetyl-3-deoxy-3-fluoro-6-thio-β-d-glucopyranosyl)thymine (5a)
The tosylate 4a (1.20 g, 2.30 mmol) was heated with potassium thioacetate (0.36 g, 3.10 mmol) in DMF (8.70 mL) at 100 °C for 2 h. The reaction mixture was neutralized with aqueous NaHCO3. After that, the mixture was concentrated under high vacuum pump to eliminate the DMF. The residue was partitioned between water and EtOAc, the organic extract was dried over anhydrous sodium sulfate, filtered, and evaporated to dryness. The residue was purified by flash chromatography (hexane/AcOEt, 4:6) to give compound 5a (0.75 g, 78%, R f = 0.4 in hexane/AcOEt, 4:6) as white solid. m.p. 204–206 °C; + 5.21 (c 0.1, CHCl3), λ max 260 nm (ε 9219); IR (Nujol): 1697 (SAc) cm−1; 1H NMR (CDCl3): δ 8.10 (br s, NH), 7.12 (s, 1H, H-6), 5.75 (d, 1H, J 1′,2′ = 9.5 Hz, H-1′), 5.29–5.18 (m, 2H, H-2′ and H-4′), 4.71 (dtr, 1H, J F,3′ = 51.8 Hz, J 2′,3′ = 9.1 J 3′,4′ = 9.0 Hz, H-3′), 3.78 (m, 1H, Η-5′), 3.30–3.11 (m, 2H, H-6a′,6b′), 2.37 (s, 3H, SAc), 2.21 and 2.08 (2s, 6H, 2OAc), 1.98 (s, 3H, 5-CH3); Anal. Calcd for C17H21FN2O8S: C, 47.22; H, 4.89; N, 6.48. Found: C, 47.39; H, 4.78; N, 6.54. ESI-MS (m/z): 433.44 (M+H+).
2.2.9. 1-(2,4-Di-O-acetyl-6-S-acetyl-3-deoxy-3-fluoro-6-thio-β-d-glucopyranosyl)uracil (5b)
Uracil derivative 5b was synthesized from 4b by the similar procedure as described for 5a. Purified by flash chromatography (hexane/AcOEt, 3:7) and obtained (0.64 g, 76%, R f = 0.45 in hexane/AcOEt, 3:7) as white solid. m.p. 158–160 °C; + 5.18 (c 0.1, CHCl3); λ max 260 nm (ε 6283); IR (Nujol): 1697 (SAc) cm−1; 1H NMR (CDCl3): δ 8.30 (br s, NH), 7.30 (d, 1H, J 5,6 = 8.3 Hz, H-6), 5.82 (d, 1H, H-5), 5.75 (d, 1H, J 1′,2′ = 9.5 Hz, H-1′), 5.26–5.15 (m, 2H, H-2′ and H-4′), 4.69 (dtr, 1H, J F,3′ = 51.8 Hz, J 2′,3′ = J 3′,4′ = 9.1 Hz, H-3′), 3.80 (m, 1H, Η-5′), 3.27–3.11 (m, 2H, H-6a′,6b′), 2.35 (s, 3H, SAc), 2.19 and 2.07 (2s, 6H, 2OAc); Anal. Calcd for C16H19FN2O8S: C, 45.93; H, 4.58; N, 6.70. Found: C, 45.89; H, 4.64; N, 6.82. ESI-MS (m/z): 419.41 (M+H+).
2.2.10. 1-(2,4-Di-O-acetyl-6-S-acetyl-3-deoxy-3-fluoro-6-thio-β-d-glucopyranosyl)5-fluorouracil (5c)
5-Fluorouracil derivative 5c was synthesized from 4c by the similar procedure as described for 5a. Purified by flash chromatography (hexane/AcOEt, 4:6) and obtained (0.65 g, 75%, R f = 0.47 in hexane/AcOEt, 4:6) as white solid. m.p. 200–202 °C; + 5.26 (c 0.1, CHCl3); λ max 260 nm (ε 8232); IR (Nujol): 1697 (SAc) cm−1; 1H NMR (CDCl3): δ 8.28 (br s, NH), 7.36 (d, 1H, J 6,F5 = 5.3 Hz, H-6), 5.71 (d, 1H, J 1′,2′ = 9.3 Hz, H-1′), 5.23–5.11 (m, 2H, H-2′ and H-4′), 4.70 (dtr, 1H, J F,3′ = 51.5 Hz, J 2′,3′ = 9.2, J 3′,4′ = 9.0 Hz, H-3′), 3.80 (m, 1H, Η-5′), 3.29–3.10 (m, 2H, H-6a′,6b′), 2.36 (s, 3H, SAc), 2.19 and 2.08 (2s, 6H, 2OAc); Anal. Calcd for C16H18F2N2O8S: C, 44.04; H, 4.16; N, 6.42. Found: C, 44.28; H, 4.29; N, 6.34. ESI-MS (m/z): 437.41 (M+H+).
2.2.11. 1-(2,4-Di-O-acetyl-6-S-acetyl-3-deoxy-3-fluoro-6-thio-β-d-glucopyranosyl)-N4-benzoyl cytosine (5d)
N 4-benzoyl cytosine derivative 5d was synthesized from 4d by the similar procedure as described for 5a. Purified by flash chromatography (hexane/AcOEt, 2:8) to give compound 5d (0.82 g, 68%, R f = 0.32 in hexane/AcOEt, 2:8) as solid. m.p. 277–279 °C; + 16.00 (c 0.1, CHCl3); λ max 263 nm (ε 20,626); IR (Nujol): 1697 (SAc) cm−1; 1H NMR (CDCl3): δ 7.88 (d, 1H, J 5,6 = 7.2 Hz, H-6), 7.84–7.48 (m, 6H, Bz and H-5), 6.03 (d, 1H, J 1′,2′ = 9.4 Hz, H-1′), 5.27–5.17 (m, 2H, H-2′ and H-4′), 4.75 (dtr, 1H, J F,3′ = 51.8 Hz, J 2′,3′ = 9.1 Hz, J 3′,4′ = 9.0 Hz, H-3′), 3.83–3.77 (m, 1H, H-5′), 3.29–3.11 (m, 2H, H-6a′,6b′), 2.35 (s, 3H, SAc), 2.20 and 2.05 (2s, 6H, 2OAc); Anal. Calcd for C23H24FN3O8S: C, 52.97; H, 4.64; N, 8.06. Found: C, 52.70; H, 4.80; N, 8.22. ESI-MS (m/z): 522.54 (M+H+).
2.2.12. 9-(2,4-Di-O-acetyl-6-S-acetyl-3-deoxy-3-fluoro-6-thio-β-d-glucopyranosyl)-N6-benzoyl adenine (5e)
N 6-benzoyl adenine derivative 5e was synthesized from 4e by the similar procedure as described for 5a. Finally it was purified by flash chromatography (hexane/AcOEt, 2:8) to give compound 5e (0.61 g, 60%, R f = 0.24 in hexane/AcOEt, 2:8) as solid. m.p. 118–120 °C; + 4.00 (c 0.1, CHCl3); λ max 279 nm (ε 12,567); IR (Nujol): 1697 (SAc) cm−1; 1H NMR (CDCl3): δ 9.07 (br s, NH), 8.77 and 8.19 (2s, 2H, H-2,8), 7.98–7.42 (m, 5H, Bz), 5.88 (d, 1H, J 1′,2′ = 9.5 Hz, H-1′), 5.70–5.61 (m, 1H, H-2′), 5.40–5.31 (m, 1H, H-4′), 4.81 (dtr, 1H, J F,3′ = 51.7 Hz, J 2′,3′ = 9.3 Hz, J 3′,4′ = 8.9 Hz), 3.95–3.89 (m, 1H, H-5′), 3.32–3.15 (m, 2H, H-6a′,6b′), 2.35 (s, 3H, SAc), 2.23 and 1.84 (2s, 6H, 2OAc); Anal. Calcd for C24H24FN5O7S: C, 52.84; H, 4.43; N, 12.84. Found: C, 52.68; H, 4.38; N, 13.09. ESI-MS (m/z): 546.56 (M+H+).
2.2.13. 1-[3-Deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-β-d-glucopyranosyl]thymine (7a) from 5a
To a solution of thioacetate 5a (1.10 g, 2.50 mmol) in methanolic ammonia (104 mL), 2,2-dipyridyl-disulfide (DTDP) (2.60 g, 12 mmol) dissolved in MeOH/H2O (1:1, 22 mL) was added dropwise and the mixture was stirred at room temperature. After stirring for 20 h, the reaction mixture was concentrated. The crude was finally purified by flash chromatography (AcOEt) to give compound 7a (0.71 g, 68%, R f = 0.30 in AcOEt) as white foam. + 16.00 (c 0.5, MeOH); λ max 266 nm (ε 9093); 1H NMR (CD3OD): δ 8.09 (br s, NH), 8.41 (d, H-6 of pyridine), 7.83–7.58 (m, H-4 and H-5 of pyridine), 7.29–7.12 (m, H-3 of pyridine and H-6), 5.40 (d, 1H, J 1′,2′ = 9.4 Hz, H-1′), 4.29 (dtr, 1H, J F,3′ = 52.3 Hz, J 2′,3′ = 8.5 Hz, J 3′,4′ = 8.4 Hz, H-3′), 3.92–3.79 (m, 1H, Η-2′), 3.56–3.45 (m, 2H, H-6a′ and H-4′), 3.44–3.35 (m, 1H, 6b′), 2.98–2.89 (m, 1H, H-5′); 19F NMR: δ −65.00; 13C NMR (CD3OD): δ 163.73, 160.52, 150.11, 148.90, 136.95, 132.92, 121.11, 119.18, 110.23, 92.18, 85.49, 72.36, 70.12, 67.95, 33.63, 12.41; Anal. Calcd for C16H18FN3O5S2: C, 46.26; H, 4.37; N, 10.11. Found: C, 46.41; H, 4.51; N, 10.32. ESI-MS (m/z): 416.47 (M+H+).
2.2.14. 1-[3-Deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-β-d-glucopyranosyl]uracil (7b) from 5b
Uracil derivative 7b was synthesized from thioacetate 5b by the similar procedure as described for 7a. Purified by flash chromatography (AcOEt) and obtained (0.70 g, 70%, R f = 0.27 in AcOEt) as white foam. + 6.00 (c 0.1, MeOH); λ max 262 nm (ε 6478); 1H NMR (CD3OD): δ 8.53 (br s, NH), 8.32 (dd, H-6 of pyridine), 7.76–7.73 (m, H-4 and H-5 of pyridine), 7.66 (m, H-3 of pyridine), 7.57 (d, 1H, J 5,6 = 8.1 Hz, H-6), 5.68 (d, 1H, H-5), 5.46 (d, 1H, J 1′,2′ = 9.4 Hz, H-1′), 4.29 (dtr, 1H, J F,3′ = 52.3 Hz, J 2′,3′ = 8.7, J 3′,4′ = 8.6 Hz, H-3′), 3.83–3.77 (m, 1H, Η-2′), 3.64–3.60 (m, 1H, H-6a′), 3.57–3.52 (m, 1H, H-4′), 3.42–3.39 (m, 1H, 6b′), 3.01–2.96 (m, 1H, H-5′); 19F NMR: δ −63.20; 13C NMR (CD3OD): δ 163.34, 160.43, 150.19, 147.96, 142.56, 132.74, 120.95, 119.26, 101.76, 91.98, 84.77, 71.83, 70.31, 67.62, 33.64; Anal. Calcd for C15H16FN3O5S2: C, 44.88; H, 4.02; N, 10.47. Found: C, 44.71; H, 4.22; N, 10.52. ESI-MS (m/z): 402.46 (M+H+).
2.2.15. 1-[3-Deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-β-d-glucopyranosyl]5-fluorouracil (7c) from 5c
5-Fluorouracil derivative 7c was synthesized from corresponding thioacetate 5c by the similar procedure as described for 7a. Purified by flash chromatography (AcOEt) and obtained (0.72 g, 69%, R f = 0.25 in AcOEt) as white foam. + 2.00 (c 0.5, CHCl3); λ max 263 nm (ε 5885); 1H NMR (CD3OD): δ 8.34–8.32 (m, H-6 of pyridine), 8.28 (br s, NH), 7.83–7.78 (m, H-4 and H-5 of pyridine), 7.17–7.13 (m, H-3 of pyridine), 5.44 (d, 1H, J 1′,2′ = 9.0 Hz, H-1′), 4.28 (dtr, 1H, J F,3′ = 52.3 Hz, J 2′,3′ = 8.7, J 3′,4′ = 8.6 Hz, H-3′), 3.82–3.76 (m, 1H, Η-2′), 3.64–3.60 (m, 1H, H-6a′), 3.56–3.51 (m, 1H, H-4′), 3.43–3.39 (m, 1H, H-6b′), 3.01–2.96 (m, 1H, H-5′); 19F NMR: δ −64.30, −63.20; 13C NMR (CD3OD): δ 162.34, 157.89, 150.41, 148.76, 138.11, 132.91, 127.45, , 120.32, 119.07, 94.41, 85.29, 72.37, 70.11, 68.65, 33.47; Anal. Calcd for C15H15F2N3O5S2: C, 42.95; H, 3.60; N, 10.02. Found: C, 42.81; H, 3.48; N, 10.28. ESI-MS (m/z): 420.44.
2.2.16. Bis-[1-(3-deoxy-3-fluoro-6-thio-β-d-glucopyranosyl)thymine]-6,6-disulfide (6a)
A mixture of methanolic ammonia (72.30 mL) and thionucleoside 5a (0.75 g, 1.73 mmol) stirred for 4 h at room temperature. The reaction mixture was concentrated and disulfide 6a was obtained (0.95 g, 90%, R f = 0.20 in AcOEt) as white foam. + 2.00 (c 0.5, MeOH); λ max 262 nm (ε 19,126); 1H NMR (CD3OD): δ 7.88 (br s, NH), 7.49 (s, 1H, H-6), 5.55 (d, 1H, J 1′,2′ = 9.4 Hz, H-1′), 4.37 (dtr, 1H, J F,3′ = 52.3 Hz, J 2′,3′ = 8.5, J 3′,4′ = 8.3 Hz, H-3′), 4.14–4.04 (m, 1H, Η-2′), 3.72–3.55 (m, 1H, H-4′), 3.35–3.23 (m, 2H, H-6a′,6b′), 2.92–2.80 (m, 1H, H-5′), 1.89 (s, 3H, 5-CH3); Anal. Calcd for C22H28F2N4O10S2: C, 43.27; H, 4.62; N, 9.18. Found: C, 43.18; H, 4.84; N, 9.07. ESI-MS (m/z): 611.64.
2.2.17. Bis-[1-(3-deoxy-3-fluoro-6-thio-β-d-glucopyranosyl)uracil]-6,6-disulfide (6b)
Uracil derivative 6b was synthesized from thionucleoside 5b following similar procedure as described for 6a. Disulfide 6b was obtained (0.89 g, 88%, R f = 0.16 in AcOEt) as white foam. + 3.00 (c 0.5, MeOH); λ max 258 nm (ε 14,339); 1H NMR (CD3OD): δ 7.66 (d, 1H, J 5,6 = 8.2 Hz, H-6), 7.24 (br s, NH), 5.64 (d, 1H, H-5), 5.56 (d, 1H, J 1′,2′ = 9.5 Hz, H-1′), 4.38 (dtr, 1H, J F,3′ = 52.0 Hz, J 2′,3′ = 8.6 Hz, J 3′,4′ = 8.1 Hz, H-3′), 3.95–3.81 (m, 1H, Η-2′), 3.74–3.53 (m, 1H, H-4′), 3.34–3.22 (m, 2H, H-6a′,6b′), 2.92–2.81 (m, 1H, H-5′); Anal. Calcd for C20H24F2N4O10S2: C, 41.23; H, 4.15; N, 9.62. Found: C, 41.38; H, 4.34; N, 9.78. ESI-MS (m/z): 583.57.
2.2.18. Bis-[1-(3-deoxy-3-fluoro-6-thio-β-d-glucopyranosyl)5-fluorouracil]-6,6-disulfide (6c)
5-Fluorouracil derivative 6c was synthesized from thionucleoside 5c by the similar procedure as described for 6a. Compound 6c was obtained (0.95 g, 89%, R f = 0.13 in AcOEt) as white foam. + 2.00 (c 0.5, MeOH); λ max 263 nm (ε 15,056); 1H NMR (CD3OD): δ 7.89 (d, 1H, J 6,F5 = 6.4 Hz, H-6), 7.35 (br s, NH), 5.56 (d, 1H, J 1′,2′ = 9.3 Hz, H-1′), 4.38 (dtr, 1H, J F,3′ = 52.5 Hz, J 2′,3′ = 8.7, J 3′,4′ = 8.6 Hz, H-3′), 3.90–3.84 (m, 1H, H-2′), 3.70–3.66 (m, 1H, H-6a′), 3.61–3.56 (m, 1H, H-4′), 3.29–3.26 (m, 1H, H-6b′), 2.88–2.83 (m, 1H, Η-5′); Anal. Calcd for C20H22F4N4O10S2: C, 38.84; H, 3.59; N, 9.06. Found: C, 38.72; H, 3.47; N, 9.12. ESI-MS (m/z): 619.56.
2.2.19. 1-[3-Deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-β-d-glucopyranosyl]thymine (7a) from 6a
To a solution of 6a (1.83 g, 3.00 mmol) in MeOH (26 mL), DTDP (3.16 g, 14.46 mmol) dissolved in MeOH/H2O (1:1, 26 mL) was added dropwise. The reaction was carried out at room temperature and within 12 h. After disappearance of the starting material (TLC), the solvents were removed under diminished pressure and the yellow residue was purified by flash column chromatography (AcOEt) to give compound 7a (0.88 g, 71%, R f = 0.30 in AcOEt) as white foam.
2.2.20. 1-[3-Deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-β-d-glucopyranosyl]uracil (7b) from 6b
Uracil derivative 7b was synthesized from 6b by the similar procedure as described for 7a. Purified by flash chromatography (AcOEt) and obtained (0.83 g, 69%, R f = 0.27 in AcOEt) as white foam.
2.2.21. 1-[3-Deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-β-d-glucopyranosyl]5-fluorouracil (7c) from 6c
5-Fluorouracil derivative 7c was synthesized from 6c by the similar procedure as described for 7a. Purified by flash chromatography (AcOEt) and obtained (0.86 g, 68%, R f = 0.25 in AcOEt) as white foam.
2.3. Synthesis of 1-[3-deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-β-d-glucopyranosyl] nucleosides (7a-c,f,g) from glycosyl donor 13
2.3.1. 3-Deoxy-3-fluoro-1,2-O-isopropylidene-6-O-p-toluenesulfonyl-a-d-glucofuranose (9)
To a solution of diol 8 [33] (4.00 g, 18 mmol) in dry pyridine (46.20 mL) was added p-toluenesulfonyl chloride (4.60 g, 24 mmol) and kept for 2 h at room temperature. After neutralization (NaHCO3) and extraction with ethyl acetate (4 × 500 mL), the combined extracts were dried over anhydrous sodium sulfate and evaporated to dryness. Purification of the residue by flash chromatography (hexane/AcOEt, 1:1) yielded the title compound (5.08 g, 74%, R f = 0.45 in hexane/AcOEt, 1:1) as colorless oil. − 8.00 (c 0.5, CHCl3); 1H NMR (CDCl3): δ 7.38 and 7.82 (dd, 4H, tosyl group), 5.92 (d, 1H, J 1′,2′ = 3.2 Hz, H-1′), 5.09 (dd, 1H, J 3′,F = 49.3 Hz, H-3′), 4.70 (dd, 1H, J 2′,F = 10.3 Hz, H-2′), 4.34 (d, 1H, H-4′), 4.13–4.06 (m, 2H, H-5a′,5b′), 2.47 (s, 3H, ArCH3), 1.48 (s, 3H, CH3), 1.33 (s, 3H, CH3); Anal. Calcd for C16H21FO7S: C, 51.06; H, 5.62. Found: C, 51.14; H, 5.46. ESI-MS (m/z): 377.42 (M+H+).
2.3.2. 3-Deoxy-3-fluoro-1,2-O-isopropylidene-5-O-tetrahydropyranyl-6-O-p-toluenesulfonyl-a-d-glucofuranose (10)
To a 0 °C solution of 9 (5.00 g, 13.30 mmol) and dried p-toluenesulfonic acid (0.25 g, 1.33 mmol) in anhydrous CH2Cl2 (26.60 mL) was added 3,4-dihydro-2H-pyran (1.80 mL, 19.90 mmol). After being kept for 2 h in room temperature, the reaction mixture was neutralized with aqueous NaHCO3 and extracted with CH2Cl2 (2 × 1 L). The organic layers were combined, dried over Na2SO4 and concentrated in vacuo. The residue was chromatographed through a column of silica gel (hexane/AcOEt, 3:7). Fractions containing the product were combined and concentrated to afford 10 (4.78 g, 78%, R f = 0.48 in hexane/AcOEt, 3:7) as off-white oil. − 6.00 (c 0.5, CHCl3); 1H NMR (CDCl3): δ 7.84–7.80 (four separate s, 2H, HC8 + HC2), 7.35–7.28 (m, 5H, Ph), 5.98 (d, 1/2H, J 1′,2′ = 2.8 Hz, H-1′), 5.89 (d, 1/2H, J 1′,2′ = 2.9 Hz, H-1′), 2.47 (s, 3H, ArCH3), 1.50 (s, 3H, CH3), 1.33 (s, 3H, CH3); Anal. Calcd for C21H29FO8S: C, 54.77; H, 6.35. Found: C, 54.64; H, 6.42. ESI-MS (m/z): 461.53 (M+H+).
2.3.3. 3-Deoxy-3-fluoro-1,2-O-isopropylidene-5-O-tetrahydropyranyl-6-S-acetyl-6-thio-a-d-glucofuranose (11)
The tosylate 10 (4.50 g, 9.80 mmol) was heated with potassium thioacetate (1.50 g, 13.50 mmol) in DMF (39.20 mL) at 100 °C for 1 h. The reaction mixture was neutralized with aqueous NaHCO3. After that, the mixture was concentrated under high vacuum pump to eliminate the DMF. The residue was partitioned between water and EtOAc, the organic extract was dried over anhydrous sodium sulfate, filtered, and evaporated to dryness. The residue was purified by flash chromatography (hexane/AcOEt, 4:6) to give compound 11 (2.71 g, 76%, R f = 0.42 in hexane/AcOEt, 4:6) as yellow syrup. − 8.00 (c 0.1, CHCl3); IR (Nujol): 1697 (SAc) cm−1; 1H NMR (CDCl3): δ 7.84–7.80 (4 separate s, 2H, HC8 + HC2), 7.35–7.28 (m, 5H, Ph), 5.96 (d, 1H, J 1′,2′ = 3.7 Hz, H-1′), 2.37 (s, 3H, SAc), 1.52 (s, 3H, CH3), 1.34 (s, 3H, CH3); Anal. Calcd for C16H25FO6S: C, 52.73; H, 6.91. Found: C, 52.84; H, 6.72. ESI-MS (m/z): 365.47 (M+H+).
2.3.4. 1,2,4-Tri-O-acetyl-3-deoxy-3-fluoro-6-S-acetyl-6-thio-d-glucopyranose (12)
A solution of thioacetate 11 (2.50 g, 6.90 mmol) in 90% TFA (172.50 mL) was stirred for 20 min at room temperature. The reaction mixture was concentrated under diminished pressure and crude was obtained as a colorless oil. Acetylation of the residue with Ac2O-pyridine (1:2, 36.50 mL) and chromatography of the product (hexane/AcOEt, 4:6) gave compound 12 (1.77 g, 70%, R f = 0.45 in hexane/AcOEt, 4:6) as colorless syrup. + 12.00 (c 0.1, CHCl3); IR (Nujol): 1697 (SAc) cm−1; 1H NMR (CDCl3): δ 6.30 (br s, 1H, H-1′), 5.62 (d, 1H, H-4′), 5.33–5.04 (m, 1H, H-2′), 4.80 (dtr, 1/2H, J 3′,F = 53.3 Hz, J 3′,4′ = 9.2 Hz, H-3′), 4.58 (dtr, 1/2H, J 3′,F = 51.9 Hz, J 3′,4′ = 9.0 Hz, H-3′), 4.03–3.98 (m, 1/2H, H-5′), 3.70–3.64 (m, 1/2H, H-5′), 3.28–3.21 and 3.15–3.08 (2 m, 2H, H-6a′,6b′), 2.36 (s, 3H, SAc), 2.17, 2.14, 2.12, 2.10 (4s, 12H, 4OAc); Anal. Calcd for C14H19FO8S: C, 45.90; H, 5.23. Found: C, 46.04; H, 5.16. ESI-MS (m/z): 367.38 (M+H+).
2.3.5. 1,2,4-Tri-O-acetyl-3-deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-d-glucopyranose (13)
To a solution of thiosugar 12 (2.00 g, 5.46 mmol) in methanolic ammonia (228 mL), DTDP (9.55 g, 43.70 mmol) dissolved in MeOH:H2O (1:1, 47 mL) was added dropwise and the mixture was stirred at room temperature. After stirring for 20 h, the reaction mixture was concentrated. Acetylation of the residue with Ac2O-pyridine (1:2, 109 mL) and chromatography of the product (hexane/AcOEt, 4:6) gave compound 13 (1.67 g, 70%, R f = 0.45 in hexane/AcOEt, 4:6) as colorless syrup. + 4.00 (c 0.1, CHCl3); λ max 283 nm (ε 5510); 1H NMR (CDCl3): δ 5.62 (d, 1/2H, H-1′), 5.26–5.03 (m, 2H, H-2′ and H-4′), 4.79 (dtr, 1/2H, J 3′,F = 51.9 Hz, J 3′,4′ = 9.2 Hz, H-3′), 4.53 (dtr, 1/2H, J 3′,F = 51.7 Hz, J 3′,4′ = 9.0 Hz, H-3′), 4.19–4.14 (m, 1/2H, H-5′), 3.75–3.70 (m, 1/2H, H-5′), 3.42–3.35 (m, 2H, H-6a′,6b′), 2.14, 2.12, 2.10, 2.08, 2.07, 2.05 (6s, 18H, 6OAc); Anal. Calcd for C17H20FNO7S2: C, 47.10; H, 4.65; N, 3.23. Found: C, 47.24; H, 4.46; N, 3.35. ESI-MS (m/z): 434.49.
2.3.6. 1-[3-Deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-β-d-glucopyranosyl]thymine (7a)
A mixture of thymine (2.33 g, 18.55 mmol), HMDS (4.80 mL, 23 mmol) and saccharine (0.16 g, 0.85 mmol) in anhydrous CH3CN (69 mL) was refluxed for 30 min. Triacetylated thiopyridyl sugar 13 (5.74 g, 13.25 mmol) and tin chloride (1.94 mL, 17.22 mmol) were then added and the reaction mixture was refluxed for 5 h, cooled, neutralized with aqueous sodium bicarbonate, and extracted with CH2Cl2 (500 mL). The organic layer was washed with water (3 × 10 mL) and dried over anhydrous sodium sulfate, evaporated to dryness. The crude underwent deacetylation with methanolic ammonia (550 mL) within 4 h, concentrated and finally purified by flash chromatography (AcOEt) to give compound 7a (3.53 g, 64%, R f = 0.30 in AcOEt) as white foam.
2.3.7. 1-[3-Deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-β-d-glucopyranosyl]uracil (7b)
A mixture of uracil (2.08 g, 18.55 mmol), HMDS (4.80 mL, 23 mmol) and saccharine (0.16 g, 0.85 mmol) in anhydrous CH3CN (69 mL) was refluxed for 30 min. Triacetylated thiopyridyl sugar 13 (5.74 g, 13.25 mmol) and trimethylsilyl trifluoromethane-sulfonate (3.10 mL, 17.22 mmol) were then added and the reaction mixture was refluxed for 5 h, cooled, neutralized with aqueous sodium bicarbonate, and extracted with CH2Cl2 (500 mL). The organic layer was washed with water (3 × 10 mL) and dried over anhydrous sodium sulfate, evaporated to dryness. The crude underwent deacetylation with methanolic ammonia (550 mL) within 4 h, concentrated and finally purified by flash chromatography (AcOEt) to give compound 7b (3.29 g, 62%, R f = 0.27 in AcOEt) as white foam.
2.3.8. 1-[3-Deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-β-d-glucopyranosyl]5-fluorouracil (7c)
A mixture of 5-fluorouracil (2.41 g, 18.55 mmol), HMDS (4.80 mL, 23 mmol) and saccharine (0.16 g, 0.85 mmol) in anhydrous CH3CN (69 mL) was refluxed for 30 min. Triacetylated thiopyridyl sugar 13 (5.74 g, 13.25 mmol) and trimethylsilyl trifluoromethane-sulfonate (3.10 mL, 17.22 mmol) were then added and the reaction mixture was refluxed for 5 h, cooled, neutralized with aqueous sodium bicarbonate, and extracted with CH2Cl2 (500 mL). The organic layer was washed with water (3 × 10 mL) and dried over anhydrous sodium sulfate, evaporated to dryness. The crude underwent deacetylation with methanolic ammonia (550 mL) within 4 h, concentrated and finally purified by flash chromatography (AcOEt) to give compound 7c (3.89 g, 70%, R f = 0.25 in AcOEt) as white foam.
2.3.9. 1-[3-Deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-β-d-glucopyranosyl]cytosine (7f)
A mixture of N4-benzoyl cytosine (3.99 g, 18.55 mmol), HMDS (4.80 mL, 23 mmol) and saccharine (0.16 g, 0.85 mmol) in anhydrous CH3CN (69 mL) was refluxed for 30 min. Triacetylated thiopyridyl sugar 13 (5.74 g, 13.25 mmol) and trimethylsilyl trifluoromethane-sulfonate (3.10 mL, 17.22 mmol) were then added and the reaction mixture was refluxed for 5 h, cooled, neutralized with aqueous sodium bicarbonate, and extracted with CH2Cl2 (500 mL). The organic layer was washed with water (3 × 10 mL) and dried over anhydrous sodium sulfate, evaporated to dryness. The crude underwent deacetylation with methanolic ammonia (550 mL) within 4 h, concentrated and finally purified by flash chromatography (AcOEt) to give compound 7f (3.60 g, 68%, R f = 0.32 in AcOEt) as white foam. + 2.00 (c 0.5, MeOH); λ max 269 nm (ε 7400); 1H NMR (CD3OD): δ 8.23 (m, H-6 of pyridine), 7.78–7.69 (m, H-4 and H-5 of pyridine), 7.51 (d, 1H, H-5), 7.46 (d, 1H, J 5,6 = 7.3 Hz, H-6), 7.08–7.03 (m, H-3 of pyridine), 5.52 (d, 1H, J 1′,2′ = 9.4 Hz, H-1′), 4.22 (dtr, 1H, J F,3′ = 52.5 Hz, J 2′,3′ = 9.0 Hz, J 3′,4′ = 8.9 Hz, H-3′), 3.73–3.66 (m, 1H, Η-2′), 3.53–3.42 (m, 2H, H-4′ and H-6a′), 3.34–3.30 (m, 1H, H-6b′), 2.92–2.87 (m, 1H, H-5′); 19F NMR: δ −64.33; 13C NMR (CD3OD): δ 165.54, 160.32, 154.28, 148.46, 137.56, 132.91, 122.03, 120.12, 99.36, 92.38, 85.62, 72.39, 70.64, 70.22, 33.63; Anal. Calcd for C15H17FN4O4S2: C, 44.99; H, 4.28; N, 13.99. Found: C, 44.82; H, 4.42; N, 13.52. ESI-MS (m/z): 401.48.
2.3.10. 9-[3-Deoxy-3-fluoro-6-S-(2-S-pyridyl)-6-thio-β-d-glucopyranosyl]adenine (7g)
A mixture of N6-benzoyl adenine (2.93 g, 12.25 mmol), HMDS (3.10 mL, 15.19 mmol) and saccharine (0.10 g, 0.56 mmol) in anhydrous CH3CN (46 mL) was refluxed for 30 min. Triacetylated thiopyridyl sugar 13 (3.80 g, 8.75 mmol) and tin chloride (1.38 mL, 12.25 mmol) were then added and the reaction mixture was refluxed for 5 h, cooled, neutralized with aqueous sodium bicarbonate, and extracted with CH2Cl2 (500 mL). The organic layer was washed with water (3 × 10 mL) and dried over anhydrous sodium sulfate, evaporated to dryness. The crude underwent deacetylation with methanolic ammonia (512 mL) within 4 h, concentrated and finally purified by flash chromatography (AcOEt) to give compound 7g (3.32 g, 64%, R f = 0.32 in AcOEt) as white foam. + 4.00 (c 0.5, MeOH); λ max 263 nm (ε 9860); 1H NMR (CD3OD): δ 8.58 and 8.35 (2s, 2H, H-2,8), 8.33–8.25 (m, H-6 of pyridine), 7.63–7.18 (m, H-4, H-5, H-3 of pyridine), 6.96 (br s, NH2), 5.88 (d, 1H, J 1′,2′ = 9.1 Hz, H-1′), 4.01 (dtr, 1H, J F,3′ = 52.3 Hz, J 2′,3′ = 8.7 Hz, J 3′,4′ = 8.6 Hz, H-3′), 3.79–3.67 (m, 1H, Η-2′), 3.59–3.53 (m, 1H, H-6a′), 3.51–3.47 (m, 1H, H-4′), 3.42–3.37 (m, 1H, H-6b′), 2.96–2.81 (m, 1H, H-5′); 19F NMR: δ −63.20; 13C NMR (CD3OD): δ 160.54, 155.75, 152.31, 149.86, 147.75, 145.76, 136.71, 120.37, 120.23, 116.98, 91.76, 89.64, 72.19, 69.70, 66.64, 33.87; Anal. Calcd for C16H17FN6O3S2: C, 45.27; H, 4.04; N, 19.80. Found: C, 45.31; H, 4.12; N, 19.62. ESI-MS (m/z): 425.51.
2.4. Biological assays
The antiviral assays were based on the inhibition of virus-induced cytopathicity in confluent cell cultures, and the cytostatic assays on inhibition of tumor cell proliferation in exponentially growing tumor cell cultures according to previously described methodology [15], [32].
2.4.1. Antiviral activity assays
The antiviral assays, other than the anti-HIV assays, were based on inhibition of virus-induced cytopathicity in HEL [herpes simplex virus type 1 (HSV-1) (KOS), HSV-2 (G), vaccinia virus, vesicular stomatitis virus, cytomegalovirus (HCMV) and varicella-zoster virus (VZV)], Vero (parainfluenza-3, reovirus-1, Sindbis virus and Coxsackie B4), HeLa (vesicular stomatitis virus, Coxsackie virus B4, and respiratory syncytial virus) or MDCK [influenza A (H1N1; H3N2) and influenza B] cell cultures. Confluent cell cultures (or nearly confluent for MDCK cells) in microtiter 96-well plates were inoculated with 100 CCID50 of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures). After a 1 h virus adsorption period, residual virus was removed, and the cell cultures were incubated in the presence of varying concentrations (200, 40, 8, …μM) of the test compounds. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. The minimal cytotoxic concentration (MCC) of the compounds was defined as the compound concentration that caused a microscopically visible alteration of cell morphology. The methodology of the anti-HIV assays was as follows: human CEM (∼3 × 105 cells/cm3) cells were infected with 100 CCID50 of HIV(IIIB) or HIV-2(ROD)/mL and seeded in 200 μL wells of a microtiter plate containing appropriate dilutions of the test compounds. After 4 days of incubation at 37 °C, HIV-induced CEM giant cell formation was examined microscopically.
2.4.2. Cytostatic/toxic activity assays
Murine leukemia L1210, murine mammary carcinoma FM3A, and human lymphocyte CEM and human cervix carcinoma HeLa cells were seeded in 96-well microtiter plates at 50,000 (L1210, FM3A), 75,000 (CEM) or 20,000 (HeLa) cells per 200 μL-well in the presence of different concentrations of the test compounds. After 2 (L1210, FM3A), 3 (CEM) or 4 (HeLa) days, the viable cell number was counted using a Coulter counter apparatus. The 50% cytostatic concentration (CC50) was defined as the compound concentration required to inhibit tumor cell proliferation by 50%.
2.4.3. Thymidine and uridine phosphorylase assays
The conversion of dThd to thymine, Urd to uracil and compounds 13b and 13c to the free base by human recombinant thymidine phosphorylase and uridine phosphorylase type 1 (kindly provided by Dr. T.P. Roosild, Las Vegas, Nevada, USA) was measured by high-pressure liquid chromatography (HPLC) analysis. To determine the conversion activity, a high amount of recombinant enzyme was incubated with 100 μM of dThd or 13b and 13c in TP-buffer (10 mM Tris.HCl, pH 7.6, 1 mM EDTA, 2 mM KH2PO4/K2HPO4 and 150 mM NaCl) or UP-buffer (same as TP-buffer, but 300 mM NaCl instead of 150 mM). At 20, 40 and 60 min, 100 μL aliquots of the reaction mixtures were withdrawn and heated at 95 °C for 3 min to inactivate the enzyme. dThd and Urd were separated from thymine and uracil and 13b and 13c were separated from 5-FU and uracil on a reverse-phase RP-8 column (Merck, Darmstadt, Germany) and quantified by high-pressure liquid chromatography (HPLC) analysis (Alliance 2690, Waters, Milford, MA). The separation was performed by a linear gradient from 98% buffer B (50 mM NaH2PO4 and 5 mM heptane sulfonic acid, pH 3.2), to 20% buffer B + 80% acetonitrile (8 min 98% buffer B + 2% acetonitrile; 5 min linear gradient of 98% buffer B + 2% acetonitrile to 20% buffer B + 80% acetonitrile; 10 min 20% buffer B + 80% acetonitrile, followed by equilibration at 98% buffer B + 2% acetonitrile). Retention times for thymine and dThd were, respectively, 5.1 and 10.8 min, for uracil and Urd, respectively, 2.1 and 2.4 min and for 13b, 13c and 5-FU, respectively, 20.1, 20.1 and 2.4 min. UV-based detection was performed at 267 nm.
3. Results and discussion
3.1. Synthesis
Two main strategies were used for the synthesis of 3-fluoro-6-S-(2-S-pyridyl) nucleosides 7a-c,f,g.
Our first approach (Scheme 1 ) was focused on the preparation of 3-fluoro-6-thio-glucopyranosyl nucleosides of thymine 5a, uracil 5b, 5-fluorouracil 5c, N 4-benzoyl cytosine 5d and N 6-benzoyl adenine 5e as useful precursors toward the synthesis of 6-S-nucleosidyl S-pyridine disulfides. 1,2:5,6-di-O-isopropylidene-3-deoxy-3-fluoro-α-d-glucofuranose (1) [34] was readily transformed into the corresponding 1,2,4,6-tetra-O-acetyl-3-deoxy-3-fluoro-glucopyranose [35], which upon condensation with purine and pyrimidine bases afforded 3-deoxy-3-fluoro-β-d-glucopyranosyl nucleosides 2a-e [14]. Deprotection of nucleosides 2a-e in methanolic ammonia gave the fully unprotected derivatives 3a-c,f,g, while the base protected benzoylated derivatives 3d,e were obtained when 2d,e were treated with NaOH-ethanol-pyridine [10], [11], [14]. Treatment of 3a-e with p-toluenesulphonyl chloride in dry pyridine [35] and direct acetylation of the free hydroxyls at C-2′ and C-4′ with acetic anhydride/pyridine afforded the desired acetylated 6-O-tosylated derivatives 4a-e in very good yields. Subsequently, the tosylated derivatives 4a-e, after treatment with potassium thioacetate in hot N,N-dimethylformamide (DMF) [36], [37], [38], were converted into the corresponding thioacetates, 1-(2,4-di-O-acetyl-6-S-acetyl-3-deoxy-3-fluoro-6-thio-β-d-glucopyranosyl)thymine (5a), 1-(2,4-di-O-acetyl-6-S-acetyl-3-deoxy-3-fluoro-6-thio-β-d-glucopyranosyl)uracil (5b), 1-(2,4-di-O-acetyl-6-S-acetyl-3-deoxy-3-fluoro-6-thio-β-d-glucopyranosyl)5-fluorouracil (5c), 1-(2,4-di-O-acetyl-6-S-acetyl-3-deoxy-3-fluoro-6-thio-β-d-glucopyranosyl)-N 4-benzoyl cytosine (5d) and 9-(2,4-di-O-acetyl-6-S-acetyl-3-deoxy-3-fluoro-6-thio-β-d-glucopyranosyl)-N 6-benzoyl adenine (5e), respectively. Deprotection of the β-protected thionucleosides 5a-e by methanolic ammonia and in situ thiopyridinylation in H2O, MeOH and 2,2-dipyridyl disulfide (DTDP) at room temperature [22], led to the desired derivatives 7a-c. These reactions were completed within 20 h and the isolated yields of the 6-S-nucleosidyl S-pyridine disulfides were generally higher than 65%. Attempts to fully unprotect thionucleosides 5a-c resulted to the formation of their corresponding symmetrical disulfides 6a-c, the thiopyridinylation of which also led to the desired 6-S-nucleosidyl S-dipyridine disulfides 7a-c. It is noteworthy that only decomposition products were obtained after deprotection/thiopyridinylation of N 4-benzoyl cytosine and N 6-benzoyl adenine derivatives, 5d and 5e, respectively.
Scheme 1.
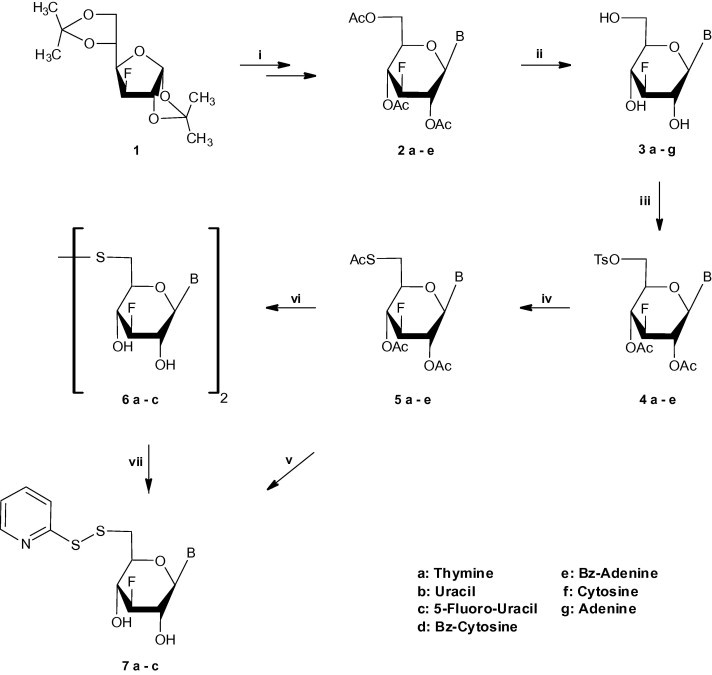
(i) (a) MeOH/H2O/amberlite IR 120(H+); (b) Ac2O/pyridine; (c) silylated base, CH3CN, trimethylsilyl trifluomethane-sulfonate or tin chloride; (ii) pyridine/MeOH/amberlite IR 120(H+)/NaOH or methanolic ammonia; (iii) (a) pyridine, p-toluenesulfonyl chloride; (b) Ac2O/Pyridine; (iv) KSAc/DMF/100 °C; (v) methanolic ammonia/H2O/MeOH/2,2-dipyridine-disulfide; (vi) methanolic ammonia; (vii) H2O/MeOH/2,2-dipyridine-disulfide.
In order to circumvent this difficulty we chose to investigate a route in which the thionucleosides could be generated from a suitable thiopyridine sugar precursor. Our alternative synthetic pathway, in which compound 12 was envisioned as an appropriate glycosyl donour for the preparation of the suitable sugar, is described in Scheme 2 . Initially, selective removal of the 5,6-O-isopropylidene group of starting material 1 [34] led to the formation of diol 8 [33]. Selective sulfonylation of 8 with p-toluenesulphonyl chloride in pyridine led to compound 9, which was treated with dihydropyran (DHP) and p-toluenesulphonic acid monohydrate in dry CH2Cl2 in order to give compound 10 in very satisfactory overall yield. Displacement of the tosyl group with the thioacetyl moiety proceeded without difficulties to afford compound 11. Precursor 12 was readily available from glucofuranose 11 through hydrolysis with 90% aqueous trifluoroacetic acid at room temperature and direct acetylation. Deprotection of the glycosyl donor 12 using methanolic ammonia, direct thiopyridinylation [22] (8 mol excess of DTDP/MeOH/H2O) followed by acetylation led to the desired thiopyridine sugar 13. In order to prepare not only the target 6-S-nucleosidyl S-dipyridine disulfides 7f,g, but the previously synthesized 6-S-(2-S-pyridyl) nucleosides 7a-c as well, glycosylation reactions with the corresponding purine and pyrimidine bases and the 6-thiopyridine sugar 13 followed by direct deacetylation were employed.
Scheme 2.
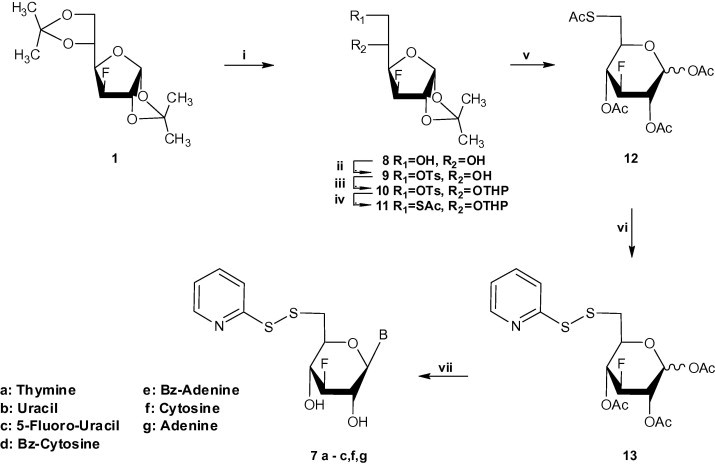
(i) 70% AcOH; (ii) pyridine, p-toluenesulfonyl chloride; (iii) CH2Cl2, p-TsOH, DHP; (iv) KSAc/DMF/100 °C; (v) (a) 90% TFA (b) Ac2O/pyridine; (vi) (a) methanolic ammonia/H2O/MeOH/2,2-dipyridine-disulfide (b) Ac2O/pyridine; (vii) (a) silylated base, CH3CN, trimethylsilyl trifluomethane-sulfonate or tin chloride, (b) methanolic ammonia.
All new compounds were well-characterized by NMR and UV spectroscopy, mass spectrometry and elemental analysis. The 1H NMR data obtained for the newly synthesized 5a-e revealed that these compounds had the β configuration (J 1′,2′ ≫ 8.0 Ηz). It must also be mentioned that the desired compounds 5a-e, showed strong infra-red absorptions at 1750 cm−1 (OAc) and 1697 cm−1 (SAc), while in their 1H NMR spectra prominent 3-proton methyl signals appeared, which were ascribed to the acetylthio moiety and 6-proton methyl peaks, which correspond to the acetoxyl group. Moreover, data in the 1H NMR spectra, obtained for the newly synthesized 6-S-(2-S-pyridyl) nucleosides 7a-c,f,g, revealed the absence of 6-proton methyl peaks, which correspond to the acetoxyl group and the presence of 2-S-pyridyl moiety, whose proton peaks appeared between 8.4 and 7.1 ppm.
The stability of the target compounds was assigned after 3 months storage, either in organic solution or in solid phase, by performing NMR spectroscopy. Comparison of the primarily NMR spectra of the target molecules with those of the same storable compounds could clearly reveal identical proton NMR spectra.
3.2. Antiviral and cytostatic activity
Compounds 6a-c and 7b,c,f,g were evaluated for their antiviral activity against a wide variety of DNA and RNA viruses, including herpes simplex virus type 1 (HSV-1) (strain KOS), HSV-2 (strain G), vaccinia virus, vesicular stomatitis virus (VSV), varicella-zoster virus (VZV) strains OKA and 07/1, and human cytomegalovirus (HCMV) strains AD-169 and Davis in HEL cell cultures, VSV, Coxsackie B4 and respiratory syncytial virus (RSV) in HeLa cell cultures, parainfluenza-3 virus, reovirus, Sindbis virus, Coxsackie virus B4 and Punta Toro virus in Vero cell cultures, influenza A (H1N1, H3N2) and B virus in MDCK cell cultures and feline corona virus (FIPV) and feline herpesvirus in CRFK cell cultures. None of the compounds tested showed an appreciable inhibitory activity at subtoxic concentrations (i.e. 4–20 μM for 7b, 20–100 μM for 7c and >100 μM for the other compounds). The new molecules were also examined for their cytostatic activity against murine leukemia L1210, murine mammary carcinoma FM3A, human lymphocyte Molt4/C8 and CEM and human cervix carcinoma HeLa cells. Compounds 3a-g (Table 1 ) and the acetyl-substituted 5a-e analogs (data not shown) did not show appreciable antiproliferative activity. Likewise, the 6,6-disulfide glucopyranosyl pyrimidine dinucleoside derivatives 6a-c did not show appreciable cytostatic action against the evaluated tumor cell lines (IC50: 71 to >500 μM) (Table 1). However, their corresponding 2-S-pyridyl 6-thioglucopyranosyl mononucleosides 7 were endowed with a more pronounced cytostatic activity in cell culture. In particular, the 5-fluorouracil (7c) and especially the uracil (7b) derivative showed considerable cytostatic potency against the murine L1210, and human Molt4/C8, CEM and HeLa cells. Also, 7c and 7b were cytostatic against proliferating human embryonic lung HEL fibroblasts (IC50: 36 μM and 9.2 μM, respectively), Crandell feline kidney cells (CRFK) (IC50: 6.2 and 39 μM) and canine Madin-Darby kidney cells (MDCK) (IC50: 50 μM and 11 μM, respectively). Conversely, the adenine (7g) and cytosine (7f) derivatives showed poor, if any cytostatic activity (192 − >500 μM) (Table 1). It is somewhat intriguing that among the 2-S-pyridyl 6-thioglucopyranosyl mononucleotides, only the derivatives containing an uracil/5-fluorouracil base were markedly more cytostatic than those, which bear a thymine, cytosine or adenine base. The fluorouracil derivatives proved equally cytostatic against murine and human cells, as well as against leukemia, mammary carcinoma, cervix carcinoma and lymphoma tumor cells. The molecular basis of the antiproliferative activity of the fluorouracil derivatives is currently unknown. When compounds 7b and 7c were exposed to purified recombinant thymidine phosphorylase (TPase) or uridine phosphorylase type I (UPase), they were not hydrolyzed to the free pyrimidine base under conditions where thymidine (exposed to TPase) and uridine (exposed to UPase) were readily converted to thymine and uracil, respectively (data not shown). These findings indicate that these molecules exert their potency via their intact nucleoside moiety (or anabolite derived thereof), rather than a catabolic metabolite (i.e. free 2-S-pyridyl 6-thioglucopyranose). Also, it is clear from the inactivity of the 3a-g series of compounds that the 2-S-pyridyl group plays an important role, in concert with the uracil base, to express pronounced cytostatic activity. In fact, Decout has recently reported on 2′,3′-dideoxyribonucleoside 3′-disulfides [22] and found that those derivatives, which contain a rather bulky moiety on the disulfide part of the molecule (i.e. nitrophenyl, butyl, hexyl, actyl) were also most cytostatic than others. This is in agreement with the presence of a rather bulky entity on the disulfide in compounds 7b and 7c as well. The exact molecular mechanism of cytostatic activity is currently subjected to further studies. Mutatis mutandis, compounds 7b and 7c could be viewed as potential lead compounds for further modification in order to redesign and synthesize more potent cytostatic agents.
Table 1.
Inhibitory effects of the test compounds on the proliferation of murine leukemia cells (L1210), murine mammary carcinoma cells (FM3A), human T-lymphocyte cells (Molt4/C8, CEM) and human cervix carcinoma (HeLa) cells.
| Compounds | IC50a (μM) |
||||
|---|---|---|---|---|---|
| L1210 | FM3A | Molt4/C8 | CEM | HeLa | |
| 3a | >200 | >200 | >200 | >200 | |
| 3b | >200 | >200 | >200 | >200 | |
| 3c | >200 | >200 | >200 | >200 | |
| 3d | >200 | >200 | >200 | >200 | |
| 3e | ⩾200 | >200 | 81 ± 7 | 120 ± 15 | |
| 3f | 159 ± 58 | >200 | 163 ± 21 | 155 ± 64 | |
| 3g | 166 ± 47 | >200 | 183 ± 24 | 124 ± 47 | |
| 6a | >500 | >500 | >500 | >500 | |
| 6b | 302 ± 36 | 123 ± 39 | >500 | 320 ± 40 | |
| 6c | 231 ± 34 | 71 ± 18 | >500 | 280 ± 0 | |
| 7a | 120 ± 56 | >200 | 84 ± 4 | 106 ± 21 | |
| 7b | 9.5 ± 0.0 | 17 ± 4 | 9.2 ± 1.1 | 8.8 ± 0.7 | |
| 7c | 54 ± 19 | 35 ± 2 | 43 ± 3 | 35 ± 3 | |
| 7f | >500 | >500 | >500 | 369 ± 7 | |
| 7g | 238 ± 41 | ⩾500 | 228 ± 36 | 192 ± 9 | |
50% Inhibitory concentration or compound concentration required to inhibit tumor cell proliferation in cell culture by 50%.
4. Conclusion
We have prepared a series of 2-S-pyridyl-6-thionucleosides, bearing natural and modified purines and pyrimidines, by developing highly efficient synthetic routes. None of the new compounds showed antiviral potency at subtoxic concentrations, but compound 7c, and in particular 7b, were endowed with significant cytostatic potency and can be viewed as novel lead compounds for further modifications. It would therefore be interesting to further explore the structure–activity relationships by modifying the base part as well as the sugar portion by incorporating different chemical entities.
Acknowledgments
This work was supported in part by the Postgraduate Programmes “Biotechnology-Quality assessment in Nutrition and the Environment”, “Application of Molecular Biology–Molecular Genetics–Molecular Markers”, Department of Biochemistry and Biotechnology, University of Thessaly. The authors are also grateful to K.U. Leuven (GOA 10/014) for financial support.
References
- 1.Plunkett W., Gandhi V. Purine and pyrimidine nucleoside analogues. In: Giaccone G., Schilsky R., Sondel P., editors. Cancer Chemotherapy and Biological Response Modifiers. Elsevier Science B.V.; 2001. pp. 21–45. [PubMed] [Google Scholar]
- 2.Robins R.K., Kini G.D. Chapman and Hall; New York: 1990. The Chemistry of Antitumor Agents. pp. 299. [Google Scholar]
- 3.MacCoss M., Robins M.J. Chapman and Hall; New York: 1990. The Chemistry of Antitumor Agents. pp. 261. [Google Scholar]
- 4.Komiotis D., Manta S., Tsoukala E., Tzioumaki N. Curr. Med. Chem.: Anti-Infect. Agents. 2008;7:219–244. [Google Scholar]
- 5.De Clercq E. Biochim. Biophys. Acta. 2002;1587:258–275. doi: 10.1016/s0925-4439(02)00089-3. [DOI] [PubMed] [Google Scholar]
- 6.Brady R.C., Bernstein D.I. Antivir. Res. 2004;61:73–81. doi: 10.1016/j.antiviral.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Papatheodoridis G.V., Dimou E., Papadimitropoulos V. Am. J. Gastroenterol. 2002;97:1618–1628. doi: 10.1111/j.1572-0241.2002.05819.x. [DOI] [PubMed] [Google Scholar]
- 8.Meng W.-D., Qing F.-L. Curr. Top. Med. Chem. 2006;6:1499–1528. doi: 10.2174/156802606777951082. [DOI] [PubMed] [Google Scholar]
- 9.Pankiewicz K.W. Carbohyd. Res. 2000;327:87–105. doi: 10.1016/s0008-6215(00)00089-6. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 10.Manta S., Agelis G., Botić T., Cencič A., Komiotis D. Bioorg. Med. Chem. 2007;15:980–987. doi: 10.1016/j.bmc.2006.10.033. [DOI] [PubMed] [Google Scholar]
- 11.Manta S., Agelis G., Botić T., Cencič A., Komiotis D. Eur. J. Med. Chem. 2008;43:420–428. doi: 10.1016/j.ejmech.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Kirk K.L. Curr. Top. Med. Chem. 2006;6:1447–1456. doi: 10.2174/156802606777951073. [DOI] [PubMed] [Google Scholar]
- 13.Clark J.L., Mason J.C., Hollecker L., Stuyver L.J., Tharnish P.M., McBrayer T.R., Otto M.J., Furman A.P., Schinazi R.F., Watanabe K.A. Bioorg. Med. Chem. Lett. 2006;16:1712–1715. doi: 10.1016/j.bmcl.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 14.Manta S., Tsoukala E., Tzioumaki N., Goropevsek A., Pamulapati R.T., Cencič A., Komiotis D. Eur. J. Med. Chem. 2009;44:2696–2704. doi: 10.1016/j.ejmech.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manta S., Tzioumaki N., Tsoukala E., Panagiotopoulou A., Pelecanou M., Balzarini J., Komiotis D. Eur. J. Med. Chem. 2009;44:4764–4771. doi: 10.1016/j.ejmech.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 16.Balatsos N.A.A., Vlachakis D., Maragozidis P., Manta S., Anastasakis D., Kyritsis A., Vlassi M., Komiotis D., Stathopoulos C. Biochemistry. 2009;48:6044–6051. doi: 10.1021/bi900236k. [DOI] [PubMed] [Google Scholar]
- 17.Yokoyama M. Synthesis. 2000:1637–1655. [Google Scholar]
- 18.Wnuk S.F. Tetrahedron. 1993;49:9877–9936. [Google Scholar]
- 19.Bacchi C.J., Sufrin J.R., Nathan H.C., Spiess A.J., Hannan T., Garofalo J., Alecia K., Katz L., Yarlett N. Antimicrob. Agents Chemother. 1991;35:1315–1320. doi: 10.1128/aac.35.7.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pignot M., Pljevaljcic G., Weinhold E. Eur. J. Org. Chem. 2000:549–555. and references cited therein. [Google Scholar]
- 21.Zheng F., Zhang X.-H., Qiu X.-L., Zhang X., Qing F.-L. Org. Lett. 2006;8:6083–6086. doi: 10.1021/ol062576k. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 22.Gerland B., Desire J., Balzarini J., Décout J.L. Bioorg. Med. Chem. 2008;16:6824–6831. doi: 10.1016/j.bmc.2008.05.065. [DOI] [PubMed] [Google Scholar]
- 23.Roy B., Chambert S., Lepoivre M., Décout J.L. Nucleos. Nucleot. Nucleic acids. 2003;22:883–885. doi: 10.1081/NCN-120022677. [DOI] [PubMed] [Google Scholar]
- 24.Roy B., Chambert S., Lepoivre M., Aubertin A., Balzarini J., Décout J.L. J. Med. Chem. 2003;46:2565–2568. doi: 10.1021/jm0256225. [DOI] [PubMed] [Google Scholar]
- 25.Elhalabi J., Rice K.G. Nucleos. Nucleot. Nucleic acids. 2004;23:195–205. doi: 10.1081/ncn-120027828. [DOI] [PubMed] [Google Scholar]
- 26.Sivapriya K., Suguna P., Shubashree S., Sridhar P.R., Chandrasekaran S. Carbohydr. Res. 2007;342:1151–1158. doi: 10.1016/j.carres.2007.02.035. [DOI] [PubMed] [Google Scholar]
- 27.Chien T.C., Chen C.S., Yu F.H., Chern J.W. Chem. Pharmaceut. Bull. 2004;52:1422–1426. doi: 10.1248/cpb.52.1422. [DOI] [PubMed] [Google Scholar]
- 28.De Clercq E. Nat. Rev. Drug Discovery. 2002;1:13–25. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
- 29.Liu S., Wolfe M.S., Borchardt R.T. Antivir. Res. 1992;19:247–265. doi: 10.1016/0166-3542(92)90083-h. [DOI] [PubMed] [Google Scholar]
- 30.Agelis G., Tzioumaki N., Botić T., Cencič A., Komiotis D. Bioorg. Med. Chem. 2007;15:5448–5456. doi: 10.1016/j.bmc.2007.05.055. [DOI] [PubMed] [Google Scholar]
- 31.Agelis G., Tzioumaki N., Tselios T., Botić T., Cencič A., Komiotis D. Eur. J. Med. Chem. 2008;43:1366–1375. doi: 10.1016/j.ejmech.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 32.Tzioumaki N., Tsoukala E., Manta S., Agelis G., Balzarini J., Komiotis D. Arch. Pharm. 2009;342:353–360. doi: 10.1002/ardp.200900004. [DOI] [PubMed] [Google Scholar]
- 33.Kamath V.P., Zhang J., Morris P.E., Babu Y.S. Bioorg. Med. Chem. Lett. 2006;16:2662–2665. doi: 10.1016/j.bmcl.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 34.Baker C.D., Horton D., Tindall G.C. Carbohydr. Res. 1972;24:192–197. doi: 10.1016/s0008-6215(00)82279-x. [DOI] [PubMed] [Google Scholar]
- 35.Foster A.B., Hems R., Webber J.M. Carbohydr. Res. 1967;5:292–301. [Google Scholar]
- 36.Hughes A.N., Munkombwe M.N. Carbohydr. Res. 1985;136:397–409. [Google Scholar]
- 37.Tsoukala E., Agelis G., Dolinšek J., Botić T., Cencič A., Komiotis D. Bioorg. Med. Chem. 2007;15:3241–3247. doi: 10.1016/j.bmc.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 38.Tsoukala E., Manta S., Tzioumaki N., Agelis G., Komiotis D. Carbohydr. Res. 2008;343:1099–1103. doi: 10.1016/j.carres.2008.02.004. [DOI] [PubMed] [Google Scholar]